

Abstract
Increasing evidence suggests that microbial species have a strong within species genetic heterogeneity. This can be problematic for the analysis of prokaryote genomes, which commonly relies on a reference genome to guide the assembly process. Differences between reference and sample genomes will therefore introduce errors in final assembly, jeopardizing the detection from structural variations to point mutations—critical for genomic surveillance of antibiotic resistance. Here we present Hound, a pipeline that integrates publicly available tools to assemble prokaryote genomes de novo, detect user-given genes by similarity to report mutations found in the coding sequence, promoter, as well as relative gene copy number within the assembly. Importantly, Hound can use the query sequence as a guide to merge contigs, and reconstruct genes that were fragmented by the assembler. To showcase Hound, we screened through 5032 bacterial whole-genome sequences isolated from farmed animals and human infections, using the amino acid sequence encoded by blaTEM-1, to detect and predict resistance to amoxicillin/clavulanate which is driven by over-expression of this gene. We believe this tool can facilitate the analysis of prokaryote species that currently lack a reference genome, and can be scaled either up to build automated systems for genomic surveillance or down to integrate into antibiotic susceptibility point-of-care diagnostics.
Keywords: genomic surveillance, antibiotic resistance, reference-free
Graphical Abstract
Graphical Abstract.

INTRODUCTION
The advent of affordable genome sequencing has exposed the wide genetic heterogeneity that exists within bacterial species [1]. With genome sizes that range between 2.69 and 2.92 Mb in Staphylococcus aureus, or between 4.66 and 5.30 Mb for Escherichia coli, it is not surprising that some begin to question the notion of species [2, 3] or even clone [4] in prokaryotes. This heterogeneity led to the concept of pan-genomes [5], but it also exposes another, more technical problem: How to study the genomes of prokaryotes without masking this genetic diversity?
Raw sequencing data are typically mapped onto a high-quality reference—whose sequence is known and resolved (i.e. circularized) [6, 7]—or databases containing them [8], to study the genetics of organisms from viruses [9] to vertebrates [10] or plants [11]. The use of reference-mapped assemblies is used in comparative genomics [12], clinical microbiology [13], public health [14, 15], and even to inform policy through the detection of specific mutations or phylogenetic analyses [16, 17]. Now, given the further reduction in sequencing costs, reference-mapped assemblies are increasingly used to predict antibiotic susceptibility in bacteria from clinical samples [18, 19]. This is driven by the ability of genomics to screen resistance to multiple antibiotics simultaneously, more than is possible with current phenotypic antibiotic sensitivity tests, improving antibiotic stewardship and patient care. But using genomics data for this can be problematic, given the limitations of these type of assemblies to detect antibiotic-resistance (ABR) genes. On one hand, reads that cannot be mapped onto the reference genome, say, because they are plasmid-borne and not part of the chromosome, are excluded from the assembly. And this loss of data hinders the detection of ABR genes [19, 20]. On the other hand, the availability of reference genomes is skewed toward the most common pathogens [13, 16], further limiting the study of rarer pathogens [21]. Consequently, the scope of tools like ResFinder [22], STARR [23], ARG-ANNOT [24], RAST [25] or ABRIcate (https://github.com/tseemann/abricate) can be limited to predict antibiotic susceptibility. Particularly, because they rely on reference genomes to report the presence—or not—of ABR genes along with mutations in the coding sequence known to be associated with specific resistant phenotypes. As we show below, mapping sequencing data onto a reference genome can artificially modify the assembly [20]. This approach is not only limited for the study of other pathogens or the finding of novel, undocumented mutations associated with important phenotypes like antibiotic resistance; but of species that may have other biological or ecological importance where reference genomes and tools are scarce [26].
Here we sought to build a pipeline to analyze bacterial genomes assembled de novo, without using a reference to guide the assembly process. De novo assemblies lack most of the limitations mentioned above, but can also introduce others. Particularly, the fragmentation of genes—whose sequences are split across multiple contigs by the assembler [19]. Hound implements an algorithm to re-purpose a query sequence as a local reference to detect and merge the relevant contigs, so that its sequence can be reconstructed unambiguously. Another issue we sought to address is that antibiotic resistance is not only caused by the presence of specific genes. The over-expression of antibiotic resistance genes, whether through specific mutations in the promoter or increase in relative gene copy number, can dramatically alter the antibiotic resistance phenotype present. For example, amoxicillin-resistant E. coli are most commonly resistant due to the production the TEM 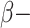 lactamase enzyme, encoded by the mobile gene blaTEM-1 [27]. Amoxicillin given in combination with clavulanate will kill amoxicillin-resistant E. coli because clavulanate inhibits TEM-1—as well as other related enzymes—explaining why this combination has been widely used in human [28, 29] and veterinary medicine [30]. However, E. coli can become resistant to amoxicillin-clavulanate by over-producing TEM-1 due to promoter mutations [28] or increased gene copy number [28, 31]. Therefore, we built into Hound the capability to retrieve sequences beyond a gene’s coding sequence and include the promoter, as well as the relative gene copy number, to allow the detection of such variants with our pipeline.
lactamase enzyme, encoded by the mobile gene blaTEM-1 [27]. Amoxicillin given in combination with clavulanate will kill amoxicillin-resistant E. coli because clavulanate inhibits TEM-1—as well as other related enzymes—explaining why this combination has been widely used in human [28, 29] and veterinary medicine [30]. However, E. coli can become resistant to amoxicillin-clavulanate by over-producing TEM-1 due to promoter mutations [28] or increased gene copy number [28, 31]. Therefore, we built into Hound the capability to retrieve sequences beyond a gene’s coding sequence and include the promoter, as well as the relative gene copy number, to allow the detection of such variants with our pipeline.
