

Abstract
Accurate identification of specific groups of proteins by their amino acid sequence is an important goal in genome research. Here we combine information theory with fuzzy logic search procedures to identify sequence signatures or predictive motifs for members of the Myc-Max-Mad transcription factor network. Myc is a well known oncoprotein, and this family is involved in cell proliferation, apoptosis, and differentiation. We describe a small set of amino acid sites from the N-terminal portion of the basic helix-loop-helix (bHLH) domain that provide very accurate sequence signatures for the Myc-Max-Mad transcription factor network and three of its member proteins. A predictive motif involving 28 contiguous bHLH sequence elements found 337 network proteins in the GenBank NR database with no mismatches or misidentifications. This motif also identifies at least one previously unknown fungal protein with strong affinity to the Myc-Max-Mad network. Another motif found 96% of known Myc protein sequences with only a single mismatch, including sequences from genomes previously not thought to contain Myc proteins. The predictive motif for Myc is very similar to the ancestral sequence for the Myc group estimated from phylogenetic analyses. Based on available crystal structure studies, this motif is discussed in terms of its functional consequences. Our results provide insight into evolutionary diversification of DNA binding and dimerization in a well characterized family of regulatory proteins and provide a method of identifying signature motifs in protein families.
Keywords: Myc, predictive motif, transcription factor, oncoprotein, molecular evolution
A sequence signature is a small set of amino acid sites that accurately identifies a specific set of proteins, such as transcription factors, or specific families of proteins like the basic helix-loop-helix (bHLH) transcriptional regulators. Sequence signatures are also known as predictive motifs (1). Use of sequences signatures can greatly facilitate database searches and, when coupled with detailed structural and functional information, provides a more detailed understanding of the sequence elements involved in evolutionary diversification of a particular set of proteins.
Although the definition of a particular protein family is ultimately made with structural, experimental, and evolutionary analyses, rapid identification of proteins through their sequence attributes remains a high priority. Typically, protein sequence identifications are achieved with alignment-based search algorithms such as blast. Although useful for a first-order approximation, this approach is insufficient, because it typically does not provide a unique sequence signature and therefore does not meaningfully enhance the biological basis for specific protein classification.
Herein, we combine information theory, fuzzy logic search algorithms, and protein structure to produce a probabilistic identification scheme for the Myc-Max-Mad transcription factor network. Specifically, we ask, “What unique set of amino acids, when considered simultaneously, will accurately define (identify) the Myc-Max-Mad network of proteins and its constituent members?” Ideally, a predictive motif should contain a small number of highly informative amino acid sites that can be defined in terms of important biological structure and function.
Importance of Myc, Max, and Mad Proteins
The Myc-Max-Mad transcription network of bHLH proteins is essential for control of cell growth, proliferation, differentiation, and apoptosis (2-6). Myc is a well established oncogene whose deregulated expression is responsible for a wide range of human cancers. Approximately 70,000 cancer deaths in the U.S. each year arise from misregulation of Myc. Protein-protein interactions with Max are a key element in proper functioning of the Myc-Max-Mad transcription factor network. Mad-Max heterodimers repress the expression of Myc and initiate differentiation. Although capable of weak homodimerization, proper Myc function requires heterodimerization with Max (7). Extensive efforts have attempted to isolate these oncoproteins in a wide variety of organisms by using molecular and computational approaches. Indeed, development of a predictive motif for bHLH proteins (1) has been very successful when applied to diverse groups such as Ascidians, Drosophila, worms, and plants (8-12).
At least six types of Myc protein reflect separate evolutionary lineages (W.R.A., unpublished data). Most widely studied is c-Myc, the cellular homologue to the viral oncoprotein (v-Myc) of the avian myelocytomatosis retrovirus (13). Additionally, the Myc family includes L-Myc, N-Myc, S-Myc, and B-Myc, which are expressed in a tissue-specific manner (5). L-Myc is associated with lung carcinoma, whereas N-Myc is associated with neuroblastomas (13). B- and S-Myc exhibit significantly more sequence and functional divergence than c-, L-, and N-Myc. B-Myc is homologous to the N-terminal transactivation domain but lacks the bHLH dimerization domain. We consider Myc from protostomes (Drosophila and Anopheles) as a separate clade from the deuterostome lineage because of each group's divergent sequence attributes (14).
