

Abstract
Interspecies comparisons of gene expression levels will increase our understanding of the evolution of transcriptional mechanisms and help to identify targets of natural selection. This approach holds particular promise for apes, as many human-specific adaptations are thought to result from differences in gene expression rather than in coding sequence. To date, however, all studies directly comparing interspecies gene expression have been performed on single-species arrays, so that it has been impossible to distinguish differential hybridization due to sequence mismatches from underlying expression differences. To evaluate the severity of this potential problem, we constructed a new multiprimate cDNA array using probes from human, chimpanzee, orangutan, and rhesus. We find a large effect of sequence divergence on hybridization signal, even in the closest pair of species, human and chimpanzee. By comparing single-species array analyses with results from multispecies arrays, we examine how estimates of differential gene expression are affected by sequence divergence. Our results indicate that naive use of single-species arrays in direct interspecies comparisons can yield spurious results.
DNA arrays make it possible to study the expression levels of tens of thousands of genes simultaneously (Bowtell 1999; Eisen and Brown 1999; White 2001; Chen et al. 2002; Reinke 2002). Recent studies have used DNA arrays to compare patterns of expression between closely related species (Enard et al. 2002; Caceres et al. 2003; Karaman et al. 2003; Meiklejohn et al. 2003; Ranz et al. 2003; Fortna et al. 2004; Khaitovich et al. 2004; Nuzhdin et al. 2004; Saetre et al. 2004). Such multiple-species expression studies can shed light on the evolution of expression and help to identify genes that evolve under selective pressures (Rifkin et al. 2003; Khaitovich et al. 2004).
DNA arrays are currently available for only a limited number of species. Thus, while existing interspecies expression studies have used different platforms (oligo or cDNA arrays), they have all assayed multiple species using arrays that were designed based on the sequence (or cDNA) of only a single species. For example, human arrays have been used to compare human expression patterns to those of chimpanzee, orangutan, and rhesus macaque (Enard et al. 2002; Caceres et al. 2003; Fortna et al. 2004; Khaitovich et al. 2004). Similarly, Drosophila melanogaster arrays have been used to directly compare expression levels in D. melanogaster and D. simulans (Meiklejohn et al. 2003; Ranz et al. 2003; Nuzhdin et al. 2004).
A possible problem with gene-expression measurements in different species using single-species arrays is sequence mismatches. Because the probes on most arrays are designed using sequence from only one species, they can differ from the target cDNA derived from the other species at many base pairs. In the presence of such sequence mismatches, relative hybridization intensities will reflect both differences in transcript abundance levels (the object of interest), as well as differences in hybridization kinetics.
Two studies tried to address this potential problem. Ranz et al. (2003) estimated the effect of sequence divergence between D. melanogaster and D. simulans by hybridizing genomic DNA from both species to the array. However, conventional normalization procedures used to correct for intensity differences between dyes mask the possible effects of sequence mismatches, making it difficult to distinguish the two (see below). In turn, in a study using Affymetrix oligo arrays, Khaitovich et al. (2004) accounted for sequence mismatches by excluding probes where the human and chimpanzee sequences were not identical. This approach is only feasible when both genome sequences are available (Nagpal et al. 2004) and only practical for closely related species, lest too few probes remain (e.g., only for chimpanzee in Khaitovich et al. 2004). In summary, previous studies have not had an effective way to estimate or correct for the effect of sequence mismatches on array hybridization. Here, we do so by constructing a novel multispecies cDNA array. We find that sequence divergence can have a substantial effect on estimates of expression levels, even for human–chimpanzee comparisons, so cannot be safely ignored in direct cross-species comparisons.
