

Abstract
Utilization of tumor-only sequencing has expanded in pediatric cancer patients, which can lead to identification of pathogenic variants in genes that may be germline and/or have uncertain relevance to the tumor in question, such as the homologous recombination (HR) pathway genes BRCA1/2. We identified patients with pathogenic BRCA1/2 mutations from somatic tumor sequencing and performed additional germline sequencing to assess for the presence of loss of heterozygosity (LOH). Of seven patients identified, four (57.1%) mutations were found in the germline and none had associated LOH. Our data suggests that BRCA1/2 mutations identified in this context are likely incidental findings.
Keywords: pediatric oncology, BRCA, pediatric oncology, cancer, genomics, homologous recombination
1. Introduction
Somatic tumor sequencing has led to a vast increase in our understanding of pediatric cancer pathogenesis, and in many cases offers clinically significant information related to diagnosis, prognosis and treatment selection.1–4 While pediatric cancers compared to adult cancers have lower mutational rates and harbor less frequent targetable kinase alterations (such as EGFR or HER2 in adult lung or breast cancer, respectively), they tend to be enriched in targetable gene fusions such as NTRK (infantile fibrosarcoma, other solid and CNS tumors) and BRAF (low grade glioma).5–7 Additionally, paired sequencing of germline can reveal information with wide-ranging clinical implications as pediatric cancer patients have been found to have a high rate of mutations in cancer susceptibility genes which can inform treatment decisions for the patient and guide surveillance recommendations for the patient and their family.8,9
Homologous recombination (HR) is a high-fidelity repair mechanism that is critical in repairing DNA double-stranding breaks.10 Germline genomic alterations of HR pathway components, mainly BRCA1/2, have been associated with predisposition to adult-onset breast and ovarian cancers which develop when the other BRCA1/2 allele is lost in the tumor (second hit).11,12 The resulting tumors are BRCA1/2 deficient which leads to sensitivity to PARP inhibitors through a “synthetic lethality” interaction, which has been exploited with encouraging results in BRCA1/2 mutated breast, ovarian, and prostate cancer patients.13–15 Biallelic inactivation of BRCA1/2 is associated with abnormal DNA repair which leads to a mutational signature known as “signature 3” or “BRCAness”.16 Germline pathogenic BRCA1/2 mutations have been reported in several pediatric sequencing studies, with rates variably reported higher than the baseline population and some reports suggest a higher than expected incidence of pediatric cancers in families with germline BRCA1/2 mutations.8,9,17,18 However, no studies have systematically evaluated whether pediatric cancers with germline or somatic BRCA1/2 mutations have loss of heterozygosity or signature 3 characteristics. This is an important unanswered question as it would be informative with regards to tumor pathogenesis and activity of PARP inhibitors.
2. Methods
2.1. Patients and Samples
This study was approved by the Dana-Farber Cancer Institute (DFCI; Boston, MA) Institutional Review Board. Patients were identified for inclusion in the study by querying a database of 278 pediatric patients with solid and CNS malignancies who had samples subjected to tumor only sequencing with a targeted next-generation sequencing assay, OncoPanel, as a participant in the PROFILE Cancer Research Study.19 Patients were included in this study if sufficient tumor DNA or material was available for re-sequencing and if germline DNA was available.
2.2. Next Generation Sequencing
Molecular profiling of tumor and germline DNA was achieved via massively parallel sequencing with the OncoPanel V3 platform as previously described.20 Briefly, samples were sequenced on an Illumina HiSeq 2500 with 2x100 paired end reads. Sequence reads were aligned to reference sequence b37 edition from the Human Genome Reference Consortium using bwa, duplicate reads were removed using Picard (version 1.90, http://broadinstitute.github.io/picard/) and indel sites were locally realigned using Genome Analysis Toolkit (GATK, version 1.6–5-g557da77).21 Single nucleotide variants were called using MuTect v1.1.4,insertions and deletions were called using GATK Indelocator, and variants that were also present in the matched germline were removed.22 MMR deficiency using Oncopanel data was determined using the clinically validated threshold of at least 2.5 single base pair insertions or deletion mutations per megabase sequenced, occurring in mononucleotide repeat regions of four or more nucleotides.23 Loss of heterozygosity (LOH) and copy-neutral LOH assessment were determined by analyzing genome-wide copy number calls and allelic frequencies using FACETS (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing).24 Mutational signature analysis was performed using SigMA (Signature Multivariate Analysis).25 BRCA1/2 mutations were evaluated for pathogenicity using the ClinVar database and in one case of a previously unreported variant using functional prediction models PolyPhen and SIFT and comparison to other reported pathogenic variants.26–28 Only patients with BRCA1/2 pathogenic or likely pathogenic mutations were included.
