

Abstract
The availability of the full Drosophila genomic DNA sequence prompts the development of a method to efficiently obtain mutations in genes of interest identified by their sequence homologies or biochemically. To date, molecularly characterized mutations have been generated in around 6000 of the ∼15,000 annotated fly genes, of which around one-third are essential for viability. To obtain mutations in essential and nonessential genes of interest, we took a reverse genetics approach, based on the large-scale detection of point mutations by Cel-I-mediated heteroduplex cleavage. A library of genomic DNA from 2086 EMS-mutagenized lines was established. The library was screened for mutations in three genes. A total of 6.1 Mb were screened, and 44 hits were found in two different mutagenesis conditions. Optimal conditions yielded an average of one mutation every 156 kb. For an essential gene tested, five of 25 mutations turned out to cause lethality, confirming that EMS mutagenesis leads to high frequency of gene inactivation. We thereby established that Cel-I-mediated TILLING can be used to efficiently obtain mutations in genes of interest in Drosophila.
In Drosophila, there has not yet been a generally applicable method to generate mutant strains directly from DNA sequence data. Two approaches with inherent advantages and limitations (for review, see Bellen et al. 2004) are most often used to target mutations to a known gene in Drosophila, i.e., imprecise excision of transposable elements (Robertson et al. 1988) and homologous recombination (Rong et al. 2002). Various screens provided a collection of transposable element insertions in close to 6000 genes, a number which is soon approaching saturation, since these mobile elements show different degrees of target specificity (Bellen et al. 2004). These transposon insertions can be used as direct tools to affect gene function, or they can be utilized to generate deletions by imprecise excision. On the other hand, homologous recombination is not yet a routine approach in the Drosophila community. Ethyl methanesulfonate (EMS) has been extensively used as a chemical mutagen that results in high-point mutational densities. EMS shows a low-target specificity (it generates G:C-to-A:T transitions [Loechler et al. 1984; Snow et al. 1984]), and therefore the full genome is accessible to it. Flybase reports 3323 Drosophila genes that have been mutated by EMS. For 2601 of them, no related transposon insertion has yet been described. Furthermore, EMS point mutations can generate null alleles, but they can also lead to conditional alleles (e.g., thermosensitive alleles) or affect specific protein domains. Allelic series generated this way can be very useful to elucidate the function of a gene of interest.
In Arabidopsis, zebrafish, and rat (McCallum et al. 2000; Wienholds et al. 2003; Smits et al. 2004), TILLING (targeting induced local lesions in genomes) is used successfully for high-throughput screening of libraries mutagenized with EMS or N-ethyl-N-nitroso-urea (ENU) (for review, see Henikoff et al. 2004; Stemple 2004). The method detects single-nucleotide polymorphisms (SNP) in genes of interest that were amplified by PCR. TILLING has been optimized for high throughput by using Cel-I endonuclease (Colbert et al. 2001), which cleaves at mismatches within heteroduplexes formed between mutant and wild-type strands (Oleykowski et al. 1998). We took a Cel-I-based Tilling approach to identify mutations in large scale in Drosophila.
Results and Discussion
We established a library of fly lines containing isogenized second chromosomes carrying mutations caused by a 50-mM EMS treatment, which resulted in 2.24 lethal hits per chromosome (Table 1). This frequency of lethal hits is consistent with previous mutagenesis (Grigliatti 1986). To build these mutant balanced stocks (see Methods), males and females of the proper genotypes were selected by eliminating the other genotypes through expression of the apoptotic gene hid driven by the heat-shock promotor (Moore et al. 1998). This allowed us to generate a high number of balanced mutant lines without the necessity of collecting virgin females of the right genotype (see Methods).
Table 1.
