

Abstract
Proteins play an important role in life activities and are the basic units for performing functions. Accurately annotating functions to proteins is crucial for understanding the intricate mechanisms of life and developing effective treatments for complex diseases. Traditional biological experiments struggle to keep pace with the growing number of known proteins. With the development of high-throughput sequencing technology, a wide variety of biological data provides the possibility to accurately predict protein functions by computational methods. Consequently, many computational methods have been proposed. Due to the diversity of application scenarios, it is necessary to conduct a comprehensive evaluation of these computational methods to determine the suitability of each algorithm for specific cases. In this study, we present a comprehensive benchmark, BeProf, to process data and evaluate representative computational methods. We first collect the latest datasets and analyze the data characteristics. Then, we investigate and summarize 17 state-of-the-art computational methods. Finally, we propose a novel comprehensive evaluation metric, design eight application scenarios and evaluate the performance of existing methods on these scenarios. Based on the evaluation, we provide practical recommendations for different scenarios, enabling users to select the most suitable method for their specific needs. All of these servers can be obtained from https://csuligroup.com/BEPROF and https://github.com/CSUBioGroup/BEPROF.
Keywords: protein, protein function, deep learning, benchmark
INTRODUCTION
Proteins, as essential molecules in living organisms, play a pivotal role in a wide range of biological processes, including gene regulation, cytoskeletal support and material transport [1, 2]. Accurately annotating protein functions is indispensable for understanding the nature of biological activities [3], diagnosing the etiology of diseases [4] and accelerating the development of new drugs [5]. Despite the remarkable progress made by researchers in this field, there is still a significant number of proteins whose functions remain unclear. To date, less than 1% of proteins in the UniProt [6] database have functional annotations, and most of these annotations are obtained through expensive and time-consuming biological experiments [7], leading to a large gap between the number of proteins with known sequences and those with known functions. Consequently, it is meaningful and necessary to develop computational methods for automatic protein function prediction (AFP), which will not only deepen our understanding of living cells, but also have the potential to revolutionize medical research and treatment.
In recent years, significant advancements in high-throughput sequencing technology and computational methods have facilitated extensive access to protein information, leading to the development of diverse and valuable databases. For example, the UniProt [6] database catalogs protein sequences, the STRING [8] database houses protein–protein interactions (PPIs), the MEDLINE [9] database stores literature information describing proteins and the PDB [10] database provides experimental protein structures, while the AlphaFold2 [11] database offers predicted protein structures generated by AlphaFold. By leveraging these comprehensive data resources, a wide range of computational methods have been proposed to predict protein functions.
From a computational perspective, AFP can be regarded as a large-scale sparse multi-label classification problem. In the past, the lack of uniformity in function definitions among biologists posed a challenge for researchers in efficiently searching for relevant biological information. To address this challenge, several databases have been established, the most widely used of which is Gene Ontology (GO) [12]. GO comprises three domains to describe protein functions: biological process ontology (BPO), molecular function ontology (MFO) and cellular composition ontology (CCO). Specifically, BPO represents the biological activities in which a gene or its product is involved; MFO encompasses the biochemical activities of the gene product, such as binding to a specific ligand or structure; and CCO describes the specific location of the gene product in the cell. As shown in Figure 1, a GO term node represents a specific function, and each domain consists of a series of GO terms with different relationships between these terms, such as is-a’, part-of’, has-part’ and regulates’. These relationships establish a hierarchical structure and connections between GO terms. To date, there are approximately 32 857 BPO GO terms, 4744 MFO GO terms and 13 681 CCO GO terms. The goal of computational methods is to annotate proteins with these GO terms accurately.
Figure 1.

The main processes of existing approaches in AFP (left) and the loosely hierarchical structure of GO Terms (right). The structure of GO terms can be visualized as a graph, where each node represents a specific GO term, and the edges connecting the nodes represent the relationships between these terms. In AFP, two types of relationships (’is_a’ and ’part_of’) are used. These relationships establish a hierarchy resembling a ”parent-child” relationship, allowing the transfer of functions between related terms in a reliable manner.
Although many computational methods and several reviews have been proposed for AFP [7, 13, 14], there remain some challenges. Firstly, these reviews only summarize existing methods and lack a comparison of the performance of these methods to provide researchers with choices for their usage. Secondly, there is a lack of comprehensive evaluation of these methods across diverse application scenarios. Additionally, current evaluation metrics fail to consider the structural relationships between protein functions, which prevents them from accurately accessing the significance of different functions. As a result, these metrics cannot accurately measure the overall performance of AFP methods.
To address these limitations, we propose a comprehensive computational benchmark, BeProf, to evaluate the performance of existing methods for AFP in various scenarios and provide a reference for future research in protein function prediction. Specifically, BeProf introduces a novel evaluation metric that takes into account both the depth and information content (IC) of functions. Additionally, BeProf incorporates the latest database, processes data for model input, and analyzes the distributions of proteins and functions. Furthermore, this benchmark covers 17 computational methods and designs 8 specific cases to thoroughly test their performance. Finally, we analyze and summarize the strengths of these methods, and offer practical guidance for users in different application scenarios.
SURVEY OF CURRENT METHODS
In the last few decades, a large amount of computational methods have been developed for AFP. To provide researchers with a comprehensive overview of computational methods in this field, BeProf covers 2 baseline methods and 15 recently published methods that provide accurate predictive results and enhance the development of AFP, most of which are based on deep learning techniques. Furthermore, these methods are categorized into five groups based on the type of biological data they used, as shown in Figure 1. The details are presented in Table 1.
Table 1.
The details of existing computational methods
| Data Source | Methods | Journal | Year | Citations | The limitation of predicting GO terms | The limitation of prediction protein sequences | Special feature / Algorithm | Considering relationships between GO terms | Code Available |
|---|---|---|---|---|---|---|---|---|---|
| Protein Sequence | DeepGOCNN/DeepGOPlus | Bioinformatics | 2020 | 263 | Predict GO terms with 50 or more annotations. | The maximum length of the protein is 2000. | One-hot encoding of sequences. 1D Convolution. |
√ | |
| TALE | Bioinformatics | 2021 | 39 | \ | \ | Graph structure of GO terms. Transformer Encoder. |
√ | √ | |
| DeepGOZero | Bioinformatics | 2022 | 16 | \ | Remove the proteins annotated with only ‘protein binding’. | Formal axioms of GO terms. Zero-shot. |
√ | √ | |
| ATGO | Plos Computational Biology | 2022 | 7 | \ | \ | Pre-trained protein large language model. Contrastive learning. |
√ | √ | |
| Protein Sequence and Protein Structure | DeepFRI | Nature Communications | 2021 | 374 | \ | PDB | Pre-trained protein sequence model. GCN |
√ | |
| GAT-GO | Briefings in Bioinformatics | 2022 | 31 | \ | \ | Pre-trained protein large language model. GAT. |
√ | ||
| Protein Sequence and PPI network | DeepGO | Bioinformatics | 2018 | 367 | Predict BP, CC, MF GO terms with 150,50,50 or more annotations. | Ignore proteins which contains ambiguous amino acid codes (B, O, J, U, X, Z). The maximum length of the protein is 1002. | Hierarchical classification. | √ | √ |
| DeepGOA | IEEE/ACM Transactions on Computational Biology and Bioinformatics | 2020 | 23 | Predict top 589 MF terms, 439 CC terms and 932 BP terms. | Ignore proteins which contains ambiguous amino acid codes (B, O, J, U, X, Z). The maximum length of the protein is 1000. | InterPro feature. Data Fusion. |
√ | ||
| DeepGraphGO | Bioinformatics | 2021 | 46 | \ | \ | InterPro feature. GCN. |
√ | ||
| NetQuilt | Bioinformatics | 2021 | 11 | \ | \ | BLAST. | √ | ||
| Protein Sequence and Literature | DeepText2GO | Elsevier | 2018 | 61 | \ | \ | TFIDF. D2V. D2V-TFIDF. InterPro feature. BLAST-KNN |
||
| Ensemble | GOLabeler | Bioinformatics | 2018 | 132 | \ | \ | Naïve. BLAST-KNN. LR3mer. LR-InterPro. LR-ProFET. |
||
| NetGO | Nucleic Acid Research | 2019 | 85 | \ | \ | Naïve. BLAST-KNN. LR-3mer. LR-InterPro. LR-ProFET. Net-KNN |
|||
| NetGO2.0 | Nucleic Acid Research | 2021 | 45 | \ | \ | Naïve. BLAST-KNN. LR-3mer. LR-InterPro. Net-KNN. LR-Text. Seq-RNN. |
|||
| NetGO3.0 | Genomics, Proteomics & Bioinformatics | 2023 | 2 | \ | \ | Naïve. BLAST-KNN. LR-3mer. LR-InterPro. Net-KNN. LR-Text. (LR)-ESM |
Sequence-based prediction methods
The secondary and tertiary structures of proteins are formed through the folding of protein sequences, which, in turn, determines their functions [15]. Therefore, there exists a close relationship between sequences and functions, which enables the identification of potential motifs and the prediction of functions from sequences alone. Currently, computational methods extensively exploit protein sequence information, and many methods can achieve good performance for AFP based on sequence alone.
In the early days of AFP, BlastKNN was the most widely used baseline method for AFP based on sequence similarity. Specifically, given a target protein, BlastKNN first employs the Blast [16] tool to calculate the similarities between the target protein and proteins with known functions. Then, as shown in formula (1), BlastKNN selects the 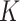 most similar proteins and merges their functions using a similarity-weighted algorithm to predict the functions of the target protein
most similar proteins and merges their functions using a similarity-weighted algorithm to predict the functions of the target protein 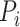 :
:
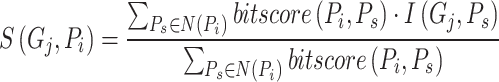 |
(1) |
where 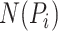 is a protein set including the
is a protein set including the 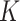 selected similar proteins for the target protein
selected similar proteins for the target protein 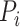 ,
, 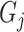 is a predicted GO term, and
is a predicted GO term, and 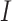 is an identity function that returns 1 if the condition is true and 0 otherwise.
is an identity function that returns 1 if the condition is true and 0 otherwise. 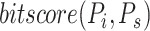 refers to the similarity score between protein
refers to the similarity score between protein 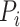 and protein
and protein 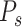 .
. 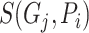 is the predicted score of GO term
is the predicted score of GO term 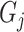 for protein
for protein 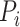 .