RESULTS
Pipeline overview
Hound integrates tools widely used to assemble Nanopore and Illumina reads de novo, and screen the resulting assemblies for user-given query sequences, into a single tool. Hound supports nucleotide and amino acid sequences, but we suggest the query to be an amino acid sequence where possible to avoid variations introduced by synonymous mutations. Our pipeline is modular as Figure 1 illustrates to allow performing only a subset of the tasks, and relies on SPAdes [32] as its backend assembler due to its combination of speed, accuracy and support for sequencing data from multiple platforms. Once assembled, Hound can optionally map the raw reads onto the assembly using Burrows-Wheeler aligner [33] to compute the coverage depth with SAMtools [34]. Following the assembly step Hound will search for the query sequences in the assembly by similarity, using the BLAST [35] algorithm, before undergoing downstream processing. At this point, Hound will integrate the data to estimate the relative gene copy number, retrieve sequences upstream of the coding sequence in the assembly, align and produce a phylogeny of all retrieved sequences, and detail the mutations with respect the query sequence. A list of the flags available in Hound, and their functionality, can be found in Table 1.
Figure 1.

Description of the Hound pipeline for the analysis of bacterial genomes assembled de novo. While Hound can be run in a single step, the user is given three steps of granularity. 1) The first step is to assemble the quality-filtered FASTQ files and depth of coverage data generated. During this step, each assembly is converted into a BLAST database to facilitate downstream analyses. Note that once assembled, there is no need to repeat the step—whence the choice of granularity. 2) In the second step, Hound will search by similarity any user-given target(s) in the assemblies generated in 1). The target sequence(s) must be in FASTA format and preferably be the amino acid sequence to avoid variations introduced by synonymous mutations. Files with multiple entries are supported. To compute the relative gene copy number (RCN), Hound will use a number of house-keeping genes (HK) to compute a baseline depth of coverage. We used four but there is no limit in number of house-keeping genes. If the identity of the sequence found in the assembly with respect to the target, translated as necessary, is above 90%, and the sequence is fragmented, Hound will use the user-given sequence as a guide to sort, de-duplicate, and merge the relevant contigs so the resulting translation is the query sequence used. All sequences are then aligned to facilitate the screening of mutations, insertions or deletions with respect to the target sequence. 3) The last step is optional, and invokes Hound to summarize all the findings from 2) into one spreadsheet for record-keeping.
Table 1.
Options available in Hound, as shown by the command HoundAnalyser–help
| Flag | Description |
|---|---|
| -h, —help | Show this help message and exit |
| —preprocess FILE DIRNAME | |
| Unzip Illumina reads and create appropriate directory structure. It requires a name to create destination directory. REQUIRED unless —project is given. | |
| —project DIR | Directory where FASTQ files can be found. It can be a directory of directories if FASTQ files are contained in a ‘reads’ directory. Maximum directory depth is 2. REQUIRED unless —preprocess is given. |
| —assemble | Assemble reads. Requires —project. |
| —reference FILE | Reference genome in FASTA format. Requires —assemble. |
| —de-novo | Assemble reads de novo. Used to assemble genomes and specify assembly type for data analysis. Requires —assemble. |
| —coverage | Compute coverage depth to estimate gene copy number. Can be used to assemble genomes or include coverage depth in the data analysis. Requires —hk-genes. |
| —hk-genes FILE | List of Multilocus sequence typing (MLST) genes, or other reference genes, in FASTA format to compute baseline coverage depth. Requires —coverage. |
| —genes FILE | List of genes to be found, in FASTA format. Requires —identity and —prefix. |
| —genes-dir DIR | Directory containing genes of interest in FASTA format. Requires —summary. |
| —nucl | Use nucleotide sequences for the search. Requires —genes. |
| —prefix NAME | Label added to all output files. Required to do multiple searches with the same assemblies. |
| —identity NUM NUM | Identity threshold required to shortlist sequences found. Two floats (min identity, max identity) between 0 and 1 are required. Requires—genes. |
| —promoter | Isolate the promoter region of the target gene(s) sequences found and ignore coding sequences. Requires —cutoff. |
| —cutoff NUM | Length of the promoter in nucleotides. Requires —promoter. |
| —phylo | Align sequences of promoter/coding sequences found, and generate the corresponding phylogeny. |
| —phylo-thres NUM | Remove sequences that are a fraction of the total size of alignment. Used to improve quality alignment. Requires a number between 0 and 1 (defaults to 0.5). |
| —plot FILE | Generate plot from the multiple alignment of sequences found, and save as FILE. |
| —roi FILE | Sequences of interest to look for in the gene(s) found, in FASTA format. Requires—plot. |
| —summary FILE | Save Hound analysis as a spreadsheet. Requires—project. |
| —labels FILE | XLS file containing assembly name (col 1), and assembly type (col 6) to label phylogeny leafs (defaults to assembly name). Requires —plot. |
| —force | Force re-generation of phylogeny and/or plot even if they already exist. |
The main drawback of de novo assemblies [19] is the fragmentation of genes, with subsets of their sequence being split across two or more contigs by the assembler. In this case, if the identity of the sequences found by Hound are at least 90% ($MIN_ID 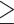 0.9) to the user-given query, and the contigs harboring subsets of the query have overlapping common sequences, Hound will shortlist the contigs for its contig-merging routine. Here, after sorting them first by identity and length, Hound will iteratively run pairwise alignments between the first pair with overlapping coordinates, discard one of the two overlapping sequences to avoid introducing duplications, and compare the resulting sequence to the user-given query. Hound will process the next contig in the list and add it to the previous merged sequence until the reconstructed sequence has, at least, equal length to the query which will have at least $MIN_ID identity. Note all these contigs will have overlapping coordinates that are located at the boundaries of the contigs, thus, only genes truncated by the assembler and not by insertion sequences—mobile elements—will be included in this analysis.
