

Abstract
Background
Numerous studies have characterized the gut microbiome (GM) in lung cancer (LC). Yet, the causality between GM and LC and its subtypes remain uncharacterized.
Methods
Two‐sample Mendelian randomization (MR) was designed to investigate the causal relationship between the GM and LC and its subtypes, using publicly available summary data of genome‐wide association studies. The researchers ran two groups of MR analyses, including the genome‐wide statistical significance threshold (5 × 10−8) and the locus‐wide significance level (1 × 10−5).
Results
Using MR analysis, we ascertained 42 groups of GM that are intimately linked to LC and its subtypes at the locus‐wide significance level. Of the 42 groups, 12 were in LC, nine in non‐small cell lung cancer (NSCLC), six in small cell lung cancer (SCLC), two in lung adenocarcinomas, and 13 in lung squamous carcinomas. After false discovery rate correction, we still found a remarkable causal interaction between the Eubacterium ruminantium group and SCLC. Moreover, five groups of GM closely linked to LC and its subtypes were recognised at the genome‐wide statistical significance threshold. This finding included one group each in LC, NSCLC and SCLC, two groups in lung adenocarcinoma and none in lung squamous carcinoma. None of the foregoing findings were heterogeneous or horizontal pleiotropy. Reverse MR revealed that genetic susceptibility to LC and its subtypes caused significant changes in three groups of GM.
Conclusion
Our findings substantiate the causality between GM and LC and its subtypes. This study offers fresh insights into the function of GM in mediating the progression of LC.
Keywords: gut microbiota, gut‐lung axis, histological subtype, lung cancer, Mendelian randomization
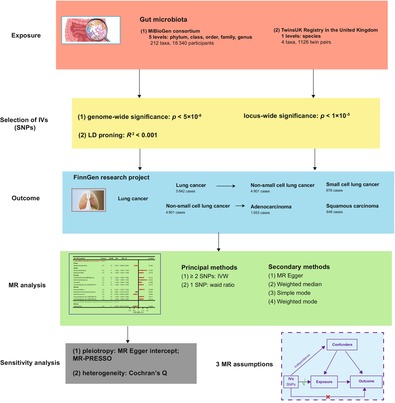
Overview of Mendelian randomization (MR) analysis process and major assumptions.
INTRODUCTION
Lung cancer (LC) is a fatal neoplasm that poses a threat to human life and well‐being. Approximately 2.2 million new cases and 1.8 million deaths from LC were reported worldwide in 2020, 1 making it the second most prevalent cancer and the chief cause of cancer deaths. Not only do the high incidence and mortality rates of LC constitute a human health hazard but they also have grave socioeconomic ramifications. Depending on the histology, LC is categorized as either small cell lung cancer (SCLC) or non‐small cell lung cancer (NSCLC). 2 The latter category mainly includes lung adenocarcinoma and lung squamous cell carcinoma. 3 Worth noting is that cigarette smoking is still perceived as the most prominent risk factor for LC. 4 However, as smoking prevalence declines and the incidence of LC among nonsmokers grows, other risk factors are gaining attention from investors. 5 , 6 These risk factors include marijuana use, biomass combustion, occupational exposure, metabolic factors, air pollution and viral infections. It is therefore essential to prioritise and define more mutable protective agents and risk factors to guard against the onset and progression of LC.
The gut microbiome (GM) is the largest microbial colony in the humankind, 7 and it is engaged in a diverse range of physiological features, from sustaining metabolic stability and mediating immune responses to combating infection. 8 In fact, some researchers have hypothesised that the GM is a novel organ that acts through the microbiota and its metabolites to exert its influence on the metabolism and other organs, such as the brain, skin and lungs. 9 With the advancement of high‐throughput sequencing technologies and platforms, several links have been identified between the GM and sophisticated disorders, including autoimmune diseases, cardiovascular diseases and malignant tumours. 10
Several studies have demonstrated an affiliation between the GM and LC risk. Gui et al. 11 and Lu et al. 12 conducted case‐control studies and detected many discrepancies between sputum and faecal bacterial types in individuals with and without NSCLC. Moreover, Liu et al. 13 observed that streptococci were mainly associated with this histological type of SCLC. Nevertheless, GM involves a wide range of types, and the causal relationship with different LC histological risks has not been adequately studied. Moreover, studies have been largely observational in nature, and the evidence of correlation is not robust. Thus, further studies are warranted to elucidate the causal relationship between distinct GMs and the risk of LC histological subtypes.
Mendelian randomization (MR) is an approach that assesses the causal relationship between risk factors and illness. 14 MR utilises genetic variants as instrumental variables (IVs). This approach allows for the effective avoidance of confounding agents, which can be elusive to control for in observational studies. 15 Since alleles that affect genetic variants are distributed randomly to offspring at gestation, regardless of environmental and other uncharted confounders, MR analyses are analogous to natural randomized controlled trials. 16 Such analyses permit the assessment of causality between exposures and outcomes at the genetic level while ruling out the likelihood of reverse causality. 17 In this study, we applied two‐sample MR analysis to assess the causal relationship between genetically predicted GM and the overall risk of LC, two LC subtypes (NSCLC and SCLC) and two NSCLC subtypes (lung adenocarcinoma and lung squamous carcinoma).
METHODS
Study design
Our study adheres to the STROBE‐MR statement, 18 which governs the reporting of MR studies. The potential causal effects of GM on LC and its subtypes were explored by means of a two‐sample MR method. Our MR was based on three key assumptions 19 : (1) IVs are firmly related to GM, (2) IVs should not be susceptible to known or unknown confounders and (3) IVs affect LC and its subtypes only through GM. This study employed publicly available data and consequently did not require ethical approval or informed consent. Figure 1 presents our study design.