Tilling results: Overview
| 125 mM EMS | 50 mM EMS | Total | |
|---|---|---|---|
| G0 mutagenized # | 10000 | 10000 | |
| G0 survivers # | ∼2500 (25%) | ∼9000 (90%) | |
| F1 #a | 700 | 3920 | |
| Number of linesb | 171 | 1915 | 2086 |
| F2 viable chromosomes (f)c | 9/171 (5.2%) | 41/384 (10.6%) | |
| Lethal hits per chrom. (λ)d | 2.94 | 2.24 | |
| Amplicons screenede | Sara-1, 2, 3, 4, 5 | Sara-1, 2, 3 (187) | |
| Alp23B-1, 2, 3 | Sara-4, 5 (1915) | ||
| Arf6-1, 2 | Alp23B-1, 3 (187) | ||
| Alp23B-2 (1915) | |||
| Arf6-1, 2 (187) | |||
| Candidates after tillingf (LI-COR hits) | 1 signal: 1 | 1 signal: 73 | |
| 2 signals: 13 | 2 signals: 33 | ||
| Total 14 | Total 106 | 120 | |
| Confirmed by sequencingf | 1 signal: 0 | 1 signal: 15 (20.5%) | |
| 2 signals: 12 (92.3%) | 2 signals: 17 (51.5%) | ||
| Total 12 | Total 32 | 44 | |
| Total screenedf | 1086.534 kb | 4977.702 kb | 6064.236 kb |
| Frequencyf | 1 in 90.5 kb | 1 in 155.6 kb | — |
| Missense SNPsf | 10 (83.3%) | 23 (71.9%) | 33 (75.0%) |
| Splice acceptorf | 0 | 1 (3.1%) | 1 (2.2%) |
| Silentf | 2 (16.7%) | 8 (25%) | 10 (22.7%) |
| C/G to T/A transitionf | 9 (75%) | 28 (87.5%) | 37 (84.1%) |
| Other mutationsf | 1 T/A to C/G | 1 T/A to G/C | 5 T/A to A/T |
| 2 T/A to A/T | 3 T/A to A/T | 1 T/A to C/G | |
| 1 T/A to G/C |
Total number of b p cn sca*/CyO, hb-lacZ F1 males.
Total number of F2 crosses that produced progeny.
Total number of lines where homozygous b p cn sca* adults survive. Denominator, total number of lines inspected.
Lethal hits per chromosome: λ = -Inf.
171 lines were screened for each amplicon in the case of the 125 mM mutagenesis. For the 50 mM mutagenesis, the number of lines screened for a particular amplicon is indicated in parenthesis.
Repeated signals due to overlap of the amplicons are only represented once.
To screen the library, we followed a “nested PCR/Cel-I strategy” similar to a recently reported Zebrafish TILLING approach (Wienholds et al. 2003; Fig. 1). In this approach, upon Cel-I heteroduplex cleavage, two fragments labeled with different fluorophores are generated with sizes that sum up to the full length of the amplicon, giving robustness to the analysis (Figs. 1, 2; see also Methods). In order to establish the genomic DNA preparation as well as the PCR and Cel-I digest conditions, we first applied the method to reidentify three known point mutations, i.e., Mad12, cniAR55, and tkv7 (Nellen et al. 1994; Roth et al. 1995; Sekelsky et al. 1995) (see Supplemental material). Then, we screened a total of 5.0 Mb in our library looking for SNPs in three genes of interest (Sara, Alp23B, and Arf6; Fig. 3, Table 1) and detected 32 novel SNPs, corresponding to a frequency of 1 in 155.6 kb.
Figure 1.

Cel-I-based Tilling strategy. We prepared a genomic DNA library from the mutagenized b pr cn sca/CyO, hb-lacZ fly lines and performed a three-step (1,2,3) nested PCR reaction (A) for a particular gene of interest. The third PCR reactions included the IRDye700 (red) and IRDye800 (blue) fluorophores bound to the forward and reverse primers, thereby differentially labeling the two ends of the amplicons (B). Then we denatured and reannealed the fragments to generate balancer/mutant heteroduplexes (C) that were digested by Cel-I (D). When a SNP is present, cleavage of the heteroduplexes generates two fragments labeled by IRDye700 and IRDye800, respectively, which can be detected upon denaturing polyacrylamide gel electrophoresis in a LI-COR setup. A typical LI-COR signal shows two fragments with complementary sizes that must sum up the original amplicon size. Subsequently, we sequenced the PCR products to confirm the molecular lesions (see Methods).
Figure 2.