Methods
Approximately 80 members of the Myc-Max-Mad transcription factor network that included a number of divergent vertebrate sequences as well as all available invertebrate sequences were chosen for analysis (15). The bHLH leucine zipper domains were aligned by using the dialign2 algorithm (16). The Boltzmann-Shannon entropy H is used to quantify sequence variability of amino acid residues at each site (1, 17) and is calculated as 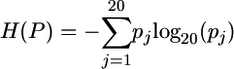 , where pj is the probability of an amino acid being of the jth kind, and 0 ≤ H(P) ≤ 1. Smaller values of E indicate a greater degree of evolutionary conservation and are used to construct the predictive motif. The final motif is verified by probing a database. All motifs were constructed via inspection and iterative trial and error to find the fewest elements that gave maximum discrimination. We then compare this procedure with results from widely used fully automatic approaches.
, where pj is the probability of an amino acid being of the jth kind, and 0 ≤ H(P) ≤ 1. Smaller values of E indicate a greater degree of evolutionary conservation and are used to construct the predictive motif. The final motif is verified by probing a database. All motifs were constructed via inspection and iterative trial and error to find the fewest elements that gave maximum discrimination. We then compare this procedure with results from widely used fully automatic approaches.
Efficacy of the predictive motif was evaluated by a dynamic programming search algorithm (i) to determine the minimum number of insertions, deletions, or point mutations (the edit distance) required to match a predictive motif to a target sequence and (ii) for a given edit distance, generate all possible edited motifs that exactly match the target sequence (18). By assigning appropriate edit distances between motif and query sequences, the algorithm permits searches with (i) a defined level of mismatch including gaps and (ii) site-specific specification of acceptable variants. This search algorithm was used to search the GenBank NR database. Although NR represents the most complete available repository of peptide sequences, it contains naturally occurring variants, sequencing errors, and pseudogene products. Such variants, if found, were removed by inspection.
Statistical significance of any particular predictive motif is implicit to the combinatorial nature of the search, with the probability of a motif/protein match being approximately the product of the probabilities for simultaneous occurrence of each element in the motif in the target protein. The actual probability depends upon the correlation among the motif elements and sequences within the database. Efficacy of the predictive motif is defined by goodness of fit of the protein sequence data to the motif. With fuzzy logic search algorithms, one defines the level of probability by controlling the numbers of mismatches permitted in the search. We will refer to this approach as the Atchley-Fernandes (AF) Method to distinguish it from other comparable methods.
Results and Discussion
Entropy profiles of the Myc-Max-Mad proteins (Fig. 1) suggest that amino acid sites in the N-terminal portion of the bHLH domain would provide accurate and specific discriminating signatures. The basic DNA-binding region and the first α-helix were chosen to build sequence signatures, because they (i) comprise a short ungapped contiguous stretch of amino acids and (ii) exhibit a number of highly conserved and potentially diagnostic sites. Table 4, which is published as supporting information on the PNAS web site, provides the amino acid composition for 28 signature sites. Sequences of non-Myc/Mad/Max network proteins are not available in sufficient quantities to adequately explore their sequence variability. The definition of the bHLH domain and the numbering scheme is that of Atchley and Fitch (19, 20) and Ferre-D'Amare et al. (21). Entropy values of each site together with the most prevalent two amino acids are provided for each protein (Table 4). Sites are simply labeled as “variable” when no amino acid had frequency >50%. Fig. 1 provides a histogram of E values over the bHLH leucine zipper domain for 80 divergent Myc-Max-Mad network proteins. A number of amino acid sites are evolutionarily highly conserved and exhibit little or no variability over extensive evolutionary time. This plot suggests a short highly specific sequence signature involving a 28-aa stretch can delimit the entire network. This network sequence signature is shown in Table 1.
Fig. 1.

Entropy (sequence variability) profile of the bHLH domain from the Myc-Mad-Mad network.
