

Abstract
The regulation of nitrate assimilation seems to follow the same pattern in all ascomycetes where this process has been studied. We show here by in vitro binding studies and a number of protection and interference techniques that the transcription factor mediating nitrate induction in Aspergillus nidulans, a protein containing a binuclear zinc cluster DNA binding domain, recognizes an asymmetrical sequence of the form CTCCGHGG. We further show that the protein binds to its consensus site as a dimer. We establish the role of the putative dimerization element by its ability to replace the analogous element of the cI protein of phage λ. Mutagenesis of crucial leucines of the dimerization element affect both the binding ability of the dimer and the conformation of the resulting protein-DNA complex. This is the first case to be described where a dimer recognizes such an asymmetrical nonrepeated sequence, presumably by each monomeric subunit making different contacts with different DNA half-sites.
A number of transcription factors of the ascomycetes contain a specific DNA binding motif, the Zn binuclear cluster. In this taxonomic group, almost all processes where a specific inducer elicits the transcription of genes involved in a single metabolic pathway are mediated by proteins comprising this motif. The Zn binuclear cluster motif has a canonical sequence Ala/Ser Cys 2X Cys 6X Cys 5–17X Cys 2X Cys 5–7X Cys. Two Zn atoms and the six cysteines form a tetrahedrally coordinate complex where cysteines 1 and 4 chelate both Zn atoms (24, 25). The third loop usually consists of five to eight residues and contains a conserved proline in the fifth position, but the thoroughly studied AlcR protein of Aspergillus nidulans has a much longer loop and lacks the canonical proline (17). A subgroup of these proteins binds as a dimer to an inverted repeated sequence of the form CGG-nX-CCG, where n is 11 for Gal4p, 10 for Put3p, and 6 for Ppr1p of Saccharomyces cerevisiae, 11 for Lac9p of Kluyveromyces lactis, and 6 for the UaY protein of A. nidulans. These proteins contain a dimerization element, separated from the DNA binding domain by a variable linker. Thus, it is the nature of this linker that determines the specificity of binding (23, 29). The differences between Gal4p and Lac9p, on the one hand, and Ppr1p and UaY, on the other, are accounted for by the fact that the linker of the latter forms a β-sheet which shortens the distance between the dimerization element and the DNA binding domain (13, 24, 25, 32). Recently, it has been shown that Cyp1p (Hap1p) recognizes a direct repeat of the same base composition as those of the above proteins and that Leu3p and probably Pdr3p recognize identical everted repeats (14, 36). However, it should be noted that an everted repeat of a CGG motif is formally identical to an inverted repeat of the complementary sequence CCG. However, the difference between everted and complementary inverted repeats is structurally important, because it implies that the two Zn clusters in the dimer structure have opposite orientations (14) and hence that the topology of dimerization is a determinant of specificity. It has been stated or implied that the CGG motif repeated in a variety of orientations and at different distances is the universal recognition motif for the binuclear Zn clusters (14, 29).
While this is correct for a number of proteins of this class, this generalization is not warranted by the data. The AlcR and NirA proteins of A. nidulans do not recognize motifs which could be reduced to the CGG sequence in any form. Furthermore, AlcR has been reported to recognize direct, inverted, and everted repeats, with the last two separated by a variable number of base pairs. The solution to this apparent paradox is simple: AlcR is able to bind as a monomer (8, 18, 19, 22).
In this publication, we describe in detail the mode of binding of NirA. This protein mediates nitrate induction of the niaD, niiA, and crnA genes of A. nidulans, which encode nitrate reductase, nitrite reductase, and a nitrate permease, respectively (4, 5, 9, 34). We have summarized (28) data that suggest strongly that this protein and its cognate DNA binding sequences are conserved among the ascomycetes that are able to assimilate nitrate. Experimental interspecies complementation data (as distinct from similarity data, which are available for many other fungi) are available for organisms as distantly related as A. nidulans, Neurospora crassa, and Fusarium oxysporum (28). Figure 1 shows the amino acid sequence of the Zn cluster and cognate putative dimerization and linker sequences of the NirA protein and the homologous NIT4 protein of N. crassa. Recently, in vivo footprinting data has proven the conservation of binding sites among A. nidulans, Penicillium chrysogenum, and Aspergillus niger (34a). We have proposed that the sequence bound by NirA is the asymmetrical sequence 5′CTCCGHGG (where H stands for A, C, or T, not G). Four such sequences are present in the intergenic region which lies between the divergently transcribed niiA and niaD genes. The physiological role of each NirA binding sequence was demonstrated by deletion and/or point mutation experiments (28).
