

Abstract
Topologically homologous four-helix-bundle heme proteins exhibit striking diversity in their refolding kinetics. Cytochrome b562 has been reported to fold on a submillisecond time scale, whereas cytochrome c′ refolding requires 10 s or more to complete. Heme dissociation in cytochrome b562 interferes with studies of folding kinetics, so a variant of cytochrome b562 (cytochrome c-b562) with a covalent c-type linkage to the heme has been expressed in Escherichia coli. Early events in the electron transfer-triggered folding of FeII-cytochrome c-b562, along with those of FeII-cytochrome c556, have been examined by using time-resolved absorption spectroscopy. Coordination of S(Met) to FeII occurs within 10 μs after reduction of the denatured FeIII-cytochromes, and shortly thereafter (100 μs) the heme spectra are indistinguishable from those of the folded proteins. Under denaturing conditions, carbon monoxide binds to the FeII-hemes in ≈15 ms. By contrast, CO binding cannot compete with refolding in the FeII-cytochromes, thereby confirming that the polypeptide encapsulates the heme in <10 ms. We suggest that Fe-S(Met) ligation facilitates refolding in these four-helix-bundle heme proteins by reducing the conformational freedom of the polypeptide chain.
Keywords: cytochrome, electron transfer, protein folding
Whether topologically similar proteins necessarily have similar folding pathways is an open question (1). With the increasing number of structures available, it has become clear that polypeptide sequences with little or no homology can assume nearly identical three-dimensional backbone architectures. Theoretical models suggest (2-4), and most experimental studies confirm (5-8), that a helical bundle is a fast folding structural motif. The presence of heme cofactors, however, can introduce new features into the helical bundle energy landscape that can greatly alter refolding pathways.
We have reported previously on the folding kinetics of two four-helix-bundle heme proteins, cytochrome b562 (cyt b562) and cytochrome c′ (cyt c′). Although the two cytochromes have nearly identical three-dimensional structures (3.4-Å rms deviation of backbone atoms), they have very low sequence identity (15%) and exhibit quite disparate folding kinetics (9-11). FeII-cyt b562 folds in less than a millisecond, whereas FeII-cyt c′ folding is quite heterogeneous, spanning time scales from milliseconds to seconds. Clearly, topology alone does not dictate these refolding rates.
The folding of cyt b562 is complicated by heme dissociation from the polypeptide, limiting the refolding yield (11). We suggested that heme dissociation could be responsible for the fast folding observed in cyt b562 by selecting against slower-folding populations in the unfolded ensemble. Indeed, a recent investigation of cyt b562 suggests that the heme in the denatured protein is bound to a native-like polypeptide conformation that is predisposed to fold rapidly (12).
To circumvent complications arising from heme dissociation, we have engineered a variant of E. coli cyt b562 (cyt c-b562) in which two thioether linkages bind the porphyrin to the polypeptide chain in the fashion of a c-type cytochrome (13-15). We also have investigated cytochrome c556 from Rhodopseudomonas palustris (16), a protein with the same four-helix-bundle fold as cyt b562 and cyt c′ (backbone atom rms deviations vs. cyt b562, 3.3 Å; vs. cyt c′, 1.5 Å) (17-20) but with low sequence identity (cyt b562, 21%; cyt c′, 34%). Here, we report early events in folding the FeII forms of c-b562 and c556.
Materials and Methods
Guanidine hydrochloride (GuHCl, Sigma, ultrapure grade), tris(2,2′-bipyridine)ruthenium(II) chloride ([Ru(bpy)3]Cl2, Strem), and NADH (Sigma) were used as received.
R. palustris cyt c556 was expressed and purified by published procedures with minor modifications (16). The N-terminal glutamine was fully cyclized by heating the protein at 50°C for 5 h in 0.5 M KH2PO4 (21). E. coli cyt c-b562 was expressed by cotransforming the construct pETcb562 (unpublished procedure) with pEC86 (22) into E. coli strain BL21 (DE3). Cyt c-b562 was purified by ion exchange chromatography on CM Sepharose Fast Flow and followed by a second purification step on a Mono S column (FPLC, Amersham Pharmacia). Proteins were judged to be pure by SDS/PAGE (PhastSystem, Amersham Pharmacia) and electrospray ionization-MS analysis (Caltech Protein/Peptide Microanalytical Laboratory).