Table 1. The Myc-Mad-Max motifs, showing positional homology.
| Amino acid number in basic and helix-1 sequence components | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Motif | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 |
| Network | KNRS | KR | AKRST | HNQST | H | N | X | ALM | E | KRQ | HKNQR | R | R | X | X | ILMV | KR | X | X | FLY | X | X | L | HKR | DEGQT | X | ILMV | P |
| Myc | KR | KR | KR | X | H | N | X | LM | E | R | X | R | R | X | DE | LM | KR | X | X | F | X | X | L | KR | DE | X | X | P |
| Max | KR | KR | A | X | H | N | X | L | E | R | X | R | R | X | H | I | K | X | X | F | X | X | L | KR | DE | X | X | P |
| Mad | X | R | EST | X | H | N? | EK | LM | E | K | X | R | R | A | X | L | RK | EL | CY | L | X | X | L | KR | X | X | X | P |
The Network motif includes Myc, Mad, Max, and other network proteins.
We use the term “mismatch” for sequence sites whose amino acid composition differs from the predictive motif. The term “misidentified” is used for sequences found by the search algorithm that do not belong to the specific protein family of the motif. The AF method applied to the 06 February 2004 version of the GenBank NR database returned 337 network members with zero mismatches and another 28 proteins with only one mismatch, showing that the network signature accurately identifies many diverse Myc-Max-Mad network proteins. There were no misidentifications of proteins; all proteins that were returned are known network members. With two mismatches, 18 additional sequences were returned, and all but two were known members of the network. Of the two misidentified sequences, both of which were bHLH proteins with mismatches at sites 1 (leucine) and 6 (lysine), one was the protein Esc1 from Schizosaccharomyces (fission yeast), and the other was a hypothetical protein from Magnaporthe (rice blast fungus). The former has been characterized as a sexual differentiation protein, and previous phylogenetic analyses suggested it was not closely related to any other known bHLH protein (20). Nothing is known about the Magnaporthe protein.
At three mismatches, 44 sequences were added, of which two were presumed misidentifications from Neurospora and Aspergillus. The Neurospora protein has the same motif sequence as the two misidentifications described above with the addition of a valine mismatch at site 3. The other misidentified sequence was a hypothetical protein from Aspergillus. The remaining 42 sequences at three mismatches were correctly identified as more divergent members of the Myc-Max-Mad network, including proteins like BigMax, Max-like interactor, and WBSCR14 (19). With four mismatches, few network proteins, but many more nonnetwork bHLH proteins, particularly those from the Hairy, SREBP, Hand2, and USF families, were obtained. Thus, the motif appears to be the smallest possible that can accurately define the entire Myc-Max-Mad network.
With regard to misidentified proteins, members of the Myc-Max-Mad network have not previously been known to occur in fungi, and little is known of the function of these presumably misidentified sequences. Fig. 2 shows an alignment of the bHLH leucine zipper regions of these four fungal sequences together with human Myc, Max, and Mad. The Magnaporthe sequence has a leucine zipper, whereas Neurospora and Aspergillus appear to have the beginning of a leucine zipper (two leucine repeats). Given such clear homology, these proteins, particularly the Magnaporthe protein, should be examined in detail experimentally.
Fig. 2.

Alignment of Myc, Max, and Mad together with four putative fungal Myc sequences, visualized by genedoc (www.psc.edu/biomed/genedoc), with conserved sites indicated by differential shading. Two gaps separate the various labeled functional domains. Heptad leucine repeats in the zipper are labeled with a bold plus (+) sign.
Knowing the amino acid sites where deviations from the predictive motif occur is helpful in understanding protein evolution. Referring to the site-numbering scheme in Table 1, the 28 sequences deviated from the predictive motif at the following sites (and number of mismatches): 1 (5), 4 (4), 5 (1), 6 (2), 23 (1), 25 (11), and 27 (2). Ten of the 11 mismatches at site 25 were valine (V). With two mismatches, the 18 sequences added had deviate residues at sites 1 (5), 2 (6), 3 (4) 4 (6), 6 (3), 8 (2) 12 (1), 13 (2), and 25 (2). At site 2, five of the six mismatches were for a G residue, whereas at site 4, all of the mismatches were a V. A predictive motif for Myc proteins alone was formulated by using 18 amino acid sites from these same first 28 amino acid sites of the DNA-binding region and helix-1 and is shown in Table 1.