FIG. 1.

Comparison of the zinc cluster and putative linker and dimerization domains of the NirA and Nit4 proteins. The NirA and Nit4 sequences were described previously (5, 35). The putative dimerization domains shown for NirA (A and B) are those predicted by Schjerling and Holmberg (30). Numbers indicate the positions of residues in the translated protein sequences in the above-mentioned references.
Here we show rigorously that the 8-bp DNA sequence bound by NirA is indeed asymmetric. Moreover, we show that the NirA protein necessarily binds as a dimer.
MATERIALS AND METHODS
Strains.
The following Escherichia coli strains were used. DH5α [F− endA1 hsdR17 (mK+ rK−) supE44 thi-1 recA1 gyrA96 relA1 ΔlacU169 (f80d-lacZΔM15)] was used for routine plasmid preparation; CSH50 [Δ(pro lac) F− (proAB lacIqZΔM15) traD36] was used in phage immunity tests; and CJ236 [dut-1 ung-1 thi-1 relA1; pCJ105 (Cmr)] was used in the in vitro mutagenesis experiments.
Recombinant plasmids.
The pNirA(1–121) plasmid was constructed by subcloning an NcoI(filled in)-EcoRI fragment from pGex2T-NirA (28) into the NdeI(filled in)-EcoRI sites of plasmid pET22-b(+) (Novagen). A second plasmid, pNirA(1–222) was generated by ligating an NcoI(filled in)-BclI nirA fragment into the NdeI(filled in)-BamHI sites of pET22-b(+).
For mutagenesis purposes, an XbaI-EcoRI fragment from pNirA(1–121) was subcloned into pBluescriptII KS(+) (Stratagene) digested with XbaI-EcoRI. The mutated fragments were reintroduced into pET22-b(+) digested with XbaI-EcoRI.
In all pET22-b(+) derivatives, the nirA fragments are under the control of the bacteriophage T7 promoter.
pC132 and pC135 (7) are plasmids that direct the synthesis of the λ repressor (cI)-Rop fusion protein under the control of the pLac promoter. pC135 differs from pC132 only in the codon encoding Asp32 of Rop, which is replaced by a TAA nonsense codon. pC132 and pC135 were kindly supplied by F. Gigliani and G. Cesareni, respectively.
The pcI-NH2 plasmid was constructed from pC132 by elimination of the rop gene (excised as a HindII-XmaI fragment) followed by filling in and religation.
The pcI-NDD plasmid was obtained from pC132 by replacing the rop gene (excised as a SalI-BamHI fragment) with the putative NirA dimerization domain-coding fragment. The nirA fragment coding for amino acid residues 78 to 148 was amplified by PCR primed by specific oligonucleotides containing SalI and BamHI sites at their 5′ and 3′ ends, respectively. The following oligonucleotides were used: 5′GCATGTCGACACACCGGCGAAAAGGAG3′ and 5′GCATGGATCCGCTATACCGCATTTGACAGCC3′.
NirA proteins used in binding and footprinting assays.
The preparation of the GST-NirA(1–125) fusion protein has been described previously (28). The NirA(1–125) peptide was prepared as follows. GST-NirA fusion protein (28) was cleaved with thrombin from human plasma (Sigma, St. Louis, Mo.). A 10-U portion of thrombin/μg of fusion protein was incubated for 30 min at 25°C in 150 mM NaCl–16 mM Na2HPO4–4 mM NaH2PO4–2.5 mM CaCl2. Cleavage products were separated on glutathione-Sepharose columns (Pharmacia, Uppsala, Sweden) as specified by the manufacturer and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
The NirA(1–121) and NirA(1–222) proteins were obtained by in vitro transcription and translation with appropriate pET22-b(+) derivative plasmids.
In vitro transcription and translation.
In vitro protein synthesis was carried out with the TNT T7 coupled reticulocyte lysate system (Promega). The reactions were carried out as recommended by the manufacturer. The amounts of NirA protein present in the transcription-translation reaction mixtures were determined, when necessary, by Western blot analysis. Translation products were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (12% polyacrylamide) as described previously (21). Prestained low-molecular-weight markers (Bio-Rad) were used as molecular weight standards. After electrophoresis, the proteins were transferred to nitrocellulose filters (pore size, 0.2 μm; Schleicher & Schuell) and immunoblotted with antiserum against the N-terminal portion of NirA (amino acids 1 to 125), kindly supplied by F. Lema. Antigen-antibody complexes were revealed by enhanced chemiluminescence detection (Amersham). The translated products were used immediately for DNA binding gel mobility shift experiments or stored at −80°C.