0.9) to the user-given query, and the contigs harboring subsets of the query have overlapping common sequences, Hound will shortlist the contigs for its contig-merging routine. Here, after sorting them first by identity and length, Hound will iteratively run pairwise alignments between the first pair with overlapping coordinates, discard one of the two overlapping sequences to avoid introducing duplications, and compare the resulting sequence to the user-given query. Hound will process the next contig in the list and add it to the previous merged sequence until the reconstructed sequence has, at least, equal length to the query which will have at least $MIN_ID identity. Note all these contigs will have overlapping coordinates that are located at the boundaries of the contigs, thus, only genes truncated by the assembler and not by insertion sequences—mobile elements—will be included in this analysis.
At this point, once the query sequence can be reconstructed, the assembly is re-written with the new contig name being a concatenation of all the founding contigs (i.e. >NODES_1 + 43 + 24). If coverage data exists, the coordinates used earlier by the merging routine are applied to this dataset to preserve coverage in the new, merged contig.
Reference-mapped versus de novo assemblies
The first step in screening recently acquired genomes of our 5032 bacterial isolates from farmed animals and human clinical samples, purified prior to sequencing, is the assembly de novo of the reads. A first look at the output of the pipeline reveals the assemblies seldom have the same size (Figure 2A). Now, while E. coli is by far the most abundant species in our dataset, as identified by Kraken 2 [36], the dataset also contains isolates of Klebsiella pneumoniae (15.7% of the total), and one isolate of Pseudomonas aeruginosa (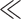 1%) and Salmonella enterica (
1%) and Salmonella enterica (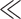 1%) among the human clinical isolates. This means multiple species can be monitored depending on each use case.
1%) among the human clinical isolates. This means multiple species can be monitored depending on each use case.
Figure 2.
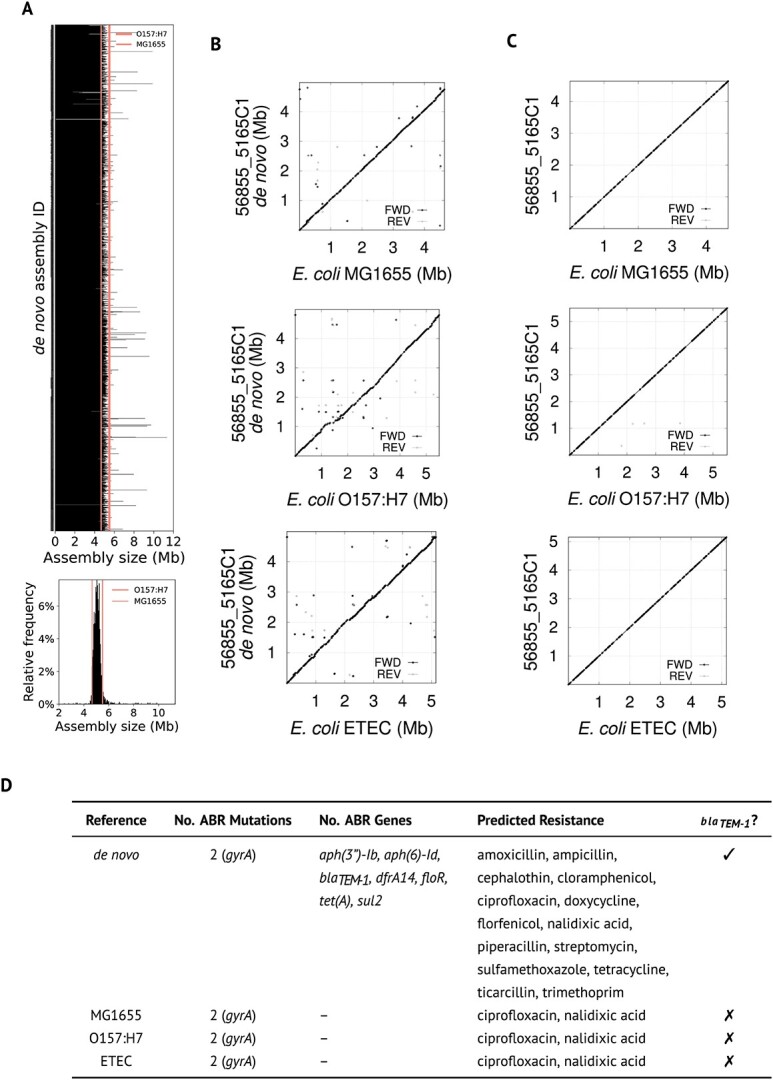
Variability of within species genome size is not captured by the canonical use of reference genomes. (A) Histogram (top) and distribution (bottom) of genome sizes across 3562 farm and clinical E. coli isolates. The canonical size of the reference wild-type (K-12 substr. MG1655, genome GCF_000005845.2 in the NCBI) and Shiga toxin-producing strain (0157:H7 str. Sakai, genome GCA_000008865.2 in the NCBI) are marked at ~4.64 Mb and 5.59 Mb by vertical lines. (B) and (C) Dotplots of one representative isolate to visualize the genome–genome sequence alignment between de novo (B) or reference-mapped (C) assemblies and three different known genomes: MG1655, O157:H7, and enterotoxigenic E. coli (ETEC). Sequences present in these references, but not in out isolate, are noted by a horizontal gap in the dotplot, whereas the converse is noted by a vertical gap. Note that in C) the reported assembly size of our isolate, in the y-axis, varies depending on the choice of reference—size is constant in those genomes assembled de novo. (D) Exemplar report from ResFinder when used against the isolate 56855_5165C1 from B to C assembled de novo, and mapped to different references. This tool reports documented ABR mutations and genes, as well as predicting resistance to certain antibiotics as reported by the literature. Note that, for the de novo assembly, ResFinder reports resistance to ampicillin and amoxicillin. The number of copies of blaTEM-1 reported by Hound is 2–3, thus, the isolate would also be resistant to amoxicillin/clavulanate [28] which is missed by ResFinder. The full report can be found in the supplementary data.