A total of 308 known Myc sequences occur in the 06 February 2004 version of GenBank NR database. The AF algorithm returned 287 sequences of these that fit the predictive motif exactly. These sequences included c-, v-, L-, and N-Myc, and all vertebrate groups were represented. S- and B-Myc were not found at the zero mismatch level. Thus, simultaneous consideration of the amino acid composition of these 18 amino acid sites from the DNA-binding domain and first α-helix produced a 93% level for correct identifications for Myc proteins. Eleven additional sequences fit the motif with one mismatch and were all Myc proteins, including c-Myc proteins from pig (1); bird (2); sea star, sea urchin, and S-Myc (2); and N-Myc (2). Permitting only a single mismatch identifies 96% of currently known Myc proteins. With two mismatches, five additional sequences were added, including two v-Myc sequences and three N-Myc sequences from canary.
Forty-one sequences were returned with three motif mismatches, including the protostome (Drosophila and Anopheles) forms of Myc and trout Myc-2. The remaining sequences were Max, the dimerization partner of Myc. Insect sequences of Myc have an N substituted for K or R at site 3, G for D or E at site 25, and K for D or E at site 25. It is unclear whether Myc-2 in trout is a pseudogene product. No Myc or Max protein sequences were returned for four or more mismatches. However, a number of other members of the Myc-Max-Mad network family were returned with five mismatches, including Mxi1, Rox, Mnt, and BigMax.
Interestingly, the sequence signature for Myc is identical to the ancestral sequence for the entire L-, N-, S-, and c-Myc group, as estimated by a parsimony analysis (W.R.A., unpublished data). This finding is significant with respect to the biological relationship among these motifs, as described later. The large number of Max sequences with three or more mismatches suggested that an accurate predictive motif could be constructed for Max and Mad. Max differed from Myc almost uniformly at three sites in the motif (sites 3, 15, and 16). Thus, a new Max family motif was generated (Table 1) consisting of 18 amino acids that matched 36 sequences with no mismatches or misidentifications. No sequences were returned with one motif mismatch, and three were returned with two mismatches, the latter including a Mxil1 sequence and the Myc sequences for sea star and sea urchin. Two hundred ninety sequences were returned with three mismatches, and all of these were Myc sequences. Thus, the Max motif is very accurate and specific. Finally, considering sequence variability in Mad (Table 1), marked differences occur between Mad and Myc and Max at sites 3, 7, 10, 14, 20, and 21. A predictive motif for Mad was generated, as shown in Table 1. The AF search algorithm returned 28 sequences with no mismatches, 5 with one mismatch, 13 with two mismatches, and 2 with three mismatches. All 48 sequences are from the Mad family, with most mismatches for Mnt or Rox proteins. Myc and Mad are similar in terms of the number of sequence mismatches, whereas Max is much more conserved (19) and, as a consequence, has fewer mismatches. It is suggested elsewhere (19) that Max is more highly conserved, because it must serve as the dimerization partner for the entire Myc-Max-Mad network. As a consequence, selection intensity is very high to preserve a canonical binding surface in Max that could interact effectively with all of the other network proteins.
One obvious extension of this procedure would be to construct a two-pass algorithm to identify sequences with regard to the entire Myc-Max-Mad network on the first pass and then take sequences that fit the motif and use the more restrictive motifs to assign the sequences to the proper protein.
Structural and Functional Significance of Motifs. A sequence signature or predictive motif is most useful when its individual elements can be shown to have specific structural or functional properties. Such information is important to understand the structural and functional basis of evolutionary diversification among closely related and interacting proteins. For the Myc-Mad-Max network, it helps to partition the domain into the basic DNA-binding component (sites 1-13) and the protein interaction region (sites 14-28). Within the Myc motif, mismatches tend to be more frequent in the DNA-binding component. There are 10 sequences exhibiting one mismatch, and seven of these mismatches occur in the basic region. Using Table 1 as the numbering guide, the variant amino acid sites (and the number of sequences with mismatches) are 2, 3 (2), 5, 6, 12 (2), and 15 (2). For sequences with two mismatches, six mismatches are found in the basic region, whereas mismatches found are in two sites in the helix region. Amino acid sites where mismatches occurred (and the number of mismatches) are 6 (3), 8, 13 (2), 16 (2), and 28 (2). Detailed crystal structure information about Max is available (21) showing the role of each amino acid in the motif (Table 2).