3. Results
3.1. Study Population and Clinical Characteristics
From the PROFILE database (n=278 at the time of initial query), 7 patients (2.5%) were identified with a likely pathogenic or pathogenic mutation in either BRCA1 or BRCA2. All patients had sufficient tumor tissue and available germline DNA to undergo targeted somatic and germline sequencing. Patients with BRCA1/2 pathogenic mutations had glioblastoma (1), low-grade glioma (1), ganglioglioma (1), Wilm’s tumor (2) and hepatocellular carcinoma (2). Clinical characteristics of the seven patients in the study cohort are summarized in Supplementary Table 1.
3.2. Identified pathogenic BRCA1/2 variants
Of the 7 patients with BRCA1/2 mutations there were 4 patients with germline mutations for a rate of germline BRCA1/2 mutations of 1.4% in the 278 patient study population (Table 1). One patient had a BRCA1 germline mutation (0.4%) and three had a germline BRCA2 mutation (1.1%). There were three somatic mutations (rate of 1.1% in the study population); one in BRCA1 and two in BRCA2. All pathogenic BRCA1/2 mutations except for the somatic BRCA1 mutation were nonsense or frameshift mutations. Of note, one patient with a high-grade glioma (referred to as P1 hereafter) had a high tumor mutational burden (TMB, 302 mutations per megabase) with a mutational signature that was indicative of underlying mismatch repair (MMR) deficiency. No other patients were found to be MMR deficient. Analysis of P1’s germline sequencing results uncovered a homozygous PMS2 missense mutation (c.133A>G, p.N45D). This amino acid change occurs at a highly conserved residue, and is predicted to affect protein function using in-silico modeling (PolyPhen-2).26 It has not been identified in cancer databases such as PeCan, COSMIC, or cBioPortal; however, other variants involving this codon (p.N45T and p.N45S) have been reported as either likely pathogenic or variants of unknown significance.29
TABLE 1:
Clinical and molecular characteristics of patients with pathogenic BRCA1/2 mutations
| Patient # | Age | Sex | Diagnosis | Gene | Variant c. | Variant p. | Germline | VAF (%) | LOH | TMB | MMR | SNV# |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 13 | M | Glioblastoma Multiforme | BRCA2 | c.7480C>T | p.R2494* | No | 46 | No | 302 | deficient | 421 |
| P2 | 3 | M | Wilms Tumor | BRCA1 | c.5363G>T | p.G1788V | No | 45 | No | 3 | proficient | 3 |
| P3 | 15 | M | Ganglioglioma | BRCA2 | c.5128–5131delTATG | p.Y1710fs | Yes | 39 | No | 11 | proficient | 1 |
| P4 | 1 | M | Low Grade Glioma | BRCA1 | c.4136–4137delCT | p.S1379fs | Yes | 41 | No | 10 | proficient | 2 |
| P5 | 24 | F | Hepatocellular Carcinoma | BRCA2 | c.2581C>T | p.Q861* | No | 23 | No | 9 | proficient | 6 |
| P6 | 4 | M | Wilms Tumor | BRCA2 | c.5946delT | p.S1982fs | Yes | 47 | No | 7 | proficient | 3 |
| P7 | 14 | F | Hepatocellular Carcinoma | BRCA2 | c.3167–3171delAAAAG | p.K1057Tfs | Yes | 36 | No | 8 | proficient | 9 |
Nonsense mutation
TMB= tumor mutational burden (per megabase); MMR = mismatch repair; VAF = Variant Allele Frequency; SNV = Single nucleotide variant
LOH = Loss of heterozygosity
3.3. Determination of LOH and Mutational Signature Analysis
Gene-level copy number analysis of somatic samples did not reveal LOH associated with any of the identified pathogenic alterations. Using both the tumor and matched normal sequencing data, we applied an allele-specific copy number analysis pipeline to identify potential copy-neutral LOH, which was not seen in any cases (example shown in Supplementary Figure 1). To detect signature 3 (so called “BRCAness”) we used a computational method termed Signature Multivariate Analysis (SigMA). This validated technique uses a likelihood-based multivariate approach and machine learning that enables signature calling from low mutation counts, broadening its’ application to targeted gene panels which are much more commonly utilized in clinical practice than whole-exome or genome sequencing. The algorithm was unable to reliably call signatures in our samples, in large part due to the small number of exonic mutations in most samples (Supplementary Table 2). This finding was not unexpected given the known lower mutational burden of pediatric compared to adult cancers.30
4. Discussion