When we removed all non-E. coli from the dataset and compared the assembly sizes, we found the variation with respect to the available reference genomes—K-12 MG1655 and 0157:h7 Sakai—to be substantial as the histogram in Figure 2A shows, with most assemblies having sizes in-between these references. These two genomes are the only ones validated by the National Center for Biotechnology Information (NCBI) and flagged as reference genomes accordingly. The variability that we observed in genome sizes is consistent with the aforementioned notion of pan-genomes. Hound supports the assembly of reads based on a reference genome. So, next, we compared reference-mapped assemblies using to different E. coli genomes using Hound with flags—assemble—reference $REF_GENOME with their respective reference—beyond the above MG1655 and 0157:h7 genomes we also used that for enterotoxigenic E. coli (ETEC)—as well as the de novo assemblies, using pairwise alignments with MUMmer [37]. Note that reference-based assemblies rely on SAMtools and the Burrows-Wheeler aligner, which are implemented in Hound, sharing the downstream analysis pipeline.
When comparing the de novo assemblies to the references, this alignment revealed signatures of small deletions, insertions and repeated regions that were different with each reference used (Figure 2B). However, the signatures vanished when we used reference-mapped assemblies (Figure 2C). This means the use of references discards or includes details from the final assembly that can ultimately alter its size—unique and robust when assembled de novo—and help explain the inconsistent results [38] that occur when comparing different tools.
Monitoring blaTEM-1 in farm and clinical isolates
We used this tool to screen through our 5032 isolate whole-genome sequencing datasets, from farmed animals (2494) and human clinical samples (2538), to detect blaTEM-1 (including non-synonymous variants), its promoter region, and relative copy number (Figure 3). Among the output files generated by Hound, there is a figure with the multiple alignment and phylogeny of the sequences, containing the aforementioned metadata depending upon the flags provided, that can be produced with the flag—plot $FILENAME. This is an exemplar of how this tool can improve the prediction and, therefore, the surveillance of antibiotic resistance with complex phenotypes that are notoriously difficult to predict from mutations in the coding sequence alone.
Figure 3.
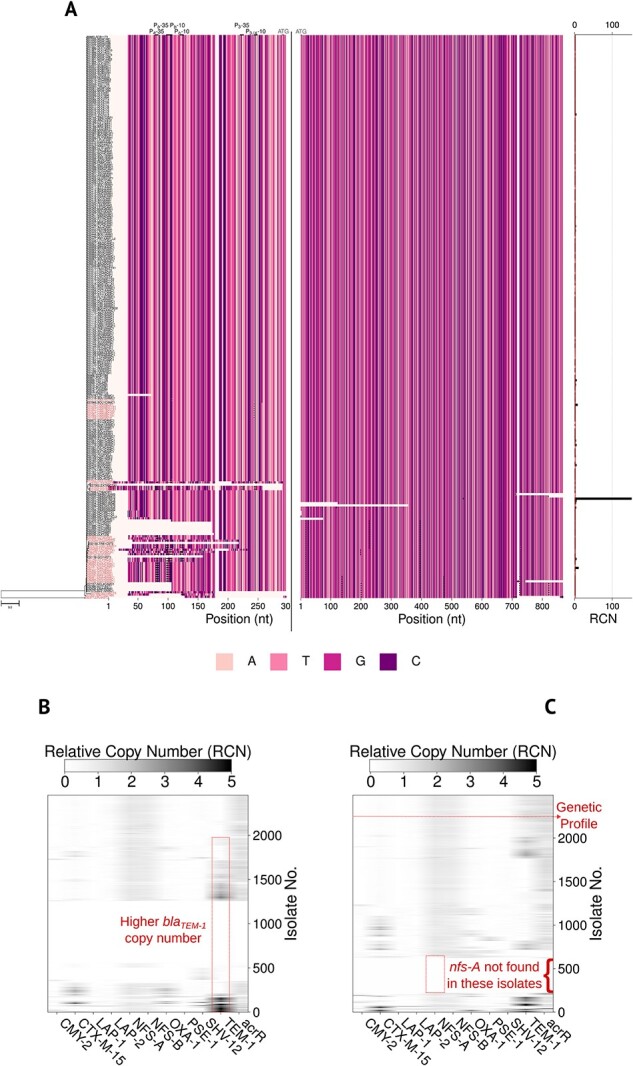
Screening blaTEM-1 across thousands of bacterial isolates. (A) Exemplar of a summary plot generated by Hound to show phylogeny (left), multiple alignment of the promoter and coding sequence (center), and relative copy number (RCN, right) of a subset of the farmed animals data positive for blaTEM-1. This is to help visualize details of the plot. A dot is placed on positions where the nucleotide sequence deviates from the consensus sequence (present in at least 80% of the isolates). Note these are mutations within the multiple alignment, the mutations reported in Hound’s spreadsheet are those from pairwise alignments between the sequence in each isolate and the user-given target sequence and the resulting change in amino acid. Isolates with mutations are highlighted in red by default. When regions of interest have been given alongside the promoter flag, they are added at the top of the alignment (here are promoter regions Pa-35, Pb-10, P3–35, P3/4–10), and start codon. (B) and (C) Heatmap created ad hoc to summarize our search of blaTEM-1 in all 5032 isolates with Hound. The presence or absence is noted by a shaded horizontal line in the heatmap, and the relative copy number is noted by different shade intensities—with darker denoting isolates with more copies and lighter those with fewer copies. Note the absence of genes such as nfs-A and nfs-B exposes the of that are not E. coli. Similarly, heatmaps reveal that genes like lap-1 or lap-2 are more frequent in farm isolates. In general, the horizontal of these heatmaps represent the genetic profile of a given isolate—here to infer resistance to the combination of amoxicillin/clavulanate.