Table 2. Structural attributes of predictive motif elements.
| Site no. | Max no. | Site | Motif? | Max | Structural explanation | Myc | Mismatch | Site no. | Mad | Site no. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 24 | B1 | Y | K | Contacts DNA (A7'); beginning of Basic region | K | 355 | S | 57 | |
| 2 | 25 | B2 | Y | R | Contacts phosphate backbone | R | 356 | R | 58 | |
| 3 | 26 | B3 | Y | A | R | 2 | 357 | S | 59 | |
| 4 | 27 | B4 | H | T | 358 | T | 60 | |||
| 5 | 28 | B5 | Y | H | Contacts DNA (G3'); bHLH group designation | H | 1 | 359 | H | 61 |
| 6 | 29 | B6 | Y | N | Contacts phosphate backbone | N | 4 | 360 | N | 62 |
| 7 | 30 | B7 | A | V | 361 | E | 63 | |||
| 8 | 31 | B8 | Y | L | L | 1 | 362 | M | 64 | |
| 9 | 32 | B9 | Y | E | Contact with DNA (C3 and A2) | E | 363 | E | 65 | |
| 10 | 33 | B10 | Y | R | Contacts phosphate backbone | R | 364 | K | 66 | |
| 11 | 34 | B11 | K | Q | 365 | N | 67 | |||
| 12 | 35 | B12 | Y | R | Contacts phosphate backbone | R | 2 | 366 | R | 68 |
| 13 | 36 | B13 | Y | R | Contacts DNA and phosphate backbone | R | 3 | 367 | R | 69 |
| 14 | 37 | H1 | D | N | 368 | A | 70 | |||
| 15 | 38 | H2 | Y | H | E | 369 | H | 71 | ||
| 16 | 39 | H3 | Y | I | Side chain packs against F43; buried site | L | 2 | 370 | L | 72 |
| 17 | 40 | H4 | Y | K | K | 371 | R | 73 | ||
| 18 | 41 | H5 | D | R | 372 | L | 74 | |||
| 19 | 42 | H6 | S | S | 373 | C | 75 | |||
| 20 | 43 | H7 | Y | F | Many Van der Waals contacts with H2 side chains; buried site | F | 374 | L | 76 | |
| 21 | 44 | H8 | H | F | 375 | E | 77 | |||
| 22 | 45 | H9 | S | A | 376 | K | 78 | |||
| 23 | 46 | H10 | Y | L | Packs against 63 and 67; buried site | L | 377 | L | 79 | |
| 24 | 47 | H11 | Y | R | R | 378 | K | 80 | ||
| 25 | 48 | H12 | Y | D | D | 379 | G | 81 | ||
| 26 | 49 | H13 | S | Q | 380 | L | 82 | |||
| 27 | 50 | H14 | V | Packs against 70 | 1 | 381 | V | 83 | ||
| 28 | 51 | H15 | Y | P | End of Helix 1; P residue turns strand; packs against 70; buried site | P | 2 | 382 | P | 84 |
Explanations are based on Ferre-D'Amare et al. (21) for Max. The numbering schemes are based on the human sequences for the Max, Myc, and Mad proteins.
The bHLH domain consists of a basic DNA-binding region, two amphipathic α-helices separated by a loop of variable length. The DNA-binding region binds to a consensus hexanucleotide 5′-CANNTG E-Box. At least five groups of bHLH proteins (A-E) are defined by the nature of the E-Box binding (20, 21). As Group B bHLH proteins, the Myc-Max-Mad network proteins bind to the CACGTG E-Box. Crystal structure studies of Max homodimers and Myc-Max dimers (7, 21) provide detailed information about the structure and function of the bHLH leucine zipper domain. The α-helical region of both Myc and Max makes four specific contacts with the DNA bases within the E-Box (Table 2) involving three amino acids: B5(H), B9(E), and B13(R). The basic region also makes phosphate backbone contacts that span the entire backbone of the recognition region (21). These latter contacts include sites B2(R), B6(N), B10(R), B12(R), and B13(R). A number of buried sites in the α-helix interact with other amino acid sites, including H3(I), H7, H10(L), H14(V), and H15(P). Additionally, H15(P) functions to break the α-helix and turn the strand as the loop region appears.
