

Abstract
Although aminoacyl-tRNA synthetases (ARSs) are essential for protein synthesis, they also function as regulators and signaling molecules in diverse biological processes. Here, we screened 11 different human ARSs to identify the enzyme that is secreted as a signaling molecule. Among them, we found that lysyl-tRNA synthetase (KRS) was secreted from intact human cells, and its secretion was induced by TNF-α. The secreted KRS bound to macrophages and peripheral blood mononuclear cells to enhance the TNF-α production and their migration. The mitogen-activated protein kinases, extracellular signal-regulated kinase and p38 mitogen-activated protein kinase, and Gαi were determined to be involved in the signal transduction triggered by KRS. All of these activities demonstrate that human KRS may work as a previously uncharacterized signaling molecule, inducing immune response through the activation of monocyte/macrophages.
Keywords: aminoacyl-tRNA synthetase, cytokine, TNF-α, immune response, cell migration
Aminoacyl-tRNA synthetases (ARSs) ligate specific amino acids to their cognate tRNAs for protein synthesis. However, they are also pleiotropic proteins regulating various biological processes (1, 2). The noncanonical activities of these enzymes appear to be more prevalent in mammalian systems. For instance, human methionyl-tRNA synthetase is translocated to nucleoli to stimulate rRNA biogenesis under proliferative conditions (3), and glutaminyl-tRNA synthetase blocks apoptosis through a glutamine-dependent interaction with apoptosis signal-regulating kinase 1 (4). Human tyrosyl-tRNA synthetase is converted into two distinct cytokines (5, 6), whereas tryptophanyl-tRNA synthetase (WRS) is processed to a cytokine through alternative splicing (7, 8). Recently, glutamyl-prolyl-tRNA synthetase has been identified as a component of the IFN-γ-activated inhibitor of translation complex involved in gene-specific silencing of translation (9). In this work, we identified human lysyl-tRNA synthetase (KRS) as a previously uncharacterized signaling molecule that activates immune cells and determined its signal mediators.
KRS is distinguished from other ARSs in several aspects. First, it has a unique evolutionary pathway. ARSs are separated into two classes depending on their structural features (10). Although most ARSs belong to either one of the two classes, both forms exist in KRS (11, 12). Second, it can synthesize diadenosine polyphosphates in addition to its aminoacylation activity (13, 14). This activity was recently shown to play a role in transcription control through microphthalmia transcription factor (15). Third, mammalian KRS is a component of the multi-ARS complex (16), specifically interacting with an auxiliary factor, p38 (17, 18), and contains a lysine-rich N-terminal extension that binds tRNA and enhances the catalytic efficiency (19, 20). Fourth, a single KRS gene encodes both of the cytoplasmic and mitochondrial forms by alternative splicing (21). Fifth, it is associated with various human diseases. For instance, patients with inflammatory myopathies have an autoantibody against KRS (22), and some pathogenic mutants of superoxide dismutase 1, causing amyotrophic lateral sclerosis, binds to KRS (23). Sixth, human KRS and its cognate tRNA are incorporated into HIV type 1 (24, 25), and KRS interacts with HIV1-gag during viral assembly (26). The extracellular signaling activity of KRS identified in this work further expands its functional versatility and provides insight into its relationship with the associated human diseases.
Materials and Methods
Plasmid Construction. Human LRS cDNA was generated by the cleavage of pM370 (pλDR2 containing cDNA for human LRS) with BamHI and XhoI, VRS cDNA by partial digestion of pG7a-1 (27) with EcoRI and XhoI, AlaRS cDNA by the digestion of pKS370 (pTZ19R containing cDNA for human AlaRS) with EcoRI and NotI, MRS cDNA by the cleavage of pM184 (pλDR2 containing cDNA for human MRS) with EcoRI and XhoI, TRS cDNA by partial digestion of pM271 (pET-3a containing cDNA for human TRS) with EcoRI and XhoI, GRS cDNA by partial digestion of pM99 (pET-3a containing cDNA encoding human GRS) with EcoRI and NotI, arginyl-tRNA synthetase (RRS) cDNA by the cleavage of pM255 (pTZ19R containing cDNA for human RRS) with EcoRI and SalI, KRS cDNA by the cleavage of pM116 (pET-3a containing cDNA for human KRS) with EcoRI and XhoI, SRS cDNA by the cleavage of pM149 (pBlueScriptKS containing human SRS) with EcoRI and NotI, and HRS cDNA by partial digestion of pM204 (pTZ19R containing human HRS) with EcoRI and XhoI. Human DRS cDNA was obtained by PCR with specific primers from pM258 (pTZ19R containing cDNA for human DRS). All of these cDNAs were ligated into the corresponding sites in pcDNA3 (Invitrogen) containing Myc tag. The original ARS plasmids were kindly provided by K. Shiba (Cancer Institute of the Japanese Foundation for Cancer Research, Tokyo).
KRS Preparation. Human KRS cDNA was subcloned into pET-28a (Novagen) with EcoRI and SalI and overexpressed in Escherichia coli BL21 (DE3). The his-tagged KRS was then purified by using nickel affinity (Invitrogen) and Mono Q ion-exchange chromatography by following the manufacturer's instructions. To remove lipopolysaccharide (LPS), the KRS solution was dialyzed in pyrogen-free buffer (10 mM potassium phosphate buffer, pH 6.0/100 mM NaCl) and passed through polymyxin resin (Bio-Rad) equilibrated with pyrogen-free buffer. To further remove residual LPS, the solution was dialyzed against PBS containing 20% glycerol and filtered through Posidyne membrane (Pall Gelman Laboratory).