The result shows 39.16% (n = 994) of clinical isolates were blaTEM-1 positive compared to 51.84% (n = 1283) in those from farm animals (Figure 3B and C). It is noteworthy to mention that human clinical isolates largely came from routine surveillance of Gram-negative bacteria and include multiple species beyond E. coli that less commonly carry blaTEM-1, and would also include isolates resistant to very few antibiotics, whereas those from farmed animals were E. coli isolates sequenced due to their resistant phenotype as recently reported [39]. Interestingly however, only 22.52% (n = 289) of the isolates from farmed animals harbored two or more copies of the gene. This contrast with data from the clinical isolates, where 39.33% of the blaTEM-1 positive isolates (n = 391) harbored two or more copies. Since increased gene copy number is associated with amoxicillin/clavulanate resistance [31], this would fit with a higher rate of resistance to this combination given its more widespread use in the clinic to treat humans. Moreover, as Figure 3A and B illustrate, in some isolates blaTEM-1 copy number is in the hundreds. While their occurrence is rare—n = 6 in farm isolates, n = 31 in clinical isolates—it suggests the circulation of one or more blaTEM-1-encoding plasmids with very high copy number. Indeed, multicopy plasmids with hundreds of copies are not unheard of [40]. Using the flag—promoter—cutoff 250 we used Hound to retrieve the promoter of blaTEM-1 and flag any mutation found. Again, mutations associated with over-expression, annotated in the figure produced as black dots, were more common in isolates from humans than farmed animals as Figure 2B and C illustrate. Mutations found by Hound with respect to the query sequence used, as well as relative copy number and other metadata can be exported into a spreadsheet by using the flag—summary $SPREADSHEET, which can then be parsed to highlight isolates with specific mutations. An illustrative spreadsheet is included as a Supplementary Table 1.
A useful feature of Hound is its ability to populate iteratively the same assemblies to look for different genes, allowing the simultaneous detection of genes, calculation of their relative copy number, or re-analysis of prior data. Figure 2D shows two heatmaps illustrating the detection of different 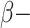 lactamases as well as nitroreductases, and efflux pumps—where mutations affecting their production can cause resistance to multiple antibiotics [41]—in both farm and clinical isolates as well as any mutations found in their coding sequence and relative copy number.
lactamases as well as nitroreductases, and efflux pumps—where mutations affecting their production can cause resistance to multiple antibiotics [41]—in both farm and clinical isolates as well as any mutations found in their coding sequence and relative copy number.
DISCUSSION
An increasingly problematic issue, particularly in the detection of ABR genes, is the lack of reproducibility [42, 43]. The choice of reference genomes is typically opaque to the user beyond the species, being notoriously difficult to underpin the exact assembly used. Along with the variety of existing pipelines that yield inconsistent results between laboratories [38], this problem led to the suggestion of standardized, ISO-certified pipelines [38]. Here we argue that they still fail to detect antibiotic resistance driven by the over-expression of enzymes like 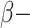 lactamases given their inability to account for gene over-expression caused by promoter mutations and, particularly, increase in gene copy number. Now, beyond the detection of antibiotic resistance mutants, if we found different genetic signatures when comparing our assemblies to different references it is not unreasonable to think this will also be the case for other reference-mapped assemblies. Thus, using a standardized reference is unlikely to avoid this problem—exacerbated by the scarcity of tools to analyze de novo assemblies.
lactamases given their inability to account for gene over-expression caused by promoter mutations and, particularly, increase in gene copy number. Now, beyond the detection of antibiotic resistance mutants, if we found different genetic signatures when comparing our assemblies to different references it is not unreasonable to think this will also be the case for other reference-mapped assemblies. Thus, using a standardized reference is unlikely to avoid this problem—exacerbated by the scarcity of tools to analyze de novo assemblies.