Tilling screen. Example of a gel used for mutation detection within the Sara-4 amplicon in 96 balanced mutant lines; 700 (left) and 800 nm (right) emission images showing the fragments generated by Cel-I-based heteroduplex cleavage. (Arrowheads) Fragments generated by cleavage at 14 different silent background polymorphisms in the balancer that could be reidentified by sequencing; (circles) two novel mutations. Notice that both the polymorphisms and the novel mutations appear in both channels with complementary sizes. In the case of the second mutation, the longer fragment (800-nm channel) is less distinguishable (asterisk). After sequencing, we could, however, reconfirm that this signal corresponds to a bona fide mutation. Note also that the silent polymorphisms are present in all 96 lines, reflecting the fact that the balancer is itself isogenic. New mutations appear only in individual lines. Full-length product, 747 bp (arrow). Cleavage of the two new mutants yields 440 bp (700 nm) + 315 bp (800 nm) (lane 4) and 90 bp (700 nm) and 660 bp (800 nm) (lane 80).
Figure 3.

Mutation spectrum in three genes. Genomic organization of Sara, Alp23B, and Arf6. White boxes represent the ORFs. Black boxes below (Sara-1 to 5, Alp23B-1 to 3, and Arf6-1,2) correspond to the position of the amplicons considered in this study. Total DNA screened, number of SNPs, missense, nonsense, and lethal mutations are listed. Lines within the ORF and introns represent the position of the balancer polymorphisms.
In order to study the effect of the EMS dosage on the frequency of SNPs, we considered a 1.5-fold higher EMS concentration treatment (125 mM) and generated a pilot library of 171 lines. A concentration of 125 mM lead to an average number of lethal mutations per chromosome of 2.94 and a frequency of SNPs of 1 in 90.5 kb (12 SNPs in 1.1 Mb; see Table 1). Therefore, a 1.5-fold higher concentration of EMS causes around a 1.5-fold increase in the frequency of SNPs and lethal hits, implying that the effects of EMS increase linearly with the concentration. However, we could only obtain few lines (171) after feeding 125 mM EMS to 10000 G0 males (Table 1). In addition, the effects of 125 mM EMS on the health of the G0 and F1 progeny were very variable and typically more deleterious in subsequent repeats (data not shown). In contrast, the fertility and viability of the progeny from the 50 mM treated males were robust and reproducible (Table 1). Therefore, we decided to take 50 mM EMS as our standard condition.
We also studied the effect of the type of genomic DNA library (isogenic/balanced), the type of LI-COR signal (single/double channel), and the pooling factor on the SNP-detection efficiency (see Supplemental material). Based on these studies, we decided (1) to use genomic DNA libraries from the balanced fly stocks, without outcrossing the lines to the original isogenized chromosomes (see Methods), (2) to consider both single and double LI-COR signals (i.e., those signals in which only one or the two complementary amplicon fragments labeled by the different fluorophores are detected) and to subsequently reconfirm the mutants by sequencing (see Supplemental material for further details), and (3) to pool in the future the genomic DNA preparations in pairs before the PCR reactions.
The Cel-I-based screen of our mutant library detected SNPs with a relatively high frequency (Table 1). We wondered whether the SNP frequency is consistent with known frequencies of mutations previously reported for EMS.
Given (1) the average size of Drosophila ORFs (1.69 kb) (Misra et al. 2002), (2) the approximate number of genes in the second chromosome according to flybase (5065 genes; ORFsII), (3) the ratio of essential genes in Drosophila (Miklos and Rubin 1996) (around one in three), (4) the frequency of SNPs generated by EMS (1 in 156 kb; Table 1), and (5) the number of lethal hits per chromosome II in our mutagenesis (2.24 hits/chromosome; Table 1), we can estimate, (a) the average number of total SNPs in ORFs per mutagenized chromosome II in our mutagenesis
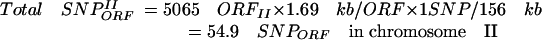 |
(b) among these 54.9 SNPs, the approximate number located in ORFs of essential genes (one-third) (Miklos and Rubin 1996)
 |
(c) the percentage of these 18.3 SNPs in ORFs that cause gene inactivation (ri), estimated after the frequency of hits per chromosome that lead to lethality in an essential gene (2.24 lethals/chromosome II; see Methods), and assuming that most inactivating base-pair changes affect ORFs
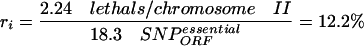 |
which corresponds to one inactivating hit in 8.2 SNPs (100/12.2).