Circular dichroism (CD) spectra were recorded by using an Aviv 62ADS spectropolarimeter; Trp fluorescence spectra were recorded on a Jobin Yvon SPEX Fluorolog-3 spectrofluorimeter (λex = 290 nm; λem = 300-500 nm). Steady-state absorption spectra were recorded on a Hewlett-Packard HP-8453 or HP-8452 diode array spectrometer. Protein (cyt c-b562) concentrations were determined by using the extinction coefficients reported for cyt b562 (23, 24).
Transient absorption kinetics measurements were made as described in refs. 11 and 25. Folding measurements were performed with solutions buffered to pH 7 with cyt c556 and pH 5 with c-b562 to inhibit misligation of His-63. Samples for folding kinetics measurements [cyt c556 or c-b562 (100 μM);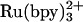 (50-150 μM) or NADH (≈200 μM); GuHCl (1-6 M)] were sealed in 1-mm cuvettes and deoxygenated by repeated evacuation/Ar or CO backfill cycles. GuHCl concentrations were determined after laser experiments from refractive index measurements (Abbe-3L refractometer, Milton Ray, Rochester, NY) (26).
(50-150 μM) or NADH (≈200 μM); GuHCl (1-6 M)] were sealed in 1-mm cuvettes and deoxygenated by repeated evacuation/Ar or CO backfill cycles. GuHCl concentrations were determined after laser experiments from refractive index measurements (Abbe-3L refractometer, Milton Ray, Rochester, NY) (26).
Results and Discussion
Equilibrium Unfolding. Cyt c556 and c-b562 are soluble proteins (molecular masses of 14.7 and 12.3 kDa, respectively). Each folded structure contains four antiparallel α-helices in a left-handed bundle with a heme group located in a hydrophobic pocket near the C and N termini; the porphyrin is covalently bound to the polypeptide chain through thioether linkages with two C-terminal cysteine residues, and the iron center is axially ligated by Met-12 and His-121 (cyt c556) or Met-7 and His-102 (cyt c-b562) (18). GuHCl titrations, monitored by absorbance, CD, and tryptophan fluorescence spectra, reveal cooperative unfolding transitions (Fig. 1), and, as expected on the basis of their high reduction potentials, the FeIII forms are less stable than the FeII proteins (27, 28). It is interesting to note that the introduction of two thioether linkages to the heme in cyt c-b562 leads to a substantial stabilization of the folded protein (the denaturation midpoint, [GuHCl]1/2, increases by ≈2 M).
Fig. 1.

Denaturant-induced unfolding of FeIII (green) and FeII (red) c556 (A) and c-b562 (B). Data points are from CD measurements; data from with heme absorption and Trp fluorescence are virtually the same.
The absorption spectra of native and denatured FeIII/II-cyt c556 and FeIII/II-cyt c-b562 (pH 5) are shown in Fig. 2. In both FeIII and FeII oxidation states, the heme becomes exposed to the solvent upon unfolding, leading to distinct shifts in absorption spectra, including variations in extinction coefficients, which can be exploited to monitor folding kinetics. The blue shift in the Soret absorption upon denaturation of each FeIII protein indicates that the heme has undergone a low- to high-spin transition, likely the result of replacement of the axial methionine ligand with a water molecule. In denatured FeII forms, however, both Soret and Q-band absorptions suggest that the heme remains low-spin. In contrast to the behavior of unfolded cytochrome c, it is not likely that nonnative His coordination accounts for the low-spin hemes in denatured FeII-cyt c556 and FeII-cyt c-b562. Cyt c556 has no available His residues, and denaturation of FeII-cyt c-b562 was performed at pH 5 to inhibit His-63 misligation. As was suggested for unfolded FeII-cyt c′, methionine residues apparently compete for the sixth FeII coordination site in the unfolded proteins. In FeII-cyt c-b562, both Met-7 and Met-58 are likely to be involved; Met-12, Met-19, and Met-20, and possibly Met-110, are the candidates in FeII-cyt c556. Analogous behavior was found in denatured R. palustris FeII-cyt c′ where Met-15 and Met-25 are potential ligands (9, 10). Although the absorption spectra of denatured FeII-cyt c556 and FeII-cyt c-b562 resemble those of the folded proteins, CD spectra clearly indicate that the native helical secondary structures are substantially disrupted (12).