Hound is a step toward facilitating the analysis of these assemblies, not only by addressing a key limitation—gene fragmentation—but also by reducing the knowledge and technological burdens. The fact that we assembled and analyzed ˃5000 genomes on an 8-core, 16GB RAM laptop over the span of 9–10 days shows the potential for Hound to be implemented in larger and more powerful infrastructures for surveillance and diagnostic purposes. Now, Hound has some limitations. For example, it cannot report whether a gene of interest sits within a genomic island, but it can be used to detect whether genes associated to such islands are in the same contig as the gene of interest—complementing other bespoke analyses. Another limitation is that it currently only supports short-read Illumina sequencing data due to its availability during the development of this tool. However, given our use of SPAdes as backend assembler it is possible to add support for long-read nanopore and PacBio sequencing data in future releases. A similar argument can be for metagenomic datasets. While here all isolates were purified, SPAdes supports metagenomic data, only requiring the corresponding implementation in Hound. Given our limited access to such datasets, we could not test this implementation.
The use of de novo assemblies means Hound is not only agnostic with respect to which genes can be monitored. It is also independent of the microbial species analyzed—not possible with the use of reference genomes. With the majority of prokaryote diversity still being unknown and unsequenced [44, 45], we believe that Hound can be a useful tool to study non-model microorganisms that lack any reference—and help build them iteratively thanks to its contig-merging routine when sequencing costs in other platforms increase.
METHODS
Bacterial isolation
Prior to screening bla, we grew all samples (clinical or from farm sources) in chromogenic agar (Chromagar, Paris) at 37°C for 24 h, using Tryptone Bile X-Glucunoride (TBX) to aid purification and identification of E. coli. We confirmed speciation of clinical isolates with MALDI-TOF mass spectrometry (Bruker Microflex). Prior to sequencing, we grew E. coli in TBX agar and all other species in Nutrient Agar (Oxoid).
Genome sequencing and assembly
Genomic DNA libraries from isolate bacteria were prepared using the Nextera XT Library Prep Kit (Illumina, San Diego, USA) following the manufacturer’s protocol with the following modifications: The input DNA was increased 2-fold with respect to the manufacturer’s protocol, and the Polymerase Chain Reaction (PCR) elongation time was increased to 45 s. DNA quantification and library preparation were carried out on a Hamilton Microlab STAR automated liquid handling system (Hamilton Bonaduz AG, Switzerland), and the libraries sequenced on a Illumina NovaSeq 6000 (Illumina, San Diego, USA) using a 250 paired-end protocol by MicrobesNG. Reads were adapter-trimmed using Trimmomatic 0.30 [46] with a sliding window quality cutoff of Q15.
The flag—preprocess reads.zip processes the reads provider by MicrobesNG to create the directory structure required by Hound, with paired reads being stored in $DIR/reads/, and assemblies in $DIR/assemblies/de_novo/ for reads assembled de novo or $DIR/assemblies/reference-mapped/ for those mapped to a reference. This will depend on whether—assembly—de-novo or—assembly—reference $REF_GENOME have been passed. Hound then assembles de novo the resulting reads using SPAdes with the—isolate flag and 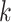 -mer size of 127, given the sequencing platform and protocol. For reference-mapped assemblies, Hound aligns the reads to a user-given reference using the Burrows-Wheeler Aligner and SAMtools with standard parameters. The coverage depth for all assemblies is then calculated with SAMtools if the flags—coverage and—hk-genes $HK_GENES are given, the baseline depth being the median coverage depth of all loci included in $HK_GENES, faster than computing the median coverage of the whole assembly to avoid any bias introduced by plasmid carriage. The relative gene copy number (RCN) is then calculated as the coverage depth of all loci in $TARGET_GENES divided by the baseline coverage [47].
-mer size of 127, given the sequencing platform and protocol. For reference-mapped assemblies, Hound aligns the reads to a user-given reference using the Burrows-Wheeler Aligner and SAMtools with standard parameters. The coverage depth for all assemblies is then calculated with SAMtools if the flags—coverage and—hk-genes $HK_GENES are given, the baseline depth being the median coverage depth of all loci included in $HK_GENES, faster than computing the median coverage of the whole assembly to avoid any bias introduced by plasmid carriage. The relative gene copy number (RCN) is then calculated as the coverage depth of all loci in $TARGET_GENES divided by the baseline coverage [47].
Indexing of assemblies
The resulting assemblies are indexed using makeblastdb from BLAST+, with flags -parse_seqids and -dbtype nucl, to facilitate the search of the sequences in file $TARGET_GENES. The search is run with blastn, which uses nucleotide sequences, or tblastn depending on whether the flag —nucl is passed to Hound. Without this flag, Hound assumes that the file $TARGET_GENES contains amino acid sequences and will therefore use tblastn.
Multiple alignment and phylogeny
When the flag—phylo is passed, Hound will use muscle 3.80 to align the target gene sequences found in all assemblies given its accuracy and speed [48]. Penalties for the introduction and extension of gaps are pre-set with a value of −9950.0 to avoid excessive fragmentation of the alignment. This alignment is then used by Hound to generate a phylogeny with PhyML [49] with seed 100 100 for repeatability.