Results and Discussion
In order to characterize the effect of sequence mismatches on hybridization more directly, we built a cDNA array that contains probes from multiple species. We amplified and spotted 350–600 bp products of 1056 genes from each of four primate species, i.e., human, chimpanzee, orangutan, and rhesus macaque. We then performed competitive hybridizations between reverse-transcribed human liver RNA and liver RNA from each of the other three primates (see Methods). There were four replicates of each comparison, two with each dye combination. In each comparison of a pair of species, we focused on results from the probe sets derived from cDNA of the two species that we assayed and ignored the probes from the other two species on the array. In addition, we labeled a human RNA sample with two dyes independently, and hybridized it together, a control that will henceforth be referred to as “self-hybridization.”
Our first goal was to examine the effect of sequence mismatches on hybridization. Given our choice of species, we could examine the effects of average nucleotide sequence divergence of ∼0.8%–1% (human–chimpanzee) (Ebersberger et al. 2002; Hellmann et al. 2003), 3% (human–orangutan) (Chen and Li 2000), and 5% (human–rhesus) (Gilad et al. 2003; Wall et al. 2003). For each pair of species, we estimated the effect of sequence divergence on the log ratios of the intensities of each spot using a linear model (see Methods). If sequence divergence does not affect hybridization—for example, if chimpanzee and human target cDNA bind equally well to chimpanzee probe cDNA—then these estimated sequence effects should be due solely to experimental error, and the log ratios of the human and chimpanzee probes should be similar.
The experimental error was estimated from the four replicate self-hybridizations, for which we compared the distribution of log ratios obtained from human probes with those obtained from each of the three other species probe sets (Fig. 1). Next, we analyzed the chimpanzee–human hybridizations. With no sequence effect, the chimpanzee/human log2 ratio from either a human or a chimpanzee probe for the same gene should be very similar, since both reflect only the difference in expression level (and the experimental error). Consequently, we expect the distributions of chimpanzee/human log2 ratio from the human and the chimpanzee probe sets to be close to identical (within the limits of experimental error, as estimated in Fig. 1). However, we found that the difference between the two distributions was significantly larger than expected from the estimated experimental error (t-test, P <10–4, Fig. 2A), with 577 of 912 genes showing a significant sequence effect (for an FDR of 0.05). Thus, even a mean sequence divergence of 1% (Chen et al. 2001; Ebersberger et al. 2002) has a detectable effect on hybridization. As expected, we observed even more pronounced effects of sequence mismatches in the human–orangutan (661/826 genes with a significant sequence effect, Fig. 2B) or the human–rhesus comparisons (759/851 genes with a significant sequence effect, Fig. 2C).
Figure 1.

Experimental error was estimated using four replicates of a human self-hybridization. The number of probes (y-axis) with a given Cy5/Cy3 log2 ratio (x-axis) is shown with black bars for the human probes and with clear bars for the (A) chimpanzee probes (mean difference 0.03), (B) orangutan probes (mean difference 0.05), and (C) rhesus probes (mean difference 0.04). The difference between the distributions of log2 ratios from different probe sets reflects the experimental error due to the hybridization to different probes on the array.
Figure 2.

Results for interspecies competitive hybridization. The number of probes (y-axis) with a given nonhuman/human log2 ratio (x-axis) is shown with black bars for the human probes and with clear bars for (A) chimpanzee probes (mean difference 0.50), (B) orangutan probes (mean difference 0.78), and (C) rhesus probes (mean difference 1.15). All three values are significantly higher than the experimental error estimated from the self-hybridization (P <10–4), indicating that sequence differences affect hybridization intensity. Note that the normalization based on both species probe sets leads to a symmetric distribution (see Methods).
We investigated the effect of sequence divergence on the detection of differential expression by performing two types of analyses as follows: (1) We considered only the human probes, thereby mimicking a single-species array, and (2) we considered probes from the two species that were hybridized, and estimated the average log ratio over the two for each gene. When we considered only the human probes, we followed a standard post-scanning normalization approach taken in single-species array analyses, i.e., mean centering each channel on each array; mean centering the log ratios; checking for scale differences between the arrays and nonlinear dye effects on each array (see Methods). One important assumption of this procedure is that the overall intensities between the two samples are very similar, so that the distribution of their log ratios will be symmetric (Yang et al. 2002; Bolstad et al. 2003; Smyth and Speed 2003). When using single-species array for multispecies comparisons, this assumption is equivalent to positing a priori that there is no effect of sequence mismatches on hybridization, because any systematic difference will be attributed to differences between the dyes and will be adjusted. This assumption presents a problem if there is an effect due to sequence divergence. Using single-species arrays, this problem is in fact inescapable, because the intensity of spots depends upon the settings of the scanner and only become meaningful when compared with other intensities in the experiment, after experiment-appropriate normalization.