FIGURE 1.
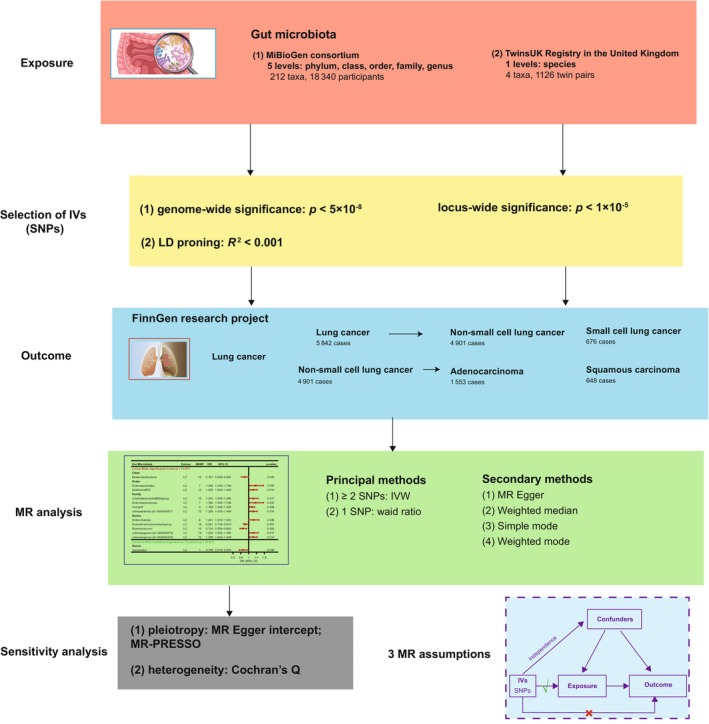
Overview of Mendelian randomization (MR) analysis process and major assumptions.
Data sources for gut microbiota
Single‐nucleotide polymorphisms (SNPs) relevant to the composition of human GM were chosen as IVs from the genome‐wide association study (GWAS) dataset of the international consortium MiBioGen. 10 This is a multiethnic large‐scale GWAS that aggregated 16S ribosomal RNA gene sequencing profiles and genotyping data from 18 340 attendees from 24 cohorts to probe for connections between autosomal human genetic variants and GM. Altogether, 211 taxa (131 genera, 35 families, 20 orders, 16 phyla and nine phyla) were enrolled.
Furthermore, given that the data captured from the MiBioGen consortium is not profiled at the species level, we pulled IVs of GM at the species level from the TwinsUK Registry's GWAS study. 20 The 16S rRNA sequencing data from 1126 bipartite pairs was secured by Goodrich et al.20 and ultimately defined four qualified species. The scientific names and the number of SNPs associated with all the above 215 candidate GMs are displayed in Table S1.
Selection of genetic instruments
Rigorous quality control procedures were followed to filter the SNPs related to GM and yield the optimal IVs needed. 21 , 22 , 23 First, SNPs prominently associated with GM were picked as IVs. Two thresholds were employed to elect IVs. A panel of SNPs below the genome‐wide statistical significance threshold (5 × 10−8) became available as IVs. To attain fuller results, another set of SNPs smaller than the locus‐wide significance level (1 × 10−5) were simultaneously chosen as IVs. Second, the linkage disequilibrium between SNPs was computed using European sample data from the 1000 Genomes Project as a reference panel. Only SNPs with the smallest p‐value among all SNPs with R 2 < 0.001 (clumping window size = 10 000 kb) are retained. Third, SNPs with an allele frequency ≤0.01 were excluded. Fourth, allele frequency information was ascribed to the forward strand alleles when palindromic SNPs were present. Fifth, the F statistic for each SNP is utilised to gauge the strength of the collinearity between IV and exposure and to preclude bias from weak IVs. In this case, weak IVs were deemed unbiased when the F value was greater than 10.
Data sources for lung cancer and its subtypes
GWAS summary statistics for LC and its subtypes were retrieved from the most recent release of version R9 (https://r9.finngen.fi/) from the FinnGen Research Project. 24 All samples were of European ancestry, and both males and females were included. The study further granularized the pathological categories of LC to include LC (5842 cases), NSCLC (4901 cases), SCLC (676 cases), lung adenocarcinoma (1553 cases) and lung squamous carcinoma (1413 cases). Data was normalized by age, sex, genetic correlation, genotyping batch and the top 10 principal components.
Mendelian randomization analysis
MR analysis was performed across five practises, including inverse variance weighting (IVW), 25 multiplicative random effects (MR‐Egger), 26 weighted median, 27 simple mode 28 and weighted mode. 29 IVW is the most used and validated method and was voted the major analytical option for this study because it delivers estimates with the utmost power reliant on the assumption that all SNPs are valid IVs. 28 In situations where the assumption of no horizontal pleiotropy is breached, MR‐Egger affords a more formidable alternative. 26 The weighted median, simple mode and weighted mode each have their virtues and constraints and are likely to be potentially useful depending on the circumstances of the specific research issue and the available data. 21 , 27 To certify the stabilization of the outcome, supplementary analyses were carried out simultaneously, utilizing these three methods. Furthermore, aiming for more robust and reliable results, we performed a reverse MR analysis with LC and its pathological subtypes as exposures and all statistically significant GMs as outcomes.