Implementation details
Hound was developed on a laptop with an x86–64 processor (AMD Ryzen 6900HS, 8-cores/16-threads) and 16GB of DDR5 Random Access Memory (RAM), running ArchLinux and using GNOME Builder. The software is written in Python3, requiring at least Python v3.9 and the libraries numpy [50], matplotlib [51], biopython [52]. For plotting the phylogenies, we used ete3 [53] through its Python API. We installed and tested Hound on Intel-(4-cores) and M1-powered (8-cores) Apple Macbook Pro to confirm compatibility with macOS. The code available through the gitlab repository below produces a Wheel file (.whl) that will automatically retrieve all Python dependencies upon installation with Python’s pip package manager. External software such as SAMtools, Burrows-Wheeler aligner, SPAdes, the BLAST suite and muscle, must be provided independently and added to the system $PATH.
Key Points
Standard tools and databases associate genotypes with the presence/absence of specific genes, or mutations in their coding sequence, failing to detect those caused by the amplification or mutations that lead to over-expression of such genes. Hound can help detect such mutants by reporting promoter mutations, along with the relative abundance of the user-given sequence within an assembly. Importantly, Hound requires amino acid sequences to only report those mutations, in the coding sequence, that could lead to a change in function.
Reference-guided assemblies mask the rich genetic diversity of bacteria, and we showed that the final assembly can change only by virtue of the reference chosen
The main drawback of de novo assemblies is the fragmentation of genens during the assembly process. To circumvent this problem, Hound uses use the user-given query as a template to aid its contig-merging routine. This occurs when the identity between user-given query and hit within the assembly have an identity of 90% or higher. The result is fewer, but larger contigs where the amino acid sequence entered by the user can be unequivocally reconstructed.
Hound produces a spreadsheet summary that can be screened to automatically highlight mutations of interest, depending on the genotype sought after—here being the increase copy number of blaTEM-1 and specific promoter mutations.
Supplementary Material
ACKNOWLEDGMENTS
Whole genome sequencing was performed by MicrobesNG. This work was funded by Medical Research Council grant MR/T005408/1 and Biotechnology and Biological Sciences Research Council Grant BB/X012670/1, and by grants MR/S004769/1, BB/T004592/1 and NE/N01961X/1 from the Antimicrobial Resistance Cross Council Initiative supported by the seven UK research councils, the Department of Health and Social Care and the National Institute for Health Research. It was further funded by grant 82459 from the Welsh Government Rural Communities—Rural Development Program 2014–2020 supported by the European Union and the Welsh Government. N.S. was supported by a postgraduate scholarship from the University of Bristol.
Author Biographies
Carlos Reding is a senior research associate at the University of Bristol. His research focuses on microbial evolution and development of quantitative methods.
Naphat Satapoomin is a PhD candidate at the University of Bristol supervised by Prof. Matthew Avison. Her research focuses on the characterization of resistance to 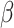 -lactam/
-lactam/ -lactamase inhibitor combinations.
-lactamase inhibitor combinations.
Matthew B. Avison is a professor at the University of Bristol. His research aims to characterize multi-drug resistance mechanisms in Gram-negative bacteria.
Contributor Information
Carlos Reding, University of Bristol School of Cellular and Molecular Medicine, University Walk, Bristol, BS8 1TD Bristol, UK.
Naphat Satapoomin, University of Bristol School of Cellular and Molecular Medicine, University Walk, Bristol, BS8 1TD Bristol, UK.
Matthew B Avison, University of Bristol School of Cellular and Molecular Medicine, University Walk, Bristol, BS8 1TD Bristol, UK.
COMPETING INTERESTS
The authors have declared no competing interests.
DATA AVAILABILITY
Whole-genome sequencing data have been deposited in the European Nucleotide Archive under project accession numbers PRJEB45949, PRJEB70722 and PRJEB72122.
CODE AVAILABILITY STATEMENT
A Python implementation of Hound, licensed under the GNU Lesser General Public License (LGPL) 2.1, can be found in https://gitlab.com/rc-reding/software/
References
- 1. McInerney JO, McNally A, O’connell MJ. Why prokaryotes have pangenomes. Nat Microbiol 2017;2:1–5. [DOI] [PubMed] [Google Scholar]
- 2. Lan R, Reeves PR. Intraspecies variation in bacterial genomes: the need for a species genome concept. Trends Microbiol 2000;8:396–401. [DOI] [PubMed] [Google Scholar]
- 3. Venter SN, Palmer M, Steenkamp ET. Relevance of prokakryotic subspecies in the age of genomics. New Microbe and New Infect 2022;48:101024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andam CP. Clonal yet different: understanding the causes of genomic heterogeneity in microbial species and impacts on public health. mSystems 2019;4:e00097–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tettelin H, Masignani V, Cieslewicz MJ, et al. Proc Natl Acad Sci USA 2005;102:13950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tatusova T, Ciufo S, Fedorov B, et al. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res 2013;42:D553–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaye AM, Wasserman WW. The genome atlas: navigating a new era of reference genomes. Trends Genet 2021;37:807–18. [DOI] [PubMed] [Google Scholar]
- 8. Clausen PT, Aarestrup FM, Lund O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform 2018;19:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo J, Bolduc B, Zayer AA, et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021;9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhie A, McCarthy SA, Fedrigo O, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 2021;592:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schneeberger K, Ossowski S, Ott F, et al. Reference-guided assembly of four diverse arabidopsis thaliana genomes. Proc Natl Acad Sci USA 2011;108:10249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Formenti G, Theissinger K, Fernandes C, et al. The era of reference genomes in conservation genomics. Trends Ecol Evol 2022;37:197–202. [DOI] [PubMed] [Google Scholar]
- 13. Didelot X, Bowden R, Wilson DJ, et al. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 2012;13:601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pankhurst LJ, del Ojo EliasC, Votintseva AA, et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: a prospective study. Lancet Respir Med 2016;4:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hendriksen RS, Bortolaia V, Tate H, et al. Using genomics to track global antimicrobial resistance. Front Public Health 2019;7:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sichtig H, Minogue T, Yan Y, et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat Commun 2019;10:3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Global Antimicrobial Resistance Surveillance System (GLASS): Whole-genome sequencing for surveillance of antimicrobial resistance . World Health Organization, 2020.