Somatic pathogenic mutations in BRCA1/2 are identified rarely in pediatric cancers, with an estimated incidence of 0.66% (BRCA1) and 1.1% (BRCA2) from the St. Jude’s PeCAN data portal (data accumulated from PCGP, TARGET, and DKFZ sequencing studies with total n = 3047).31 Several studies have investigated the specific incidence of germline mutations of BRCA1/2 in this population, and in some cases identified an incidence higher than the general population which is estimated at 0.16% and 0.25%, for BRCA1 and 2 respectively in the gnomAD database.32 Specifically, sequencing studies of pediatric patients with medulloblastoma (MB) and rhabdomyosarcoma (RMS) have identified relatively high rates of germline BRCA2 pathogenic mutations giving an estimated relative risk of >4 for MB, and an odds ratio of 3.6 for RMS.33,34 Recently, Ewing sarcoma was also found to have higher than expected rates of germline BRCA mutations.35 A recent meta-analysis of 11 studies totaling 3795 children and adolescents with cancer and germline genomic data suggested that BRCA2 (odds ratio 3.81 (1.97 to 7.10)) but not BRCA1 (odds ratio 1.83 (0.77 to 3.91)) pathogenic variants were associated with statistically significant increased cancer risk when compared to two control populations.36 These studies suggest a possible link between germline pathogenic variants and pediatric cancer development; however, proof of causality through the “second hit” mechanism observed in breast cancer has not been reported.
Although our study population was small, we identified a similar rate of germline (1.4%) and somatic (0.4%) BRCA1/2 mutations as previous studies. We did not observe LOH in patients with germline BRCA1/2 mutations. Lending further credence to our findings, a recent case-control study investigated the association of high- and moderate-penetrance germline pathogenic variants in associated vs. non-associated pediatric and adult-onset cancers.37 This study identified 5 patients with BRCA1, BRCA2 or PALB2 pathogenic germline mutations and none had LOH or a second hit in the tumor.
It remains unclear why an increased frequency of BRCA1/2 germline mutations has been identified in pediatric cancer populations. Possible hypotheses include an alternative mechanism of germline BRCA1/2-induced oncogenesis in pediatric patients. Another possibility is the comparison of a pediatric cancer population to an unaffected adult population where some BRCA1/2 carriers are likely to have already presented with an early-onset cancer is not appropriate. Indeed, two published responses to the Kratz et al study disputed their findings by pointing out biases present within both control populations, and propose that only a true age-matched control population could be used to properly determine risk.38.39
There are several limitations to ourstudy. Because a targeted sequencing approach was used, there may be some localized LOH regions that could not be detected at the resolution of SNP sites on the panel. It’s also possible that loss of the second allele could have occurred through mechanisms unable to be detected through targeted sequencing such as epigenetic silencing or enhancer hijacking. While we attempted to perform signature 3 analysis using the machine-learning predicter SigMA to address these issues, this did not result in any high-confidence results, likely due to the low mutational burden of most pediatric tumor samples and further limitations of panel sequencing coverage.
A recent paper from the SickKids Cancer Sequencing (KiCS) program suggests further work is needed to evaluate the performance of HR deficiency signature analysis in pediatric cancers.40 Out of 293 patients with whole-genome somatic sequencing data, 76 (25.9%) were identified as having signature 3, which was enriched in the 25 patients with germline pathogenic/likely-pathogenic variants affecting the HR pathway (12/25, 48%). Similar to our study, LOH of the germline variant was found in a minority of pediatric tumors and did not always correlate with the presence of signature 3. Furthermore, the HRDetect algorithm was used as a parallel method and did not show meaningful correlation with samples the authors had determined to harbor signature 3, which further suggests biological differences between homologous recombination proficiency in pediatric vs. adult cancers.
In summary, BRCA1/2 pathogenic germline mutations, while identified more often in pediatric solid and CNS cancer patients than in unaffected adults, likely do not give rise to cancer through a two-hit mechanism and may not be responsible for tumorigenesis in the younger population. These findings should be validated in larger studies ideally using whole genome and whole transcriptome sequencing data.