In contrast, when we analyzed probes from both species, we expected the log-ratio distribution to be symmetric because we normalized over both probe sets. For each gene, we used the mean of the log ratios from probes of both species as the measure of relative expression level. We made the assumption that sequence divergence has an equal, but opposite effect on the log ratio results from each probe set, so that taking the average cancels the effect of sequence divergence. Thus, in a multispecies comparison context, array-specific normalization corrects for dye effects, and averaging the signal from both probe sets corrects for the gene-specific effect of sequence divergence, while on a single-species array, the two sources of variation are completely confounded.
We compared results obtained by averaging the signal from both probe sets to those obtained from our single-species analysis. In order to estimate experimental error due to probe effects, we performed a similar comparison for the human self-hybridization (Fig 3A). We found that, for a given gene, the estimated differential expression levels from single and multispecies arrays rarely agree (Fig. 3B–D; the discrepancy is significantly greater than expected from the estimated experimental error, t-test, P <10–10). The likely explanation for these discrepancies is that normalization of the two channels using a single-species probe set corrects for the average effect of the sequence mismatches. However, there is a distribution of divergence values around that mean, which, in turn, leads to a distribution of effects on hybridization. Hence, the normalization procedure tends to overcompensate for genes with lower than average divergence and undercompensate for genes with relatively higher divergence. As long as the exact pattern of divergence between each probe and the corresponding RNA from each species is unknown, the contribution of sequence mismatches on the apparent levels of expression cannot be taken into account.
Figure 3.

The difference between gene-expression estimates from multi- and single-species array analyses is plotted on the y-axis. Thus, positive values indicate that a greater difference was found in the multispecies analysis than in the single species one, while negative values point to the reverse. Probes are ordered by their log2 expression difference, as estimated from the multispecies analysis (x-axis). (A) Human–human (for which the human and chimpanzee probes were used—mean absolute log2 difference 0.07 ± 0.06) (B) Human–chimp (mean absolute log2 difference 0.18 ± 0.16) (C) Human–orangutan (mean absolute log2 difference 0.24 ± 0.21) (D) Human–rhesus (mean absolute log2 difference 0.25 ± 0.20)
To examine the difference between the single and multi–species array analysis in more detail, we identified genes that appear to be differentially expressed between our human and chimpanzee samples, by mimicking a single-species array analysis and using an FDR value of 0.05 as our cutoff (Table 1). We then used the same criteria, but relied on probes from both species. In the comparison, 72/344 (20.9%) of the genes were identified as differentially expressed by the single-species array, but not the multispecies array analysis. Moreover, 151/423 (35.7%) of the genes were found to be differentially expressed when probe sets from both species were considered, but not by the single-species array analysis (Table 1). These numbers are slightly higher when we focus on the human–orangutan and human–rhesus comparisons (Table 1).
Table 1.
Numbers of differentially expressed genes
| Species comparison | Single-species analysisb | Not confirmedc | Multi-species analysisd | Not found by single speciese |
|---|---|---|---|---|
| Human—Humana | 0 | n.a | 0 | n.a |
| Human—Chimpanzee | 344 (37.4%) | 72 | 423 | 151 |
| Human—Orangutan | 480 (58.1%) | 79 | 582 | 181 |
| Human—Rhesus | 554 (65.1%) | 96 | 629 | 171 |
This is a self—hybridization, which serves as a measure of experimental error (see Methods).