- 18. Walker TM, Kohl TA, Omar SV, et al. Whole-genome sequencing for prediction of mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis 2015;15:1193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Su M, Satola SW, Read TD. Genome-based prediction of bacterial antibiotic resistance. J Clin Microbiol 2019;57:e01405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Valiente-Mullor C, Beamud B, Ansari I, et al. One is not enough: on the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput Biol 2021;17:e1008678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mukherjee S, Seshadri R, Varghese NJ, et al. 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat Biotechnol 2017;35:676–83. [DOI] [PubMed] [Google Scholar]
- 22. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 2012;67:2640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de ManTJ, Limbago BM. SSTAR, a stand-alone easy-to-use antimicrobial resistance gene predictor. mSphere 2016;1:10–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gupta SK, Padmanabhan BR, Diene SM, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother 2014;58:212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis JJ, Boisvert S, Brettin T, et al. Antimicrobial resistance prediction in PATRIC and RAST. Sci Rep 2016;6:27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuentes-Pardo AP, Ruzzante DE. Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol Ecol 2017;26:5369–406. [DOI] [PubMed] [Google Scholar]
- 27. Bailey JK, Pinyon JL, Anantham S, Hall RM. Distribution of the blaTEM gene and blaTEM-containing transposons in commensal escherichia coli. J Antimicrob Chemother 2011;66:745–51. [DOI] [PubMed] [Google Scholar]
- 28. Davies TJ, Stoesser N, Sheppard AE, et al. Reconciling the potentially irreconcilable? Genotypic and phenotypic amoxicillin-clavulanate resistance in escherichia coli. Antimicrob Agents Chemother 2020;64:10–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruffles TJ, Goyal V, Marchant JM, et al. Duration of amoxicillin-clavulanate for protracted bacterial bronchitis in children (DACS): a multi-Centre, double blind, randomised controlled trial. Lancet Respir Med 2021;9:1121–9. [DOI] [PubMed] [Google Scholar]
- 30. KuKanich K, Lubbers B, Salgado B. Amoxicillin and amoxicillin-clavulanate resistance in urinary escherichia coli antibiograms of cats and dogs from the midwestern United States. J Vet Intern Med 2020;34:227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hansen KH, Andreasen MR, Pedersen MS, et al. Resistance to piperacillin/tazobactam in escherichia coli resulting from extensive IS 26-associated gene amplification of blaTEM-1. J Antimicrob Chemother 2019;74:3179–83. [DOI] [PubMed] [Google Scholar]
- 32. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinform 2009;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with kraken 2. Genome Biol 2019;20:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 2002;30:2478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sherry NL, Horan KA, Ballard SA, et al. An ISO-certified genomics workflow for identification and surveillance of antimicrobial resistance. Nat Commun 2023;14:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mounsey O, Marchetti L, Parada J, et al. Genomic epidemiology of third-generation cephalosporin-resistant escherichia coli from argentinian pig and dairy farms reveals animal-specific patterns of co-resistance and resistance mechanisms. Appl Env Microbiol 2024;90(1):e01791–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. San Millan A, Escudero JA, Gifford DR, et al. Multicopy plasmids potentiate the evolution of antibiotic resistance in bacteria. Nat Ecol Evol 2016;1:0010. [DOI] [PubMed] [Google Scholar]
- 41. Blair JM, Bavro VN, Ricci V, et al. AcrB drug-binding pocket substitution confers clinically relevant resistance and altered substrate specificity. Proc Natl Acad Sci USA 2015;112:3511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Doyle RM, O’sullivan DM, Aller SD, et al. Discordant bioinformatic predictions of antimicrobial resistance from whole-genome sequencing data of bacterial isolates: an inter-laboratory study. Microbial Genom 2020;6:e000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coolen JP, Jamin C, Savelkoul PH, et al. Centre-specific bacterial pathogen typing affects infection-control decision making. Microb Genom 2021;7:000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol 2003;57:369–94. [DOI] [PubMed] [Google Scholar]
- 45. Locey KJ, Lennon JT. Scaling laws predict global microbial diversity. Proc Natl Acad Sci USA 2016;113:5970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reding C, Catalán P, Jansen G, et al. The antibiotic dosage of fastest resistance evolution: gene amplifications underpinning the inverted-u. Mol Biol Evol 2021;9:3847–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 2004;5:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 2003;52:696–704. [DOI] [PubMed] [Google Scholar]
- 50. Harris CR, Millman KJ, van der WaltSJ, et al. Array programming with NumPy. Nature 2020;585:357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng 2007;9:90–5. [Google Scholar]
- 52. Cock PJ, Antao T, Chang JT, et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 2009;25:1422–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 2016;33:1635–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole-genome sequencing data have been deposited in the European Nucleotide Archive under project accession numbers PRJEB45949, PRJEB70722 and PRJEB72122.