Entropy calculations (Fig. 1) indicate that all of these sites have greatly reduced variability, and amino acid composition at these sites contains significant phylogenetic information about group identity. All of the functionally and structurally significant sites described above are included in the predictive motif. Examination of Table 1 shows some sites are highly conserved with the same or very similar amino acid throughout the Myc-Max-Mad network (sites 2, 5, 6, 8-10, 12, 13, etc.). Other sites are highly discriminatory among Myc, Max, and Mad (sites 3, 4, 7, 11, 14, 15, 21, 22, and 26). Consideration of Tables 1 and 2 shows that one set of amino acid sites is highly conserved across the network and serves to delimit the Myc-Mac-Mad network from all other proteins. A second set of amino acids is highly diagnostic of the individual proteins. The first set includes those with specific functions with regard to DNA-binding or dimerization. The second set of individually diagnostic sites does not have general structural or functional attributes. Thus, at site 3, Myc proteins have a basic and polar R or K amino acid. Max has an uncharged nonpolar A (or polar E), whereas Mad contains an uncharged S or T. At site 15, Myc has a negatively charged D or E, whereas Max, its dimerization partner, has a negatively charged H or uncharged N residue. At site 14, however, Myc and Mad primarily have the neutral N or A residues, whereas Max has a charged D residue. Such interesting contrasts are available at a number of other sites within this region of amino acids. These findings help to clarify many aspects of the dimerization behavior of these proteins, their respective functions, and the biological bases for their evolutionary divergence.
Comparison with Other Approaches. How does the AF method compare with other motif-finding approaches? The AF approach requires a priori biologically insightful input from the researcher in terms of a high-quality sequence alignment and selection of motif elements based upon entropy scores and structural data. To assess its efficacy, we compared the AF method against meme/mast (22, 23) and hmmer (24). meme automatically deduces conserved motifs without need of either multiple sequence alignment or resolved phylogeny. mast is meme's associated search algorithm. hmmer is an established motif generation and search tool that uses a hidden Markov strategy and, like our method, requires an a priori multiple alignment as input. In these comparisons, we used the same Myc input sequences (not the entire network) and the default meme and hmmer parameter settings.
meme found three position-specific scoring matrices diagnostic for Myc proteins. However, because two meme motifs were contiguous, meme effectively located only two different motifs. The first meme motif substantially overlaps our predictive motif but extends it significantly by adding 21 additional amino acid sites past the end of our motif (past amino acid 28 in Table 1), giving an overall motif length of 80 amino acids. The second meme motif was much shorter but involved amino acids outside the bHLH domain. mast searches meme-produced motifs and assigns E scores to each putative match. Interpretation of the E score is similar to that of blast (25), corresponding roughly to the probability of making a Type I (false-positive) error in classification. This interpretation is not strictly accurate when searching most databases, because the definition of E score assumes that the database contents are random and independent. Sequences within GenBank are not independent, because families of proteins, genomes, or phyla tend to be sequenced in clusters, resulting in highly correlated database entries. Thus the E score does not directly indicate the fraction of false positives we might expect.
A better interpretation of E score values is to treat them as random variables drawn from the database. The cumulative distribution function (CDF) of this random variable reveals the true distribution of motif matches within the database. Such a CDF is shown (Fig. 3) for both meme and hmmer motifs. The meme CDF is fairly smooth from ≈10-92 to 10-70, at which point there is a small jump to a plateau at ≈10-53. This jump and plateau correspond to Myc fragments that could not match all meme motifs because of missing sequence components and hence could not minimize their E score. Importantly, this very low E score region corresponded to the region of zero, one, or two predictive motif mismatches. The second large increase in E score occurred around 10-45 (corresponding to three mismatches) and denotes the point where both methods begin to be find Max sequences. These sudden changes in E scores tend to be diagnostic of family inclusion/exclusion. However, it is not clear whether presence or absence of an obviously sharp end-point, as seen in Fig. 3, necessarily needs to be correlated generally with the number of mismatches or family membership. With six exceptions, every sequence discovered by our predictive motif was discovered by mast. Two of those exceptions were short Myc sequence fragments, one was a fragment of Max, and the remaining two were bHLH proteins that had five motif mismatches. Thus it would seem that meme's Type II (false-negative) error rate is not appreciably different from the AF results.
