

Abstract
The human AlkB family proteins, such as FTO and ALKBH5, are known to mediate RNA m6A demethylation. However, although ALKBH7 localizes in mitochondria and affects metabolism, the detailed biological function and mechanism have remained unknown for years. We developed Demethylation-Assisted Multiple Methylation sequencing (DAMM-seq) to simultaneously detect N1-methyladenosine (m1A), N3-methylcytidine (m3C), N1-methylguanosine (m1G) and N2,N2-dimethylguanosine methylations in both steady-state RNA and nascent RNA, and discovered that human ALKBH7 demethylates and m1A within mt-Ile and mt-Leu1 pre-tRNA regions, respectively, in mitochondrial polycistronic RNA. DAMM-seq quantitatively and sensitively monitors the methylation stoichiometry change at pre-tRNA junctions within nascent mt-RNA, revealing the target region where ALKBH7 regulates RNA processing and local structural switch of polycistronic mt-RNAs. A new RNA demethylase in human cells was characterized through the base-resolution quantification of multiple RNA methylations in nascent mt-RNA, resolving the long-standing question about the functional substrate of ALKBH7.
1. Introduction
Posttranscriptional RNA modifications occur in almost all types of RNA in a living cell (Frye, Harada, Behm, & He, 2018; Roundtree, Evans, Pan, & He, 2017). Compared with the well-studied RNA modifications on high-abundance rRNA and tRNA, RNA modifications present in low-abundance messenger RNA (mRNA) have revealed broad regulatory functions in gene expression regulation. The recent discovery of mRNA m6A ‘writer’, ‘eraser’, and ‘reader’ proteins initiated the emerging area of reversible RNA methylation (Zhao, Roundtree, & He, 2017), exhibiting the impact on numerous research areas such as epigenetics, cancer biology, neuroscience, development, immunology, aging, etc. Dated back to 2010, Prof. Chuan He’s lab discovered the first RNA m6A demethylase FTO (Jia et al., 2011) (Fig. 1), setting the theoretical basis for reversible RNA methylation and RNA epigenetics. The other two ALKBH family proteins, ALKBH5 (Zheng et al., 2013) and ALKBH1 (Liu et al., 2016), were sequentially characterized as RNA m6A and m1A demethylases (Fig. 1) in 2013 and 2016, respectively. However, for other members of human ALKBH protein family, except ALKBH2 and ALKBH3 mediate DNA demethylation (Zheng, Fu, & He, 2014), the molecular substrates of ALKBH6 (Ma et al., 2022) and ALKBH7 (Wang et al., 2014) remained unclear as a long-standing scientific question for many years; Notably, ALKBH7 specifically localizes inside mitochondria and displays a strong phenotype of obesity in Alkbh7 knockout mice (Solberg et al., 2013).
Fig. 1.
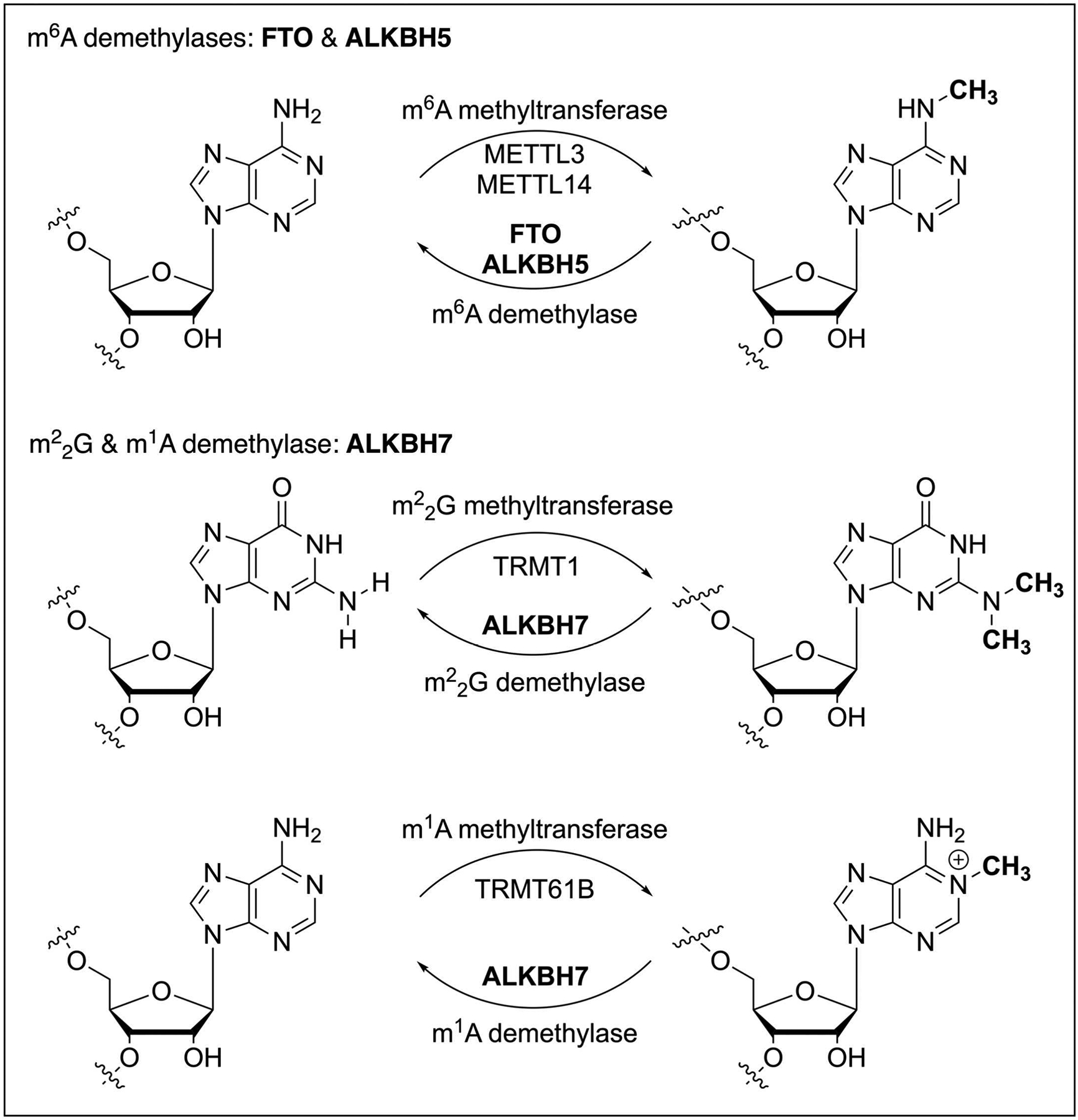
RNA demethylases FTO, ALKBH5, and ALKBH7 in human cells.