Secretion Test. HEK 293 cells were cultivated in DMEM containing 10% FBS (GIBCO) to ≈50% confluency. Then, each of ARS-expressing vectors was transfected into the cells by using geneporter (Gene Therapy Systems, San Diego) according to the manufacturer's instructions and incubated for 24 h. The cells were washed twice and further cultivated in serum-free DMEM for 6 h. The culture supernatants were carefully collected, centrifuged at 1,500 × g for 3 min, and the supernatants were centrifuged again at 26,000 × g to further remove debris. The supernatants were then concentrated by using VIVAspin filters (10-kDa cutoff) (Viva-science, Hannover, Germany). The concentrated proteins were separated by SDS/PAGE, and the secreted ARSs were identified by Western blotting with anti-Myc antibody. To determine whether the endogenous KRS was secreted, different cells cultivated in the complete medium were transferred to serum-free medium, and treated with 10 ng/ml TNF-α or 2 ng/ml TGF-β for 24 h. The culture supernatants were harvested and the proteins were precipitated with 50% ethanol, separated by SDS/PAGE, and subjected to Western blotting with an anti-KRS antibody.
Cell Binding Assay. RAW264.7 cells (3 × 105) were seeded onto six-well dishes and cultured in DMEM with 10% FBS and 1% antibiotics. After the biotinylated KRS was added to the culture medium at the indicated concentrations, the cells were harvested, washed three times with cold PBS, lysed in lysis buffer (25 mM Tris·HCl, pH 7.4/150 mM NaCl/1 mM EDTA/1 mM sodium orthovanadate/10 mM sodium fluoride/12 mM β-glycerophosphate/1 mM DTT/1% Triton X-100/1% sodium deoxycholate/0.1% SDS/0.1 mM phenylmethylsulfonyl fluoride) containing protease inhibitors (Roche Molecular Biochemicals), and centrifuged at 26,000 × g for 15 min. The extracted proteins (40 μg) were resolved by SDS/PAGE, and both of the exogenously added and endogenous KRS were detected with a polyclonal anti-KRS antibody. For biotinylation, recombinant KRS (3 mg) was incubated with 0.1 mg/ml sulfo-NHS-SS-biotin (Pierce) in PBS on ice for 2 h. The remaining biotin was quenched with 100 mM Tris buffer (pH 7.5), and the reaction was dialyzed against PBS. RAW264.7 cells (1 × 105) were cultured on 22 × 22 mm cover glasses in DMEM with 10% FBS and 1% antibiotics for 12 h. The culture plates were incubated at room temperature for 30 min, and each of 50 nM biotin-labeled KRS, WRS, and BSA was added and further incubated for 20 min. The cells were fixed with 5% formalin for 10 min and washed with PBS three times. The cover glasses were incubated with 2% BSA in PBS for 30 min to inhibit nonspecific binding, and then the bound biotin-labeled KRS was captured with FITC-conjugated streptavidin. The biotin-labeled KRS was visualized by confocal immunofluorescence microscopy (×60, μ-Radiance, Bio-Rad). To determine the specificity of cell binding, cells were treated with 1 μM unlabeled KRS or BSA for 20 min before the treatment with 50 nM biotinylated KRS.
TNF-α Secretion Assay. RAW264.7 cells (2 × 104) were cultured on 24-well plates containing DMEM with 10% FBS and 1% antibiotics for 5 h. KRS, p43, and WRS each were added at the indicated concentrations for 2 h, and the medium was harvested after centrifugation at 3,000 × g for 5 min. The secreted TNF-α was detected by using a TNF-α ELISA kit following the manufacturer's instructions (Pharmingen).
RT-PCR. Cells (2 × 105) were cultivated on six-well plates for 12 h, starved in serum-free media for 2 h, and stimulated with TNF-α (10 ng/ml) for the indicated times. Total RNAs were isolated with TRIzol (Invitrogen), and RT-PCR was performed with the primers specific to the KRS or GAPDH cDNA.
Cell Migration Assay. Cell migration was determined by using 24-well Transwell chambers with polycarbonate membranes (5.0-μm pore size, Costar) as described in ref. 28. The wells were coated with 0.5 mg/ml gelatin (Sigma) in PBS and allowed to air dry. RAW264.7 cells or peripheral blood mononuclear cells (PB-MCs) were suspended in serum-free DMEM and added to the upper chamber at 1 × 105 cells per well. Phorbol 12-myristate 13-acetate (16 nM), KRS (10 nM), or WRS (10 nM) was placed in the lower well, and the cells were allowed to migrate for 7 h at 37°C in CO2 incubator. The cells were then fixed with 5% formalin in PBS for 30 min and washed with PBS three times. The cells were stained with hematoxylin (Sigma) for 30 min and washed with distilled water. Nonmigrant cells were removed from the upper face of the membrane with a cotton swab. The membranes were excised from the chamber and mounted with Gel/Mount (Biomeda, Foster City, CA). The migrant cells (those attached to the lower face of the membrane) were counted in high power fields (×20).