.
Compared to Blast [16], Diamond [17] offers faster speed. Instead of choosing 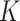 proteins, the strategy based on Diamond considers all available proteins to annotate target proteins, as shown in formula (2):
proteins, the strategy based on Diamond considers all available proteins to annotate target proteins, as shown in formula (2):
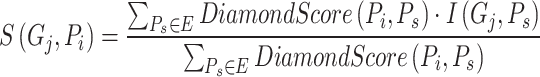 |
(2) |
where 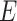 represents the corresponding similar protein set of the target protein
represents the corresponding similar protein set of the target protein 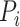 calculated by Diamond with an
calculated by Diamond with an  -value of 0.001. Furthermore, the Diamond-based method considers the relationships between GO terms when assigning values to each predicted GO term. This approach ensures that the predicted value for an ancestor GO term is always greater than or equal to its child GO terms, which is a common post-process procedure and widely used in later studies [18, 19].
-value of 0.001. Furthermore, the Diamond-based method considers the relationships between GO terms when assigning values to each predicted GO term. This approach ensures that the predicted value for an ancestor GO term is always greater than or equal to its child GO terms, which is a common post-process procedure and widely used in later studies [18, 19].
Recently, deep learning [20] has demonstrated robust capabilities in feature extraction from various types of data modalities [21]. Researchers have also begun to utilize deep learning to extract features from protein sequences for AFP.
DeepGOCNN [14] is the first sequence-based model using deep learning. It employs one-hot encoding to represent protein sequences and then uses a stack of CNN layers with varying receptive fields to extract features. Finally, the extracted features are concatenated to predict protein functions. However, it is worth noting that DeepGOCNN is limited in its ability to predict GO terms with more than 50 occurrences in the training set, which restricts its applicability to rare GO terms with few samples. To overcome this limitation, DeepGOPlus [14] combines the results from DeepGOCNN and a homology-based method (Diamond) via a weighted fusion approach. It integrates homologous protein similarity information, reducing the impact of the limitation on rare GO terms, increasing the number of predicted GO terms and achieving higher predictive performance.
To date, self-attention and transformer technology [22] have achieved impressive performance in extracting features from long sequences. TALE [23] is a transformer-based method that combines protein sequences with hierarchical GO labels. First, TALE employs an embedding layer to encode protein sequences and a transformer block to extract sequence features. It then constructs a hierarchical GO label matrix with dimensions of 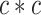 , where
, where 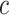 represents the total number of GO labels. For the
represents the total number of GO labels. For the 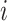 th GO label’s feature, both the label itself and its ancestors are assigned a value of 1. Additionally, ancestor nodes must always precede their child nodes. Therefore, TALE introduces a constrained loss function to ensure that the predicted scores for ancestor nodes surpass those of their corresponding child nodes. Finally, TALE integrates sequence- and label-based features to learn the contribution of each amino acid to individual GO labels. Additionally, the weighted fusion of TALE predictions and Diamond predictions further improves the model performance.
th GO label’s feature, both the label itself and its ancestors are assigned a value of 1. Additionally, ancestor nodes must always precede their child nodes. Therefore, TALE introduces a constrained loss function to ensure that the predicted scores for ancestor nodes surpass those of their corresponding child nodes. Finally, TALE integrates sequence- and label-based features to learn the contribution of each amino acid to individual GO labels. Additionally, the weighted fusion of TALE predictions and Diamond predictions further improves the model performance.
The zero-shot technique can predict labels that have not appeared in the training data, and thus can be used to predict new functions for AFP, which is a limitation of existing methods. DeepGOZero [24] is a sequence-based method that can predict unseen functions via zero-shot learning. It also uses InterPro binary features as input and generates dense features through two MLP layers. Most remarkably, DeepGOZero can leverage GO formal axioms to predict new functions via zero-shot learning. These GO formal axioms, represented by EL Embeddings [25], denote classes as n-balls and relationships as vectors to embed ontology semantics into geometric models. During the zero-shot process, DeepGOZero integrates model theory with neural networks to predict protein functions. Specifically, it computes the binary cross-entropy losses between predicted results and labels, and then optimizes them with losses derived from the ontology axiom specified by EL Embeddings. Moreover, DeepGOZero integrates homologous protein similarity information to further enhance prediction performance.
Recently, large language models have shown significant potential in various downstream tasks [11, 26]. Inspired by this, ATGO [18] has been proposed as a state-of-the-art method for AFP. ATGO extracts sequence features from the last three layers of the pre-trained protein language model ESM-1b [26]. These features from different layers are then concatenated to generate a comprehensive feature representation through several MLP layers. Subsequently, ATGO designs a triplet network based on contrastive learning. Within this architecture, the similarity between proteins is measured via the Euclidean distance between their features. The primary objective of the triplet network is to drive proteins with similar functions closer together in the latent embedding space, which can further improve performance. Additionally, ATGO integrates its results with those of homology-based methods as the final prediction results.
Since the motifs and the large language models of protein sequences have been demonstrated to be closely related to protein functions, as well as the basic and abundance of protein sequences, predicting protein functions based on protein sequences is of great promise and significance.
Sequence- and structure-based prediction methods
Protein sequences fold into three-dimensional structures to perform their functions. Therefore, protein structures exhibit a closer correlation with functions than sequences. However, experimentally obtained protein structures account for only a small fraction of known protein sequences, posting a limitation for the development of structure-based algorithms. With the advancement of deep learning technology, AlphaFold2 [11] has been proposed, which can predict protein structures with high reliability based on protein sequences. This advancement makes it possible to predict protein functions based on structural information. Nowadays, more and more structure-based methods have been proposed.
DeepFRI [27] is a graph convolutional network [28] (GCN)-based model that combines protein sequences and structures to predict protein functions. For protein sequences, it utilizes a self-supervised model with recurrent neural networks and long short-term memory networks [29], which is trained to predict residues in the sequences. Then, residue-level features can be obtained from the trained model. For protein structures, DeepFRI constructs an alpha carbon (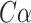 ) contact graph from coordinates of residues, treating residues as nodes and calculating distances between their
) contact graph from coordinates of residues, treating residues as nodes and calculating distances between their 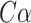 atoms. Residues with distances less than 10 Å are considered in contact. Given the residue-level features and corresponding contact graphs, DeepFRI uses several GCN layers and a mean-pooling layer to learn the features of the entire protein. Finally, these features are fed into two fully connected layers, which makes the prediction of protein function.
atoms. Residues with distances less than 10 Å are considered in contact. Given the residue-level features and corresponding contact graphs, DeepFRI uses several GCN layers and a mean-pooling layer to learn the features of the entire protein. Finally, these features are fed into two fully connected layers, which makes the prediction of protein function.
Unlike DeepFRI, GAT-GO [30] is a graph attention network [31] (GAT)-based method that leverages a pre-trained protein sequence language model. GAT-GO takes a protein sequence as input and extracts three types of features: sequential features, residue-level features and structure features. The sequential features are generated by HHBlits, including the one-hot encoded residues, corresponding solvent accessibility (SA), secondary structure (SS) and position specific scoring matrix (PSSM). The residue-level embeddings are derived from the pre-trained language model, ESM-1b. Meanwhile, Raptor X [32] predicts the  distance and generates the contact graphs. When the
distance and generates the contact graphs. When the  distance is less than 8 Å, an edge between corresponding residues is added. Then, the sequential features and residue-level embeddings serve as node-level features, and several GAT layers are applied to learn structural information. This GAT-based encoder integrates distant residue interactions and node features to produce more informative protein-level representations. The final model predicts functions based on the combination of protein-level representations and residue-level embeddings. Experimental results demonstrate that GAT-GO achieves state-of-the-art performance.
distance is less than 8 Å, an edge between corresponding residues is added. Then, the sequential features and residue-level embeddings serve as node-level features, and several GAT layers are applied to learn structural information. This GAT-based encoder integrates distant residue interactions and node features to produce more informative protein-level representations. The final model predicts functions based on the combination of protein-level representations and residue-level embeddings. Experimental results demonstrate that GAT-GO achieves state-of-the-art performance.
In conclusion, the predicted protein structures with high confidence provide a new perspective to predict protein functions, and several models have been proposed and get comparable performance. However, the advancements of protein structures have still not been highlighted. How to effectively detect the important motifs in the structures and generate the corresponding features remains the key to improving the performance for AFP.
Sequence- and PPI network-based prediction methods
Since proteins interact to perform functions together [33–35], PPI information also plays an important role in AFP. In a PPI network, nodes represent proteins, and edges represent their interactions. These data reflect the complex biological processes in which proteins are involved, making them widely utilized in AFP [36].
DeepGO [13] is the first deep learning-based model for AFP, integrating both protein sequences and PPI networks. It first uses 3-mer to encode protein sequences, and then learns their latent embeddings with an embedding layer. Subsequently, the sequential features are extracted through 1D convolution and max pooling layers. For the PPI network, DeepGO utilizes DeepWalk [37] to generate a 256-dimensional network topology feature for each protein. After combining the sequential and network features, prediction scores for individual GO terms are calculated through a fully connected layer. Additionally, DeepGO designs a hierarchical classification neural network model to predict functions, ensuring that the results satisfy the GO taxonomic structure for is-a’ relations. Experimental results demonstrate that DeepGO surpasses traditional methods that only depend on sequence similarity, especially in CCO. Although DeepGO provides valuable insights for AFP based on deep learning, it also has limitations. Its hierarchical classification neural network requires huge memory resources and is difficult to apply to large-scale labels.
Inspired by DeepGO, DeepGOA [19] extracts sequential features more efficiently and robustly. It first uses Word2vec to generate residue-level embeddings of sequences, which are then fed into Bi-LSTM [38] and multi-scale CNN layers to extract global and local features. On the other hand, DeepGOA uses InterProScan [39] tool to extract specific protein domains, motifs and family information. For the PPI network, DeepGOA also uses the features generated by DeepWalk [37]. These three types of features are then concatenated to form the final features, which encompass comprehensive protein information and facilitate the improvement of AFP. The probabilities for all GO terms are determined using a fully connected layer. DeepGOA directly uses MLP layers to predict functions, which requires less memory than DeepGO and can predict a broad range of functions. Experimental results further prove the effectiveness of DeepGOA.