Supplementary Material
Supplementary Figure 1: Representative FACETS sample plot for Patient P2
Representative allele-specific copy number plot from FACETs for patient P2. The panels show log2-based copy number ratios and based on read counts from tumor and normal sequencing, the allelic log-odds-ratio of variant allele read counts, and the integer copy number calls. Copy number segments are shown as red horizontal lines in the top two panels, and were used to determine copy number aberrant or loss of heterozygosity regions of the genome.
Figure 1:
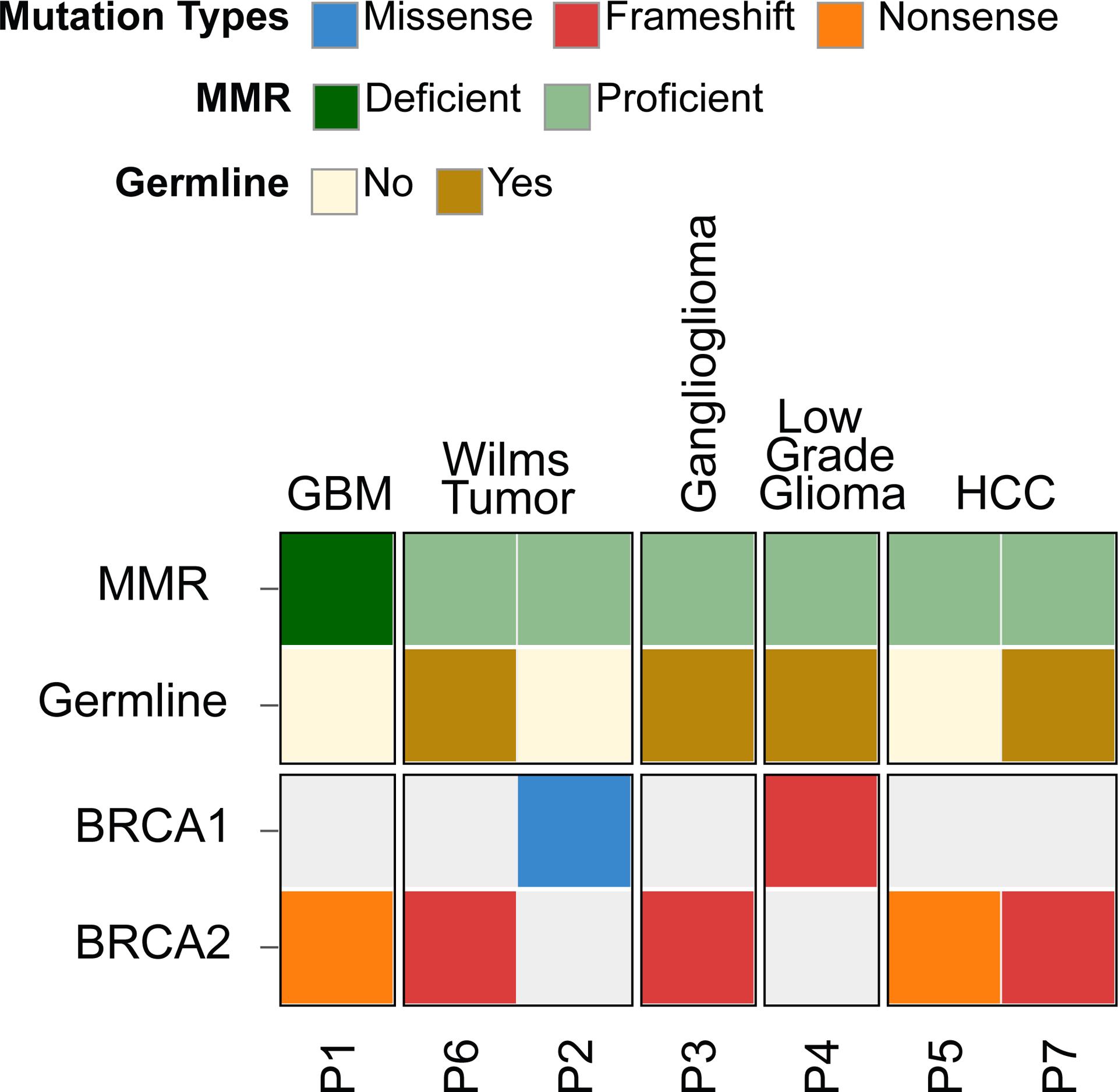
Heatmap of genomic features of patients with pathogenic BRCA1 and BRCA2 alterations
Acknowledgements
We would like to thank the patients and families who participated in the Profile study. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516. We would like to acknowledge the Profile study at Dana-Farber/Brigham and Women’s Cancer Center and Dana-Farber/Boston Children’s Cancer and Blood Disorders Center for generating the sequencing data for the Profile cohort. The authors would like to acknowledge the DFCI Oncology Data Retrieval System (OncDRS) for the aggregation, management, and delivery of the clinical and operational research data used in this project for the Profile cohort. Support for this study was provided by the Lamb Family Fund (KA Janeway), the Precision for Kids Pan Mass Challenge Team (KA Janeway) and the 4C’s Fund (KA Janeway).