Fig. 2.

Absorption spectra of unfolded FeIII (green dashed line), folded FeII (red dashed line), and unfolded FeII (blue dashed line) cyt c556 (A) and cyt c-b562 (B).
Electron Transfer-Triggered Refolding. At suitable denaturant concentrations (cyt c556, 1.5-2.5 M; cyt c-b562 3.8-4.8 M), electron injection into the denatured FeIII protein will initiate folding of the FeII form (27). In prior work, we have demonstrated that 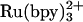 and NADH are useful sensitizers for photochemical triggering of FeII-cytochrome refolding (27, 29). Electronically excited
and NADH are useful sensitizers for photochemical triggering of FeII-cytochrome refolding (27, 29). Electronically excited 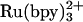 [E°(Ru3+/*2+) = -0.85 V vs. normal hydrogen electrode] injects an electron into the unfolded protein within a few microseconds. Subsequent charge recombination regenerates the initial reagent in a few milliseconds, allowing extensive signal averaging but limiting the observable window for folding to ≈1 ms. Two-photon excitation (355 nm) of NADH produces two reductants (
[E°(Ru3+/*2+) = -0.85 V vs. normal hydrogen electrode] injects an electron into the unfolded protein within a few microseconds. Subsequent charge recombination regenerates the initial reagent in a few milliseconds, allowing extensive signal averaging but limiting the observable window for folding to ≈1 ms. Two-photon excitation (355 nm) of NADH produces two reductants ( and
and 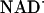 ) that irreversibly reduce the heme group in ≈100 μs (≈100 μM protein) (30). Therefore, with NADH it is possible to expand the observation time window to seconds and longer.
) that irreversibly reduce the heme group in ≈100 μs (≈100 μM protein) (30). Therefore, with NADH it is possible to expand the observation time window to seconds and longer.
We have used both 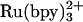 and NADH to trigger FeII-cyt c556 and FeII-cyt c-b556 refolding. In experiments with
and NADH to trigger FeII-cyt c556 and FeII-cyt c-b556 refolding. In experiments with 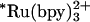 as the photoreductant, the observed transient absorption kinetics depend on denaturant concentration. These data are adequately described by a biexponential function. The faster rate is independent of [GuHCl] and corresponds to decay of
as the photoreductant, the observed transient absorption kinetics depend on denaturant concentration. These data are adequately described by a biexponential function. The faster rate is independent of [GuHCl] and corresponds to decay of 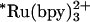 (k1 ∼ 1.6 × 106 s-1) with parallel reduction of the FeIII protein. The rate constant for the slower phase varies with [GuHCl]; this reaction channel represents early events in the folding of the protein around the heme (Fe-cyt c556, k2 = 4× 105 to 8× 105 s-1, [GuHCl] = 3-2 M; Fe-cyt c-b562, k2 = 5× 105 to 2× 106 s-1, [GuHCl] = 5.5-4.5 M). The transient spectra measured at the end of the slower phase (t ∼ 100 μs) are consistent with the formation of reduced folded protein.
(k1 ∼ 1.6 × 106 s-1) with parallel reduction of the FeIII protein. The rate constant for the slower phase varies with [GuHCl]; this reaction channel represents early events in the folding of the protein around the heme (Fe-cyt c556, k2 = 4× 105 to 8× 105 s-1, [GuHCl] = 3-2 M; Fe-cyt c-b562, k2 = 5× 105 to 2× 106 s-1, [GuHCl] = 5.5-4.5 M). The transient spectra measured at the end of the slower phase (t ∼ 100 μs) are consistent with the formation of reduced folded protein.