Sensitivity analysis
We performed sensitivity analyses to help us assess the robustness of our findings. Two methods were used to assay and address horizontal pleiotropy 30 : MR‐Egger regression and MR pleiotropy residual sum and outlier (MR‐PRESSO). Furthermore, Cochran's Q test was enlisted to gather the heterogeneity among the SNPs relevant to each GM taxa. 16
Statistical analysis
Statistical analysis was performed using R software (version 4.3.1). The two sample MR R package (version 0.5.7, Stephen Burgess, Chicago, IL, USA) was used to perform MR analysis of the causal effects between GM and LC and its subtypes. A two‐sided p < 0.05 was regarded as statistically significant. Moreover, we corrected for false discovery rate (FDR), using a threshold for the p‐value. Causality was assumed to be significant at p < 0.05 and FDR < 0.05. Of note, when p < 0.05 but FDR > 0.05, causality was seen as only potentially significant.
RESULTS
Data sources
GWAS summary statistics for GM were collected from the MiBioGen and TwinsUK Registry consortium, which incorporate 18 340 and 1126 participants, respectively. In addition, GWAS summary statistics for LC and its subtypes were sourced from the FinnGen consortium, which contains 5842 cases of LC, 4902 cases of NSCLC, 676 cases of SCLC, 1553 cases of lung adenocarcinoma and 1413 cases of lung squamous carcinoma. Table 1 presents the breakdown of the two datasets pertaining to GM and the five datasets concerning LC and its subtypes.
TABLE 1.
Description of gut microbiota and lung cancer.
| Traits | Consortium | Sample size | Populations | Year | Journal |
|---|---|---|---|---|---|
| Gut | |||||
| Gut microbiota | MiBioGen | 18 340 individuals | European | 2021 | Nature genetics |
| TwinsUK Registry | 1126 individuals | European | 2016 | Cell host and microbe | |
| Lung cancer | |||||
| LC | FinnGen (R9) | 5842 cases | European | 2022 | Nature |
| NSCLC | FinnGen (R9) | 4901 cases | European | 2022 | Nature |
| SCLC | FinnGen (R9) | 676 cases | European | 2022 | Nature |
| NSCLC, adenocarcinoma | FinnGen (R9) | 1553 cases | European | 2022 | Nature |
| NSCLC, squamous carcinoma | FinnGen (R9) | 1413 cases | European | 2022 | Nature |
Abbreviations: LC, lung cancer; NSCLC, non‐small cell lung cancer; SCLC, small cell lung cancer.
Selection of instrumental variables
Following strict quality control and other steps, a grand total of 2242 SNPs were neutralised as IVs related to 213 GMs in LC at the locus‐wide significance level (p < 1 × 10−5). In addition, an aggregate of 2061 SNPs in NSCLC was mapped to 195 GM‐associated IVs, and a total of 2226 SNPs in SCLC was characterized as 211 GM‐associated IVs. In parallel, 2215 SNPs in lung adenocarcinoma were programmed to identify IVs that correlate with 211 GMs, and 2260 SNPs in lung squamous carcinoma were profiled to indicate IVs that correlate with 215 GMs. In all types, each SNP is devoid of weak instrumental bias (between 14.59 and 88.43, all F > 10; Table S2). Table S3 lists in detail the information of the IVs.
Furthermore, at the genome‐wide statistical significance threshold (p < 5 × 10−8), only 22 SNPs were identified as IVs linked to 26 GMs, irrespective of the LC type, and each SNP exhibited adequate validity (between 29.35 and 88.43, with all F > 10; Table S2). Table S4 elaborates on the primary messages of the IVs.
Causal effects of gut microbiota and lung cancer
Based on the locus‐wide significance level, Figure 2 shows the relationship between GM and LC by MR analysis. In addition, comprehensive results are shown in Table S5. Among the MR results, our findings suggest that three groups of GM are protective factors for LC (Class Betaproteobacteria, Genus Eubacterium ruminantium group, and Genus Ruminococcus 1), whereas nine groups of GMs are risk factors for LC (Order Enterobacteriales, Order Mollicutes RF9, Family ClostridialesvadinBB60group, Family Enterobacteriaceae, Family FamilyXI, Family.unknownfamily.id.1000005471, Genus Enterorhabdus, Genus unknowngenus.id.1000000073, and Genus unknowngenus.id.1000005472). Correction for all p‐values does not detect significant differences in GM, meaning that the FDRs are all greater than 0.05 (Table S6). The sensitivity analysis stated that none of the mentioned outcomes had heterogeneity or horizontal pleiotropy (Table S7).
FIGURE 2.
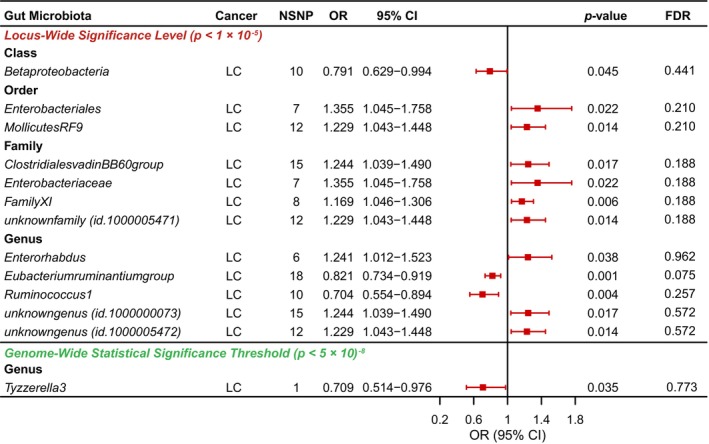
Causal analysis of gut microbiota (GM) and lung cancer (LC).