The success of graph neural networks [40] (GNNs) provides more strategies in AFP. DeepGraphGO [41] is an end-to-end model that leverages GNNs to extract information from PPI networks to predict protein functions. Initially, DeepGraphGO integrates PPI networks of 17 species to form a multi-species PPI network. Simultaneously, it generates the binary features of InterPro from protein sequences. Considering the high dimension and sparsity of binary features, DeepGraphGO employs an embedding layer to learn the dense features for each property. These dense features are then fed into two GCN layers, which process protein networks across all species and update each protein node based on its connected nodes. This operation is repeated twice to ensure that the neighbor information is sufficiently captured, enabling the model to extract the higher-order structure of the PPI network. Experimental results highlight both the effectiveness of the PPI network and the feasibility of the multi-species strategy employed by DeepGraphGO.
NetQuilt [42] introduces a novel approach to seamlessly integrate sequence and network information of multiple species. By utilizing IsoRank [43] similarity scores, it constructs comprehensive meta-network maps of proteins across different species, aiming to establish relationships between different species. NetQuilt commences by sourcing data from the STRING database using specific species IDs. Then, it employs Blast to quantify sequence similarity between proteins, both within a single species and across different species. Building upon this foundation, NetQuilt computes IsoRank alignment scores, producing a weighted adjacency matrix and a sequence identity matrix. Various information matrices are ultimately combined to form a dense matrix 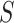 , which includes both intra-species and inter-species information. Finally, this matrix
, which includes both intra-species and inter-species information. Finally, this matrix 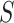 serves as training input for a maxout neural network. The results demonstrate that NetQuilt achieves commendable performance on new species without the target PPI network.
serves as training input for a maxout neural network. The results demonstrate that NetQuilt achieves commendable performance on new species without the target PPI network.
These studies have demonstrated that PPIs are closely related to function, but such approaches are also facing several limitations. A certain percentage of proteins are lacking in interaction relationships, and this phenomenon is particularly common in new species [44]. Consequently, it is important to consider how to apply the known PPI information to newly sequenced organisms.
Sequence- and literature-based prediction methods
To date, some relevant literature sources have been published, providing descriptions of protein functions and offering insights into AFP. However, due to the complexity of the descriptions and the challenges with gathering pertinent data, it is difficult to predict protein function on large-scale data. Consequently, only a limited number of methods have been proposed.
DeepText2GO [45] is a representative method using biomedical literature. In contrast to traditional bag-of-words representations, DeepText2GO incorporates deep semantic text representation with a variety of protein information sources. For instance, it contains sequence homology, protein families, domains and motifs, which can be obtained by Blast and InterProScan. For literature-based data, DeepText2GO extracts PubMed identifiers from UniProtKB/SwissProt and retrieves the corresponding abstracts. Subsequently, algorithms such as TFIDF, Document2Vec [46] and D2V-TFIDF are then applied to the literature information to generate embeddings. Then, sequence and literature features are merged and classified using an LR model. Finally, the predictions from the LR model are combined with BlastKNN to generate the final predicted scores for GO terms. Experimental results prove the superior effectiveness of DeepText2GO.
Ensemble prediction methods
Ensemble models can often achieve superior results via integrating multiple individual predictors, leading to significant advancements in various tasks. Given the multifaceted nature of protein data, encompassing sequences, structures and interactions, it is necessary to develop methods that effectively integrate these diverse data sources to enhance the accuracy of protein function prediction.
GOLabeler [47] is a machine learning method for integrating five component classifiers that leverages the learning-to-rank (LTR) [48] paradigm to integrate different sequence-based features. It selects five distinct sequential information to generate corresponding classifiers, including Naive (GO term frequency), BlastKNN (K-nearest neighbor based on BLAST results), LR-3mer (logistic regression (LR) for 3-mer frequency of sequence), LR-InterPro (LR for InterPro features) and LR-ProFET (LR for ProFET [49] features). Unlike regular classification models that treat positive examples equally, LTR models penalize lower-ranked positive examples more heavily. GOLabeler effectively integrates multiple sequence-based predictions from diverse classifiers, where all information is generated from the sequence alone. Overall, GOLabeler presents a robust framework for combining various sequence-based tools for AFP, with the potential to enhance their performance.
NetGO [50] is an effective ensemble method that integrates extensive PPI network information. It covers a large amount of PPI data from over 2000 species in the STRING database. For the sub-predictors, NetGO covers five existing methods based on the sequences and a newly proposed method based on the PPI network, namely Naive, Blast-KNN, LR-3mer, LR-InterPro, LR-ProFET and Net-KNN, where the first five methods are used in GOLabeler. Net-KNN is a newly proposed method, which is similar to Blast-KNN and replaces sequence similarities with interaction scores in the PPI network. Notably, even unseen proteins not present in the PPI network can also be annotated via homology relationships. Similar to GOLabeler, NetGO also utilizes LTR framework to incorporate the outputs from these predictors. The efficacy of NetGO is convincingly proved by its significant performance improvements over other methods, including GOLabeler.
Later, NetGO 2.0 [51] is an upgraded version of the original NetGO. In comparison to its predecessor, NetGO2.0 replaces the LR-ProFET component with two novel components, named LR-text and Seq-RNN. The procedure of LR-text is the same as DeepText2GO, while Seq-RNN uses a Bi-LSTM network and an MLP layer to annotate proteins from their sequences, which is similar to the process in DeepGOA. The final results demonstrate that NetGO 2.0 surpasses its predecessor, particularly in terms of BPO and CCO. Recently, inspired by protein language models (PLMs), NetGO 3.0 [52] was proposed. It discards the Seq-RNN component and incorporates an LR-ESM model, which uses logistic regression to predict protein functions from protein embeddings generated by the pre-trained PLM, ESM-1b. Impressively, the performance of LR-ESM achieves comparable performance with the top-performing components in NetGO 2.0.
PROTEIN AND FUNCTION DATA
Data processing
Following the Critical Assessment of Function Annotation challenge [53] (CAFA), protein information and their corresponding functions are collected from the UniProt/Swiss-Prot [54] database (released on April 2022, published on December 2022). Then, 23 specials (10090’, 223283’, 273057’, 559292’, 85962’, 10116’, 224308’, 284812’, 7227’, ’9606’, 160488’, 237561’, 321314’, 7955’, 99287’, 170187’, 243232’, 3702’, 83333’, 208963’, 243273’, 44689’, 8355’) and experimental annotations are considered, including EXP’, IDA’, IPI’, IMP’, IGI’, IEP’, TAS’ and IC’. After filtering out proteins without GO annotations, the final protein set contains 60 121 proteins.
The hierarchical structure of the GO terms is retrieved from GO (released on 4 December 2022). GO establishes three ontologies to describe protein functions: BPO, MFO and CCO. Each ontology encompasses a set of GO terms and the relationships between them. These relationships primarily include is-a’, part-of’, has part’ and regulates’, with only the is-a’ and part-of’ relationships safely transferable to functions, forming a parent-child hierarchy. In other words, if a protein is annotated with GO term A and GO term A is-a’ or part-of’ GO term B, it can be deduced that the protein is also annotated with GO term B.
As for PPI networks, they are downloaded from the STRING database (full links v11.5). Additionally, orthology relationships are extracted from the eggNOG database [55]. Finally, 115 706 292 PPIs and 1 473 178 orthology relations between 214 050 proteins are collected.
Statistical information of datasets
In this study, following previous studies [14, 27, 30], we adopt two approaches to generate datasets, and the statistical information is shown in Table 2:
Table 2.
The statistical information of datasets generated by different rules
| Dataset | Data Split | Statistics | BP | CC | MF | All |
|---|---|---|---|---|---|---|
| Dataset1 | Time stamps | Training | 47,830 | 42,891 | 31,756 | 57,254 |
| Validation | 777 | 717 | 684 | 1432 | ||
| Test | 1070 | 898 | 408 | 1435 | ||
| GO | 19,717 | 2506 | 6095 | 28,318 | ||
| Dataset2 | Sequence identity | Training | 44,827 | 40,235 | 29,745 | 54,302 |
| Validation | 2378 | 2091 | 1510 | 2823 | ||
| Test | 2472 | 2180 | 1593 | 2996 | ||
| GO | 19,450 | 2515 | 5914 | 27,879 |
Dataset1: the original data are split into three distinct subsets by different time stamps. The first subset serves as the training set, which contains the proteins in the UniProt202004. The second subset forms the validation set, including the proteins in the UniProt202104 but not in the UniProt202004. Finally, the last subset, designated as the test set, comprises the proteins in the UniProt202204 but not in the UniProt202004 or UniProt202104.
Dataset2: the raw protein data undergo an initial clustering step based on sequence similarity. To ensure the distinctiveness among the resulting clusters, the sequence similarity between any two clusters is restricted to a maximum of 30%. Subsequently, the training, validation and test sets are divided according to a ratio of 18:1:1, respectively.
Data characteristics of proteins and functions
To achieve a more comprehensive evaluation of computational methods, we perform an analysis of the raw data from several aspects, including the distribution of protein sequence length, the number of GO terms annotated per protein, the number of annotations per GO term and the IC values. These analyses shed light on the characteristics of the raw data and provide a foundation for evaluating the performance of computational methods for AFP.
As illustrated in Figure 2, approximately 89.16% proteins in the dataset have sequence lengths shorter than 1000 amino acids, while only 338 proteins (about 0.56%) with sequence lengths greater than 3000. According to Table 1, certain methods, such as DeepGO and DeepGOA, are incapable of handling proteins with sequence lengths greater than 1000, while DeepGOPlus also has limitations in protein sequence lengths. Therefore, it is essential for predictors to consider how to effectively annotate these long proteins. Additionally, evaluating the performance of these methods specifically on long proteins is crucial to assess their effectiveness.
Figure 2.
Distribution of protein sequence lengths. Most of protein sequence lengths are less than 1000 (89%), while only 0.56% protein sequence lengths are larger than 3000.
Figure 3 represents the correlations between proteins and their corresponding functions. It can be obtained that the number of known GO terms associated with each protein is significantly smaller than the total number of GO terms, indicating a highly sparse and unbalanced distribution of labels. Furthermore, it also reveals that most GO terms exhibit low frequency. Specifically, a majority of GO terms annotate less than 200 proteins for BPO, less than 100 proteins for MFO, and less than 250 proteins for CCO. Additionally, it is worth noting that some high-frequency GO terms are not shown in Figure 3. For instance, only above 1.85% of these GO terms for BPO have more than 1000 annotations, 1.82% of these GO terms for MFO have more than 500 annotations, while 2.08% of the GO terms for CCO exceed 1500 annotations. Above all, predicting protein functions is an unbalanced multi-label classification problem, where each label has a limited number of available samples, presenting challenges for AFP.