Fig. 3.

A comparison of the AF method, meme, and hmmer (discussed in the text).
In contrast, hmmer search results show an almost uniform E score of ≈10-45 for sequences with zero, one, or two mismatches, followed by a sharp jump to 10-5 and/or greater for three or more mismatches. Results returned by hmmer were surprisingly bimodal, categorizing query sequences as either strong matches or strong mismatches, with numerous Type I and II errors of classification in each case. Manual reexamination of the hmmer search results found at least 24 false positives with E score of <10-3. In each case, these false positives were non-Myc bHLH transcription factors, many of which were from the higher vascular plants.
The most interesting contrast among the three methods occurs in the boundary between “clearly Myc” and “clearly not Myc” where predominantly Max sequences exist. When three mismatches are allowed, the AF method finds mainly Max interspersed with the occasional other member of the Myc-Max-Mad family. In contrast, at a mast E score corresponding to three mismatches, >1 order of magnitude more false-positive (non-Myc-Max-Mad) family members are found, suggesting that meme has substantially less discriminatory power than the AF method at moderate evolutionary distance despite including two and one-half times more putative diagnostic amino acid sites. The relative merits of these three approaches to motif building and developing sequence signatures are summarized in Table 3. The AF method is more laborious, because it requires a priori knowledge to be input from the researcher in terms of a manually curated alignment and decisions about amino acid sites to be included. However, our method generates much shorter and more highly discriminatory motifs. In this particular instance, the motif did not involve the loop portion of the bHLH domain. The loop in bHLH proteins is often of variable length and difficult to accurately align, necessitating the introduction of gaps (20). Our method also permits information to be included about covariances among elements in the predictive motif and permits construction of motifs based upon specific components of the proteins, making effective use of a priori knowledge.
Table 3. A comparison of the meme, hmmer, and AF methods for motif building.
| Action | meme | hmmbuild | AF method |
|---|---|---|---|
| Input | An unaligned set of sequences. Motif region (if any) in each sequence may be unknown. | An aligned set of sequences. Nonhomologous sites may be removed prior to model building. | An aligned set of sequences. Entropy is used to differentiate con served from nonconserved sites. |
| Output | A set of PSSMs, one for each motif found by the algorithm. | Markov transition matrix specific to hmmer model. | A motif pattern that is mathematically very similar to a thresholded pssm. |
| Interpretability of the output | Not readily interpretable unless entries are thresholded or compared statistically. | Relatively uninterpretable. hmm is a nonconstructive statistical null hypothesis. | A readily interpretable motif pattern. |
| Strengths and weaknesses | Does not need an initial alignment to find or create motifs. | Requires initial sequence alignment. | Requires an initial sequence alignment. |
| Search algorithm gives an estimate of how well the motif fits a test sequence. | Picking conserved regions to train model is subjective. | A priori biological knowledge can be included. | |
| Search algorithm gives an estimate of how well the motif fits a test sequence. | Mismatch count is correlated with probability of motif family membership. |
PSSM, position-specific scoring matrix; thresholding refers to mapping 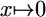 if x < ε and
if x < ε and 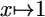 otherwise.
otherwise.
Conclusion
Sequence signatures consisting of 18 of the first 28 elements of the basic DNA-binding region and first α-helix of the bHLH domain provide a highly accurate and powerful identification scheme for the Myc-Max-Mad transcription factor network. Further, these signatures incorporate structural and functional data and therefore provide useful information about the biological basis for these unique signatures. Structural and evolutionary studies can incorporate these findings to better understand the origin, evolution, and function of these important proteins.
Supplementary Material
Acknowledgments
We thank Dr. Bonnie Deroo for her comments. This work is supported by the National Institutes of Health (Grant GM45344), North Carolina State University, and the Alexander von Humboldt Stiftung.