The number of differentially expressed genes with an FDR of 0.05 according to the single-species array analysis (in parentheses, the percentage from all genes studied).
The number of genes that were found to be differentially expressed by the single-species analysis, but not when probe sets from both species were considered.
The number of differentially expressed genes with an FDR of 0.05 according to the multi-species array analysis.
The number of genes that were found to be differentially expressed when probe sets from both species were considered, but not by the single-species array analysis.
Finally, we wanted to verify that the differences that we observed between probe sets are indeed due to the effect of sequence mismatches. To do so, we first considered the mean difference in log ratios between the human and chimpanzee probes for each gene, using four replicates. We identified and sequenced the five gene probes with the lowest (SSR1, RODH, SPP2, WDR7, DDB2) and the highest (KIAA0116, ATP5C, H2FY, KNG, RAP140) P-values obtained from testing the hypothesis of no-sequence mismatch effect (using a t-test). As predicted, human–chimpanzee divergence was significantly higher (1.52% ± 0.51%) for genes that showed a significant difference between the human and chimpanzee probes, compared with the genes with no apparent difference (0.62% ± 0.28%, t-test P = 0.012). To confirm this result, we used BLAT (Kent 2002) to identify the orthologous chimpanzee sequences for the 100 genes with the lowest and the 100 genes with the highest P-values (see Methods; Supplemental Table 1). As expected, human–chimpanzee divergence for the 100 genes with lowest P-values was significantly higher (0.61% ± 0.39%) than for the 100 genes with highest P-values (0.42% ± 0.32%, t-test, P = 0.008). Hence, our estimated sequence-mismatch effect partly reflects the total sequence divergence at a given gene. Overall divergence level is unlikely to be the sole factor influencing hybridization; other aspects, such as GC content, the length of the probe and the position of the mismatches, are likely to play an important role. If single-species arrays are to be used for multispecies comparisons, there will be a need for a statistical model that quantifies these relationships and corrects for their effects on estimates of expression intensities.
There is one context, however, in which single-species arrays may suffice for reliable cross-species comparisons; if the goal is solely to identify some genes with large changes in expression between closely related species. For example, if we impose an expression-change cutoff of 1.5-fold, almost all of the genes that were identified as differentially expressed by the single-species array are confirmed by the multispecies array analysis (96.9% for the human–chimpanzee comparison, 100% for the human–orangutan, and 94.5% for the human–rhesus comparison). This approach presents two disadvantages, however; first, we lose approximately two-thirds of the genes that were originally identified as differentially expressed between species. Many of these small, but consistent expression differences may be of functional importance; second, a very large proportion of genes that are above the 1.5 cutoff and are significantly differentially expressed in the multispecies analysis fall below the 1.5-fold cutoff in the single-species array and, are therefore missed (45%–60% in all species comparisons). Thus, by using a combination of fold-change cutoff and statistical analyses in a single-species array analysis, we are able to reliably identify genes that are differentially expressed between closely related species, but at the cost of missing a large proportion of potentially interesting genes. It should also be kept in mind that these figures are specific to this particular array; different genes, probes, or experimental design may affect estimates of overlap between the analyses of single- and multispecies arrays.
In conclusion, there is a marked effect of sequence mismatches on array measurements, even between organisms that are only ∼1% diverged, on average. Yet compared with other species, primates have relatively low-sequence divergence; 1% diversity is no more than observed within natural populations of D. melanogaster (Andolfatto and Przeworski 2000). This suggests that sequence divergence can also affect intraspecific comparisons of expression patterns. It may therefore be necessary to use more conservative cutoffs to identify differentially expressed genes or, when possible, to explicitly design experiments to eliminate the sequence effect (Rifkin et al. 2003). More generally, our findings indicate that the naive use of single species arrays for comparisons of expression profiles between species may be problematic. As we have shown, this difficulty can be circumvented by the use of species-specific arrays; alternatively, novel statistical approaches could be developed to permit reliable interspecies comparisons of expression patterns.