This percentage of 12.2% inactivating mutations by EMS in the ORF is consistent with the expected average frequency of inactivating mutations predicted by the PARSESNP algorithm (http://www.proweb.org/parsesnp/) based on the codon composition of Drosophila genes, taking into account that EMS causes G:C to T:A transitions. This implies an average recovery of at least 2.8 inactivating mutations for the typical Drosophila gene with 1.69 kb ORF in our library of 2086 fly lines (1915 + 171 from the two EMS concentration experiments):
 |
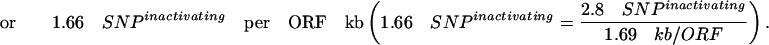 |
This corresponds to a frequency of 1.3 × 10–3 inactivating mutations in a typical Drosophila gene per haploid genome
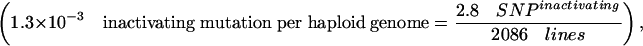 |
which is consistent with the average number of one inactivating hit per gene every 1000 lines upon EMS mutagenesis in our own experience and previous reports (Grigliatti 1986). Therefore, for a typical gene of interest, the probability of recovering at least one inactivating mutation in our library can be estimated, assuming a Poisson distribution, as
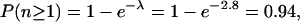 |
i.e., our library is 94% saturated. In order to reach 98% saturation, we are currently extending our library to 3000 lines.
We then studied whether the mutations obtained for Sara, Alp23B, and Arf6 represent loss of functions in the respective genes (Fig. 3). In the case of Sara, we previously knew that it is an essential gene (C. Bökel and M. González-Gaitán, unpubl.) whose mutations lead to embryonic lethality when removing the maternal and zygotic components. Five of 21 missense Sara mutations cause semilethality when the maternal and zygotic components are removed (see Methods). This represents an efficiency of 20% of the total number of 25 Sara SNPs after screening 3.2 Mb (Fig. 3). For Alp23B, a Drosophila TGF-β homolog, it was not known whether it is an essential gene. We identified 14 Alp23B SNPs (2.5 Mb screened), of which nine are missense alleles and one a nonsense mutation. The nonsense Alp23B mutation leads to a stop codon in the prodomain region, resulting in a truncated protein that lacks the mature, secreted TGF-β peptide. Therefore, this mutation represents a “signaling null” allele in Alp23B. Using deletions for the Alp23B genomic region (see Methods), we performed complementation tests with this null allele in order to address whether Alp23B is an essential gene. This mutation leads to 85%–100% lethality, depending on the genetic background, indicating that Alp23B is essential for viability (see Methods). In the case of Arf6, we determined that it is not a gene essential for viability, but for fertility in the males (M. González-Gaitán, unpubl.). After a pilot screening of 0.4 Mb, we identified five Arf6 SNPs, of which two were missense mutations and one was a mutation in a conserved residue of a splice acceptor. None of the missense mutations seems to overtly affect Arf6 function on male spermatogenesis. These results indicate that our Tilling approach can be used to obtain inactivating mutations in genes of interest. Of course, the missense mutations are candidates for hypomorphic or conditional alleles.
EMS is reported to alkylate guanine residues, producing O6-ethylguanine, which pairs with T, but not with C, leading to the replacement of the G/C base pair by A/T (Snow et al. 1984). This predicts a strong G/C-to-A/T bias in EMS-induced mutations as observed in numerous mutagenesis studies. Indeed, 37 (84%) of the 44 mutations observed correspond to G/C-to-A/T transitions, confirming this prediction (Table 1). Another five mutations are A/T-to-T/A transversions and two are other kinds of changes. This result is in contrast with a previous report where 100% were G/C-to-A/T transitions, based on 16 novel EMS mutations in Drosophila detected by DHPLC (Bentley et al. 2000). This could be due to a different sensitivity to detect the mutations by these two methods (Cel-I vs. DHPLC). Among the 44 mutations obtained, 33 represent missense mutations, one is nonsense, and 10 are silent or map in noncoding regions. Only one represented a mutation affecting a splice acceptor. This spectrum corresponds roughly to the frequency of mutations expected upon EMS treatment as predicted by an analysis with the PARSESNP algorithm on the amplicons considered (data not shown).