Depending on the genome‐wide statistical significance threshold, Figure 2 further shows the relationship between GM and LC by MR analysis. In addition, comprehensive results are shown in Table S8. However, it was noteworthy that only one GM, Genus Tyzzerella3, acted as a protective factor for LC occurrence (odds ratio [OR], 0.709; 95% confidence interval [CI], 0.514–0.976; p = 0.035). The results of the sensitivity analysis are presented in Table S9.
Causal effects of gut microbiota and non‐small cell lung cancer and small cell lung cancer
Based on the locus‐wide significance level, Figure 3 and Table S10 show the relationship between GM and two LC subtypes (NSCLC and SCLC) by MR analysis. First, in NSCLC, we identified four groups of GMs that were protective factors (Class Betaproteobacteria, Family Clostridiaceae1, Genus Ruminococcus1, and Genus Ruminococcusgauvreauiigroup) and five groups that were risk factors (Class Coriobacteriia, Family ClostridialevadinBB60group, Family Coriobacteriaceae, Family FamilyXI, and Genus unknownfamily.id. 1000000073; Figure 3a). In addition, in SCLC, we detected only one group of GM that offered protection, namely the Genus Eubacterium ruminantium group. Furthermore, our findings suggest that six groups of GMs are risk factors for SCLC (Family Alcaligenaceae, Family Rikenellaceae, Genus Adlercreutzia, Genus Eubacteriumbrachygroup, and Genus Intestinibacter; Figure 3b). Calibrating the p‐value for all results for NSCLC and SCLC, we found that genus levels of Eubacterium ruminantium group had a significant negative correlation in SCLC (FDR = 0.017; Table S11). Sensitivity analysis found no heterogeneity or horizontal pleiotropy in any of the above results (Table S12).
FIGURE 3.
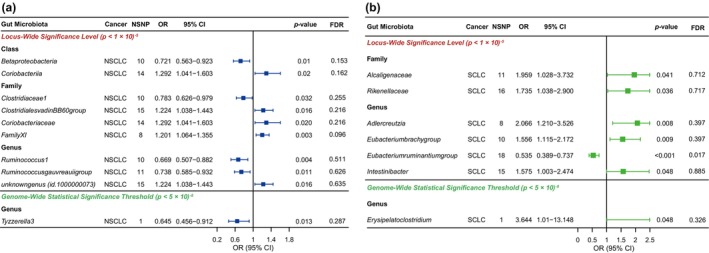
Causal analysis of gut microbiota (GM) and (a) non‐small cell lung cancer (NSCLC) and (b) small cell lung cancer (SCLC).
Depending on the genome‐wide statistical significance threshold, Figure 3 and Table S13 further show the relationship between GM and two LC subtypes (NSCLC and SCLC) by MR analysis. We found that Genus Tyzzerella3 (OR, 0.709; 95% CI: 0.514–0.976; p = 0.035) was a protective factor for the onset of NSCLC (Figure 3a), whereas Genus Erysipelatoclostridium (OR, 3.644; 95% CI: 1.010–13.148; p = 0.048) was a risk factor for SCLC occurrence (Figure 3b). The results of the sensitivity analysis are presented in Table S14.
Causal effect of gut microbiota on lung adenocarcinoma and lung squamous carcinoma
Based on the locus‐wide significance level, Figure 4 and Table S15 show the relationship between GM and two NSCLC subtypes (lung adenocarcinoma and lung squamous carcinoma) by MR analysis. First, for lung adenocarcinoma, we found that only Genus Oscillibacter was a protective factor, and only Genus Intestinibacter was a risk factor (Figure 4a). Then, for lung squamous carcinoma, nine groups of GM were risk factors (Phylum Lentisphaerae, Class Betaproteobacteria, Class Lentisphaeria, Class Negativicutes, Order Burkholderiales, Order Selenomonadales, Order Victivallales. Genus LachnospiraceaeUCG004 and Genus Ruminococcus1). In addition, four groups of GM were risk factors for lung squamous carcinoma (Phylum Tenericutes, Class Mollicutes, Family FamilyXI and Genus Parasutterella; Figure 4b). Adjustment of p‐values for all results for adenocarcinomas and squamous carcinomas yields no significant correlation with GM (Table S16). Sensitivity analysis expressed the absence of heterogeneity and horizontal pleiotropy for all the described results (Table S17).
FIGURE 4.
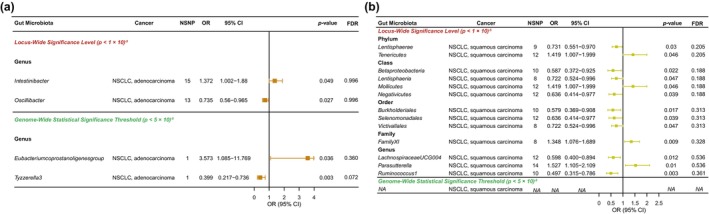
Causal analysis of gut microbiota (GM) and (a) lung adenocarcinoma and (b) lung squamous cell carcinoma.