Figure 3.

Distribution of the number of known functions per protein in three ontologies (top) and distribution of the number of known proteins per GO term in three ontologies (bottom).
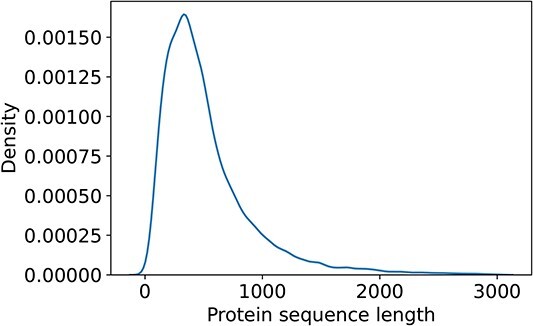
IC is a basic metric in information theory, quantifying the probability of an event occurring randomly. In AFP, we evaluate the IC values of all GO terms, as shown in formula (3):
 |
(3) |
where 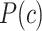 is the superclass set of class
is the superclass set of class 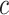 , and
, and 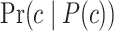 denotes the probability of GO term
denotes the probability of GO term 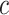 appearing simultaneously with its ancestors. Figure 4 (top) shows the distribution of IC values across three ontologies. It can be seen that most GO terms exhibit low IC values. Consequently, it is particularly meaningful to predict GO terms with high IC values, since these terms are more informative and valuable.
appearing simultaneously with its ancestors. Figure 4 (top) shows the distribution of IC values across three ontologies. It can be seen that most GO terms exhibit low IC values. Consequently, it is particularly meaningful to predict GO terms with high IC values, since these terms are more informative and valuable.
Figure 4.

Distribution of IC values of GO terms in three ontologies (top) and the depths of GO terms in three ontologies (bottom).
According to previous studies [14], the structure of GO terms exhibits a loosely hierarchical organization. Within this structure, functions become more generalized as they approach the root node, while functions become more specific as they descend to deeper depths. Consequently, it is more practical to predict functions located at deeper depths. As shown in Figure 4 (bottom), it can be concluded that the number of functions diminishes as one traverses to deeper depths, and the majority of functions are in the middle region of the whole structure.
EVALUATION METRICS
In this study, we evaluate the performance of these models using five existing metrics and a novel proposed metrics: 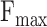 ,
, 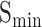 , AUPR, IC weighted AUPR [53], Depth weighted AUPR and M-AUPR.
, AUPR, IC weighted AUPR [53], Depth weighted AUPR and M-AUPR.
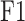 and
and 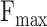 (Protein-centric) are basic metrics used to evaluate the performance of binary classification models.
(Protein-centric) are basic metrics used to evaluate the performance of binary classification models. 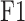 considers both precision and recall, particularly suitable for evaluating models on unbalanced data. For the prediction results,
considers both precision and recall, particularly suitable for evaluating models on unbalanced data. For the prediction results, 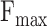 is the maximum
is the maximum 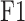 score at different thresholds.
score at different thresholds.
Given the threshold 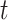 , corresponding precision and recall can be calculated as follows:
, corresponding precision and recall can be calculated as follows:
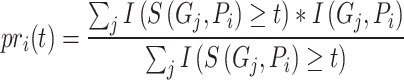 |
(4) |
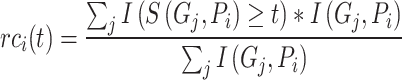 |
(5) |
where 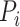 is the
is the 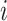 th protein and
th protein and 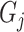 is the
is the 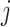 th GO term.
th GO term. 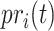 is the precision of protein
is the precision of protein 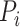 at threshold
at threshold 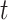 ,
, 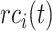 is the corresponding recall value.
is the corresponding recall value. 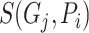 represents the predicted probability of function
represents the predicted probability of function 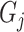 for protein
for protein 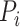 .
. 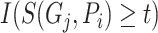 determines whether the probability is greater than or equal to the threshold
determines whether the probability is greater than or equal to the threshold 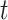 , which is 1 if it does, and 0 otherwise.
, which is 1 if it does, and 0 otherwise. 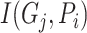 determines whether protein
determines whether protein 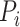 is annotated by function
is annotated by function 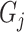 , which is 1 if it does, and 0 otherwise.
, which is 1 if it does, and 0 otherwise.
Then, the average precision and recall of all proteins can be calculated as follows:
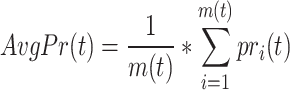 |
(6) |
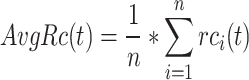 |
(7) |
where 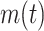 refers to the number of proteins that contain at least one predicted GO term, and
refers to the number of proteins that contain at least one predicted GO term, and 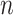 is the number of all proteins in the test set. Finally, we calculate
is the number of all proteins in the test set. Finally, we calculate 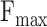 as follows:
as follows:
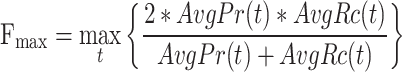 |
(8) |
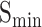 (protein-centered) measures the semantic distance between the real and predicted annotations based on their IC values. The calculation process is as follows:
(protein-centered) measures the semantic distance between the real and predicted annotations based on their IC values. The calculation process is as follows:
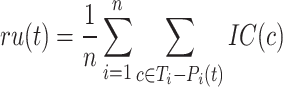 |
(9) |
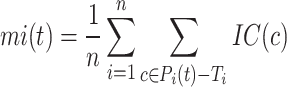 |
(10) |
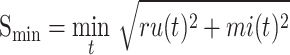 |
(11) |
where 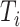 is the true labels of protein
is the true labels of protein 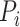 ,
, 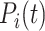 is the predicted labels of protein
is the predicted labels of protein 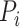 with threshold
with threshold 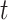 ,
, 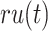 is the sum of IC values of the functions that failed to be predicted, while
is the sum of IC values of the functions that failed to be predicted, while 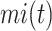 is the sum of IC values of false positive labels.
is the sum of IC values of false positive labels.
AUPR (protein-centric) is another common evaluation metric based on precision and recall, which is determined by calculating the area under the precision-recall curve. Above all, for the predicted results, higher values of 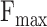 , AUPR values and smaller values of
, AUPR values and smaller values of 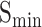 indicate better performance of models.
indicate better performance of models.
To achieve a more comprehensive evaluation of model performance, IC-weighted AUPR (IC_AUPR) was first introduced by the CAFA challenge [53]. Different from previous precision and recall, IC_AUPR considers the weighted of GO terms based on their IC values:
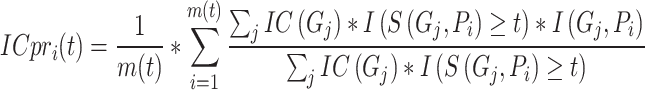 |
(12) |
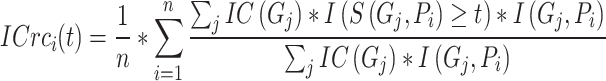 |
(13) |
Inspired by IC_AUPR, we propose a novel metric named depth weighted AUPR (DP_AUPR), which can consider both the IC values and the depths of GO terms in the whole GO structure. Similar to IC_AUPR, DP_AUPR calculates precision and recall as follows:
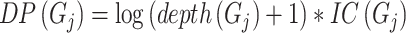 |
(14) |
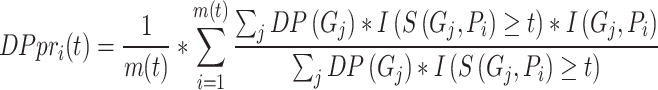 |
(15) |
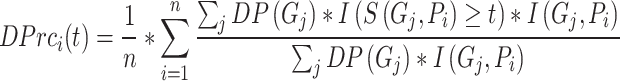 |
(16) |
where 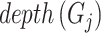 represents the distance of the shortest path from GO term
represents the distance of the shortest path from GO term 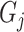 to the root node. The further
to the root node. The further 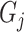 is from the root node and the larger its
is from the root node and the larger its 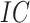 value, the more meaningful it is to predict it correctly.
value, the more meaningful it is to predict it correctly.
M-AUPR (GO-centric) evaluates the performance of models on each label separately and then calculates the average of these values:
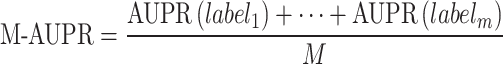 |
(17) |
where 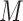 represents the total number of labels, and
represents the total number of labels, and 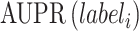 represents the value of AUPR calculated on the
represents the value of AUPR calculated on the 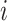 -th label. Consistent with AUPR, higher values of IC_AUPR, DP_AUPR and M-AUPR indicate better performance of models.
-th label. Consistent with AUPR, higher values of IC_AUPR, DP_AUPR and M-AUPR indicate better performance of models.
COMPARISON AND ANALYSIS
Taking into account the data characteristics, in this section, we collect 14 computational methods mentioned before (Dimond, BlastKNN, DeepGO, DeepGOA, DeepGOCNN, NetQuilt, TALE, DeepGOZero, ATGO, DeepGraphGO, DeepGOPlus, TALE+, DeepGOZero+, ATGO+) and design 8 cases to evaluate their performance. These cases can be categorized into two groups: protein- and GO term-centered. The protein-centered cases encompass several aspects, including a comprehensive performance comparison based on timestamps, performance evaluation on long or normal proteins, assessment of performance on difficult proteins with low sequence similarities, performance evaluation on disorder proteins, and examination of generality to new species. On the other hand, the GO term-centered cases encompass the performance on different depths of GO terms, the performance on rare GO terms with limited samples, and the performance on GO terms grouped by IC values. Each case provides valuable insights into the strengths of different methods, thus offering algorithm recommendations tailored to specific application scenarios.