Being rapid and cost-effective, the generation of mutants by Tilling will, in the future, be a routine approach in reverse genetics for the fly community. Indeed, we are currently generating mutant libraries for the X and third chromosome. Much like the Arabidopsis field, our results enable fly researchers to organize the generation of mutants by Tilling screens as an efficient resource for the entire community.
Methods
Isogenization
The b pr cn sca chromosome to be mutagenized was isogenized according to standard procedures. In short, a single male was crossed to CyO/In(2L)nocSco females and 10 single F1 b pr cn sca/CyO males were crossed independently to CyO/Df(2L) females to amplify different individual b pr cn sca chromosomes. F3 b pr cn sca/CyO males and virgin females were crossed to generate 10 stocks, which were scored for viability of the homozygous bprcn sca chromosome.
Mutagenesis
Mutagenesis was performed according to standard protocols (Grigliatti 1986). Isogenized b pr cn sca males (100 per vial) were kept at 25°C overnight in vials containing a paper tissue soaked in water and then transferred to vials containing a paper tissue soaked in 10 mL of a 40% sucrose solution containing 50 or 125 mM EMS (Sigma), where they were kept for 24 h. Afterward, they were transferred into empty vials to let them recover and defecate for 30 min, and finally transferred to food vials in groups of 30 together with 30 CyO, hb-lacZ/Sp, hs-hid virgin females (G0 cross) and maintained at 25°C. After 4 d, all the G0 males were discarded to avoid mutation clustering, and the G0 females transferred to a new food vial. The F1 b p cn sca/CyO, hb-lacZ males containing the mutagenized chromosome were collected and crossed individually to three CyO, hb-lacZ/Sp, hs-hid virgin females (F1 cross) at room temperature. After 4 d, the crosses were heat shocked by incubating the vials in a water bath at 38.3°C for 75 min. If only a few dead larvae were observed in a vial, the heat treatment was prolonged for 10 more minutes in the water bath. Individual mutant fly stocks are maintained at 18°C, and transferred to a new food vial once a month. A total of 25 flies from each fly stock were frozen to be processed for genomic DNA extraction.
To determine the number of lethal hits per chromosome II, the frequency of stocks containing a homozygous viable b pr cn sca* chromosome (f) was determined. From the null term of the Poisson distribution, f = P(0) = e–λ, where λ is the hit frequency per chromosome. Therefore, λ = –lnf.
To compare the detection efficiency in flies containing and not containing the polymorphisms of the balancer, b p cn sca*/CyO, hb-lacZ males from each of the 125 mM EMS fly lines (171 lines) were crossed to virgins from the original bprcnsca isogenic stock and b pr cn sca*/b pr cn sca F1 flies were selected to extract genomic DNA and prepare an “isogenic library” as described below.
Drosophila strains and lethality tests
The alleles used in this work are described in reference (Lindsley and Zimm 1992) and flybase (http://flybase.bio.indiana.edu/). The Sara– allele is a deletion caused by imprecise excision of a transposable element.
To test the lethality of the Sara alleles, Sara–/Sara– virgins were crossed to Sarax/CyO, hb-lacZ males containing the allele of interest. The number of Sara–/Sarax versus Sara–/CyO, hb-lacZ flies was recorded to assess the lethality/semilethality associated with the allele. To test the lethality of the Alp23B alleles, complementation tests were performed using two deletions of the Alp23B genomic region, i.e., Df(2)C144 (Lindsley and Zimm 1992) and a deletion generated by imprecise excision of a P-element (Df(2)) which uncovers Alp23B-flanking genes. The Alp23B stop codon mutation leads to lethality (0 Df(2)/b pr cn sca* vs. 59 CyO/b pr cn sca* progeny) or semilethality (five Df(2)C144/b pr cn sca* vs. 36 CyO/b pr cn sca*) depending on the background. Semilethality in the case of the Df(2)C144 might be due to a known dpp second hit on the Defeciency chromosome, which might behave as a dominant suppressor mutation (M. González-Gaitán and M. Fürthauer, unpubl.).
The Arf6 alleles were tested by monitoring male sterility in a complementation test against Arf6 null alleles generated in the lab.