Abbreviation Table
- HR
Homologous Recombination
- FFPE
Formalin fixed paraffin-embedded
- LOH
Loss of heterozygosity
- TMB
Tumor mutational burden
- MMR
Mismatch repair
- VAF
Variant Allele Frequency
- SNV
Single Nucleotide Variant
Footnotes
Conflict of Interest Statement
KAJ has received honoraria from Takeda and Foundation Medicine and performed consulting for Bayer, Ipsen and Illumina.
References
- 1.Harris MH, DuBois SG, Glade Bender JL, et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA Oncol. May 1 2016;2(5):608–615. doi: 10.1001/jamaoncol.2015.5689 [DOI] [PubMed] [Google Scholar]
- 2.Mody RJ, Wu YM, Lonigro RJ, et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA. Sep 1 2015;314(9):913–25. doi: 10.1001/jama.2015.10080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsons DW, Roy A, Yang Y, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. May 1 2016;2(5):616–624. doi: 10.1001/jamaoncol.2015.5699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Worst BC, van Tilburg CM, Balasubramanian GP, et al. Next-generation personalised medicine for high-risk paediatric cancer patients - The INFORM pilot study. Eur J Cancer. Sep 2016;65:91–101. doi: 10.1016/j.ejca.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science. Mar 29 2013;339(6127):1546–58. doi: 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. May 2018;19(5):705–714. doi: 10.1016/S1470-2045(18)30119-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. Nov 1 2008;68(21):8673–7. doi: 10.1158/0008-5472.CAN-08-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. Mar 15 2018;555(7696):321–327. doi: 10.1038/nature25480 [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Walsh MF, Wu G, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med. Dec 10 2015;373(24):2336–2346. doi: 10.1056/NEJMoa1508054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. Jan 25 2002;108(2):171–82. doi: 10.1016/s0092-8674(02)00615-3 [DOI] [PubMed] [Google Scholar]
- 11.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. Oct 7 1994;266(5182):66–71. doi: 10.1126/science.7545954 [DOI] [PubMed] [Google Scholar]
- 12.Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. Dec 21-28 1995;378(6559):789–92. doi: 10.1038/378789a0 [DOI] [PubMed] [Google Scholar]
- 13.Robson M, Im SA, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. Aug 10 2017;377(6):523–533. doi: 10.1056/NEJMoa1706450 [DOI] [PubMed] [Google Scholar]
- 14.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. Jul 24 2010;376(9737):245–51. doi: 10.1016/S0140-6736(10)60893-8 [DOI] [PubMed] [Google Scholar]
- 15.de Bono J, Mateo J, Fizazi K, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. May 28 2020;382(22):2091–2102. doi: 10.1056/NEJMoa1911440 [DOI] [PubMed] [Google Scholar]
- 16.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. Aug 22 2013;500(7463):415–21. doi: 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magnusson S, Borg A, Kristoffersson U, Nilbert M, Wiebe T, Olsson H. Higher occurrence of childhood cancer in families with germline mutations in BRCA2, MMR and CDKN2A genes. Fam Cancer. 2008;7(4):331–7. doi: 10.1007/s10689-008-9195-7 [DOI] [PubMed] [Google Scholar]
- 18.Brooks GA, Stopfer JE, Erlichman J, Davidson R, Nathanson KL, Domchek SM. Childhood cancer in families with and without BRCA1 or BRCA2 mutations ascertained at a high-risk breast cancer clinic. Cancer Biol Ther. Sep 2006;5(9):1098–102. doi: 10.4161/cbt.5.9.3167 [DOI] [PubMed] [Google Scholar]
- 19.Sholl LM, Do K, Shivdasani P, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. Nov 17 2016;1(19):e87062. doi: 10.1172/jci.insight.87062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia EP, Minkovsky A, Jia Y, et al. Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of Somatic Variants in Cancer. Arch Pathol Lab Med. Jun 2017;141(6):751–758. doi: 10.5858/arpa.2016-0527-OA [DOI] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. Sep 2010;20(9):1297–303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. Mar 2013;31(3):213–9. doi: 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papke DJ Jr., Nowak JA, Yurgelun MB, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod Pathol. Dec 2018;31(12):1882–1890. doi: 10.1038/s41379-018-0091-x [DOI] [PubMed] [Google Scholar]