Comprehensive predictive performance comparison
Dataset1 is generated by different timestamps, which serves as a fundamental approach to evaluate the performance of methods in CAFA. As shown in Table 3, although DeepGOZero+ achieves the highest 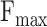 for all three ontologies, it falls short of the best performance in
for all three ontologies, it falls short of the best performance in 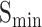 . In contrast, ATGO+ shows comparable performance to the top values in both
. In contrast, ATGO+ shows comparable performance to the top values in both 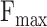 and
and 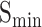 for BPO, MFO and CCO. Meanwhile, Table 4 shows the performance of these methods in terms of AUPR, IC_AUPR and DP_AUPR, which are more effective in evaluating the performance of prediction results on unbalanced multi-label data. Obviously, DeepGOZero+ shows significant improvements for MFO and BPO, while ATGO+ consistently outperforms other methods and achieves the best AUPR on CCO, substantiating its potential for accurate protein function prediction. Notably, in terms of AUPR, DeepGraphGO underperforms DeepGOPlus on CCO, while it surpasses DeepGOPlus in IC_AUPR and DP_AUPR, which suggests that DeepGraphGO exhibits superior performance on specific GO terms with high IC values or deep depths. In addition, comparing DeepGOCNN and DeepGOPlus, DeepGOZero and DeepGOZero+, TALE and TALE+, ATGO and ATGO+, it can be found that the performance of these methods is significantly enhanced after combining the prediction scores based on sequence similarity, indicating the close correlation between sequence similarity and functions.
for BPO, MFO and CCO. Meanwhile, Table 4 shows the performance of these methods in terms of AUPR, IC_AUPR and DP_AUPR, which are more effective in evaluating the performance of prediction results on unbalanced multi-label data. Obviously, DeepGOZero+ shows significant improvements for MFO and BPO, while ATGO+ consistently outperforms other methods and achieves the best AUPR on CCO, substantiating its potential for accurate protein function prediction. Notably, in terms of AUPR, DeepGraphGO underperforms DeepGOPlus on CCO, while it surpasses DeepGOPlus in IC_AUPR and DP_AUPR, which suggests that DeepGraphGO exhibits superior performance on specific GO terms with high IC values or deep depths. In addition, comparing DeepGOCNN and DeepGOPlus, DeepGOZero and DeepGOZero+, TALE and TALE+, ATGO and ATGO+, it can be found that the performance of these methods is significantly enhanced after combining the prediction scores based on sequence similarity, indicating the close correlation between sequence similarity and functions.
Table 3.
Fmax and Smin performance comparison on the first dataset generated by a time-based split
| Methods | Fmax | Smin | |||||
|---|---|---|---|---|---|---|---|
| MF | CC | BP | MF | CC | BP | ||
| Single algorithms | Diamond | 0.589 | 0.572 | 0.426 | 8.303 | 11.548 | 31.502 |
| BlastKNN | 0.614 | 0.595 | 0.443 | 7.852 | 10.182 | 31.319 | |
| DeepGO | 0.352 | 0.579 | 0.321 | 12.442 | 10.646 | 36.028 | |
| DeepGOA | 0.500 | 0.621 | 0.393 | 10.331 | 9.829 | 31.323 | |
| DeepGOCNN | 0.367 | 0.563 | 0.323 | 12.070 | 11.092 | 34.986 | |
| NetQuilt | 0.358 | 0.438 | 0.316 | 12.610 | 15.536 | 38.374 | |
| TALE | 0.258 | 0.549 | 0.256 | 13.495 | 11.546 | 35.548 | |
| DeepGOZero | 0.600 | 0.613 | 0.443 | 8.790 | 10.451 | 30.330 | |
| ATGO | 0.455 | 0.599 | 0.395 | 11.375 | 10.192 | 30.893 | |
| DeepGraphGO | 0.548 | 0.633 | 0.427 | 9.492 | 9.295 | 29.988 | |
| Composite algorithms | DeepGOPlus | 0.586 | 0.630 | 0.437 | 8.515 | 10.021 | 32.419 |
| TALE+ | 0.597 | 0.607 | 0.426 | 8.375 | 10.019 | 30.354 | |
| DeepGOZero+ | 0.623 | 0.633 | 0.463 | 8.193 | 10.062 | 31.687 | |
| ATGO+ | 0.619 | 0.630 | 0.454 | 8.048 | 9.671 | 29.667 |
Note: The best performance values are highlighted in bold and the next best performance are underlining.
Table 4.
AUPR, IC_AUPR and DP_AUPR performance comparison on the first dataset generated by a time-based split
| Methods | AUPR | IC_AUPR | DP_AUPR | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MF | CC | BP | MF | CC | BP | MF | CC | BP | ||
| Single | Diamond | 0.384 | 0.281 | 0.195 | 0.360 | 0.229 | 0.165 | 0.345 | 0.227 | 0.154 |
| algorithms | BlastKNN | 0.482 | 0.383 | 0.257 | 0.457 | 0.308 | 0.218 | 0.436 | 0.303 | 0.202 |
| DeepGO | 0.265 | 0.583 | 0.251 | 0.215 | 0.384 | 0.185 | 0.171 | 0.367 | 0.154 | |
| DeepGOA | 0.444 | 0.610 | 0.327 | 0.376 | 0.452 | 0.252 | 0.323 | 0.438 | 0.218 | |
| DeepGOCNN | 0.302 | 0.570 | 0.251 | 0.244 | 0.360 | 0.175 | 0.195 | 0.342 | 0.144 | |
| NetQuilt | 0.245 | 0.287 | 0.191 | 0.212 | 0.164 | 0.150 | 0.183 | 0.156 | 0.134 | |
| TALE | 0.160 | 0.476 | 0.155 | 0.112 | 0.287 | 0.089 | 0.078 | 0.268 | 0.063 | |
| DeepGOZero | 0.576 | 0.572 | 0.393 | 0.538 | 0.392 | 0.325 | 0.510 | 0.384 | 0.298 | |
| ATGO | 0.437 | 0.596 | 0.338 | 0.387 | 0.423 | 0.265 | 0.341 | 0.409 | 0.232 | |
| DeepGraphGO | 0.515 | 0.586 | 0.381 | 0.458 | 0.479 | 0.314 | 0.412 | 0.468 | 0.286 | |
| Composite algorithms |
DeepGOPlus | 0.548 | 0.625 | 0.366 | 0.508 | 0.467 | 0.303 | 0.478 | 0.455 | 0.277 |
| TALE+ | 0.545 | 0.598 | 0.327 | 0.497 | 0.440 | 0.261 | 0.468 | 0.425 | 0.234 | |
| DeepGOZero+ | 0.618 | 0.592 | 0.412 | 0.581 | 0.446 | 0.349 | 0.554 | 0.437 | 0.324 | |
| ATGO+ | 0.593 | 0.634 | 0.396 | 0.555 | 0.478 | 0.328 | 0.522 | 0.466 | 0.299 | |
Note: The best performance values are highlighted in bold and the next best performance are underlining.
Performance comparison on long proteins
As shown in Table 1, these methods all rely on protein sequence information, and several methods ignore or truncate long proteins. For example, both DeepGO and DeepGOA ignore protein sequences exceeding a length of 1000 amino acids, ATGO(+) ignores sequences longer than 1022 amino acids, while DeepGOCNN and DeepGOPlus ignore sequences surpassing 2000 amino acids in length. In this section, we explore the performance of these methods on these long proteins and other normal proteins. Specifically, as shown in Table 5, we split the test data into two subsets based on sequence lengths, and the corresponding results are displayed in Table 6 and 7. Due to the consideration of the entire sequence information, DeepGraphGO achieves the best performance among single algorithms. Moreover, it is evident that the performance gap between DeepGOA and ATGO for long proteins is significantly larger compared to the gap for normal proteins, indicating the crucial role of the ignored amino acids. On the other hand, as DeepGOPlus covers a maximum protein length of 2000, it achieves comparable performance on long proteins, further substantiating the significance of ignored amino acids. Furthermore, when incorporating the results based on sequence similarity, ATGO+ exhibits a substantial improvement, providing a new perspective for addressing long proteins. Above all, it is recommended to consider sequences that are as long as possible, as it tends to yield better performance.
Table 5.
The statistical information of test dataset extracted from Dataset1 based on protein sequence length
| Protein Type | Protein number | Percentage | Protein sequence length range |
|---|---|---|---|
| Normal protein set | 1301 | 90.7% | <=1000 |
| Long protein set | 134 | 9.3% | >1000 |
Table 6.
Performance comparison of these methods on long proteins
| Methods | Fmax | Smin | AUPR | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MF | CC | BP | MF | CC | BP | MF | CC | BP | ||
| Single | Diamond | 0.600 | 0.567 | 0.359 | 9.442 | 11.720 | 48.347 | 0.452 | 0.359 | 0.194 |
| algorithms | BlastKNN | 0.606 | 0.586 | 0.372 | 9.730 | 11.001 | 42.290 | 0.527 | 0.458 | 0.238 |
| DeepGO | 0.378 | 0.544 | 0.313 | 15.331 | 12.765 | 38.549 | 0.291 | 0.545 | 0.252 | |
| DeepGOA | 0.460 | 0.573 | 0.372 | 13.314 | 11.971 | 37.661 | 0.404 | 0.563 | 0.282 | |
| DeepGOCNN | 0.339 | 0.540 | 0.332 | 16.374 | 12.660 | 45.936 | 0.283 | 0.536 | 0.247 | |
| NetQuilt | 0.212 | 0.390 | 0.245 | 18.352 | 17.093 | 77.704 | 0.077 | 0.198 | 0.119 | |
| TALE | 0.296 | 0.516 | 0.282 | 16.206 | 13.554 | 38.100 | 0.182 | 0.435 | 0.156 | |
| DeepGOZero | 0.561 | 0.592 | 0.374 | 11.442 | 12.569 | 49.218 | 0.450 | 0.529 | 0.323 | |
| ATGO | 0.392 | 0.556 | 0.358 | 19.405 | 12.457 | 37.290 | 0.293 | 0.553 | 0.284 | |
| DeepGraphGO | 0.569 | 0.615 | 0.389 | 11.683 | 10.771 | 45.848 | 0.528 | 0.587 | 0.326 | |
| Composite | DeepGOPlus | 0.617 | 0.607 | 0.389 | 9.642 | 10.709 | 46.557 | 0.557 | 0.601 | 0.308 |
| Algorithms | TALE+ | 0.613 | 0.576 | 0.385 | 11.018 | 11.237 | 40.116 | 0.534 | 0.564 | 0.271 |
| DeepGOZero+ | 0.590 | 0.618 | 0.387 | 11.199 | 11.877 | 52.550 | 0.586 | 0.573 | 0.325 | |
| ATGO+ | 0.566 | 0.587 | 0.414 | 10.890 | 11.094 | 37.796 | 0.484 | 0.605 | 0.330 |
Note: The best performance values are highlighted in bold and the next best performance are underlining.