Genomic DNA preparation and construction of the Drosophila DNA library
A total of 10 (Mad12, cniAR55, and tkv7) or 25 flies (50 mM, 125 mM EMS library) were anesthetized, collected in Eppendorf tubes and frozen at –80°C for 20 min. Flies were ground in 100 (10 flies), or 200 μL (25 flies) of lysis buffer (100 mM Tris-Hcl at pH 7.5, 100 mM EDTA, 100 mM NaCl, 0.5% SDS) with a disposable tissue grinder (Eppendorf) and incubated at 65°C for 30 min. A total of 200 (10) or 400 (25) μL of 1.4 M KAc, 4.3 M LiCl solution was added, followed by incubation on ice for 10 min. Cell debris was precipitated by spinning at 13,000 rpm for 15 min at room temperature. Supernatant (0.5 mL) was transferred to either Eppendorf tubes (Mad12, cniAR55, and tkv7 and primary hits for genotyping) or deep 96-well plates (1.1 mL cavities, 50 mM, 125 mM EMS library). A total of 200 (10) or 400 (25) μL of Isopropanol was added, mixed, and DNA was precipitated by centrifugation at 3671g for 1 h at room temperature. The DNA pellets were washed with 70% ethanol, dried, and resuspended in 50 (10 flies) or 150 μL (25 flies) TE (pH 7.5). Aliquots of genomic DNA were separated on 0.7% agarose gels to check for yield and variability of the DNA preparation. Genomic DNA was diluted 1:50 in ultrapure H2O, and 5-μL aliquots were arrayed in 384-well PCR plates using a Tecan Genesis workstation 200. The plates were sealed with aluminum foil tape (3MM) and stored at –20°C. A copy of the original DNA preparation (masterplate) was stored independently at –20°C.
Primer design
Nested primer sets that span the respective genomic region were designed automatically by using a modified PRIMER3-based application (http://primers.niob.knaw.nl/; Wienholds et al. 2003). This application searches for gene-specific outer primers, as well as corresponding inner primers with universal 5′ overhangs (M13 forward: TGTAAAACGACGGCCAGT, and M13 TILLING: AGGA AACAGCTATGACCAT). A third set of primers comprises the M13-forward and M13-TILLING sequence, and each primer is linked to IRDye700 and IRDye800 dyes, respectively.
In total, 10 sets of primers were automatically designed against three different genes (Sara, Alp23B, Arf6) to screen the Drosophila TILLING library for new mutations (Fig. 3; Table 1; Supplemental Table 2).
Cel-I-mediated screen for mutations
DNA of interest was amplified by a nested PCR approach as described in detail in Wienholds et al. (2003). The first PCR was performed with gene-specific primers. The first PCR product served as a template for a reaction using a mix of gene-specific inner primers that are linked to universal 5′ overhangs (second PCR), as well as IRDye-labeled primers as described above (third PCR). The resulting labeled PCR product was denatured and reannealed to allow heteroduplex formation, digested by Cel-I-mediated cleavage, and fragments were visualized on LI-COR 4200 DNA sequencers.
Pipeting steps were performed by a Tecan Genesis workstation 200 or manually with multichannel pipettes (Eppendorf). PCRs were amplified with a Multicycler PTC 225 Tetrad with four MotoBlocks384 (MJ Research) that was integrated into the Tecan Genesis workstation, or alternatively with a GeneAmp9700 (Applied Biosystems).