Methods
cDNA microarrays
PCR primers for 1056 human genes were designed based on human sequences obtained from the NCBI reference sequence database (http://www.ncbi.nlm.nih.gov/RefSeq/). Primers were designed based on the longest mRNA sequence available for each gene, and were located within the last 1.5 kb of the mRNA sequence. The size of the amplified product was 300–600 bp. We blasted all predicted amplicons against the human genome (July 2003) (http://genome.ucsc.edu/) in order to verify that they are unique sequences in the human genome (no more than a single >100-bp segment with identity cutoff of 85% is found). The same PCR primers were used for all species. PCR was performed in a total volume of 50 μL containing 0.2 μM of each deoxynucleotide (Promeg), 50 pmol of each primer, 1.5 mM MgCl2, 50 mM KCl, 10 mM Tris (pH 8.3), 2 U of Taq DNA polymerase, and 500 ng of liver cDNA as template. Conditions for the PCR amplification from all species were as follows: 10 cycles of denaturation at 94°C, annealing at 55°C, and extension at 72°C, each step for 1 min, followed by 33 cycles denaturation at 94°C, annealing at 53°C, and extension at 72°C, each step for 1 min. The first step of denaturation and the last step of extension were 5 and 7 min, respectively. PCR products were analyzed by gel electrophoresis. Successful PCR products of the expected size were obtained for 1021 (96.7%) genes in human, 1025 (97%) genes in chimp, 1003 (95%) genes in orangutan, and 973 (92.1%) genes in rhesus macaque. PCR products were purified and spotted onto polylysine-coated glass slides using a GeneMachines arrayer with 16 pins. After printing, the slides were post-processed with 1,2-dichloroethane and N-methylimidazol (Diehl et al. 2001) and stored under low humidity until use.
Samples and hybridizations
Total RNA was extracted from liver tissues of one adult male of each species using Trizol (Invitrogen) as follows: human, chimpanzee (Pan troglodytes), orangutan (Pongo pygmaeus), and rhesus macaque (Macaca mulatta). First-strand cDNA was synthesized using a T7-poly-T oligo and the superscript kit (Invitrogen). The second-strand was synthesized using DNA Pol I enzyme (Invitrogen). cDNA was subjected to linear amplification using MEGAscript (Ambion). RNA was purified using the RNeasy kit (Qiagen). For each hybridization, 4 μg of amplified RNA (Perou et al. 2000; Ranz et al. 2003; Rifkin et al. 2003) were used for amino-allele labeling (BD Bioscience) with either Cy5 or Cy3 dyes (Amersham). The labeled RNA was purified (Qiagen), and the hybridization and washes were carried out as described in Eisen and Brown (1999).
Sequencing
PCR products were sequenced directly. The PCR primers were also used for sequencing. Sequencing reactions were performed using a dye-terminator cycle sequencing kit (Perkin Elmer) on an ABI 3100 automated sequencer (Perkin Elmer). After base calling with the ABI Analysis Software (version 3.0), the data were edited and assembled using the Sequencher program, version 4.0 (GeneCodes Corp.).
Data acquisition
After hybridization, we scanned the slides using a GenePix 4000 series scanner (Axon), flagged bad spots, and used the background-subtracted median foreground values as the intensity levels in the ensuing analysis. During scanning, we matched the intensity distributions between channels on each array. Because of the phylogenetic relationships between the four species, this will tend to decrease intensities for the human samples in the human–orangutan and human–rhesus hybridizations. We corrected for this bias later in the analysis.
All subsequent analyses were coded in Python (Ascher et al. 2001; Magwene 2002; van Rossum 2003) with calls to the R statistical package (Team 2003). For each experiment, we only used spots (DNA probes printed on the array) that were not flagged on any of the replicate arrays for either of the target species in the experiment. Although sequences from four species are on each array, only data from two of the species are relevant for each experiment. In the following discussion, `all of the spots' refers to the relevant half of the sequences on each array. All measurements are log2 transformed.