Biexponential kinetics also are observed when NADH is used as the photoreductant, with rate constants independent of [GuHCl] that are attributable to the reduction of the protein by 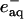 (4-5 × 104 s-1) and
(4-5 × 104 s-1) and 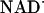 (9 × 103 s-1). The relative signal amplitudes observed after excitation of samples at low and high [GuHCl] (Fe-cyt c556, 1.5 and 4 M; Fe-cyt c-b562, 4 and 7 M) suggest that folded reduced protein is formed 300 μs after excitation (Fig. 3). No additional changes in absorption were detected on time scales as long as several seconds; the formation of folded reduced protein appears to be limited by the rate of reduction by
(9 × 103 s-1). The relative signal amplitudes observed after excitation of samples at low and high [GuHCl] (Fe-cyt c556, 1.5 and 4 M; Fe-cyt c-b562, 4 and 7 M) suggest that folded reduced protein is formed 300 μs after excitation (Fig. 3). No additional changes in absorption were detected on time scales as long as several seconds; the formation of folded reduced protein appears to be limited by the rate of reduction by 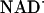 . Moreover, steady-state UV-visible and CD spectra recorded after laser excitation of NADH-containing samples confirm that the photochemically reduced proteins adopt native folds.
. Moreover, steady-state UV-visible and CD spectra recorded after laser excitation of NADH-containing samples confirm that the photochemically reduced proteins adopt native folds.
Fig. 3.

ET-triggered refolding of FeII-cyt c-b562. (A) Steady-state difference spectra between folded FeII and unfolded FeIII (red) and between unfolded FeII and unfolded FeIII (blue) c-b562. Symbols indicate transient absorption changes measured 300 μs after 355-nm pulsed-laser excitation of NADH in the presence of denatured FeIII-cyt c-b562 (red squares, [GuHCl] = 4 M; blue circles, [GuHCl] = 7 M). (B) Transient absorption kinetics observed at 556 nm upon reduction with NADH of c-b562 at 4 M GuHCl (red) and at 7 M GuHCl (blue). (C) Transient absorption kinetics observed at 548 nm.
The transient absorption data suggest that FeII-cyt c556 and FeII-cyt c-b562 refolding rates are faster than 104 s-1. UV-visible spectra provide information about the immediate environment of the heme cofactor but do not directly report on the conformation of the polypeptide. To gain more insight into the submillisecond FeII-cyt c556 and FeII-cyt c-b562 folding dynamics, we repeated the photoinitiated refolding experiments in the presence of CO. The deeply buried iron centers of native c-b562 and c556 are six-coordinate and therefore not available for CO binding. Under denaturing conditions, the ferroheme remains low-spin, likely due to Met ligation in the sixth coordination site. Nevertheless, CO will replace Met as an FeII ligand in denatured c-b562 and c556. We have examined the kinetics of CO rebinding to the heme after photo-dissociation from denatured FeII(CO)cyt c-b562 ([GuHCl] = 7 M) and FeII(CO)cyt c556 ([GuHCl] = 4 M). Under 1 atm (1 atm = 101.3 kPa) of CO, the rate constant for CO rebinding to the FeII heme is ≈65 s-1. We have used CO ligation to the heme as a probe of the extent of heme protection afforded by the polypeptide during electron transfer-triggered refolding. If the polypeptide wraps around the heme rapidly (>102 s-1), little CO should bind to the heme. On the other hand, if folding is slower than ≈102 s-1, substantial CO binding is expected. We find that at 7 M GuHCl, where formation of native, reduced c-b562 is disfavored, there is a major change in absorbance consistent with CO binding after reduction of the FeIII protein. In contrast, very little CO binding is apparent under conditions favoring formation of the folded FeII-protein (4 M GuHCl) (Fig. 4).
Fig. 4.

ET-triggered refolding of FeII-cyt c-b562 in the presence of CO. (A) Transient absorption kinetics observed at 415 nm upon photochemical reduction (NADH) of c-b562 at 4 M GuHCl (red) and at 7 M GuHCl (pink) (1 atm CO). (B) Difference spectra between folded FeII and unfolded FeIII (red), between unfolded FeII and unfolded FeIII (blue), and between CO-FeII and unfolded FeIII c-b562 (pink).