Depending on the genome‐wide statistical significance threshold, Figure 4 and Table S18 further show the relationship between GM and two NSCLC subtypes (lung adenocarcinoma and lung squamous carcinoma) by MR analysis. The results indicated that significant GM was found only in adenocarcinomas. Among them, at the genus level, Tyzzerella3 (OR, 0.399; 95% CI: 0.217–0.736; p = 0.003) was a protective factor for adenocarcinoma, whereas the Eubacterium coprostanoligenes group (OR, 3.573; 95% CI: 1.085–11.769; p = 0.003) was a risk factor (Figure 4a). The results of the sensitivity analysis are presented in Table S19.
Reverse MR analysis
Reverse MR illustrated that among the 47 GM groups, genetically predicted NSCLC risk was associated with decreased abundance of Family ClostridialevadinBB60group and Genus unknownfamily.id.1000000073 (p < 0.05, FDR > 0.05; Figure 5). Meanwhile, genetically predicted risk of lung adenocarcinoma was related to lower levels of Genus Tyzzerella3, and this association persisted significantly after correction (p < 0.05, FDR < 0.05; Figure 5). Furthermore, the genetic susceptibility of LC and its subtypes was irrelevant to GM abundance in the other 44 groups.
FIGURE 5.
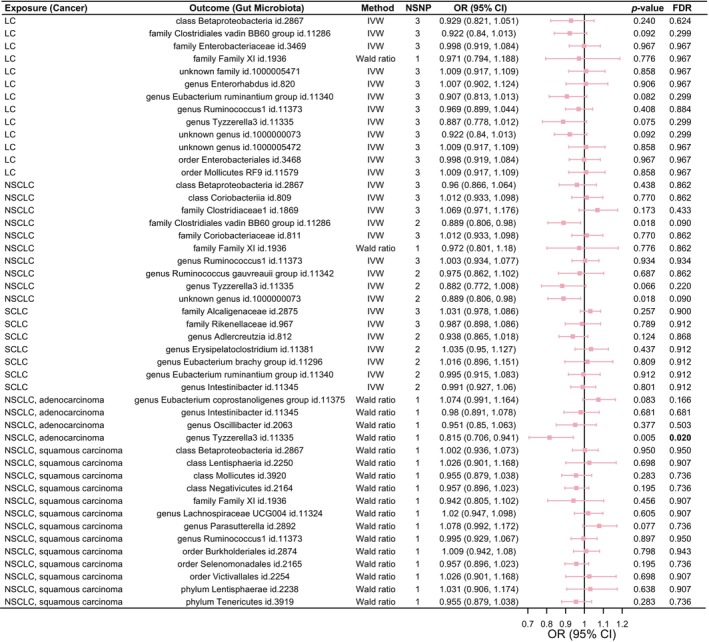
Causal analysis of genetic susceptibility to lung cancer (LC) and its subtypes with the gut microbiota (GM).
DISCUSSION
Although existing research has found evidence of a multifaceted and intricate nexus between GM and LC, most studies have been observational. An observational study is a traditional epidemiological approach that does not settle confounds arising from diverse biases and is vulnerable to the constraints of small sample sizes. Consequently, the evidence for causal associations from observational studies is not robust. Most studies measured GM changes in patients with a final diagnosis of LC and its subtypes and then speculated on the underlying carcinogenicity of GM. This approach lacks causal predictive evidence in the right direction. To overcome these shortcomings, we can judge the potential GM that can trigger the risk of LC and its subtypes by the MR method, which can minimally discard the interference of latent factors as well as reverse causality and hence draw more precise conclusions. In this study, the researchers utilized the largest and most up‐to‐date GWAS data on both GM and LC and its subtypes to identify closely matched SNPs as IVs. By means of MR analysis, we first determined 42 groups of GMs closely connected with LC and its subtypes at the locus‐wide significance level. We identified 12 groups in LC, nine groups in NSCLC, nine groups in LC, six groups in SCLC, two groups in lung adenocarcinoma, and 13 groups in lung squamous carcinoma. After FDR correction, we found that a significant causal relationship still existed between the Eubacterium ruminantium group and SCLC. Moreover, five groups of GM closely associated with LC and its subtypes were characterized at the genome‐wide statistical significance threshold, including one group each in LC, NSCLC and SCLC, two groups in lung adenocarcinoma and none in lung squamous carcinoma. Sensitivity analysis showed that none of the above‐mentioned results were heterogeneous or horizontally pleiotropic, and the results were plausible. Reverse MR indicated that among the 47 statistically significant GM groups, the genetic susceptibility of LC and its subtypes exhibited the reverse causality with GM abundance in three of the groups.
A comprehensive population‐based study 31 found that exposure to specific antibiotics, including penicillin, cephalosporins and macrolides, was associated with an increased risk of LC. This finding suggests a strong link between GM and LC. Zhuang et al. 32 reported that raised Enterococcus levels in GM were implicated in LC. Furthermore, LC patients have been found to have a general decline in gut microbial fitness, and Enterococcus and Bifidobacterium have been cited as potential biomarkers of LC. 32 Early LC was remarkably correlated with a diminished relative abundance of GM, encompassing three phyla, 13 genera and 20 species, while enrichment happened in four phyla, 11 genera and 15 species. 33 In addition, high levels of Bacillus and Akkermansia muciniphila can promote the development of LC. Similarly, Liu et al. 34 conducted a study involving 30 LC patients and observed a reduced abundance of gut microbial communities and a lower biodiversity of microbial ecosystems. These findings were characterized by a pathogenic microbiome that was both diverse and specific, along with a decreased presence of probiotic genera. In our results, some GMs have similarities for the pathogenic feature of LC, such as Enterobacteriales. The results of our study also implicate certain GMs that have never been implicated in LC before, such as Tyzzerella3 and MollicutesRF9. This finding lays the foundation for further studies in the future.