Normalization
We performed two separate analyses for each experiment, one where we accounted for the effect of sequence mismatches, and one on the human sequences alone, to mimic the use of human arrays for cross-species comparisons. For the first analysis, we subtracted the average Cy3 and Cy5 from the spots on an array that corresponded to the two species that were hybridized (e.g., only human and orangutan spots, when human and orangutan mRNA were cohybridized), accounted for nonlinear, intensity-dependent dye effects by subtracting the local regression of the log-ratio on the average intensity of a spot (loess span = 2/3) (Yang et al. 2001), and subtracted the mean log-ratio across the array. The distributions of log-ratios for each array in an experiment were similar (Supplemental Fig. 1), so we did not adjust for scaling differences. We used the log-ratios for each spot as our measurements. We took the same normalization steps for the second analysis, but only considered the human spots on each array.
Analysis of variance
We fit the following linear models to the data:
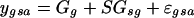 |
(1) |
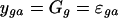 |
(2) |
where ygsa is the nonhuman/human log-ratio for the spot on array a with gene g in species s, Gg is the average across all measurements of gene-specific effect, SGsg is the gene-specific sequence mismatch effect, and εgsa is the spot-specific error. Because the design is balanced across species, we can separate the contribution of sequence (SG) from that of differential expression (G). In Model 2—used for the second analysis—we only consider the data from the human sequences, and so the term involving species-specific sequences drops out.
For Model 1, we estimated each effect using a standard two-way analysis of variance. We evaluated the significance of the effects by a bootstrap-randomization procedure (Kerr et al. 2002). For each gene, we computed a gene-specific standard deviation of the errors and placed all standardized errors in a common pool. We stepped gene-by-gene, recreating the data and re-estimating the remaining part of the Model 2 (number of genes/FDR level) times to create distributions for Gg and SGsg. We used these distributions to derive P-values for the estimated parameters and assessed significance by a step-up false discovery rate procedure using FDR level of 0.05 (Benjamini and Hochberg 1995). We performed the analogous estimation and bootstrapping procedure for Model 2).
As a control, we mimicked the comparison between Models 1 and 2 using the human–human self hybridizations and the human and chimpanzee probes on the arrays. We arbitrarily designated one human sample on each array to be a mock-chimpanzee sample. Given the goal of estimating differential expression and the constraint of maintaining balance between the dyes, there are three nonequivalent arrangements of the samples on the four arrays into pairs of human and mock-chimpanzee. We call a gene significantly differentially expressed in Model 1 (Model 2) if its P-value is below the 0.05 FDR cutoff for the data sets from each of the three arrangements, based on bootstrapping Model 1 (Model 2) as described above.
Data mining
This analysis yields a P-value for each gene, obtained from testing the hypothesis of no sequence mismatch. We considered the first 100 genes from each tail of the distribution of P-values, specifying that the percent identity scores be equal or above 97% (4.5% of all queries were excluded in this process). These human cDNA sequences were then used as queries in a BLAT (Kent 2002) search of the November 2004 build of the chimpanzee genome sequence at http://genome.ucsc.edu/. The percent sequence identity of the top score results were recorded for each search. We did not use sequence-quality cutoff for the chimpanzee genomic sequence. However, assuming a random distribution of sequencing errors, this should have no effect on our conclusion.
Electronic database information
All expression data was submitted to the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the series GSE2009, with sample accession numbers GSM35572, GSM35573, GSM35574, GSM35575, GSM35576, GSM35577, GSM35578, GSM35579, GSM35580, GSM35581, GSM35582, GSM35583, GSM35584, GSM35585, GSM35586, GSM35587.
Acknowledgments
We thank S. Pääbo and P. Khaitovich for providing the nonhuman primate samples, and D. Hartl, C. Meiklejohn, M. Przeworski, and J. M. Ranz for helpful discussions, as well as M. Przeworski, T. Speed, and three anonymous reviewers for comments on the manuscript. Y.G. is a supported by a postdoctoral fellowship of the European Molecular Biology Organization. K.P.W. is supported by the Arnold and Mabel Beckman Foundation and by the W.M. Keck Foundation. This research was supported by a grant to K.P.W. from the NIH National Human Genome Research Institute.