In exploring the molecular substrates of ALKBH7 to investigate its biological functions, the experimental evidence suggested that ALKBH7 could likely regulate RNA demethylation in mitochondrial nascent RNA species. However, the traditional molecular biology approaches, such as mass spectrometry, met the difficulty in studying how ALKBH7 regulates the methylation profiles at specific sites within nascent mt-RNA, due to the ultra-low amount of nascent RNAs. Therefore, the scientific question about ALKBH7 molecular substrate turned out to be a biotech challenge, since there were no quantitative tools to measure the methylation stoichiometry on RNA species of an ultra-low amount. To quantitatively monitor the RNA methylation dynamics at specific sites within nascent RNA, we developed Demethylation-Assisted Multiple Methylation sequencing (DAMM-seq) (Zhang et al., 2021), which simultaneously detects N1-methyladenosine (m1A), N3-methylcytidine (m3C), N1-methylguanosine (m1G), and N2,N2-dimethylguanosine methylations in mitochondrial nascent RNA. Quantitative DAMM-seq demonstrated human ALKBH7 as a new RNA demethylase to regulate reversible and m1A methylations in polycistronic mitochondrial RNA (Fig. 1), impacting nascent mt-RNA processing, structural dynamics, and mitochondrial activity (Zhang et al., 2021).
DAMM-seq briefly presents two versions, a demethylation-based version for bulk RNA samples starting with ~400 ng input RNA and an ultra-low-input version for studying RNA methylations in nascent RNA species. For instance, with a selected cell line, the demethylation-based version of DAMM-seq starts with ~400 ng cellular small RNA (less than 200 nt), and maps m1A, m3C, m1G, and in ‘Input’ libraries as misincorporation signatures in the presence of HIV reverse transcriptase (RT), which enables the excellent readthrough and mutation readout at the methylated sites; Meanwhile, the side-by-side ‘Demethylation’ libraries could validate whether the misincorporations observed at A, C or G sites respond to in vitro AlkB (D135S mutant) treatment (Zhang et al., 2021). Different from the previously reported methods (Zheng et al., 2015), DAMM-seq achieves the complete demethylation of m1A, m3C, m1G, and in one pot, per its well-optimized reaction condition in the demethylation step (Fig. 2A). Only the A, C, or G sites displaying a notable misincorporation level in ‘Input’ library and a dramatic drop in misincorporation ratios after in vitro demethylation treatment are identified as candidate sites for m1A, m3C, m1G, and , but not other RNA modifications. For m1G and methylations, considering their different locations on cytoplasmic tRNA and mitochondrial tRNA in mammals, the G site with a notable misincorporation at position 9 and 37 of tRNAs are assigned as m1G sites, with highly mutated G site at position 26 of tRNA identified as sites. Interestingly, for the six mitochondrial tRNAs (mt-tRNA) containing G at position 26, only mt-Ile was proven to be -modified with an observation of high misincorporation ratio, while G26 of the other five tRNAs display low mutation rates and indicate m2G methylations (Suzuki & Suzuki, 2014).