Cycling conditions for the first PCR were as follows: 94°C for 1 min, 35 cycles of 94°C for 20 sec, 57°C for 30 sec, 72°C for 1 min, followed by a final extension for 3 min at 72°C (volume: 10 μL). The second/third, nested PCR was done in a final volume of 10 μL (cycling conditions: 94°C for 1 min, 30 cycles of 94°C for 20 sec, 57°C for 30 sec, 72°C for 1 min, and a final extension for 3 min at 72°C). Heteroduplex formation was performed by denaturing and slow reannealing of PCR fragments (conditions: 95°C for 3 min, 30 cycles of 95°C for 20 sec with a decrement of 0.5°C per cycle, 50 cycles of 80°C for 1 min with a decrement of 0.5°C per cycle, 55°C for 20 min, 20 cycles of 55°C for 1 min with a decrement of 0.5°C per cycle, and finally, 40 cycles of 45°C for 30 sec with a decrement of 0.5°C per cycle). A total of 5 μL of the processed PCR fragments were digested with a 1:1000 dilution of Cel-I enzyme. Cel-I was isolated as described (Wienholds et al. 2003), tested for activity, and titrated by a dilution series on proof of principle samples. The Cel-I digest was stopped by adding 5 μL of 75 mM EDTA to each sample. Sample purification was done using Sephadex G50 (medium coarse) columns, and samples were concentrated to a final volume of 2 μL for 35 min at 85°C. In total, 0.5 μL were loaded on 5.5% polyacrylamide gels (25 cm) on LI-COR 4200 DNA sequencers. The LI-COR slab gel sequencers were used to perform high-resolution DNA electrophoresis in combination with two-channel infrared detection of the fluorescently labeled (700 and 800 nm emission) DNA fragments. Potential mutations were detected and scored on gel pictures that were produced from raw TIFF images using Adobe Photoshop.
DNA sequencing
Presumptive SNPs that were detected by Cel-I-mediated digestion were reamplified from the respective first PCR product for verification. The second PCR was done in the same way as described before, but without fluorescently labeled primers. A total of 8 μL of the second PCR product was digested with 0.3 U Exonuclease 1 and 1.6 U Shrimp Alkaline Phosphatase for 15 min at 37°C, followed by 15 min at 72°C to inactivate the enzymes. A total of 0.5–1 μL (about 50 ng of DNA) of the enzyme-purified PCR products were used as a template for sequencing. Sequencing was performed with universal sequencing primers (M13 forward, M13 TILLING) or gene-specific internal primers to cover complete amplicons (sequencing mix: 0.5 pmol primer, 1 μL BigDye terminator version 3.1 Sequencing kit, 2 μL 5× Sequencing buffer [Applied Biosystems] in a total volume of 10 μL cycling conditions, i.e., 96°C for 1 min, 30 cycles of 96°C for 10 sec, 52°C for 5 sec, and 60°C for 3 min). After purification by ethanol precipitation, sequencing products were analyzed on a 3730 DNA Analyzer (Applied Biosystems) in the DNA sequencing facility at the MPI-CBG. Eventually, sequence traces were screened for heterozygous SNPs using Polyphred (http://droog.mbt.washington.edu/PolyPhred.html).
To reconfirm presumptive mutants, genomic DNA was isolated from the corresponding fly stock as described before (10 flies preparation), diluted 1:20 in ultra pure water, and used as template for the nested PCR approach. Verification of hits was always done by resequencing to sort out false-positive hits that resulted from presumptive Taq-polymerase errors.
Acknowledgments
We thank A. Hyman and C.-P. Heisenberg for their support in building up the TILLING project at the MPI-CBG, E. Cuppen for sharing protocols and reagents and supporting our TILLING project, B. Habermann and A. Henschel for setting up an internal Polyphred Server and helping us with the bioinformatics, D. Drechsel for the purification of the Cel-I enzyme, G. Wiebe and the sequencing facility at the MPI-CBG for optimization of sequencing conditions, S. Kutscherjawy and U. Yalzin for help with the construction of the fly library, J. Wagner and H. Grabner for setting up the robotics and H. Bellen, K. Klämbt, A. Oates, and C. González for critical reading of the manuscript. Work by S.W., C.S., S.B., and M.B. is supported by the Max-Planck Society, the European Union (ZF Models), and the University of Technology, Dresden, and A.S., D.B., C.B., M.F., A.K., L.C., and M.G.G. by the Max-Planck Society, DFG, HFSP, and an EMBO fellowship.
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3721805.
References
- Bellen, H.J., Levis, R.W., Liao, G., He, Y., Carlson, J.W., Tsang, G., Evans-Holm, M., Hiesinger, P.R., Schulze, K.L., Rubin, G.M., et al. 2004. The BDGP gene disruption project: Single transposon insertions associated with 40% of Drosophila genes. Genetics 167: 761–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, A., MacLennan, B., Calvo, J., and Dearolf, C.R. 2000. Targeted recovery of mutations in Drosophila. Genetics 156: 1169–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert, T., Till, B.J., Tompa, R., Reynolds, S., Steine, M.N., Yeung, A.T., McCallum, C.M., Comai, L., and Henikoff, S. 2001. High-throughput screening for induced point mutations. Plant Physiol. 126: 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigliatti, T. 1986. Mutagenesis. In Drosophila. A practical approach (ed. D.B. Roberts), pp. 39–59. IRL Press Ltd., Oxford, UK.