[Supplemental material is available online at www.genome.org. All expression data was submitted to the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the series GSE2009, with sample accession numbers GSM35572, GSM35573, GSM35574, GSM35575, GSM35576, GSM35577, GSM35578, GSM35579, GSM35580, GSM35581, GSM35582, GSM35583, GSM35584, GSM35585, GSM35586, GSM35587. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: S. Pääbo and P. Khaitovich.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3335705.
References
- Andolfatto, P. and Przeworski, M. 2000. A genome-wide departure from the standard neutral model in natural populations of Drosophila. Genetics 156: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher, D., Dubois, P.F., Hinsen, K., Hugunin, J., and Oliphant, T. 2001. Numerical python. Lawrence Livermore National Laboratory, Livermore, CA.
- Benjamini, Y. and Hochberg, Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Royal Stat. Soc. B 57: 289–300. [Google Scholar]
- Bolstad, B.M., Irizarry, R.A., Astrand, M., and Speed, T.P. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193. [DOI] [PubMed] [Google Scholar]
- Bowtell, D.D. 1999. Options available—from start to finish—for obtaining expression data by microarray. Nat. Genet. 21: 25–32. [DOI] [PubMed] [Google Scholar]
- Caceres, M., Lachuer, J., Zapala, M.A., Redmond, J.C., Kudo, L., Geschwind, D.H., Lockhart, D.J., Preuss, T.M., and Barlow, C. 2003. Elevated gene expression levels distinguish human from non-human primate brains. Proc. Natl. Acad. Sci. 100: 13030–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F.C. and Li, W.H. 2000. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F.C., Vallender, E.J., Wang, H., Tzeng, C.S., and Li, W.H. 2001. Genomic divergence between human and chimpanzee estimated from large-scale alignments of genomic sequences. J. Hered. 92: 481–489. [DOI] [PubMed] [Google Scholar]
- Chen, X., Cheung, S.T., So, S., Fan, S.T., Barry, C., Higgins, J., Lai, K.M., Ji, J., Dudoit, S., Ng, I.O., et al. 2002. Gene expression patterns in human liver cancers. Mol. Biol. Cell 13: 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl, F., Grahlmann, S., Beier, M., and Hoheisel, J.D. 2001. Manufacturing DNA microarrays of high spot homogeneity and reduced background signal. Nucleic Acids Res. 29: E38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger, I., Metzler, D., Schwarz, C., and Pääbo, S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70: 1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen, M.B. and Brown, P.O. 1999. DNA arrays for analysis of gene expression. Methods Enzymol. 303: 179–205. [DOI] [PubMed] [Google Scholar]
- Enard, W., Khaitovich, P., Klose, J., Zollner, S., Heissig, F., Giavalisco, P., Nieselt-Struwe, K., Muchmore, E., Varki, A., Ravid, R., et al. 2002. Intra- and interspecific variation in primate gene expression patterns. Science 296: 340–343. [DOI] [PubMed] [Google Scholar]
- Fortna, A., Kim, Y., MacLaren, E., Marshall, K., Hahn, G., Meltesen, L., Brenton, M., Hink, R., Burgers, S., Hernandez-Boussard, T., et al. 2004. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2: E207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad, Y., Man, O., Pääbo, S., and Lancet, D. 2003. Human specific loss of olfactory receptor genes. Proc. Natl. Acad. Sci. 100: 3324–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann, I., Zollner, S., Enard, W., Ebersberger, I., Nickel, B., and Pääbo, S. 2003. Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res. 13: 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman, M.W., Houck, M.L., Chemnick, L.G., Nagpal, S., Chawannakul, D., Sudano, D., Pike, B.L., Ho, V.V., Ryder, O.A., and Hacia, J.G. 2003. Comparative analysis of gene-expression patterns in human and African great ape cultured fibroblasts. Genome Res. 13: 1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. 2002. BLAT–the BLAST-like alignment tool. Genome Res. 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, M.K., Afshari, C.A., Bennett, L., Bushel, P., Martinez, J., Walker, N.J., and Churchill, G.A. 2002. Statistical analysis of a gene expression microarray experiment with replication. Stat. Sin. 12: 203–217. [Google Scholar]