Fig. 2.
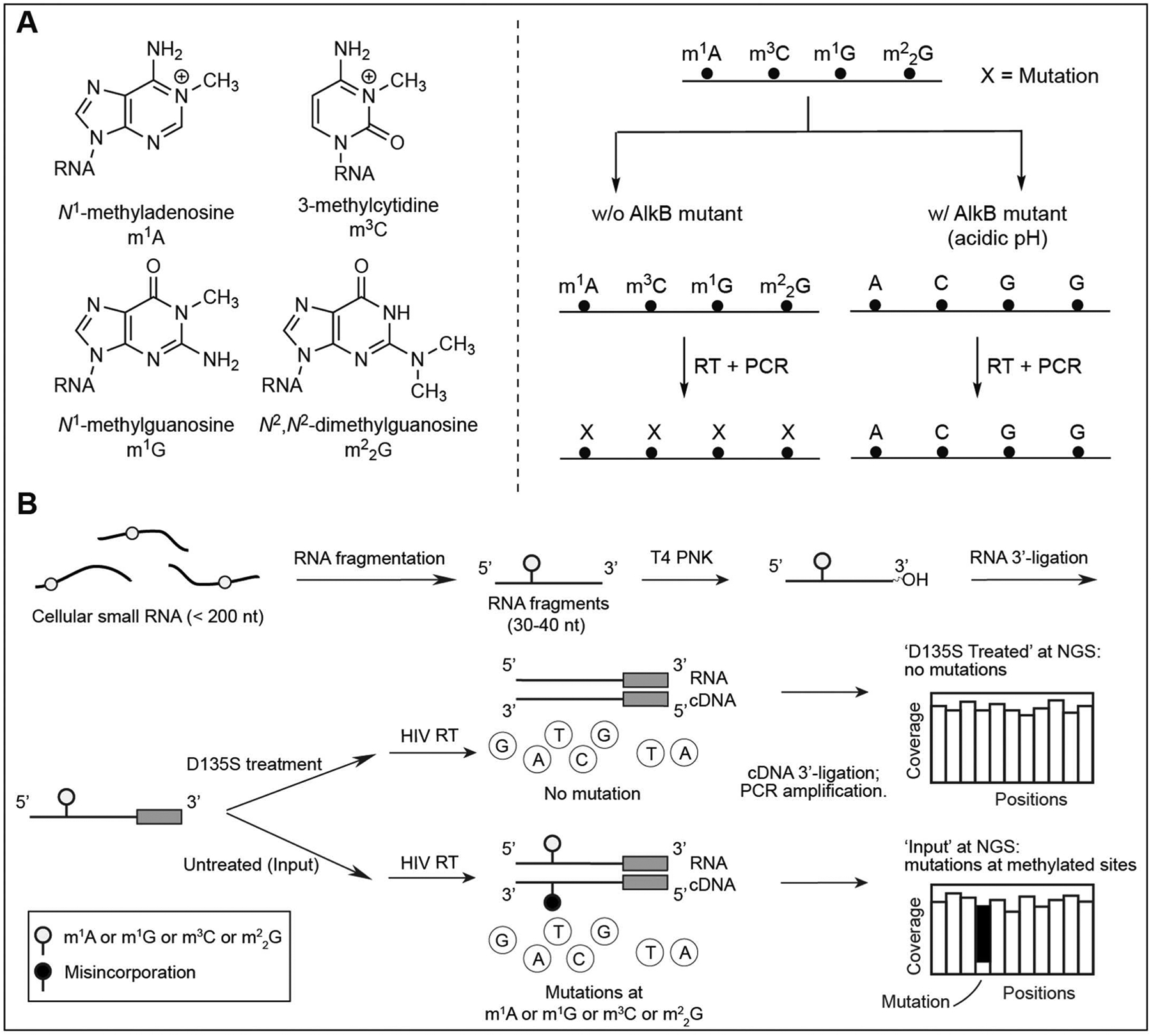
DAMM-seq quantitatively detects four RNA methylations in one pot.