Our electron transfer-triggered experiments reveal that early events (t < 100 μs) in FeII-cyt c556 and FeII-cyt c-b562 refolding involve formation of a low-spin heme and some degree of heme encapsulation by the polypeptide. A lower limit to the time required for the heme to ligate a Met residue can be estimated from studies of tertiary contact dynamics in unfolded proteins and peptides (31-33). Met ligation to the heme in FeII-cyt c556 will produce polypeptide loops comprised of 96, 97, and 104 residues, and loop sizes of 39 and 90 residues in FeII-cyt c-b562 are possible. Energy-transfer quenching studies in synthetic polypeptides suggest that tertiary contact rate constants (kc) for 90- to 100-residue loops are 106 to 107 s-1 (32). These values compare with our observed rates of 105 to 106 s-1 for formation of native heme absorption spectra in electron transfer-triggered FeII-cyt c556 and FeII-cyt c-b562 refolding experiments. The variation of these rate constants with [GuHCl] concentration is likely due in part to the viscosity dependence of the intrachain diffusion dynamics that lead to tertiary contacts. In synthetic peptides, the logarithms of tertiary contact rates exhibit a linear dependence on [GuHCl] with mc values {mc = -RT∂(ln(kc))/∂[GuHCl] ∼ 0.5 (kJ/mol)/M} (34) that are substantially smaller than the mk values {mk = -RT∂(ln(kobs))/∂[GuHCl]} found for the early kinetics phases of FeII-cyt c556 [≈2.5 (kJ/mol)/M] and FeII-cyt c-b562 [≈2.1 (kJ/mol)/M] refolding (Fig. 5). Fast-folding small proteins have been reported to have mk values of 1-2 (kJ/mol)/M (34). The mk values for larger proteins displaying two-state refolding kinetics are larger, 5-10 (kJ/mol)/M (34). These denaturant dependences observed for FeII-cyt c556 and FeII-cyt c-b562 refolding suggest that the early kinetics phases involve more than intrachain diffusion leading to Met-Fe ligation. Rapid Met-Fe ligation could facilitate refolding of FeII-cyt c556 and FeII-cyt c-b562, because formation of one or more strong native tertiary contacts will substantially reduce the size of the conformational space available to the polypeptide (35). Constraining the folding energy landscape in this manner could lead to a substantial reduction in the time required to find the native structure (36).
Fig. 5.

Folding rates as a function of [GuHCl]. Red, FeII-cyt c-b562; blue, FeII-cyt c556; green, FeII-cyt b562 (11); yellow, FeII-cyt c′ (the vertical bar reflects the range of observed rate constants) (10).
After reduction of the unfolded oxidized proteins, CO will bind to the ferroheme under solution conditions where formation of native structure is disfavored. The observed rate constant for CO binding to denatured FeII-cyt c556 and FeII-cyt c-b562 (65 s-1) is ≈10 times smaller than the corresponding rate found for FeII-cyt c′ (10), and ≈20 times smaller than the rate for FeII-cyt c (37). The smaller CO binding rate constant for FeII-cyt c556 and FeII-cyt c-b562 may be a consequence of stronger Met binding to the reduced denatured proteins. Indeed, the similarity in the absorption spectra of folded and denatured reduced proteins (Fig. 2) suggests that Met is completely bound in the denatured forms. The Soret absorption of denatured FeII-cyt c′ is less intense than that of denatured FeII-cyt c556 and FeII-cyt c-b562, possibly because of less complete Met binding.
Our finding that CO does not bind to the FeII-cyt c556 and FeII-cyt c-b562 hemes under GuHCl conditions favoring folding points to encapsulation of the heme by the polypeptide in <10 ms. The spectroscopic data do not reveal whether the polypeptide has developed secondary and tertiary structure by this time. Ultrafast mixing measurements on a F65W mutant of apo-cyt b562, which adopts a three-helix-bundle fold (19, 38), are consistent with a refolding rate constant of 2,600 s-1 in the absence of denaturant (12). This value represents a reasonable upper limit to the folding rate for the holoprotein and is in line with the lower limit indicated by our CO ligation measurements.
The refolding of FeII-cyt c556 and FeII-cyt c-b562 clearly begins from an extensively denatured state. This finding contrasts with recent results on FeII-cyt b562 (12) where, as we had suggested earlier (11), heme dissociation preselects fast-folding members of the denatured ensemble. The only events revealed by changes in heme absorption spectra in FeII-cyt c556 and FeII-cyt c-b562 occur on very early time scales (1-10 μs) and involve Met-Fe ligation processes. The absence of separate kinetics phases attributable to heme encapsulation by the polypeptide, and the observation that the heme is protected from CO binding, confirm that a substantial degree of refolding occurs on submillisecond time scales. This behavior contrasts sharply with results from FeII-cyt c′ experiments in which changes in heme spectra were observed on time scales from 10-4 s to 101 s after reduction of the unfolded oxidized protein. We suggest that formation of one persistent native contact in the early stages of FeII-cyt c556 and FeII-cyt c-b562 refolding puts each polypeptide on a fast track to its native structure (36). The slower and more complex refolding kinetics found for FeII-cyt c′ may be a consequence of weaker S(Met)-Fe bonding in the denatured protein.