Preparation of PBMCs. We collected fresh blood from four healthy donors into heparin-coated blood collection tubes and immediately diluted the samples with prewarmed RPMI medium 1640 at 1:1 ratio. The diluted bloods were loaded onto a Ficoll cushion and centrifuged at 1,200 × g for 40 min. We then pooled PBMCs without disturbing the other layer, diluted 3-fold with RPMI medium 1640, and centrifuged at 1,800 × g for 15 min. This procedure was repeated three times, and PBMCs were finally resuspended in RPMI medium 1640.
Zymographic Assay. RAW264.7 cells (1 × 105) were cultured on 12-well plates for 12 h. The cells were then washed twice and starved for 3 h in serum-free medium. KRS, WRS, and phorbol 12-myristate 13-acetate were added at the indicated concentrations for 20 h. A 15-μl aliquot of the medium was mixed with 5× FOD buffer, and proteins were separated by electrophoresis on 8% SDS-gel-containing gelatin as described in ref. 28.
Mitogen-Activated Protein Kinase (MAPK) Assay. RAW264.7 cells (3 × 105) were cultured on six-well plates for 7 h, washed twice, and starved in serum-free DMEM for 3 h. The KRS-treated RAW264.7 cells were washed twice with cold PBS, and proteins were extracted with the lysis buffer used above, resolved by 10% SDS/PAGE, and transferred onto a polyvinylidene difluoride membrane (Millipore). Total and phosphorylated MAPKs were immunoblotted with their specific antibodies.
Chemical Inhibitor Treatment. RAW264.7 cells were pretreated with U0126 (50 μM), SB202190 (10 μM), SB203580 (10 μM) and LY294002 (10 μM) for 1 h and subsequently with KRS (10 nM) for 0.5 h (to see MAPK activation), for 2 h (TNF-α production), for 12 h (cell migration), and for 20 h (matrix metalloproteinase-9 induction). To determine the involvement of G proteins in the KRS signal, the cells were pretreated with cholera or pertussis toxin for 1 h to inhibit stimulatory G protein and inhibitory G protein (Gαi), respectively.
Results
TNF-α-Induced Secretion of KRS. To identify secretable ARSs, we cloned the cDNAs encoding 17 different human ARSs in a mammalian expression vector and transfected them into HEK293 cells. Among them, 11 ARSs that were well expressed have been used for the secretion tests. The cells transfected with each ARS were cultivated, and the culture media were collected without disrupting the cell membranes. Among the tested ARSs, full-size KRS and AlaRS were observed in the culture media, whereas no tubulin was leaked to the culture media (Fig. 1A). Because the secretion of KRS was apparent, we further investigated its secretion in more detail. We then expressed Myc-KRS in HEK293 cells and collected the culture medium at time intervals. KRS was detected from 6 h after transfection in the culture medium (Fig. 1B). To see whether the endogenous KRS can be secreted in a signal-dependent manner, we treated different cell lines with cytokines, such as TNF-α and TGF-β. In HCT116 colon cancer cells, KRS secretion was strongly increased by treatment with TNF-α but not with TGF-β (Fig. 1C). TNF-α-induced KRS secretion was also observed in DU145, SK-N-SH, and MCF-7 cells but not in several other cancer and normal cell lines (Table 1, which is published as supporting information on the PNAS web site). The time-dependent secretion of KRS was observed from HCT116 and other KRS-secreting cells after TNF-α treatment (Fig. 1D and data not shown). Quantitative analysis of the secreted KRS from four different KRS-secreting cell lines determined that 0.4-1% of total cellular KRS is secreted (data not shown). Thus, the secretion of the endogenous KRS appears to be a cell-specific and signal-dependent process.
Fig. 1.

TNF-α-dependent secretion of KRS. (A) Each of 11 different human ARSs was expressed in HEK293 cells by transfection and by their expression in whole-cell lysates (WCL), and secretion into the culture media was determined by Western blotting with an anti-Myc antibody. The expected molecular masses of ARSs, in kDa, are as follows: L, 138; V, 140; A, 107; M, 101; T, 82; D, 57; G, 78; R, 75; K, 68; S, 59; HRS, 57. LRS contains ≈4 kDa extra peptide originating from the vector, KRS runs slightly bigger than the expected size in the gel for unknown reason, and VRS lacking its N-terminal 300 amino acids was used because its full-size version was not expressed well. All of the other ARSs are expressed as full length. (B) HEK293 cells transfected with human KRS were incubated, and the culture medium was harvested at the indicated times. The lack of tubulin in the culture medium indicates the absence of cell lysis resulting from physical damage. (C) The macrophage cell line, RAW264.7, and the colon cancer cell line, HCT116, were treated with TNF-α (10 ng/ml) or TGF-β (2 ng/ml), and the secretion of KRS into the culture medium was determined with its specific antibody. (D) Time course of TNF-α-induced secretion of KRS from HCT116 cells.
KRS Binding to Immune Cells. Because KRS secretion was induced by proinflammatory cytokine, TNF-α, we assumed that the secreted KRS may act on immune cells. We examined whether KRS would bind to RAW264.7 cells. The recombinant human KRS was biotinylated, incubated with RAW264.7, and the cell-bound KRS was stained with FITC-conjugated streptavidin. KRS gradually accumulated on the cell membrane (Fig. 2A). The biotinylated KRS signal did not appear when the cells were pretreated with unlabeled KRS, and no FITC signal was observed when the cells were incubated with FITC-streptavidin alone or biotinylated WRS (Fig. 2B), confirming the specific cell binding of KRS. The cell binding of KRS was also verified by Western blotting of the protein extracts. Total proteins were extracted from RAW264.7 and THP-1 after treatment with His-KRS and subjected to Western blotting with anti-KRS antibody. The amount of exogenously treated KRS increased in a dose-dependent manner (Fig. 2C), whereas WRS did not bind to the cells (data not shown). For both cell types, the KRS binding became saturated at ≈200 nM, with ED at ≈105 nM (Fig. 2D). The saturable cell binding of KRS was also
Fig. 2.