With the confirmed locations of m1A, m3C, m1G, and sites on tRNA, the ultra-low-input version of DAMM-seq starts with < 10 ng input RNA, and quantitatively maps these four methylations in nascent RNA species, such as mitochondrial polycistronic RNA. DAMM-seq utilizes the misincorporation ratios obtained at each methylated site for estimating the methylation stoichiometry, enabling the quantitative characterization of m1A, m3C, m1G, and in nascent RNAs. Under different cellular treatments, such as ALKBH7 depletion and overexpression, DAMM-seq sensitively monitored the methylation level changes at m1A, m3C, m1G, and sites within mitochondrial polycistronic RNA and uncovered ALKBH7 demethylation effect at m1A and sites within pre-tRNA regions of mt-Leu1 and mt-Ile (Zhang et al., 2021), respectively; For the first time, we have this approach for quantitative investigation of RNA methylations within nascent mt-RNA. The quantitative base-resolution sequencing technology has proven its potential to characterize the molecular substrates of other ALKBH family proteins, and to promote the comprehensive mechanistic study of the reversible RNA methylation in mammals.
Collectively, DAMM-seq facilitated the identification of ALKBH7 as an RNA demethylase that mediates unprecedented and m1A demethylation in nascent mt-RNA, which regulates nascent polycistronic mt-RNA processing, local structural switch, and the subsequent mitochondrial gene expression (Zhang et al., 2021). Further functional investigation revealed the mechanistic features of ALKBH7 demethylation, and its depletion led to reduced mt-tRNA, reduced protein levels, and attenuated mitochondrial activity, impacting fat storage and obesity phenotype. The dynamics of ALKBH7 function in diverse biological and physiological processes will be further studied in the future, particularly its regulatory roles in cellular metabolism. In our recent publication of DAMM-seq (Zhang et al., 2021), although we described the procedures of library preparation, a step-by-step workflow of DAMM-seq could be better for people to reproduce and apply this quantitative sequencing technology in their own research projects. In this chapter, we included the detailed protocol of DAMM-seq in this edition of Methods in Enzymology.
2. Materials
2.1. Preparation of RNA samples
TRIzol Reagent (Invitrogen, cat. no. 15596026).
mirVana miRNA Isolation Kit (Invitrogen, cat. no. AM1561)
Chloroform
2-Propanol
Protein G Dynabeads (Invitrogen, cat. no. 10004D).
Anti-dsRNA (J2) antibody (Scions, cat. no. 10010500).
TURBO DNase (Invitrogen, cat. no. AM2238).
SUPERase•In RNase Inhibitor (Invitrogen, cat. no. AM2696).
Pierce™ Protease Inhibitor Tablets, EDTA-free (Thermo Scientific™, cat. no. A32965).
RNA Clean & Concentrator-5 (Zymo Research, cat. no. R1016).
Qubit RNA HS Assay Kit (Invitrogen, cat. no. Q32852).
UltraPure DEPC-treated water.
2.2. RNA fragmentation
UltraPure DEPC-treated water.
Ethyl alcohol
RNA Fragmentation Reagents (Invitrogen, cat. no. AM8740).
Oligo Clean & Concentrator-5 (Zymo Research, cat. no. D4061).
2.3. Demethylation
UltraPure DEPC-treated water.
Ethyl alcohol
Oligo Clean & Concentrator-5 (Zymo Research, cat. no. D4061).
SUPERase•In RNase Inhibitor (Invitrogen, cat. no. AM2696).
2.4. RNA 3′-end repair
UltraPure DEPC-treated water.
Ethyl alcohol
Oligo Clean & Concentrator-5 (Zymo Research, cat. no. D4061).
10× T4 Polynucleotide Kinase Reaction Buffer (700 mM Tris–HCl, pH 7.6 100 mM MgCl2, 50 mM DTT; New England BioLabs, cat. no. B0201S).
T4 Polynucleotide Kinase (10 U/μL) (Thermo Scientific, cat. no. EK0032).
SUPERase•In RNase Inhibitor (Invitrogen, cat. no. AM2696).
2.5. RNA 3′-adaptor ligation
UltraPure DEPC-treated water.
Ethyl alcohol
Oligo Clean & Concentrator-5 (Zymo Research, cat. no. D4061).
T4 RNA Ligase 2, truncated KQ (New England BioLabs, cat. no. M0373L).
SUPERase•In RNase Inhibitor (Invitrogen, cat. no. AM2696).
5′ Deadenylase (New England BioLabs, cat. no. M0331S).
RecJf (New England BioLabs, cat. no. M0264L).
2.6. Reverse transcription
UltraPure DEPC-treated water.
Ethyl alcohol
HIV Reverse Transcriptase (Worthington, cat. no. LS05006).
10× AMV Reverse Transcriptase Reaction Buffer (Included in New England BioLabs, cat. no. M0277S).
Deoxynucleotide (dNTP) Solution Mix (10 mM)
RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen, cat. no. 10777019).
RNase H (New England BioLabs, cat. no. M0297L).