Table 7.
Performance comparison of these methods on normal proteins
| Methods | Fmax | Smin | AUPR | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MF | CC | BP | MF | CC | BP | MF | CC | BP | ||
| Single | Diamond | 0.588 | 0.574 | 0.434 | 8.228 | 11.370 | 30.574 | 0.379 | 0.274 | 0.195 |
| algorithms | BlastKNN | 0.616 | 0.597 | 0.451 | 8.120 | 10.306 | 30.477 | 0.478 | 0.376 | 0.258 |
| DeepGO | 0.350 | 0.583 | 0.322 | 12.718 | 10.412 | 35.217 | 0.262 | 0.586 | 0.251 | |
| DeepGOA | 0.504 | 0.628 | 0.395 | 10.071 | 9.590 | 30.492 | 0.447 | 0.614 | 0.332 | |
| DeepGOCNN | 0.370 | 0.566 | 0.321 | 11.832 | 10.911 | 33.959 | 0.303 | 0.573 | 0.251 | |
| NetQuilt | 0.370 | 0.445 | 0.324 | 12.188 | 18.504 | 41.494 | 0.255 | 0.292 | 0.199 | |
| TALE | 0.256 | 0.554 | 0.254 | 13.278 | 11.338 | 35.108 | 0.157 | 0.480 | 0.155 | |
| DeepGOZero | 0.605 | 0.616 | 0.452 | 8.455 | 10.239 | 29.206 | 0.587 | 0.576 | 0.401 | |
| ATGO | 0.464 | 0.604 | 0.399 | 11.035 | 9.955 | 30.185 | 0.448 | 0.600 | 0.344 | |
| DeepGraphGO | 0.547 | 0.635 | 0.431 | 9.314 | 9.179 | 29.189 | 0.514 | 0.585 | 0.387 | |
| Composite | DeepGOPlus | 0.585 | 0.632 | 0.442 | 8.347 | 9.639 | 31.266 | 0.548 | 0.628 | 0.373 |
| algorithms | TALE+ | 0.597 | 0.610 | 0.431 | 8.188 | 9.903 | 29.566 | 0.545 | 0.602 | 0.332 |
| DeepGOZero+ | 0.627 | 0.635 | 0.472 | 7.932 | 9.874 | 28.948 | 0.621 | 0.594 | 0.422 | |
| ATGO+ | 0.623 | 0.634 | 0.459 | 7.821 | 9.532 | 28.959 | 0.601 | 0.637 | 0.404 |
Note: The best performance values are highlighted in bold and the next best performance are underlining.
Performance comparison on difficult proteins
To mitigate the potential impact of similar proteins present in both the training and test data, which could bias the results, we create a difficult protein set specifically for evaluating model performance on previously unseen proteins. In this set, all proteins have a sequence similarity of no more than 60% to the proteins in the training data. This challenging test set comprises 135, 334 and 295 difficult proteins for MFO, BPO and CCO, respectively. As shown in Figure 5, among single algorithms, DeepGOA and DeepGraphGO demonstrate superior performance for BPO and CCO, indicating the importance of incorporating PPI information for BPO and CCO. Notably, BlastKNN, a method based on sequence similarity, always achieves comparable performance, and most composite algorithms show significant improvements. These observations collectively indicate that these individual methods cannot capture the sequence similarity effectively and integrating sequence-based methods is still a helpful strategy.
Figure 5.

Predictive performance comparison on difficult proteins (low similarity with training proteins) in terms of Fmax and Smin.
To gain a comprehensive understanding of model generalizability, we test existing models on Dataset2, which is split by sequence similarity. The results are shown in Table 8 and Table 9. Remarkably, in terms of 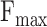 ,
, 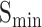 and AUPR, DeepGOA and DeepGraphGO consistently achieve the best performance, even outperforming the composite algorithms. This demonstrates that PPI information between proteins can mitigate the challenges posed by low sequence similarity. Furthermore, the advantages of DeepGraphGO are even more pronounced for IC_AUPR and DP_AUPR, especially for BPO. Specifically, DeepGraphGO improves the IC_AUPR by 16% compared to DeepGOA, and even by 20% in terms of DP_AUPR. Additionally, due to the lower sequence similarities between the test data and training data, it is hard for Diamond and BlastKNN to find similar sequences to annotate target proteins, significantly reducing their performance. Interestingly, it is worth noting that almost all methods show good performance for CCO, except for BlastKNN and Diamond, proving that the hidden motifs within single sequences may be more informative for CCO than similarities between proteins.
and AUPR, DeepGOA and DeepGraphGO consistently achieve the best performance, even outperforming the composite algorithms. This demonstrates that PPI information between proteins can mitigate the challenges posed by low sequence similarity. Furthermore, the advantages of DeepGraphGO are even more pronounced for IC_AUPR and DP_AUPR, especially for BPO. Specifically, DeepGraphGO improves the IC_AUPR by 16% compared to DeepGOA, and even by 20% in terms of DP_AUPR. Additionally, due to the lower sequence similarities between the test data and training data, it is hard for Diamond and BlastKNN to find similar sequences to annotate target proteins, significantly reducing their performance. Interestingly, it is worth noting that almost all methods show good performance for CCO, except for BlastKNN and Diamond, proving that the hidden motifs within single sequences may be more informative for CCO than similarities between proteins.
Table 8.
Fmax and Smin performance comparison on the second dataset generated by sequence identity
| Methods | Fmax | Smin | |||||
|---|---|---|---|---|---|---|---|
| MF | CC | BP | MF | CC | BP | ||
| Single algorithms | Diamond | 0.467 | 0.454 | 0.309 | 11.600 | 12.932 | 49.172 |
| BlastKNN | 0.535 | 0.550 | 0.383 | 11.747 | 11.951 | 45.470 | |
| DeepGO | 0.436 | 0.653 | 0.404 | 13.242 | 10.728 | 44.024 | |
| DeepGOA | 0.585 | 0.696 | 0.440 | 10.099 | 9.903 | 42.308 | |
| DeepGOCNN | 0.386 | 0.609 | 0.334 | 13.263 | 12.160 | 49.271 | |
| TALE | 0.288 | 0.587 | 0.300 | 13.947 | 12.856 | 48.802 | |
| DeepGOZero | 0.561 | 0.618 | 0.407 | 10.206 | 11.690 | 45.226 | |
| ATGO | 0.499 | 0.663 | 0.406 | 11.465 | 10.932 | 43.887 | |
| DeepGraphGO | 0.579 | 0.671 | 0.460 | 9.956 | 9.960 | 41.373 | |
| Composite algorithms | DeepGOPlus | 0.521 | 0.626 | 0.383 | 11.054 | 11.868 | 48.316 |
| TALE+ | 0.501 | 0.615 | 0.366 | 11.051 | 11.962 | 45.110 | |
| DeepGOZero+ | 0.570 | 0.624 | 0.414 | 11.198 | 11.506 | 45.055 | |
| ATGO+ | 0.577 | 0.665 | 0.435 | 10.408 | 10.638 | 43.556 |
Note: The best performance values are highlighted in bold and the next best performance are underlining.
Table 9.
AUPR, IC_AUPR and DP_AUPR performance comparison on the second dataset generated by sequence identity
| Methods | AUPR | IC_AUPR | DP_AUPR | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MF | CC | BP | MF | CC | BP | MF | CC | BP | ||
| Single | Diamond | 0.282 | 0.265 | 0.147 | 0.254 | 0.195 | 0.115 | 0.237 | 0.190 | 0.104 |
| algorithms | BlastKNN | 0.403 | 0.403 | 0.242 | 0.363 | 0.302 | 0.189 | 0.338 | 0.295 | 0.170 |
| DeepGO | 0.385 | 0.693 | 0.356 | 0.325 | 0.505 | 0.266 | 0.279 | 0.492 | 0.228 | |
| DeepGOA | 0.533 | 0.696 | 0.392 | 0.469 | 0.579 | 0.297 | 0.424 | 0.567 | 0.256 | |
| DeepGOCNN | 0.330 | 0.620 | 0.270 | 0.266 | 0.418 | 0.184 | 0.220 | 0.403 | 0.149 | |
| TALE | 0.210 | 0.573 | 0.191 | 0.156 | 0.353 | 0.109 | 0.118 | 0.335 | 0.078 | |
| DeepGOZero | 0.533 | 0.622 | 0.357 | 0.479 | 0.407 | 0.270 | 0.444 | 0.397 | 0.239 | |
| ATGO | 0.467 | 0.681 | 0.357 | 0.410 | 0.513 | 0.268 | 0.365 | 0.499 | 0.232 | |
| DeepGraphGO | 0.569 | 0.727 | 0.431 | 0.509 | 0.564 | 0.346 | 0.467 | 0.552 | 0.313 | |
| Composite | DeepGOPlus | 0.463 | 0.636 | 0.319 | 0.404 | 0.458 | 0.239 | 0.366 | 0.445 | 0.208 |
| algorithms | TALE+ | 0.436 | 0.600 | 0.266 | 0.375 | 0.424 | 0.180 | 0.337 | 0.408 | 0.149 |
| DeepGOZero+ | 0.552 | 0.625 | 0.364 | 0.501 | 0.428 | 0.280 | 0.465 | 0.418 | 0.250 | |
| ATGO+ | 0.554 | 0.693 | 0.383 | 0.500 | 0.527 | 0.296 | 0.460 | 0.514 | 0.262 | |
Note: The best performance values are highlighted in bold and the next best performance are underlining.
Performance comparison on disorder proteins
Several proteins that lack stable structures are known as disorder proteins [56], which perform essential functions, such as display site, assembler, effector, and chaperone [57]. Consequently, in this section, we extract 16 disorder proteins from the test set and use existing methods to predict the functions of their disorder regions. As shown in Figure 6, it can be obtained that all of these methods get poor performance with low AUPR values for each GO term. Additionally, Figure 7 demonstrates that DeepGOA and DeepGOZero surpass other methods significantly in terms of M-AUPR, while other methods fail to predict the functions of disorder regions, especially homology-based methods, BlastKNN and Diamond. On the other hand, Figure 8 shows that these methods can only predict several functions with shallow depths, such as molecular function regulator activity (GO:0098772) and binding (GO:0005488). With the deeper functions, beginning from cellular component disassembly (GO:0022411) in the heat map, there are tougher challenges for these methods. Overall, DeepGOA and DeepGOZero achieve better performance than other methods. And all of these methods have limitations on disorder proteins.