Specific binding of KRS to immune cells. (A) KRS or p18 was biotinylated and incubated with RAW264.7 cells, which were harvested at the indicated times. The cell-bound KRS was reacted with FITC-conjugated streptavidin and visualized by confocal immunofluorescence microscopy. (B) The cell binding specificity of KRS. RAW264.7 cells were pretreated with unlabeled KRS or BSA, and the biotinylated KRS was added subsequently. The biotinylated WRS was also incubated with RAW246.7 cells. (C) RAW264.7 and THP-1 cells were incubated with the indicated concentrations of the His-tagged KRS, and, after they were harvested, the proteins were extracted. The exogenously added (exo) and endogenous (endo) KRS were detected by Western blotting with an anti-KRS antibody. (D) Cell binding by KRS was monitored as above and quantified with a phosphoimage analyzer (Fuji), and the amount of the cell-bound KRS was plotted.
50observed at 4°C (Fig. 6, which is published as supporting information on the PNAS web site), suggesting that the cell binding of KRS would be receptor-mediated.
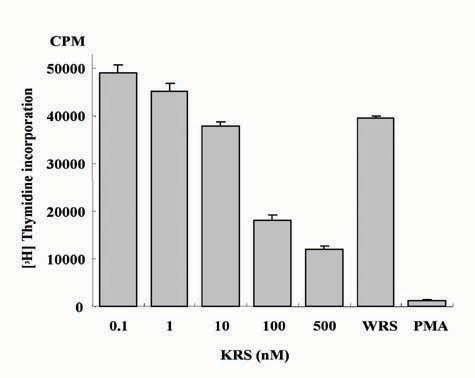
KRS Increases TNF-α Secretion from Macrophages. To determine the activity of the secreted KRS, we treated RAW264.7 and THP-1 with KRS, and its effects on cell proliferation, death, and migration were examined. The proliferation of both cells was inhibited by KRS in a dose-dependent manner (Fig. 3A; see also Fig. 7, which is published as supporting information on the PNAS web site). Phorbol 12-myristate 13-acetate, which is known to activate macrophages (29, 30), also inhibited proliferation, whereas the full-length WRS had no effect. Although KRS bound to the two cells with similar affinity, RAW264.7 cells appeared to respond more sensitively to KRS. Therefore, we used RAW264.7 for the following experiments. KRS did not induce cell death while suppressing the proliferation (data not shown), implying that it may activate macrophages. Because the activated macrophages secrete various inflammatory cytokines such as TNF-α (31), we tested whether TNF-α secretion was increased by KRS treatment by using ELISA. p43, known to induce TNF-α secretion from macrophage/monocytes (32, 33), was used as a positive control and WRS as a negative control. As expected, KRS induced TNF-α secretion from RAW264.7 cells like p43, whereas WRS had no effect (Fig. 3B). We also investigated whether KRS can induce the expression of TNF-α mRNA by RT-PCR. The expression of TNF-α was strongly enhanced by KRS in RAW264.7 but not in HCT116 cells (Fig. 3C). Conversely, we tested whether TNF-α can induce KRS expression. Although TNF-α induced p43 expression in RAW264.7 as expected (34), the transcription of KRS was not affected (Fig. 3D), implying that TNF-α would not induce de novo synthesis of KRS. Based on these results, the secreted KRS appears to work as an activating signal to macrophages.
Fig. 3.

The effect of KRS on macrophage proliferation. (A) The proliferation of RAW264.7 cells was monitored by the incorporation of tritium-labeled thymidine at different KRS concentrations. Phorbol 12-myristate 13-acetate (PMA) and WRS (7) were used as positive and negative controls, respectively. (B) The effect of KRS on the secretion of TNF-α from RAW264.7 cells. p43 (32) and WRS were used as positive and negative controls, respectively. (C) RAW264.7 and HCT116 cells were treated with 10 nM KRS for the indicated times, and the transcript levels for TNF-α and KRS were determined by RT-PCR as described in Materials and Methods.(D) The two cells were treated with 10 ng/ml TNF-α, and the expression of KRS and p43 was determined by RT-PCR. GAPDH was used as a control.