Oligo Clean & Concentrator-5 (Zymo Research, cat. no. D4061).
2.7. cDNA 3′-adaptor ligation
UltraPure DEPC-treated water.
Ethyl alcohol.
T4 RNA Ligase 1 (ssRNA Ligase), High Concentration (New England BioLabs, cat. no. M0437M).
Adenosine 5′-Triphosphate (ATP) (10 mM).
DNA Clean & Concentrator-5 (Zymo Research, cat. no. D4014).
2.8. PCR amplification
UltraPure DEPC-treated water.
Ethyl alcohol.
LongAmp® Taq 2× Master Mix (New England BioLabs, cat. no. M0287L).
NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1, 2, 3, and 4) (New England BioLabs, cat. no. E7335S, E7500S, E7710S, E7730S).
Agarose (Low-Melting, Nucleic Acid Recovery/Molecular Biology Grade) (Thermo Fisher Scientific, cat. no. BP165-25).
Gel Loading Dye (6×) (New England Biolabs, cat. no. B7021S).
pBR322 DNA-MspI Digest (New England Biolabs, cat. no. N3032S).
MinElute Gel Extraction Kit (QIAGEN).
2.9. NGS sequencing and data analysis
NovaSeq 6000 System (Illumina).
DAMM-seq analysis pipeline (Zhang et al., 2021).
3. Methods
3.1. Overview of DAMM-seq
The DAMM-seq workflow includes ten steps (Fig. 2B): (1) Preparation of RNA sample; (2) RNA fragmentation; (3) Demethylation; (4) RNA 3′-end repair; (5) RNA 3′-adaptor ligation; (6) Reverse transcription; (7) cDNA 3′-adaptor ligation; (8) PCR amplification; (9) Next-generation sequencing (NGS) and data analysis. The step-by-step procedures are described in Step 3.2–Step 3.10.
3.2. Preparation of RNA sample
For sequencing RNA methylations in steady-state tRNAs using DAMM-seq: With the cultured cells in one 6-cm plate, purify total RNA with the standard manufacturer’s protocol of TRIzol Reagent, based on isopropanol precipitation. Cellular small RNA (<200 nt) was purified from the total RNA with mirVana miRNA Isolation Kit. Elute RNA with a proper amount of RNase-free water.
For sequencing RNA methylations in cellular dsRNA using DAMM-seq: Prepare the cultured cells in five 15-cm plates, as one replicate. 250 μL of Protein G Dynabeads were washed twice and resuspended in 2.5 mL of IP wash buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM MgCl2 and 0.5% NP-40). A 25-μg quantity of monoclonal anti-dsRNA (J2) antibody (Dhir et al., 2018) was added to the beads and incubated for 1 h at room temperature, followed by another hour at 4 °C. Cultured cells were washed once with cold DPBS. The cells in each plate were soaked in 5 mL cold DPBS and collected by cell lifters, combining 5 plates for each replicate. The cell suspension was spun at 300 g at 4 °C for 2 min. Cells were resuspended in 1 mL NP-40 lysis buffer (10 mL solution: 50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM EDTA, 10 mM MgCl2 and 0.5% NP-40, 50 μL TURBO DNase (2 U/μL), 500 μL SUPERase•In RNase Inhibitor (20 U/μL) and a Pierce EDTA-free protease inhibitor tablet) and incubated at 4 °C for 30 min. The lysate was spun at 16,000 g at 4 °C for 5 min. The lysate was diluted to 4.75 mL with IP wash buffer. A 250-μL volume of J2–Dyna-beads was added to the lysate and incubated at 4 °C for 2 h. The magnetic beads were washed once with 1 mL IP wash buffer and transferred to a new Eppendorf tube. The beads were further washed 4 times with 1 mL of IP wash buffer. J2-bound dsRNA was extracted using TRIzol, followed by the RNA Clean & Concentrator-5 purification.
Measure the concentration of the purified cellular small RNA or cellular dsRNA by Qubit RNA HS Assay Kit.
3.3. RNA fragmentation
The 400–500 ng cellular small RNA (size < 200 nt) or cellular dsRNA from Step 3.2 was fragmented into ~50-nt RNAs using RNA Fragmentation Reagents. Dilute the cellular small RNA or cellular dsRNA in 14 μL RNase-free water and add 1 μL RNA Fragmentation Reagents (used as 15X). Mix well by pipetting more than ten times.
Heat the RNA samples at 70 °C for 12 min. Put the RNA sample onto ice upon the completion of incubation.
Purify the fragmented RNA with the standard protocol of Zymo Oligo Clean & Concentrator-5 Kit. Elute RNA with 21 μL RNase-free water and harvest the purified RNA in 20 μL volume.