LC is subdivided into NSCLC and SCLC based on histological subtypes, and the exploration of GM characteristics associated with the risk of these two different subtypes is particularly important. Given that NSCLC accounts for the bulk of LC caseloads, it is essential to learn the mechanisms by which the microbiome may shape its advancement to optimise patient surveillance and reaction to cure. Gui et al. 11 have described ecological dysregulation of gut butyrate‐producing bacteria in NSCLC patients. Furthermore, intestinal butyrate‐producing bacteria, such as Clostridium leptum, Clostridial cluster I spp., Ruminococcus and Faecalibacterium prausnitzii, displayed significantly low levels in NSCLC, in contrast to Clostridial Cluster XIVa and Eubacterium rectal, which exhibited no shift in abundance. Botticelli et al. 35 reported that levels of Prevotella, Lactobacillus, Streptococcus, Enterobacteriaceae, Rikenellaceae, Oscillospira and Bacteroides plebeius in the feces of individuals with NSCLC were substantially higher than levels in healthy subjects. In addition, some studies have indicated that GM also aids in the efficacy of NSCLC treatment. Among NSCLC patients undergoing programmed cell death protein 1 (PD‐1) immunotherapy, responders exhibited a higher abundance of Akkermansia muciniphila, Ruminococcus, Eubacterium and Alistipes in their gut, and simultaneously a decreased abundance of Bifidobacterium and Parabacteroides, compared to nonresponders. 36 In related results, the use of antibiotics was found to interfere with the efficacy of LC treatment. Earlier studies found that antibiotic use preceding and for the duration of antitumor therapy dramatically lowered the clinical benefit of antitumor drugs in patients with NSCLC. 37 , 38 Nevertheless, the fact that SCLC accounts for a small fraction of LC, resulting in the limited publication of studies on the causal association of GM and SCLC so far, has left a major research gap in this field. Our study completes the causal effects of GM and NSCLC to some extent and fills the gap in studying the causal relationship between GM and SCLC. The GM‐related findings of several previous studies partly supports our foregoing conclusion, including our findings on Clostridiaceae1 and Ruminococcus1, while also expanding on some of the brand‐new drawings. In addition, in SCLC, we detected the Eubacterium ruminantium group remained statistically significant after FDR correction, which indicates that this group deserves further in‐depth validation and exploration. In an animal experiment, the usage of vitamin K2 in mice with colitis led to an increase in the abundance of Eubacterium ruminantium group in the colon, thereby alleviating colitis. 39 An MR analysis demonstrated that Eubacterium ruminantium group was negatively correlated with bone mineral density. 40 Moreover, our study revealed that this GM taxon was a protective factor for SCLC. Eubacterium ruminantium group possessed the ability to produce short‐chain fatty acids that inhibit proinflammatory cytokines such as IFN‐γ, IL‐1β, IL‐6, IL‐8, and TNF‐α, as well as increase the expression of anti‐inflammatory cytokines, such as IL‐10 and TGF‐β, resulting in an overall anti‐inflammatory effect. 41 Therefore, the inhibitory effect on inflammation may be a potential mechanism to reduce tumorigenesis. We also found that the GM associated with the risk of NSCLC and SCLC are not the same. This finding indicates that these two subtypes of LC are significantly different in terms of disease development and treatment, and that it is necessary to differentiate between them.
NSCLC, the most common type of LC, can be further subdivided into lung adenocarcinoma and lung squamous carcinoma. Previous studies found that the GM characteristics of these two histological types were also different. The study by Yan et al. 42 illustrated that Capnocytophaga, Selenomonas, Veillonella and Neisseria were prominently modified in patients with squamous and adenocarcinomas when compared to controls in saliva samples. Moreover, adenocarcinomas exhibited greater phylogenetic diversity, a higher relative abundance of Thermus and a reduced relative abundance of Ralstonia in comparison to squamous carcinomas. 43 These differences suggest that microbiota play a role in cancer histology. However, studies have focused mainly on the lung microbiota, and there is a paucity of research on the causal relationship between GM and adenocarcinoma and squamous carcinoma and comparisons between the two. Given this knowledge gap, we performed MR analysis to investigate the relationship between GM and lung adenocarcinoma or lung squamous carcinoma. We also observed that the GM causally associated with lung adenocarcinoma and squamous lung cancer differed dramatically between these two LC subtypes. This finding warrants further investigation in the future.
Despite their physical proximity, the gastrointestinal and respiratory tracts possess a common embryonic origin and highly analogous structures. These correspondences mean that the two areas can interact with each other in several aspects. In fact, the interaction between the gastrointestinal and respiratory tracts, referred to as the gut‐lung axis (GLA), is characterized by significant crosstalk. 44 This study concludes that the GLA plays an instrumental role in the mechanisms of action in the pathogenesis and progression of LC. Lung bacteria are a main constituent of the lung tumour microenvironment, and the GLA enables the indirect alteration of lung bacterial composition through strategies aimed at modifying the GM. 45 For example, a previous study documented that TNF‐α triggered the process of epithelial‐to‐mesenchymal transition, thereby promoting the metastasis of LC. 46 Crosstalk between microbiota in the GLA suggests promising novel therapeutic destinations for LC. Consequently, the potential mechanisms that underlie the connectivity between the GLA and LC should be further discussed in conjunction with ongoing MR studies.