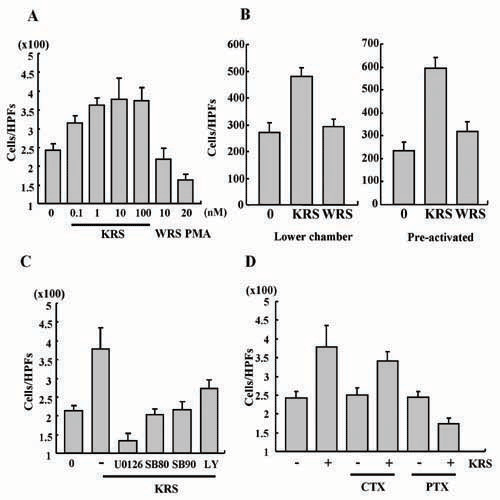
KRS Induces Migration of Macrophages. We then investigated the cell migration inducing activity of KRS by using the Transwell chamber assay. The migration of RAW264.7 cells was stimulated by KRS in a dose-dependent manner and by the N-terminal truncated minityrosyl-tRNA synthetase that is known to induce immune cell migration (6) (Fig. 4 A and B). Because gelatinases matrix metalloproteinase (MMP)-2 and -9, which degrade collagen type IV, is known to enhance cell migration (35, 36), we tested whether KRS can induce MMP-2 and -9 from RAW264.7 by using a zymography assay. The activity of MMP-9, but not MMP-2, was significantly increased by the treatment of KRS (Fig. 4C and data not shown). To determine how KRS induces the mobilization of the cells, we monitored the KRS-induced cell migration in two different ways. First, we put KRS into the lower well of Transwell chamber and RAW264.7 cells to the upper well. Second, we pretreated the cells with KRS (or WRS), washed the cells, placed them in the upper well, and no KRS (or WRS) was added to the lower well. Then, we counted the number of the cells that migrated through the membrane to the lower chamber. The number of the migrated cells also increased simply by the pretreatment with KRS even though KRS was not present in the lower well, implying that KRS stimulates chemokinesis of the target cells (Fig. 4D). To further confirm the KRS-induced chemokinesis, we first loaded KRS to the lower or upper or both wells and incubated the cells in the upper well. Then, we compared the number of cells that migrated from the upper to the lower well through the membrane. The number of migrated cells in these three cases were all increased compared with that of the case without the addition of KRS, but a similar number of cells migrated regardless of the location of KRS (Fig. 4E). We checked whether KRS also shows the activity against the primary immune cells by using PBMCs. KRS also induced the migration of PBMC (Fig. 8 A and B, which is published as supporting information on the PNAS web site). However, it did not induce the migration of endothelial cells (data not shown), indicating its specific activity to immune cells.
Fig. 4.

The effect of KRS on cell migration. (A) RAW264.7 cells (3 × 105) and KRS (10 nM) were added to the upper and lower wells of Transwell chamber, respectively, and the cells migrated through the membrane were detected by the hematoxylin staining as described in Materials and Methods. (B) Dose-dependent cell migration by KRS. (C) The KRS-dependent induction of MMP-9 was determined by a zymographic assay. RAW264.7 cells, treated as above, were lysed, and the proteins were resolved on SDS/PAGE-containing gelatin. The hydrolysis of gelatin was determined as described in ref. 28. (D Left) To determine how KRS induces cell migration, RAW264.7 cells were loaded onto the upper well, and KRS or WRS was added to the lower well and allowed them to migrate. (Right) The cells were pretreated with 10 nM KRS or WRS for 30 min and loaded to the upper well and incubated them to migrate without adding KRS or WRS to the lower well. Then, the migrated cells through the membrane were counted as above. (E) The cell migration was also determined in the Transwell chambers in which 10 nM KRS was added to the upper or lower or both wells. As the control, no KRS was added, or 10 nM WRS was added to the lower well.
KRS Signal Is Mediated by ERK, p38 MAPK, and Gαi. To identify the KRS signal mediators, we treated RAW264.7 cells with KRS, and the activation of three major MAPKs was monitored by their phosphorylation. The phosphorylation of ERK and p38 MAPK increased by KRS in dose- and time-dependent manners, whereas jun aminoterminal kinase was not affected (Fig. 5 A and B). To determine the functional significance of ERK and p38 MAPK for the KRS signaling, we pretreated the cells with U0126 that inhibits MEK, the upstream kinase for ERK, and the SB compounds that specifically block p38 MAPK (37, 38). The KRS-dependent activation of ERK and p38 MAPK was blocked with these chemical inhibitors (Fig. 5C). However, LY294002 did not show the effect, excluding the involvement of its target kinase, phosphatidylinositol 3-kinase in the KRS signaling. U0126 and the SB compounds also blocked the KRS-dependent TNF-α secretion (Fig. 5D), MMP-9 induction (Fig. 5E) and cell migration (Fig. 5F). Thus, ERK and p38 MAPK seem to mediate the KRS-dependent cytokine production and cell migration. To see whether G proteins are also involved in the KRS signal, it was added to RAW264.7 that were pretreated with cholera or pertussis toxin to inhibit Gαs and Gαi, respectively, and the effect of these toxins on the KRS-dependent growth suppression and cell migration was monitored by using RAW264.7 cells. The KRS-dependent growth suppression and cell migration were blocked by pertussis toxin but not by cholera toxin (Fig. 5 G and H). The similar results were also observed in PBMC (Fig. 8 C and D). Combined together, the Gαi and two MAPKs (ERK and p38 MAPK) appear to be involved in the signal transduction triggered by KRS.
Fig. 5.