Acknowledgments
This research was supported by National Institutes of Health Grant GM068461, Department of Energy Grant DE-FG02-02ER15359, and the Arnold and Mabel Beckman Foundation.
Author contributions: J.F.-M. and J.R.W. designed research; J.F.-M. performed research; J.F.-M., H.B.G., and J.R.W. analyzed data; and J.F.-M., H.B.G., and J.R.W. wrote the paper.
References
- 1.Plaxco, K. W., Larson, S., Ruczinski, I., Riddle, D. S., Thayer, E. C., Buchwitz, B., Davidson, A. R. & Baker, D. (2000) J. Mol. Biol. 298, 303-312. [DOI] [PubMed] [Google Scholar]
- 2.Wolynes, P. G. (1996) Proc. Natl. Acad. Sci. USA 93, 14249-14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivankov, D. N., Garbuzynskiy, S. O., Alm, E., Plaxco, K. W., Baker, D. & Finkelstein, A. V. (2003) Protein Sci. 12, 2057-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou, Y. & Karplus, M. (1999) Nature 401, 400-403. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson, N., Capaldi, A. P., James, R., Kleanthous, C. & Radford, S. E. (1999) J. Mol. Biol. 286, 1597-1608. [DOI] [PubMed] [Google Scholar]
- 6.Kragelund, B. B., Osmark, P., Neergaard, T. B., Schiødt, J., Kristiansen, K., Knudsen, J., Poulsen, F. M. (1999) Nat. Struct. Biol. 6, 594-601. [DOI] [PubMed] [Google Scholar]
- 7.Teilum, K., Kragelund, B. B., Knudsen, J. & Poulsen, F. (2000) J. Mol. Biol. 301, 1307-1314. [DOI] [PubMed] [Google Scholar]
- 8.Thomsen, J. K., Kragelund, B. B., Teilum, K., Knudsen, J. & Poulsen, F. M. (2002) J. Mol. Biol. 318, 805-814. [DOI] [PubMed] [Google Scholar]
- 9.Lee, J. C., Engman, K. C., Tezcan, F. A., Gray, H. B. & Winkler, J. R. (2002) Proc. Natl. Acad. Sci. USA 99, 14778-14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee, J. C., Gray, H. B. & Winkler, J. R. (2001) Proc. Natl. Acad. Sci. USA 98, 7760-7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wittung-Stafshede, P., Lee, J. C., Winkler, J. R. & Gray, H. B. (1999) Proc. Natl. Acad. Sci. USA 96, 6587-6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia, P., Bruix, M., Rico, M., Ciofi-Baffoni, S., Banci, L., Shastry, M. C. R., Roder, H., Woodyear, T. d. L., Johnson, C. M., Fersht, A. R. & Barker, P. D. (2005) J. Mol. Biol. 346, 331-344. [DOI] [PubMed] [Google Scholar]
- 13.Moore, G. R. & Pettigrew, G. W. (1990) Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects (Springer, New York).
- 14.Scott, R. A. & Mauk, A. G. (1996) (University Science Books, Sausalito, CA), pp. 738.