Author contributions: W.R.A. designed research; W.R.A. and A.D.F. performed research; A.D.F. contributed new reagents/analytic tools; W.R.A. and A.D.F. analyzed data; and W.R.A. and A.D.F. wrote the paper.
Abbreviations: bHLH, basic helix-loop-helix; AF, Atchley-Fernandes.
References
- 1.Atchley, W. R., Terhalle, W. & Dress, A. (1999) J. Mol. Evol. 48, 501-516. [DOI] [PubMed] [Google Scholar]
- 2.Grandori, C., Cowley, S. M., James, L. P. & Eisenman, R. N. (2000) Annu. Rev. Cell Dev. Biol. 16, 653-699. [DOI] [PubMed] [Google Scholar]
- 3.Levens, D. L. (2003) Genes Dev. 17, 1071-1077. [DOI] [PubMed] [Google Scholar]
- 4.Luscher, B. (2001) Gene 277, 1-14. [DOI] [PubMed] [Google Scholar]
- 5.Nasi, S., Ciarapica, R., Jucker, R., Rosati, J. & Soucek, L. (2001) FEBS Lett. 490, 153-162. [DOI] [PubMed] [Google Scholar]
- 6.Zhou, Z. Q. & Hurlin, P. J. (2001) Trends Cell Biol. 11, S10-S14. [DOI] [PubMed] [Google Scholar]
- 7.Nair, S. K. & Burley, S. K. (2003) Cell 112, 193-205. [DOI] [PubMed] [Google Scholar]
- 8.Buck, M. J. & Atchley, W. R. (2003) J. Mol. Evol. 56, 742-750. [DOI] [PubMed] [Google Scholar]
- 9.Moore, A. W., Barbel, S., Jan, L. Y. & Jan, Y. N. (2000) Proc. Natl. Acad. Sci. USA 97, 10436-10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyrefitte, S., Kahn, D. & Haenlin, M. (2001) Mech. Dev. 104, 99-104. [DOI] [PubMed] [Google Scholar]
- 11.Satou, Y., Imai, K. S., Levine, M., Kohara, Y., Rokhsar, D. & Satoh, N. (2003) Dev. Genes Evol. 213, 213-221. [DOI] [PubMed] [Google Scholar]
- 12.Toledo-Ortiz, G., Huq, E. & Quail, P. H. (2003) Plant Cell 15, 1749-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vennstrom, B., Sheiness, D., Zabielski, J. & Bishop, J. M. (1982) J. Virol. 42, 773-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nesbit, C. E., Grove, L. E., Yin, X. Y. & Prochownik, E. V. (1998) Cell Growth Differ. 9, 731-741. [PubMed] [Google Scholar]
- 15.Ledent, V. & Vervoort, M. (2001) Genome Res. 11, 754-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgenstern, B. (1999) Bioinformatics 15, 211-218. [DOI] [PubMed] [Google Scholar]
- 17.Atchley, W. R., Wollenberg, K. R., Fitch, W. M., Terhalle, W. & Dress, A. W. (2000) Mol. Biol. Evol. 17, 164-178. [DOI] [PubMed] [Google Scholar]
- 18.Gusfield, D. (1997) Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology (Cambridge Univ. Press, Cambridge, U.K.).
- 19.Atchley, W. R. & Fitch, W. M. (1995) Proc. Natl. Acad. Sci. USA 92, 10217-10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atchley, W. R. & Fitch, W. M. (1997) Proc. Natl. Acad. Sci. USA 94, 5172-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferre-D'Amare, A. R., Prendergast, G. C., Ziff, E. B. & Burley, S. K. (1993) Nature 363, 38-45. [DOI] [PubMed] [Google Scholar]
- 22.Bailey, T. L. & Elkan, C. (1994) in Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology (AAAI Press, Menlo Park, CA), pp. 28-36. [PubMed]
- 23.Bailey, T. L. & Gribskov, M. (1998) Bioinformatics 14, 48-54. [DOI] [PubMed] [Google Scholar]
- 24.Durbin, R., Eddy, S., Krogh, A. & Mitchison, G. (1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids (Cambridge Univ. Press, Cambridge, U.K.).
- 25.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J. H., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.