Determination of mediators for KRS signaling. RAW264.7 cells were treated for 30 min with different concentrations of KRS (A) or for different times at 10 nM KRS (B). The activation of three major MAPKs, ERK, p38 MAPK, and JNK, was determined by detection of their phosphorylation with their phosphospecific antibodies. The total amount of each kinase was determined by Western blotting with specific antibodies. (C) RAW264.7 cells were pretreated with different kinase inhibitors, 50 μM U0126 (MEK inhibitor), 10 μM SB202190 (p38 MAPK inhibitor), 10 μM SB203580 (p38 MAPK inhibitor), and 10 μM LY294002 (phosphatidylinositol 3-kinase inhibitor), and 10 nM KRS was subsequently added. The specific inhibition of ERK and p38 MAPK was confirmed by using the method above. The effects of kinase inhibitors on the KRS-dependent TNF production (D) and MMP-9 production (E) and cell migration (F) were determined. The effects of cholera toxin (CTX) and pertussis toxin (PTX) on the KRS-induced growth suppression (G) and cell migration (H) are shown as previously described.
Discussion
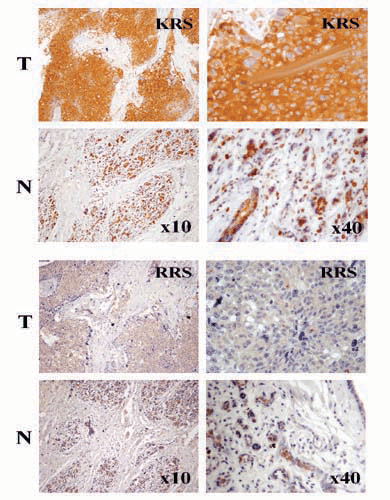
Among human ARSs, human tyrosyl-tRNA synthetase is secreted and cleaved by leukocyte elastase into two different cytokines (6). The cleaved C-terminal polypeptide shows chemotactic activity to immune cells, and induces TNF-α and tissue factor, whereas the N-terminal domain is similar to interleukin 8 (6). Human WRS (tryptophanyl-tRNA synthetase) undergoes alternative splicing to generate its N-terminal truncated form (39). Although these two enzymes need to be cleaved or truncated to become active cytokines, the full-length KRS itself works as the active cytokine (Fig. 3). In this regard, it is reminiscent of p43, a component of the multi-ARS complex, the full-length of which is secreted as an active cytokine (32, 34). Interestingly, the chemotactic activity of KRS was not observed against CD4+, CD8+ lymphocytes, interleukin-2-activated monocytes, dendritic cells, and neutrophils (40). Because the work focused on HRS (histidyl-tRNA synthetase) and NRS (asparaginyl-tRNA synthetase), the detailed information on the experimental condition for KRS is not available. Perhaps, the concentration of KRS may not have been in effective range or the tested cells may not have been sensitive enough to KRS. Although two different ARSs (tyrosyl-tRNA synthetase and WRS) and one ARS-associating factor, p43, have been reported to work as cytokines, it is not understood yet how their secretion is controlled. KRS lacks a typical signal peptide for secretion and is tightly associated with a factor, p38/JTV-1, within the multi-ARS complex in cytosol (18, 41). Because TNF-α induced the secretion but not the transcription of KRS, the preexisting KRS may be mobilized by TNF-α signal, perhaps through its posttranslational modification. Because <1% of total KRS is secreted (data not shown), the secretion would not significantly affect the cellular level of KRS. Interestingly, we found that KRS is highly expressed in the tumor regions of the breast cancer patients (Fig. 9, which is published as supporting information on the PNAS web site). In fact, overexpression of KRS in breast cancers has been also reported at Gene Expression Omnibus database (www.ncbi.nlm.gov/geo). Although it is not clear what would be the functional reason for the KRS overexpression in a tumor region, it may be related to the signaling property of KRS.
Because the secretion of KRS and TNF-α is enhanced by their counterpart cells in paracrine manner (Figs. 1C and 3), these two signaling molecules appear to form a positive feedback loop. Although both of KRS and p43 are capable of activating macrophages (33, 42), KRS does not work on endothelial cells in contrast to p43 (28) (data not shown). In addition, p43 activates all three MAPKs in immune cells (32), whereas KRS activates only ERK and p38 MAPK but not jun aminoterminal kinase (Fig. 5A). Thus, KRS appears to have its receptor and signal pathways distinguished from those of p43.
Although the physiological significance for the KRS secretion is yet to be evaluated, it may have a couple of pathophysiological implications. First, KRS may stimulate immune response against extragenous insults such as infections. Secondly, it may be related to the survival or metastasis of cancer cells. Interestingly, the secretion of KRS was detected in a few different cancer cell lines (Table 1). Perhaps, KRS secreted from these cancer cells may activate macrophages to produce TNF-α. Although TNF-α normally suppresses cell proliferation or induces cell death (43-46), the cancer cells can turn it to a proliferative signal (47). Thus, the KRS-induced secretion of TNF-α would help the growth of cancer cells. In addition, the KRS-dependent induction of MMP-9 (Fig. 4 B and C) may assist in cancer cell metastasis. Third, the extracellular KRS may be recognized as an autoantigen because some autoimmune patients suffering from myositis or transplant-associated coronary artery disease develop antiantibodies against KRS (48, 49).
Several different human ARSs and their binding factors showed previously uncharacterized regulatory activities (1, 2, 50). Using WRS as a model, we delineated how this enzyme structurally acquired its angiostatic cytokine activity (51). Here we report a signaling activity of KRS to the list of noncanonical ARS functions. The growing evidence for the multifunctionality of these enzymes strongly suggests that they have evolved to build a pluripotent functional network to coordinate various signal pathways with basic metabolic process such as protein synthesis.