- Henikoff, S., Till, B.J., and Comai, L. 2004. TILLING. Traditional mutagenesis meets functional genomics. Plant Physiol. 135: 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley, D.L. and Zimm, G.G. 1992. The genome of Drosophila. Academic Press, London, UK.
- Loechler, E.L., Green, C.L., and Essigmann, J.M. 1984. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. 81: 6271–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum, C.M., Comai, L., Greene, E.A., and Henikoff, S. 2000. Targeted screening for induced mutations. Nat. Biotechnol. 18: 455–457. [DOI] [PubMed] [Google Scholar]
- Miklos, G.L. and Rubin, G.M. 1996. The role of the genome project in determining gene function: Insights from model organisms. Cell 86: 521–529. [DOI] [PubMed] [Google Scholar]
- Misra, S., Crosby, M.A., Mungall, C.J., Matthews, B.B., Campbell, K.S., Hradecky, P., Huang, Y., Kaminker, J.S., Millburn, G.H., Prochnik, S.E., et al. 2002. Annotation of the Drosophila melanogaster euchromatic genome: A systematic review. Genome Biol. 3: 0083.0081–0083.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, L.A., Broihier, H.T., Van Doren, M., Lunsford, L.B., and Lehmann, R. 1998. Identification of genes controlling germ cell migration and embryonic gonad formation in Drosophila. Development 125: 667–678. [DOI] [PubMed] [Google Scholar]
- Nellen, D., Affolter, M., and Basler, K. 1994. Receptor serine/threonine kinases implicated in the control of Drosophila body pattern by decapentaplegic. Cell 78: 225–237. [DOI] [PubMed] [Google Scholar]
- Oleykowski, C.A., Bronson Mullins, C.R., Godwin, A.K., and Yeung, A.T. 1998. Mutation detection using a novel plant endonuclease. Nucleic Acids Res. 26: 4597–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, H.M., Preston, C.R., Phillis, R.W., Johnson-Schlitz, D.M., Benz, W.K., and Engels, W.R. 1988. A stable genomic source of P element transposase in Drosophila melanogaster. Genetics 118: 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong, Y.S., Titen, S.W., Xie, H.B., Golic, M.M., Bastiani, M., Bandyopadhyay, P., Olivera, B.M., Brodsky, M., Rubin, G.M., and Golic, K.G. 2002. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes & Dev. 16: 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, S., Neuman-Silberberg, F.S., Barcelo, G., and Schupbach, T. 1995. cornichon and the EGF receptor signaling process are necessary for both anterior-posterior and dorsal-ventral pattern formation in Drosophila. Cell 81: 967–978. [DOI] [PubMed] [Google Scholar]
- Sekelsky, J.J., Newfeld, S.J., Raftery, L.A., Chartoff, E.H., and Gelbart, W.M. 1995. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 139: 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits, B.M., Muddek, J., Plasterk, R.H., and Cuppen, E. 2004. Target-selected mutagenesis of the rat. Genomics 83: 332–334. [DOI] [PubMed] [Google Scholar]
- Snow, E.T., Foote, R.S., and Mitra, S. 1984. Base-pairing properties of O6-methylguanine in template DNA during in vitro DNA replication. J. Biol. Chem. 259: 8095–8100. [PubMed] [Google Scholar]
- Stemple, D.L. 2004. TILLING—a high-throughput harvest for functional genomics. Nat. Rev. Genet. 5: 145–150. [DOI] [PubMed] [Google Scholar]
- Wienholds, E., van Eeden, F., Kosters, M., Mudde, J., Plasterk, R.H., and Cuppen, E. 2003. Efficient target-selected mutagenesis in zebrafish. Genome Res. 13: 2700–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web site references
- http://droog.mbt.washington.edu/PolyPhred.html; polyphred.
- http://flybase.bio.indiana.edu/; flybase.
- http://www.proweb.org/parsesnp/; PARSESNP.
- http://primers.niob.knaw.nl/;