3.4. Demethylation (optional)
This demethylation step is not needed when conducting DAMM-seq for cellular dsRNA.
For cellular small RNA samples, about 2 μL RNA from Step 3.3 was saved as ‘Input’ and the rest 18 μL RNA was used as ‘Treated’ samples. Prepare a 49-μL reaction stock with 30 mM MES buffer (pH 5.5), 100 μM (NH4)2Fe(SO4)2·6H2O, 300 μM α-ketoglutarate, 2 mM l-ascorbic acid, 150 mM NaCl, 2 mM MgCl2, 40 μg/mL BSA, 2.5 μL SUPERase•In RNase Inhibitor and the RNA of ‘Treated’ samples. Add 1.0 μL of engineered AlkB (D135S, 200 pmol) and mix the 50-μL reaction mixture very well by pipetting more than fifteen times.
Incubate the reaction at 25 °C for 2 h. Put the RNA sample onto ice upon the completion of incubation.
Purify the ‘Treated’ RNA with the standard protocol of Zymo Oligo Clean & Concentrator-5 Kit. Elute RNA with 11 μL RNase-free water, and harvest the purified RNA in 10 μL volume. Dilute ‘Input’ samples into 10 μL volume with RNase-free water.
3.5. RNA 3′-end repair
For the ‘Input’ and ‘Treated’ samples of cellular small RNA or cellular dsRNA, prepare the reaction mixture with 12.5 μL RNase-free water, 3 μL 10× PNK buffer, 1.5 μL SUPERase•In™, 3 μL T4 PNK, and 10 μL RNA as a final volume of 30 μL. Mix well by pipetting more than ten times.
Heat the samples at 37 °C for 1 h.
Purify the 3′-repaired RNA with the standard protocol of Zymo Oligo Clean & Concentrator-5 Kit. Elute RNA with 11 μL RNase-free water, and harvest the purified RNA in 10 μL volume.
3.6. RNA 3′-adaptor ligation
For the ‘Input’ and ‘Treated’ samples of cellular small RNA or cellular dsRNA, mix the 10 μL 3′-repaired RNA fragments with 1.0 μL 20 μM RNA 3′ SR Adapter (5′App-NNNNNAGATCGGAAGAGCGTC GTG-3SpC3). Heat at 70 °C for 2 min and immediately move onto the ice. This RNA 3′ SR Adapter is ssDNA oligo containing a 5′ adenylation modification (/5rApp/ in IDT catalog) and a 3′ spacer (/3SpC3/ in IDT catalog), prepared with RNase-free HPLC purification. Dissolve this ssDNA oligo in Nuclease-free water and keep it as 20 μM stock solution.
Then, prepare a stock containing 2.5 μL 10× T4 RNA Ligase Reaction Buffer, 7.5 μL PEG8000 (50%), and 1 μL SUPERase•In. Mix the stock well by pipetting more than ten times.
Add the stock into the RNA–adapter mixture, and mix each sample by pipetting several times. Then add 2 μL T4 RNA Ligase 2 truncated KQ (Zhang et al., 2019), and mix each sample well by pipetting more than twenty times.
The reaction was incubated at 25 °C for 2 h followed by 16 °C for 10 h, at a PCR block.
After the ligation, dilute the reaction mixture to 47 μL with RNase-free water. Add 2 μL 5′-deadenylase and mix the samples well by pipetting more than ten times. Incubate the samples at 30 °C for 30 min.
After the 5′-deadenylation, add 1 μL RecJf for ssDNA digestion. Mix the samples well by pipetting more than ten times. Incubate the samples at 37 °C for 30 min
Purify the ligated RNA with Zymo RNA Clean & Concentrator-5 Kit according to the manufacturer’s protocol. Elute RNA with 11 μL RNase-free water. Harvest the purified RNA in 10 μL volume.
3.7. Reverse transcription
For the ‘Input’ and ‘Treated’ samples of small RNA or the sample of cellular dsRNA, mix the 10 μL RNA with 1.0 μL 2.0 μM SR RT primer (5′-ACACGACGCTCTTCCGATCT-3′). Heat at 65 °C for 2 min and immediately move onto the ice. This SR RT primer is ssDNA oligo and needs to be ordered with RNase-free HPLC purification. Dissolve this ssDNA oligo in RNase-free water to produce a 100 μM stock solution. Dilute the stock solution into 2 μM aliquots.
Add 2 μL 10× AMV Reverse Transcriptase Reaction Buffer, 2 μL 10 mM dNTP Solution Mix, 0.5 μL RNaseOUT Recombinant Ribonuclease Inhibitor, 4.5 μL RNase-free water, and 1 μL HIV Reverse Transcriptase (Worthington) to the RNA-primer mixture. Mix the samples well by pipetting more than ten times.