Supplementary Material
Acknowledgments
This work was supported by a grant of National Creative Research Initiatives from the Ministry of Science and Technology, Korea.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ARS, aminoacyl-tRNA synthetase; ERK, extracellular signal-regulated kinase; Gαi, inhibitory G protein; KRS, lysyl-tRNA synthetase; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; PBMC, peripheral blood mononuclear cell; WRS, tryptophanyl-tRNA synthetase.
References
- 1.Ko, Y. G., Park, H. & Kim, S. (2002) Proteomics 2, 1304-1310. [DOI] [PubMed] [Google Scholar]
- 2.Han, J. M., Kim, J. Y. & Kim, S. (2003) Biochem. Biophys. Res. Commun. 303, 985-993. [DOI] [PubMed] [Google Scholar]
- 3.Ko, Y. G., Kang, Y. S., Kim, E. K., Park, S. G. & Kim, S. (2000) J. Cell Biol. 149, 567-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ko, Y. G., Kang, Y. S., Park, H., Seol, W., Kim, J., Kim, T., Park, H. S., Choi, E. J. & Kim, S. (2001) J. Biol. Chem. 276, 39103-39106. [DOI] [PubMed] [Google Scholar]
- 5.Kleeman, T. A., Wei, D., Simpson, K. L. & First, E. A. (1997) J. Biol. Chem. 272, 14420-14425. [DOI] [PubMed] [Google Scholar]
- 6.Wakasugi, K. & Schimmel, P. (1999) Science 284, 147-151. [DOI] [PubMed] [Google Scholar]
- 7.Wakasugi, K., Slike, B. M., Hood, J., Otani, A., Ewalt, K. L., Friedlander, M., Cheresh, D. A. & Schimmel, P. (2002) Proc. Natl. Acad. Sci. USA 99, 173-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tolstrup, A. B., Bejder, A., Fleckner, J. & Justesen, J. (1995) J. Biol. Chem. 270, 397-403. [DOI] [PubMed] [Google Scholar]
- 9.Sampath, P., Mazumder, B., Seshadri, V., Gerber, C. A., Chavatte, L., Kinter, M., Ting, S. M., Dignam, J. D., Kim, S., Driscoll, D. M. & Fox, P. L. (2004) Cell 119, 195-208. [DOI] [PubMed] [Google Scholar]
- 10.Eriani, G., Delarue, M., Poch, O., Gangloff, J. & Moras, D. (1990) Nature 347, 203-206. [DOI] [PubMed] [Google Scholar]
- 11.Ibba, M., Morgan, S., Curnow, A. W., Pridmore, D. R., Vothknecht, U. C., Gardner, W., Lin, W., Woese, C. R. & Soll, D. (1997) Science 278, 1119-1122. [DOI] [PubMed] [Google Scholar]
- 12.Ambrogelly, A., Korencic, D. & Ibba, M. (2002) J. Bacteriol. 184, 4594-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brevet, A., Geffrotin, C. & Kellermann, O. (1982) Eur. J. Biochem. 124, 483-488. [DOI] [PubMed] [Google Scholar]
- 14.Blanquet, S., Plateau, P. & Brevet, A. (1983) Mol. Cell. Biochem. 52, 3-11. [DOI] [PubMed] [Google Scholar]
- 15.Lee, Y. N., Nechushtan, H., Figov, N. & Razin, E. (2004) Immunity 20, 145-151. [DOI] [PubMed] [Google Scholar]
- 16.Wahab, S. Z. & Yang, D. C. (1985) J. Biol. Chem. 260, 12735-12739. [PubMed] [Google Scholar]
- 17.Robinson, J. C., Kerjan, P. & Mirande, M. (2000) J. Mol. Biol. 304, 989-994. [DOI] [PubMed] [Google Scholar]
- 18.Kim, J. Y., Kang, Y.-S., Lee, J.-W., Kim, H. J., Ahn, Y. H., Park, H., Ko, Y.-G. & Kim, S. (2002) Proc. Natl. Acad. Sci. USA 99, 7912-7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corti, O., Hampe, C., Koutnikova, H., Darios, F., Jacquier, S., Prigent, A., Robinson, J. C., Pradier, L., Ruberg, M., Mirande, M., et al. (2003) Hum. Mol. Genet. 12, 1427-1437. [DOI] [PubMed] [Google Scholar]
- 20.Francin, M., Kaminska, M., Kerjan, P. & Mirande, M. (2002) J. Biol. Chem. 277, 1762-1769. [DOI] [PubMed] [Google Scholar]
- 21.Tolkunova, E., Park, H., Xia, J., King, M. P. & Davidson, E. (2000) J. Biol. Chem. 275, 35063-35069. [DOI] [PubMed] [Google Scholar]
- 22.Gelpi, C., Kanterewicz, E., Gratacos, J., Targoff, I. N. & Rodriguez-Sanchez, J. L. (1996) Arthritis Rheum. 39, 692-697. [DOI] [PubMed] [Google Scholar]
- 23.Kunst, C. B., Mezey, E., Brownstein, M. J. & Patterson, D. (1997) Nat. Genet. 15, 91-94. [DOI] [PubMed] [Google Scholar]