Certain limitations restricted the work of this study. First, we conducted reverse causality analyses only for those GMs that were statistically significantly linked to LC and its subtypes, and did not comprehensively investigate other GM alterations arising from genetic susceptibility to LC and its subtypes. Second, since GWAS covers European populations only, the findings of this study may not be transferable to other ethnic groups. Third, the multiple statistical corrections employed were overly stringent and conservative and could have resulted in the omission of GM that may be causally related to LC and its subtypes. Consequently, it is necessary to validate these results in a broader cohort. Fourth, this is the first time that we have analyzed the relation between GM and the risk of LC and its subtypes using MR studies at the species level, but we did not discover positive results at the species level. Finally, given the difficulty of accessing complete data, we failed to validate our findings using a test cohort, hoping that this can be further refined in future studies.
In conclusion, through our analysis of the causal relationship between GM and LC and its subtypes by MR, we identified 47 groups of GM that were closely linked to LC and its subtypes, including 13 groups for LC, 10 for NSCLC, seven for SCLC, four for lung adenocarcinoma, and 13 for lung squamous carcinoma. After adjustment, the Eubacterium ruminantium group remained firmly causally associated with SCLC. However, more cohorts are necessary to prove this result in the future, and experiments are required to investigate the exact mechanism.
AUTHOR CONTRIBUTIONS
Yunlei Ma and Yefeng Shen were involved in the conception and design. Yunlei Ma, Yefeng Shen, Yuqing Deng, Tingting Shao, and Yong Cui were involved in analysis and interpretation of the data. Yefeng Shen and Yong Cui were involved in the drafting of the paper or revising it critically for intellectual content. All authors agree to be accountable for all aspects of the work.
CONFLICT OF INTEREST STATEMENT
All authors declare no competing interests.
Supporting information
Data S1. Supporting information.
Ma Y, Deng Y, Shao T, Cui Y, Shen Y. Causal effects of gut microbiota in the development of lung cancer and its histological subtypes: A Mendelian randomization study. Thorac Cancer. 2024;15(6):486–495. 10.1111/1759-7714.15220
Contributor Information
Yong Cui, Email: shenyefeng555@163.com, Email: cywork1@sina.com.
Yefeng Shen, Email: shenyefeng555@163.com.
DATA AVAILABILITY STATEMENT
All data used in the present study were obtained from genome‐wide association study summary statistics which were publicly released by genetic consortia. Gut microbiota: (1) MiBioGen: https://mibiogen.gcc.rug.nl; (2) TwinsUK Registry: Information from the supplementary table of the article with doi of 10.1016/j.chom.2016.04.017. Lung cancer and its subtypes: https://r9.finngen.fi/. All datasets generated for this study are included in the article/additional files.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2. Oser MG, Niederst MJ, Sequist LV, et al. Transformation from non‐small‐cell lung cancer to small‐cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165–e172. 10.1016/s1470-2045(14)71180-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang BY, Huang JY, Chen HC, et al. The comparison between adenocarcinoma and squamous cell carcinoma in lung cancer patients. J Cancer Res Clin Oncol. 2020;146:43–52. 10.1007/s00432-019-03079-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Groot P, Munden RF. Lung cancer epidemiology, risk factors, and prevention. Radiol Clin North Am. 2012;50:863–876. 10.1016/j.rcl.2012.06.006 [DOI] [PubMed] [Google Scholar]
- 5. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;41:1–24. 10.1016/j.ccm.2019.10.001 [DOI] [PubMed] [Google Scholar]
- 6. Malhotra J, Malvezzi M, Negri E, et al. Risk factors for lung cancer worldwide. Eur Respir J. 2016;48:889–902. 10.1183/13993003.00359-2016 [DOI] [PubMed] [Google Scholar]
- 7. Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adak A, Khan MR. An insight into gut microbiota and its functionalities. Cell Mol Life Sci. 2019;76:473–493. 10.1007/s00018-018-2943-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simonyté Sjödin K, Hammarström ML, Rydén P, et al. Temporal and long‐term gut microbiota variation in allergic disease: a prospective study from infancy to school age. Allergy. 2019;74:176–185. 10.1111/all.13485 [DOI] [PubMed] [Google Scholar]
- 10. Kurilshikov A, Medina‐Gomez C, Bacigalupe R, et al. Large‐scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53:156–165. 10.1038/s41588-020-00763-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gui Q, Li H, Wang A, et al. The association between gut butyrate‐producing bacteria and non‐small‐cell lung cancer. J Clin Lab Anal. 2020;34:e23318. 10.1002/jcla.23318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu H, Gao NL, Tong F, et al. Alterations of the human lung and gut microbiomes in non‐small cell lung carcinomas and distant metastasis. Microbiol Spectr. 2021;9:e0080221. 10.1128/Spectrum.00802-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu HX, Tao LL, Zhang J, et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int J Cancer. 2018;142:769–778. 10.1002/ijc.31098 [DOI] [PubMed] [Google Scholar]
- 14. Lawlor DA, Harbord RM, Sterne JA, et al. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. 10.1002/sim.3034 [DOI] [PubMed] [Google Scholar]
- 15. Huang J, Li X, Hong J, et al. Inflammatory bowel disease increases the risk of hepatobiliary pancreatic cancer: a two‐sample Mendelian randomization analysis of European and East Asian populations. Cancer Med. 2023;12:13599–13609. 10.1002/cam4.6057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sekula P, Del Greco MF, Pattaro C, et al. Mendelian randomization as an approach to assess causality using observational data. J Am Soc Nephrol. 2016;27:3253–3265. 10.1681/asn.2016010098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith GD, Ebrahim S. 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. 10.1093/ije/dyg070 [DOI] [PubMed] [Google Scholar]