- 24.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. Sep 19 2016;44(16):e131. doi: 10.1093/nar/gkw520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. May 2019;51(5):912–919. doi: 10.1038/s41588-019-0390-2 [DOI] [PubMed] [Google Scholar]
- 26.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. Jan 4 2018;46(D1):D1062–D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. Apr 2010;7(4):248–9. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. Jan 2016;11(1):1–9. doi: 10.1038/nprot.2015.123 [DOI] [PubMed] [Google Scholar]
- 29.Picanço-Albuquerque CG, Bezerra MJB, Silva PGdB, et al. A new PMS2 gene variant of unknown significance: How pathogenic is it? Journal of Clinical Oncology. 2020;38(15_suppl):e13532–e13532. doi: 10.1200/JCO.2020.38.15_suppl.e13532 [DOI] [Google Scholar]
- 30.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science. Mar 29 2013;339(6127):1546–58. doi: 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.St Jude Children’s Research Hospital St Jude PeCan data portal. Accessed April 5, 2022. http://pecan.stjude.org/proteinpaint/study/target-tall
- 32.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. May 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waszak SM, Northcott PA, Buchhalter I, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. Jun 2018;19(6):785–798. doi: 10.1016/S1470-2045(18)30242-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Sisoudiya SD, Martin-Giacalone BA, et al. Germline Cancer Predisposition Variants in Pediatric Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J Natl Cancer Inst. Jul 1 2021;113(7):875–883. doi: 10.1093/jnci/djaa204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillani R, Camp SY, Han S, et al. Germline predisposition to pediatric Ewing sarcoma is characterized by inherited pathogenic variants in DNA damage repair genes. Am J Hum Genet. Jun 2 2022;109(6):1026–1037. doi: 10.1016/j.ajhg.2022.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kratz CP, Smirnov D, Autry R, et al. Heterozygous BRCA1 and BRCA2 and Mismatch Repair Gene Pathogenic Variants in Children and Adolescents With Cancer. J Natl Cancer Inst. Nov 14 2022;114(11):1523–1532. doi: 10.1093/jnci/djac151 [DOI] [PubMed] [Google Scholar]
- 37.Mutetwa T, Goudie C, Foulkes WD, Polak P. Companion Tumor Sequencing to Assess the Clinical Significance of Germline Sequencing in Children With Cancer. JAMA Netw Open. Nov 1 2021;4(11):e2135135. doi: 10.1001/jamanetworkopen.2021.35135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans DG, Woodward ER. RE: Heterozygous BRCA1/BRCA2 and mismatch repair gene pathogenic variants in children and adolescents with cancer. J Natl Cancer Inst. Feb 8 2023;115(2):224–225. doi: 10.1093/jnci/djac223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Nguyen-Dumont T, Southey MC, Hopper JL. RE: Heterozygous BRCA1 and BRCA2 and mismatch repair gene pathogenic variants in children and adolescents with cancer. J Natl Cancer Inst. Jun 8 2023;115(6):757–759. doi: 10.1093/jnci/djad056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villani A, Davidson S, Kanwar N, et al. The clinical utility of integrative genomics in childhood cancer extends beyond targetable mutations. Nat Cancer. Feb 2023;4(2):203–221. doi: 10.1038/s43018-022-00474-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Representative FACETS sample plot for Patient P2
Representative allele-specific copy number plot from FACETs for patient P2. The panels show log2-based copy number ratios and based on read counts from tumor and normal sequencing, the allelic log-odds-ratio of variant allele read counts, and the integer copy number calls. Copy number segments are shown as red horizontal lines in the top two panels, and were used to determine copy number aberrant or loss of heterozygosity regions of the genome.