- 24.Cen, S., Khorchid, A., Javanbakht, H., Gabor, J., Stello, T., Shiba, K., Musier-Forsyth, K. & Kleiman, L. (2001) J. Virol. 75, 5043-5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cen, S., Javanbakht, H., Niu, M. & Kleiman, L. (2004) J. Virol. 78, 1595-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Javanbakht, H., Halwani, R., Cen, S., Saadatmand, J., Musier-Forsyth, K., Gottlinger, H. & Kleiman, L. (2003) J. Biol. Chem. 278, 27644-27651. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh, S. L. & Campbell, R. D. (1991) Biochem. J. 278, 809-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park, S. G., Kang, Y. S., Ahn, Y. H., Lee, S. H., Kim, K. R., Kim, K. W., Koh, G. Y., Ko, Y. G. & Kim, S. (2002) J. Biol. Chem. 277, 45243-45248. [DOI] [PubMed] [Google Scholar]
- 29.Gieche, J., Mehlhase, J., Licht, A., Zacke, T., Sitte, N. & Grune, T. (2001) Biochim. Biophys. Acta 1538, 321-328. [DOI] [PubMed] [Google Scholar]
- 30.Huang, W., Ishii, I., Zhang, W. Y., Sonobe, M. & Kruth, H. S. (2002) J. Lipid Res. 43, 1275-1282. [PubMed] [Google Scholar]
- 31.Gonzalez, A., Sahaza, J. H., Ortiz, B. L., Restrepo, A. & Cano, L. E. (2003) Med. Mycol. 41, 391-399. [DOI] [PubMed] [Google Scholar]
- 32.Ko, Y.-G., Park, H., Kim, T., Lee, J.-W., Park, S. G., Seol, W., Kim, J. E., Lee, W.-H., Kim, S.-H., Park, J. E. & Kim, S. (2001) J. Biol. Chem. 276, 23028-32303. [DOI] [PubMed] [Google Scholar]
- 33.Park, H., Park, S. G., Kim, J., Ko, Y. G. & Kim, S. (2002) Cytokine 20, 148-153. [DOI] [PubMed] [Google Scholar]
- 34.Park, S. G., Shin, H., Shin, Y. K., Lee, Y., Choi, E. C., Park, B. J. & Kim, S. (2005) Am. J. Pathol. 166, 387-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraling, B. M., Wiederschain, D. G., Boehm, T., Rehn, M., Mulliken, J. B. & Moses, M. A. (1999) J. Cell Sci. 112, 1599-1609. [DOI] [PubMed] [Google Scholar]
- 36.Puyraimond, A., Weitzman, J. B., Babiole, E. & Menashi, S. (1999) J. Cell Sci. 112, 1283-1290. [DOI] [PubMed] [Google Scholar]
- 37.Engelman, J. A., Lisanti, M. P. & Scherer, P. E. (1998) J. Biol. Chem. 273, 32111-32120. [DOI] [PubMed] [Google Scholar]
- 38.Rottinger, E., Besnardeau, L. & Lepage, T. (2004) Development (Cambridge, U.K.) 131, 1075-1087. [DOI] [PubMed] [Google Scholar]
- 39.Liu, J., Shue, E., Ewalt, K. L. & Schimmel, P. (2004) Nucleic Acids Res. 32, 719-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howard, O. M., Dong, H. F., Yang, D., Raben, N., Nagaraju, K., Rosen, A., Casciola-Rosen, L., Hartlein, M., Kron, M., Yiadom, K., et al. (2002) J. Exp. Med. 196, 781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quevillon, S., Robinson, J.-C., Berthonneau, E., Siatecka, M. & Mirande, M. (1999) J. Mol. Biol. 285, 183-195. [DOI] [PubMed] [Google Scholar]
- 42.Park, H., Park, S. G., Lee, J.-W., Kim, T., Kim, G., Ko, Y.-G. & Kim, S. (2002) J. Leukocyte Biol. 71, 223-230. [PubMed] [Google Scholar]
- 43.Ichijo, H., Nishida, E., Irie, K., ten Dijke, P., Saitoh, M., Moriguchi, T., Takagi, M., Matsumoto, K., Miyazono, K. & Gotoh, Y. (1997) Science 275, 90-94. [DOI] [PubMed] [Google Scholar]
- 44.Tobiume, K., Matsuzawa, A., Takahashi, T., Nishitoh, H., Morita, K., Takeda, K., Minowa, O., Miyazono, K., Noda, T. & Ichijo, H. (2001) EMBO Rep. 2, 222-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu, H., Lo, C. R. & Czaja, M. J. (2002) Hepatology 35, 772-778. [DOI] [PubMed] [Google Scholar]
- 46.Baisch, H. (2002) Cell Prolif. 35, 333-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young, M. R. & Wright, M. A. (1992) Cancer Res. 52, 6335-6340. [PubMed] [Google Scholar]
- 48.Targoff, I. N., Trieu, E. P. & Miller, F. W. (1993) J. Clin. Invest. 91, 2556-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linke, A. T., Marchant, B., Marsh, P., Frampton, G., Murphy, J. & Rose, M. L. (2001) Clin. Exp. Immunol. 126, 173-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee, S. W., Cho, B. H., Park, S. G. & Kim, S. (2004) J. Cell Sci. 117, 3725-3734. [DOI] [PubMed] [Google Scholar]
- 51.Kise, Y., Lee, S. W., Park, S. G., Fukai, S., Sengoku, T., Ishii, R., Yokoyama, S., Kim, S. & Nureki, O. (2004) Nat. Struct. Mol. Biol. 11, 149-156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}