Incubate the reaction at 37 °C for 1.5 h.
After the RT reaction, 1 μL RNase H was added. Mix well by pipetting more than ten times. Incubate at 37 °C for 20 min, and then denature at 70 °C for 5 min.
Purify the cDNA with the standard protocol of Zymo Oligo Clean & Concentrator-5 kit. Elute RNA with 11 μL RNase-free water. Harvest the purified cDNA in 10 μL volume.
3.8. cDNA 3′-end ligation
For ‘Input’ and ‘Treated’ samples of small RNA or the samples of cellular dsRNA, mix the 10 μL cDNA with 1.0 μL 50 μM cDNA 3′ SR Adapter (5′Phos-NNNNNAGATCGGAAGAGCACACGTCTG-3SpC3). Heat at 70 °C for 2 min and immediately move onto the ice. This cDNA 3′ SR Adapter is ssDNA oligo containing a 5′ phosphate modification (/5Phos/ in IDT catalog) and a 3′ spacer (/3SpC3/ in IDT catalog), prepared with HPLC purification. Dissolve this ssDNA oligo in Nuclease-free water and keep it as 50 μM stock solution.
Then, prepare a stock containing 3 μL 10× T4 RNA Ligase Reaction Buffer, 15 μL PEG8000 (50%), and 3 μL 10 mM ATP. Mix the stock well by pipetting more than ten times.
Add the stock into the RNA–adapter mixture, and mix each sample by pipetting several times. Then add 1 μL T4 RNA Ligase 1(High Concentration), and mix each sample well by pipetting more than twenty times.
The reaction was incubated at 25 °C for 12 h.
Purify the ligated cDNA samples with Zymo DNA Clean & Concentrator kit. Add a 7× volume (147 μL) of DNA binding buffer (Chen et al., 2023; Dai et al., 2023), and mix by pipetting 15 times. Wash the columns according to the manufacturer’s protocol. Elute cDNA with 11 μL RNase-free water. Harvest the purified cDNA in 10 μL volume.
3.9. PCR amplification
4 μL cDNA was used for each 15-cycle PCR amplification reaction, which was performed by adding 1.5 μL NEB indexed primers and a premixed stock containing 1.5 μL SR Primer for Illumina, 25 μL 2× LongAmp Master Mix, and 18 μL RNase-free water.
- Perform PCR amplification, according to the table below.
Cycle number Denature Anneal Extend Preincubation 94 °C 30 s NA NA 15 cycles 94 °C 15 s 62 °C 30 s 70 °C 30 s Final extension NA NA 70 °C 5 min After PCR amplification, add 10 μL Gel Loading Dye (6X) to each sample and mix well by pipetting more than ten times.
All libraries were purified on a 3.5% (wt/vol) low melting point agarose gel and run at 90 V for 45 min, with pBR322 DNA-MspI Digest (NEB) as the size marker. Conduct gel-cutting to collect the band around 160 bp, according to the size marker.
Recovery of DNA from the agarose gel was enabled by MinElute Gel Extraction Kit (QIAGEN) according to the manufacturer’s protocol. Elute DNA with 15 μL RNase-free water.
3.10. NGS sequencing and data analysis
Sequence all libraries on Illumina Nova-seq 6000, with single-end 100 bp read length.
The bioinformatic pipeline for mutation calling has been included in the original publication (Zhang et al., 2021).
4. Notes
In Step 3.2 (Preparation of RNA sample), the production yield of cellular dsRNA is very low. As a result, it is necessary to prepare at least 100 million cells for each biologically independent replicate. Typically, we need to have two biological replicates for one sample, which requires five 15-cm plates per replicate and a > 70% confluency before cell lysis. When eluting cellular dsRNA from the beads, add ~300 μL TRIzol reagent to the dry beads, followed by a 30-min incubation at room temperature. Then, a standard purification with the columns from RNA Clean & Concentrator-5 Kit and the buffers from Direct-zol RNA Miniprep Kit yields cellular dsRNA in 10 μL volume, with a concentration of around 1–2 ng/μL measured by Qubit assay.
Starting from Step 3.2 (Preparation of RNA sample), please use RNase-free or nuclease-free water for all procedures related to RNA purification and library construction. If the RNA quality is low or RNA degradation occurs during RNA preparation, a much shorter band size might be observed when running an agarose gel in Step 3.9 (PCR amplification).
In Step 3.2 (Preparation of RNA sample), except for cellular small RNA (<200 nt) and cellular dsRNA, the other types of RNA samples can also be used as the input RNA in DAMM-seq, such as rRNA-depleted total RNA, chromosome-associated RNA (caRNA), EU-labeled nascent RNA, etc. DAMM-seq could be further extended to study m1A, m3C, m1G, and modifications in diverse RNA species, including mRNA, lncRNA, repeats RNA, snRNA, snoRNA, etc.