Figure 6.
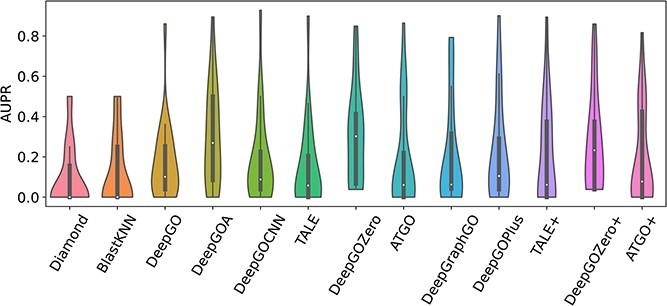
Predictive performance comparison on disorder proteins in terms of AUPR.
Figure 7.
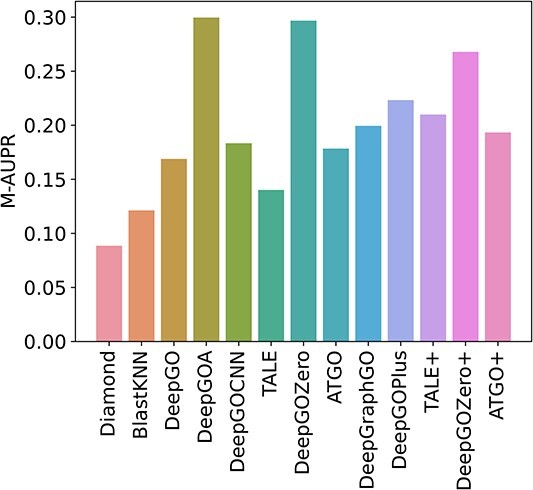
Predictive performance comparison on disorder proteins in terms of M-AUPR.
Figure 8.
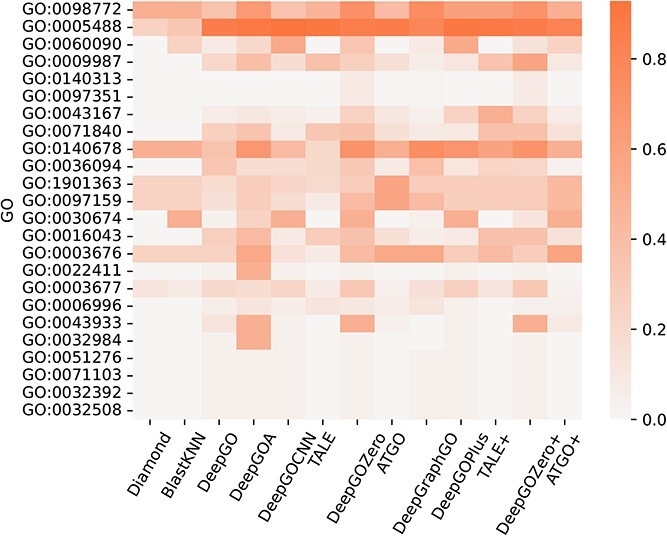
Predictive performance of existing methods on the functions of disorder regions in terms of AUPR. GO terms are sorted from top to bottom by their depths, from shallow to deep.
Generality to new specific species (HUMAN and MOUSE)
To assess the cross-species performance of existing models, we select two crucial species, HUMAN and MOUSE, for evaluation. In particular, we design two training sets: (1) the original training set of Dataset1, including the proteins from the target species. (2) an extracted training set of Dataset1, excluding the proteins from the target species. The results are shown in Figures 9 and 10. An obvious and common finding is that all methods exhibit a significant decline in performance when the proteins of the target species are removed from the training data. Notably, the effect of DeepGraphGO is much slighter than other methods, as it benefits from the cross-species strategy proposed by DeepGraphGO, where various species’ PPI networks share the same GCN layers. Furthermore, in terms of AUPR, DeepGOZero(+) shows significant advantages compared to other methods, except ATGO(+). This suggests that the approach of ontology embedding is valuable for inferring functions. Additionally, ATGO(+) consistently outperforms other methods in most cases, highlighting the effectiveness of the protein large language model, which is pre-trained on many extra proteins not covered by Dataset1.
Figure 9.

Generalizability of existing methods to HUMAN species. The blue part indicates that several HUMAN proteins are contained in the training set, while the yellow part indicates that these proteins are removed from the training set.
Figure 10.

Generalizability of existing methods to MOUSE species. The blue part indicats that several MOUSE proteins are contained in the training set, while the yellow part indicates that these proteins are removed from the training set.
Performance comparison on different depths of GO terms
The function representation of GO terms becomes more specific as their depth increases, providing researchers with a more detailed understanding of protein roles in living cells. However, these GO terms tend to have fewer associated samples, resulting in a challenge for accurately annotating these GO terms to proteins. In this section, taking into account the distribution of annotation depths (Figure 4), we classify these GO terms into three subgroups based on their depths: 0–3, 4–6 and >6 for BPO and MFO, while 0–2, 3–5 and >5 for CCO. The results are presented in the form of box plots in Figure 11. Obviously, the M-AUPR values exhibit a gradual downward trend from left to right, as expected. Notably, as the depth increases, the size of some boxes significantly reduces, and in some cases, certain boxes are even missing. For instance, DeepGO, DeepGOA and TALE are absent, which can be attributed to the limitations of these methods, like not being able to predict GO terms with few samples.
Figure 11.

Distribution of AUPR on different GO terms grouped by different depths, as calculated by existing methods.
Performance comparison on GO terms grouped by different frequencies
Learning with limited samples remains a persistent challenge in deep learning. This challenge is particularly acute in AFP, where many GO terms annotate only a small number of proteins, making it difficult for models to accurately annotate these GO terms to proteins. In this section, we evaluate the performance of these methods on rare GO terms with different frequencies (samples). Based on the frequency distribution of GO terms (Figure 3), we divide rare GO terms into three groups via their frequencies: 0–30, 30–60 and 60–100. As shown in Figure 12, consistent with our expectations, all methods perform poorly on GO terms with small sample sizes (frequencies < 30). Among these methods, DeepGOZero+ stands out from the rest, due to its utilization of zero-shot learning, which can learn from zero or a few samples. In contrast, methods that solely rely on learning the relationships between sequence motifs and functions, such as DeepGO and DeepGOCNN, both perform poorly on rare GO terms, due to the lack of learning samples, which is also a common challenge faced by traditional deep learning-based methods.
Figure 12.

Performance comparison of AUPR on different GO terms grouped by different frequencies.
Performance comparison on GO terms grouped by IC
Accurately predicting informative and valuable functions has always been a crucial criterion for evaluating the practical applicability of AFP methods. Since IC values can reflect the information contained in GO terms, previous studies [30] have often evaluated their methods on GO terms grouped by IC values. Following this approach, we divide the GO terms into distinct groups based on their IC values and evaluate the performance of these methods on each group. As shown in Figure 13, all methods encounter challenges in predicting functions with rich information. Specifically, for GO terms with IC values between 4 and 7, many single methods can only predict a small portion of the functions, i.e. the corresponding AUPR > 0, except for DeepGOZero, ATGO and DeepGraphGO. This observation is more pronounced in the most difficult groups with IC values exceeding 7. For instance, among the single algorithms, only DeepGOZero predicts these functions with a small median AUPR for MFO. On the other hand, for CCO, DeepGOZero consistently outperforms other methods, while ATGO and DeepGraphGO can predict a limited number of functions. For BPO, none of these methods can accurately predict the target functions, while only sequence similarity-based methods and composite methods achieve minimal scores. Above all, integrating sequence similarity-based methods and considering the relationships between functions can facilitate AFP, such as DeepGOZero and composite algorithms. Despite these advancements, there are still significant limitations in accurately predicting rare functions with high IC values, and existing methods cannot achieve satisfactory results.
Figure 13.

Distribution of AUPR on different GO terms grouped by different IC values.
CONCLUSION
Advances in high-throughput technologies have facilitated the prediction of protein functions using computational methods based on massive biological data. Many computational methods have been proposed to tackle this task. In this study, we first collect and analyze the latest protein and function data, demonstrating that protein function prediction is an unbalanced multi-label classification problem, where more specific and informative functions are more challenging to predict. Then, we investigate existing computational methods and classify them into sequence-based methods, sequence- and structure-based methods, sequence- and PPI network-based methods, sequence- and literature-based methods, and ensemble prediction methods. To evaluate these methods, we introduce a novel evaluation metric that considers the informativeness and depths of functions. Additionally, we also design eight application cases in terms of different properties of proteins and functions and evaluated these methods in specific cases. Each method shows different strengths in different application scenarios. Specifically, for regular cases, we recommend ATGO+ and DeepGOZero+, as they consistently demonstrate stable and outstanding performance. In the case of long proteins, DeepGOPlus and DeepGraphGO are recommended for MFO and CCO, while ATGO+ is advised for BPO. For proteins with low similarity to known proteins, DeepGOA and DeepGraphGO are recommended due to their effectiveness in handling this challenge. For proteins with disorder regions, DeepGOA and DeepGOZero are recommended. When predicting functions for proteins of new species, DeepGOZero+ and ATGO+ are suitable choices. In conclusion, accurately predicting deep and informative protein functions remains a significant challenge for all current methods. On the other hand, pre-trained large language models have shown great potential in AFP. Additionally, with the advancements in structural biology, leveraging predicted protein structure holds substantial potential for future protein function prediction.
Key Points
We survey a comprehensive collection of 17 computational approaches for protein function prediction, focusing on their input data characteristics.
We collect protein information and their function data, and analyze the data characters of proteins and functions.
We propose a new evaluation metric to consider the performance of existing methods comprehensively, which considers both informative values and depths of functions.
We provide a comprehensive comparison of these methods under eight application scenarios, such as the performance on long proteins, the performance on disorder proteins and the performance on different GO terms. Finally, we provide practical observations and discuss the suitability of each method for specific scenarios.
Author Biographies
Wenkang Wang received his BS degree from Central South University, China, in 2020. He is working toward the PhD degree in computer science and engineering at Central South University, China. His currently research interests include bioinformatics, complex network analysis, protein functions and interactions, machine learning and deep learning.
Yunyan Shuai received her BS degree in Computer Science from Central South University, China. She is currently a master student at the School of Computer Science and Engineering, Central South University. Her current research interests include bioinformatics and protein functions.