- Khaitovich, P., Weiss, G., Lachmann, M., Hellmann, I., Enard, W., Muetzel, B., Wirkner, U., Ansorge, W., and Pääbo, S. 2004. A neutral model of transcriptome evolution. PLoS Biol. 2: E132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwene, P. 2002. disipyl.py—An object-oriented Python interface to DISLIN. http://biology.duke.edu/magwenelab/disipyl.html
- Meiklejohn, C.D., Parsch, J., Ranz, J.M., and Hartl, D.L. 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. 100: 9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal, S., Karaman, M.W., Timmerman, M.M., Ho, V.V., Pike, B.L., and Hacia, J.G. 2004. Improving the sensitivity and specificity of gene expression analysis in highly related organisms through the use of electronic masks. Nucleic Acids Res. 32: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin, S.V., Wayne, M.L., Harmon, K.L., and McIntyre, L.M. 2004. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol. Biol. Evol. 21: 1308–1317. [DOI] [PubMed] [Google Scholar]
- Perou, C.M., Sorlie, T., Eisen, M.B., van de Rijn, M., Jeffrey, S.S., Rees, C.A., Pollack, J.R., Ross, D.T., Johnsen, H., Akslen, L.A., et al. 2000. Molecular portraits of human breast tumours. Nature 406: 747–752. [DOI] [PubMed] [Google Scholar]
- Ranz, J.M., Castillo-Davis, C.I., Meiklejohn, C.D., and Hartl, D.L. 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300: 1742–1745. [DOI] [PubMed] [Google Scholar]
- Reinke, V. 2002. Functional exploration of the C. elegans genome using DNA microarrays. Nat. Genet. 32: 541–546. [DOI] [PubMed] [Google Scholar]
- Rifkin, S.A., Kim, J., and White, K.P. 2003. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat. Genet. 33: 138–144. [DOI] [PubMed] [Google Scholar]
- Saetre, P., Lindberg, J., Leonard, J.A., Olsson, K., Pettersson, U., Ellegren, H., Bergstrom, T.F., Vila, C., and Jazin, E. 2004. From wild wolf to domestic dog: Gene expression changes in the brain. Brain Res. Mol. Brain Res. 126: 198–206. [DOI] [PubMed] [Google Scholar]
- Smyth, G.K. and Speed, T. 2003. Normalization of cDNA microarray data. Methods 31: 265–273. [DOI] [PubMed] [Google Scholar]
- Team, R.D.C. 2003. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- van Rossum, G. 2003. python. http://www.python.org/.
- Wall, J.D., Frisse, L.A., Hudson, R.R., and Di Rienzo, A. 2003. Comparative linkage-disequilibrium analysis of the β-globin hotspot in primates. Am. J. Hum. Genet. 73: 1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, K.P. 2001. Functional genomics and the study of development, variation and evolution. Nat. Rev. Genet. 2: 528–537. [DOI] [PubMed] [Google Scholar]
- Yang, Y.H., Dudoit, S., Luu, P., and Speed, T.P. 2001. Normalization for cDNA microarray data. In Proceedings of SPIE. Microarrays: Optical technologies and informatics (eds. M.L. Bittner et al.), pp. 141–152. Society for Optical Engineering, San Jose, CA.
- Yang, Y.H., Dudoit, S., Luu, P., Lin, D.M., Peng, V., Ngai, J., and Speed, T.P. 2002. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web site references
- http://genome.ucsc.edu/; Human Genome.
- http://www.ncbi.nlm.nih.gov/RefSeq/; NCBI Sequence Database.