In Step 3.8 (cDNA 3′-adaptor ligation), please mix each sample immediately after adding T4 RNA Ligase 1. It is not recommended to add ligase enzyme to all samples and then start mixing all samples. An irreversible white precipitation would be seen in the ligation mixture, if the sample is not mixed immediately after adding the ligase.
In Step 3.8 (cDNA 3′-adaptor ligation), with the Zymo DNA Clean & Concentrator kit, a 7× volume of DNA binding buffer was used to remove short oligos such as RT primers and ligation adapters. The elimination of RT primers and ligation adapters is necessary to avoid the consumption of PCR primers in Step 3.9 (PCR amplification). The 7× volume of DNA binding buffer can safely keep the cDNA above ~100 nt (Chen et al., 2023; Dai et al., 2023).
In Step 3.9 (PCR amplification), the PCR cycle number is typically no more than 15 cycles. DAMM-seq protocol has been optimized to start with input RNA of several nanograms (ng), and the 15-cycle PCR amplification can robustly generate enough dsDNA for NGS sequencing. The overamplification leads to unexpected PCR duplicates in NGS data and should be avoided.
In Step 3.10 (NGS sequencing and data analysis), representative m1A sites on cellular RNA (like rRNA) are employed as the benchmark for DAMM-seq quality control, to exclude batch variations. In DAMM-seq, since a small portion of rRNA always remains in the purified cellular small RNA or mitochondrial nascent RNA, the misincorporation profile at 28S rRNA m1A site can be used as the benchmark for monitoring DAMM-seq library quality. Typically, the misincorporation rates observed at the rRNA m1A site are highly consistent among the sequencing samples in one sequencing run.
Funding
We thank the support from National Institutes of Health (NIH) Grants RM1 HG008935 (C.H.) and R01 ES030546 (C.H.).
References
- Chen L, Zhang L-S, Ye C, Zhou H, Liu B, Gao B, … Dickinson B (2023). Nm-Mut-seq: A base-resolution quantitative method for mapping transcriptome-wide 2′-O-methylation. Cell Research. 10.1038/s41422-023-00836-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q, Zhang L-S, Sun H-L, Pajdzik K, Yang L, Ye C, … He C (2023). Quantitative sequencing using BID-seq uncovers abundant pseudouridines in mammalian mRNA at base resolution. Nature Biotechnology, 41, 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, … Proudfoot NJ (2018). Mitochondrial double-stranded RNA triggers antiviral signaling in humans. Nature, 560, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Harada BT, Behm M, & He C (2018). RNA modifications modulate gene expression during development. Science (New York, N. Y.), 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, … He C (2011). N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature Chemical Biology, 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, … He C (2016). ALKBH1-mediated tRNA demethylation regulates translation. Cell, 167, 816–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lu H, Tian Z, Yang M, Ma J, Shang G, … Chen Z (2022). Structural insights into the interactions and epigenetic functions of human nucleic acid repair protein ALKBH6. The Journal of Biological Chemistry, 298, 101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, & He C (2017). Dynamic RNA modifications in gene expression regulation. Cell, 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg A, Robertson AB, Aronsen JM, Rognmo Ø, Sjaastad I, Wisløff U, & Klungland A (2013). Deletion of mouse Alkbh7 leads to obesity. Journal of Molecular Cell Biology, 5, 194–203. [DOI] [PubMed] [Google Scholar]
- Suzuki T, & Suzuki T (2014). A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Research, 42, 7346–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, He Q, Feng C, Liu Y, Deng Z, Qi X, … Chen Z (2014). The atomic resolution structure of human AlkB homolog 7 (ALKBH7), a key protein for programmed necrosis and fat metabolism. The Journal of Biological Chemistry, 289, 27924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L-S, Liu C, Ma H, Dai Q, Sun H-L, Luo G, … He C (2019). Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Molecular Cell, 74, 1304–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L-S, Xiong Q-P, Perez SP, Liu C, Wei J, Le C, … He C (2021). ALKBH7-mediated demethylation regulates mitochondrial polycistronic RNA processing. Nature Cell Biology, 23, 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, & He C (2017). Post-transcriptional gene regulation by mRNA modifications. Nature Reviews. Molecular Cell Biology, 18, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, … He C (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell, 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Fu Y, & He C (2014). Nucleic acid oxidation in DNA damage repair and epigenetics. Chemical Reviews, 114, 4602–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, … Pan T (2015). Efficient and quantitative high-throughput tRNA sequencing. Nature Methods, 12, 835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]