- 15.Allen, J. W. A., Barker, P. D. & Ferguson, S. J. (2003) J. Biol. Chem. 278, 52075-52083. [DOI] [PubMed] [Google Scholar]
- 16.McGuirl, M. A., Lee, J. C., Lyubovitsky, J. G., Thanyakoop, C., Richards, J. H., Gray, H. B. & Winkler, J. R. (2003) Biochim. Biophys. Acta 1619, 23-28. [DOI] [PubMed] [Google Scholar]
- 17.Lederer, F., Glatigny, A., Bethge, P. H., Bellamy, H. D. & Mathews, F. S. (1981) J. Mol. Biol. 148, 427-448. [DOI] [PubMed] [Google Scholar]
- 18.Bertini, I., Faraone-Mennella, J., Gray, H. B., Luchinat, C., Parigi, G. & Winkler, J. R. (2004) J. Biol. Inorg. Chem. 9, 224-230. [DOI] [PubMed] [Google Scholar]
- 19.Arnesano, F., Banci, L., Bertini, I., Faraone-Mennella, J., Rosato, A., Barker, P. D. & Fersht, A. R. (1999) Biochemistry 38, 8657-8670. [DOI] [PubMed] [Google Scholar]
- 20.Shibata, N., Iba, S., Misaki, S., Meyer, T. E., Bartsch, R. G., Cusanovich, M. A., Morimoto, Y., Higuchi, Y. & Yasuoka, N. (1998) J. Mol. Biol. 284, 751-760. [DOI] [PubMed] [Google Scholar]
- 21.Khandke, K. M., Fairwell, T., Chait, B. T. & Manjula, B. N. (1989) Int. J. Pept. Protein Res. 34, 118-123. [DOI] [PubMed] [Google Scholar]
- 22.Arslan, E., Schulz, H., Zufferey, R., Kunzler, P. & Thony-Meyer, L. (1998) Biochem. Biophys. Res. Commun. 251, 744-747. [DOI] [PubMed] [Google Scholar]
- 23.Moore, G. R., Williams, R. J. P., Peterson, J., Thomson, A. J. & Mathews, F. S. (1985) Biochim. Biophys. Acta 829, 83-96. [DOI] [PubMed] [Google Scholar]
- 24.Itagaki, E. & Hager, L. P. (1966) J. Biol. Chem. 241, 3687-3695. [PubMed] [Google Scholar]
- 25.Stowell, M. H. B., Larsen, R. W., Winkler, J. R., Rees, D. C. & Chan, S. I. (1993) J. Phys. Chem. 97, 3054-3057. [Google Scholar]
- 26.Nozaki, Y. (1972) Methods Enzymol. 26, 43-50. [DOI] [PubMed] [Google Scholar]
- 27.Pascher, T., Chesick, J. P., Winkler, J. R. & Gray, H. B. (1996) Science 271, 1558-1560. [DOI] [PubMed] [Google Scholar]
- 28.Telford, J. R., Wittung-Stafshede, P., Gray, H. B. & Winkler, J. R. (1998) Acc. Chem. Res. 31, 755-763. [Google Scholar]
- 29.Telford, J. R., Tezcan, F. A., Gray, H. B. & Winkler, J. R. (1999) Biochemistry 38, 1944-1949. [DOI] [PubMed] [Google Scholar]
- 30.Orii, Y. (1993) Biochemistry 32, 11910-11914. [DOI] [PubMed] [Google Scholar]
- 31.Bieri, O., Wirz, J., Hellrung, B., Schutkowski, M., Drewello, M. & Kiefhaber, T. (1999) Proc. Natl. Acad. Sci. USA 96, 9597-9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieger, F., Fierz, B., Bieri, O., Drewello, M. & Kiefhaber, T. (2003) J. Mol. Biol. 332, 265-274. [DOI] [PubMed] [Google Scholar]
- 33.Chang, I.-J., Lee, J. C., Winkler, J. R. & Gray, H. B. (2003) Proc. Natl. Acad. Sci. USA 100, 3838-3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Möglich, A., Krieger, F. & Kiefhaber, T. (2005) J. Mol. Biol. 345, 153-162. [DOI] [PubMed] [Google Scholar]
- 35.Dyer, R. B., Maness, S. J., Peterson, E. S., Franzen, S., Fesinmeyer, R. M. & Andersen, N. (2004) Biochemistry 43, 11560-11566. [DOI] [PubMed] [Google Scholar]
- 36.Ittah, V. & Haas, E. (1995) Biochemistry 34, 4493-4506. [DOI] [PubMed] [Google Scholar]
- 37.Arcovito, A., Gianni, S., Brunori, M., Travaglini-Allocatelli, C. & Bellelli, A. (2001) J. Mol. Biol. 276, 41073-41078. [DOI] [PubMed] [Google Scholar]
- 38.Laidig, K. E. & Daggett, V. (1996) Folding Des. 1, 335-346. [DOI] [PubMed] [Google Scholar]