Qiurong Yang is currently a bachelor student at the School of Computer Science and Engineering, Central South University. Her current research interests include bioinformatics and protein functions.
Fuhao Zhang received his PhD degree at the School of Computer Science and Engineering, Central South University, China. His research focuses on computational prediction and characterization of protein structure and function.
Min Zeng is currently an assistant professor in the School of Computer Science and Engineering, Central South University. His main research interests include bioinformatics, machine learning and deep learning.
Min Li received her BS degree in Communication Engineering and MS and PhD degrees in Computer Science from Central South University, Changsha, China, in2001, 2004 and 2008, respectively. She is currently a Professor at the School of Computer Science and Engineering, Central South University. Her main research interests include bioinformatics and system biology.
Contributor Information
Wenkang Wang, School of Computer Science and Engineering, Central South University, 932 South Lushan Road, Yuelu District, Changsha 410083, China.
Yunyan Shuai, School of Computer Science and Engineering, Central South University, 932 South Lushan Road, Yuelu District, Changsha 410083, China.
Qiurong Yang, School of Computer Science and Engineering, Central South University, 932 South Lushan Road, Yuelu District, Changsha 410083, China.
Fuhao Zhang, School of Computer Science and Engineering, Central South University, 932 South Lushan Road, Yuelu District, Changsha 410083, China.
Min Zeng, School of Computer Science and Engineering, Central South University, 932 South Lushan Road, Yuelu District, Changsha 410083, China.
Min Li, School of Computer Science and Engineering, Central South University, 932 South Lushan Road, Yuelu District, Changsha 410083, China.
FUNDING
This work is supported in part by the High Performance Computing Center of Central South University, the National Natural Science Foundation of China under Grants (No. 61832019, No. 61622213 and No. 61728211), the Project (G20190018001), the Hunan Provincial Science and Technology Program (2018WK4001), and the Hunan Graduate Research and Innovation Project (CX20190082).
References
- 1. Li M, Ni P, Chen X, et al. Construction of refined protein interaction network for predicting essential proteins. IEEE/ACM Trans Comput Biol Bioinform 2017;16(4):1386–97. [DOI] [PubMed] [Google Scholar]
- 2. Zeng M, Li M, Wu FX, et al. DeepEP: a deep learning framework for identifying essential proteins. BMC Bioinform 2019;20:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang W, Meng X, Xiang J, et al. CACO: a core-attachment method with cross-species functional ortholog information to detect human protein complexes. IEEE J Biomed Health Inform 2023;27:4569–78. [DOI] [PubMed] [Google Scholar]
- 4. Uhlén M, Fagerberg L, Hallström BM, et al. Tissue-based map of the human proteome. Science 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 5. Lounkine E, Keiser MJ, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012;486(7403):361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. UniProt Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47(D1):D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shehu A, Barbará D, Molloy K. A survey of computational methods for protein function prediction. Big Data Anal Genom 2016;225–98. [Google Scholar]
- 8. Szklarczyk D, Gable AL, Nastou KC, et al. The string database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 2021;49(D1):D605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Motschall E, Falck-Ytter Y. Searching the MEDLINE literature database through PubMed: a short guide. Oncologie 2005;28(10):517–22. [DOI] [PubMed] [Google Scholar]
- 10. Burley SK, Berman HM, Kleywegt GJ, et al. Protein Data Bank (PDB): the single global macromolecular structure archive. Methods Mol Biol 2017;627–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with alphafold. Nature 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kulmanov M, Khan MA, Hoehndorf R, Wren J. DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 2018;34(4):660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kulmanov M, Hoehndorf R. DeepGOplus: improved protein function prediction from sequence. Bioinformatics 2020;36(2):422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koehler Leman J, Szczerbiak P, Renfrew PD, et al. Sequence-structure-function relationships in the microbial protein universe. Nat Commun 2023;14(1):2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones CE, Schwerdt J, Bretag TA, et al. Gosling: a rule-based protein annotator using blast and go. Bioinformatics 2008;24(22):2628–9. [DOI] [PubMed] [Google Scholar]
- 17. Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using diamond. Nat Methods 2021;18(4):366–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu YH, Zhang C, Yu DJ, Zhang Y. Integrating unsupervised language model with triplet neural networks for protein Gene Ontology prediction. PLoS Comput Biol 2022;18(12):e1010793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang F, Song H, Zeng M, et al. A deep learning framework for Gene Ontology annotations with sequence-and network-based information. IEEE/ACM Trans Comput Biol Bioinform 2020;18(6):2208–17. [DOI] [PubMed] [Google Scholar]
- 20. LeCun Y, Bengio Y, Hinton G. Deep learning. Deep Learn Nat 2015;521(7553):436–44. [DOI] [PubMed] [Google Scholar]
- 21. Liu W, Wang Z, Liu X, et al. A survey of deep neural network architectures and their applications. Neurocomputing 2017;234:11–26. [Google Scholar]
- 22. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. Adv Neural Inform Process Syst 2017;30. [Google Scholar]
- 23. Cao Y, Shen Y. Tale: transformer-based protein function annotation with joint sequence–label embedding. Bioinformatics 2021;37(18):2825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kulmanov M, Hoehndorf R. DeepGOZero: improving protein function prediction from sequence and zero-shot learning based on ontology axioms. Bioinformatics 2022;38:i238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kulmanov M, Liu-Wei W, Yan Y, et al. EL embeddings: geometric construction of models for the description logic EL ++. arXiv preprint arXiv:1902.10499. 2019.
- 26. Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci 2021;118(15):e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gligorijević V, Renfrew PD, Kosciolek T, et al. Structure-based protein function prediction using graph convolutional networks. Nat Commun 2021;12(1):3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kipf TN, Welling M. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907. 2016.
- 29. Graves A. Generating sequences with recurrent neural networks. arXiv preprint arXiv:1308.0850. 2013.
- 30. Lai B, Xu J. Accurate protein function prediction via graph attention networks with predicted structure information. Brief Bioinform 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Veličković P, Cucurull G, Casanova A, et al. Graph attention networks. arXiv preprint arXiv:1710.10903. 2017.
- 32. Xu J, Mcpartlon M, Li J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat Mach Intell 2021;3(7):601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang J, Ren J, Li M, et al. Identification of hierarchical and overlapping functional modules in PPI networks. IEEE Trans Nanobiosci 2012;11(4):386–93. [DOI] [PubMed] [Google Scholar]
- 34. Meng X, Li W, Peng X, et al. Protein interaction networks: centrality, modularity, dynamics, and applications. Front Comp Sci 2021;15:1–17. [Google Scholar]
- 35. Liu X, Lu Y, Wang L, et al. RF-PSSM: a combination of rotation forest algorithm and position-specific scoring matrix for improved prediction of protein-protein interactions between hepatitis C virus and human. Big Data Mining Anal 2022;6(1):1–11. [Google Scholar]
- 36. Peng W, Li M, Chen L, Wang L. Predicting protein functions by using unbalanced random walk algorithm on three biological networks. IEEE/ACM Trans Comput Biol Bioinform 2015;14(2):360–9. [DOI] [PubMed] [Google Scholar]
- 37. Perozzi B, Al-Rfou R, Skiena S. DeepWalk: online learning of social representations. In: Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Association for Computing Machinery, New York, USA, 2014, p. 701–10.
- 38. Graves A, Schmidhuber J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw 2005;18(5–6):602–10. [DOI] [PubMed] [Google Scholar]
- 39. Jones P, Binns D, Chang HY, et al. Interproscan 5: genome-scale protein function classification. Bioinformatics 2014;30(9):1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu Z, Pan S, Chen F, et al. A comprehensive survey on graph neural networks. IEEE Trans Neural Netw Learn Syst 2020;32(1):4–24. [DOI] [PubMed] [Google Scholar]
- 41. You R, Yao S, Mamitsuka H, Zhu S. DeepGraphGO: graph neural network for large-scale, multispecies protein function prediction. Bioinformatics 2021;37:i262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barot M, Gligorijević V, Cho K, Bonneau R. NetQuilt: deep multispecies network-based protein function prediction using homology-informed network similarity. Bioinformatics 2021;37(16):2414–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liao CS, Lu K, Baym M, et al. IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics 2009;25(12):i253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torres M, Yang H, Romero AE, Paccanaro A. Protein function prediction for newly sequenced organisms. Nat Mach Intell 2021;3(12):1050–60. [Google Scholar]
- 45. You R, Huang X, Zhu S. DeepText2GO: improving large-scale protein function prediction with deep semantic text representation. Methods 2018;145:82–90. [DOI] [PubMed] [Google Scholar]
- 46. Le Q, Mikolov T. Distributed representations of sentences and documents. In: International Conference on Machine Learning. PMLR, 2014, p. 1188–96. [Google Scholar]
- 47. You R, Zhang Z, Xiong Y, et al. GOLabeler: improving sequence-based large-scale protein function prediction by learning to rank. Bioinformatics 2018;34(14):2465–73. [DOI] [PubMed] [Google Scholar]
- 48. Li H. A short introduction to learning to rank. IEICE Trans Inform Syst 2011;E94-D(10):1854–62. [Google Scholar]
- 49. Ofer D, Linial M. ProFET: feature engineering captures high-level protein functions. Bioinformatics 2015;31(21):3429–36. [DOI] [PubMed] [Google Scholar]
- 50. You R, Yao S, Xiong Y, et al. NetGO: improving large-scale protein function prediction with massive network information. Nucleic Acids Res 2019;47(W1):W379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yao S, You R, Wang S, et al. NetGO 2.0: improving large-scale protein function prediction with massive sequence, text, domain, family and network information. Nucleic Acids Res 2021;49(W1):W469–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang S, You R, Liu Y, et al. NetGO 3.0: protein language model improves large-scale functional annotations. Genom Proteom Bioinform 2023;21:349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou N, Jiang Y, Bergquist TR, et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol 2019;20(1):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boutet E, Lieberherr D, Tognolli M, et al. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt knowledgebase: how to use the entry view. Methods Mol Biol 2016;23–54. [DOI] [PubMed] [Google Scholar]
- 55. Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 2019;47(D1):D309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peng Z, Yan J, Fan X, et al. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci 2015;72:137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pang Y, Liu B. DMFpred: predicting protein disorder molecular functions based on protein cubic language model. PLoS Comput Biol 2022;18(10):e1010668. [DOI] [PMC free article] [PubMed] [Google Scholar]