- 18. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE‐MR statement. JAMA. 2021;326:1614–1621. 10.1001/jama.2021.18236 [DOI] [PubMed] [Google Scholar]
- 19. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–R98. 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goodrich JK, Davenport ER, Beaumont M, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19:731–743. 10.1016/j.chom.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birney E. Mendelian randomization. Cold Spring Harb Perspect Med. 2022;12(4):a041302. 10.1101/cshperspect.a041302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755–764. 10.1093/ije/dyr036 [DOI] [PubMed] [Google Scholar]
- 23. Zheng J, Baird D, Borges MC, et al. Recent developments in mendelian randomization studies. Curr Epidemiol Rep. 2017;4:330–345. 10.1007/s40471-017-0128-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well‐phenotyped isolated population. Nature. 2023;613:508–518. 10.1038/s41586-022-05473-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665. 10.1002/gepi.21758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR‐Egger method. Eur J Epidemiol. 2017;32:377–389. 10.1007/s10654-017-0255-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bowden J, Davey Smith G, Haycock PC, et al. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res. 2017;26:2333–2355. 10.1177/0962280215597579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–1998. 10.1093/ije/dyx102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boehm FJ, Zhou X. Statistical methods for Mendelian randomization in genome‐wide association studies: a review. Computat Struct Biotechnol J. 2022;20:2338–2351. 10.1016/j.csbj.2022.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boursi B, Mamtani R, Haynes K, et al. Recurrent antibiotic exposure may promote cancer formation–another step in understanding the role of the human microbiota? Eur J Cancer. 2015;51:2655–2664. 10.1016/j.ejca.2015.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhuang H, Cheng L, Wang Y, et al. Dysbiosis of the gut microbiome in lung cancer. Front Cell Infect Microbiol. 2019;9:112. 10.3389/fcimb.2019.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng Y, Fang Z, Xue Y, et al. Specific gut microbiome signature predicts the early‐stage lung cancer. Gut Microbes. 2020;11:1030–1042. 10.1080/19490976.2020.1737487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu F, Li J, Guan Y, et al. Dysbiosis of the gut microbiome is associated with tumor biomarkers in lung cancer. Int J Biol Sci. 2019;15:2381–2392. 10.7150/ijbs.35980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Botticelli A, Putignani L, Zizzari I, et al. Changes of microbiome profile during nivolumab treatment in NSCLC patients. American Society of Clinical Oncology; 2018. J Clin Oncol. 2018;36:e15020–e15020. 10.1200/JCO.2018.36.15_suppl.e15020 [DOI] [Google Scholar]
- 36. Jin Y, Dong H, Xia L, et al. the diversity of gut microbiome is associated with favorable responses to anti‐programmed death 1 immunotherapy in Chinese patients with NSCLC. J Thorac Oncol. 2019;14:1378–1389. 10.1016/j.jtho.2019.04.007 [DOI] [PubMed] [Google Scholar]
- 37. Biancheri P, Divekar D, Watson AJM. Could fecal transplantation become part of PD‐1‐based immunotherapy, due to effects of the intestinal microbiome? Gastroenterology. 2018;154:1845–1847. 10.1053/j.gastro.2018.03.060 [DOI] [PubMed] [Google Scholar]
- 38. Hakozaki T, Okuma Y, Omori M, et al. Impact of prior antibiotic use on the efficacy of nivolumab for non‐small cell lung cancer. Oncol Lett. 2019;17:2946–2952. 10.3892/ol.2019.9899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu S, Ma Y, Xiong K, et al. Ameliorating effects of vitamin K2 on dextran sulfate sodium‐induced ulcerative colitis in mice. Int J Mol Sci. 2023;24:2986. 10.3390/ijms24032986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y, Zhang X, Tang G, et al. The causal relationship between gut microbiota and bone mineral density: a Mendelian randomization study. Front Microbiol. 2023;14:1268935. 10.3389/fmicb.2023.1268935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mukherjee A, Lordan C, Ross RP, et al. Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microb. 2020;12:1802866. 10.1080/19490976.2020.1802866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan X, Yang M, Liu J, et al. Discovery and validation of potential bacterial biomarkers for lung cancer. Am J Cancer Res. 2015;5:3111–3122. [PMC free article] [PubMed] [Google Scholar]
- 43. Yu G, Gail MH, Consonni D, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016;17:163. 10.1186/s13059-016-1021-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut‐lung axis. Nat Rev Microbiol. 2017;15:55–63. 10.1038/nrmicro.2016.142 [DOI] [PubMed] [Google Scholar]
- 45. Zhao Y, Liu Y, Li S, et al. Role of lung and gut microbiota on lung cancer pathogenesis. J Cancer Res Clin Oncol. 2021;147:2177–2186. 10.1007/s00432-021-03644-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shang GS, Liu L, Qin YW. IL‐6 and TNF‐α promote metastasis of lung cancer by inducing epithelial‐mesenchymal transition. Oncol Lett. 2017;13:4657–4660. 10.3892/ol.2017.6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information.
Data Availability Statement
All data used in the present study were obtained from genome‐wide association study summary statistics which were publicly released by genetic consortia. Gut microbiota: (1) MiBioGen: https://mibiogen.gcc.rug.nl; (2) TwinsUK Registry: Information from the supplementary table of the article with doi of 10.1016/j.chom.2016.04.017. Lung cancer and its subtypes: https://r9.finngen.fi/. All datasets generated for this study are included in the article/additional files.