Mobility shift assays.
Binding reactions for mobility shift assays were as described previously (28). For the in vitro transcription and translation products, 5 to 10 μl of the translation mixtures was used in the binding reactions.
Radiolabelled double-stranded oligonucleotides for mobility shift assays were prepared as follows. Equimolar amounts of single-stranded oligonucleotides were annealed in annealing buffer (100 mM Tris-HCl [pH 8.0], 150 mM NaCl) by heating the mixture for 2 min at 95°C and cooling it to 40°C in a thermocycler over a period of 3 h. Then the mixtures were chilled on ice. The receding ends of the double-stranded oligonucleotides were filled in with Sequenase version 2.0 (Amersham) and [α-32P]dCTP as specified by the manufacturer. The reaction products were purified on a 6% nondenaturating polyacrylamide gel, and approximately 10 pmol (104 dpm) was used together with 500 ng of purified NirA1–125 in the mobility shift assay. The single-stranded oligonucleotide used to fill in the wild-type and mutant probes for binding site 2 (WT, 1A, 2G, 3G, 4G, 5A, 6G, 7T, 8T, and HS) was 5′CTGCCTGAATTT3′. The sequence of the complementary strand for binding site 4 (WT and 6/7GA) was 5′CTGCCTTTCAGC3′. In experiments determining the relative binding efficiencies of oligonucleotides containing wild-type or mutated binding sequences, the protein concentrations were calibrated to yield an approximately 1:3 ratio of bound to free probe. The ratio between free and bound probe was determined by excising bands from the gel and Cerenkov counting the gel slices. The percentage of probe shifted was obtained by dividing the disintegrations per minute of each shifted probe by the disintegrations per minute of the corresponding free probe. The ratio obtained for the wild-type probes WT2 and WT4 was set to 100%, and the ratios for mutant probes are relative to that of the wild-type probes.
Protection and interference assays.
The restriction fragments used for the footprinting assays were as follows: for site 1, the DdeI (30)-ClaI (230) fragment [α-32P]dCTP labelled in the upper strand and [α-32P]dATP labelled in the lower strand; for site 2, (i) the ClaI (230)-DdeI (439) fragment [α-32P]dCTP labelled in both strands and then digested with HpaII (305) to obtain the HpaII (305)-DdeI (439) fragment [α-32P]dCTP labelled in the upper strand, and (ii) the HpaII (305)-BanI (471) fragment [α-32P]dCTP labelled in both strands and then digested with DdeI (439) to obtain HpaII (305)-DdeI (439) [α-32P]dCTP labelled in the lower strand; for site 3, the HinfI (731)-DdeI (948) fragment [α-32P]dATP labelled in the upper strand and [α-32P]dCTP labelled in the lower strand; and for site 4, the BamHI (927)-HpaII (1093) fragment [α-32P]dCTP labelled in the upper strand and the BamHI (927)-EcoRI (1268) fragment [α-32P]dCTP labelled in the lower strand. End-labelled DNA fragments derived from the niiA-niaD intergenic region were subjected to footprinting analysis by the different protection and interference methods described below.
Methylation protection footprinting, methylation interference, and depurination interference assays were carried out as described previously (10).
Depyrimidination mixtures (20 μl) containing 50 ng (105 dpm) of end-labelled probe and 2 μg of yeast tRNA were chilled on ice for 5 min. Hydrazine (30 μl; Sigma) was added, and the mixture was incubated for 7 min at 20°C. The reactions were stopped by adding 60 μl of 3 M sodium acetate (pH 7.0), and the DNA was ethanol precipitated. The precipitation was repeated twice, and the DNA was washed with 70% ethanol and resuspended in 20 μl of water. Binding-reaction mixtures containing 5 ng (104 dpm) of probe and 0.5 to 1 μg of purified protein were used for binding reactions as described previously (10). Free and bound probes were separated, recovered, and cleaved with piperidine (26). The reaction products were analyzed on a 6% polyacrylamide–urea sequencing gel.
Phosphate contact interference assays were carried out as follows. To obtain partially ethylated probes, end-labelled fragments (50 ng containing 105 dpm) were incubated for 15 min at 50°C in 50-μl mixtures containing 2 μl of cacodylate buffer (0.5 M sodium cacodylate, 10 mM EDTA), 2 μg of yeast tRNA, and 30 μl of a saturated solution of nitroso-ethyl-urea in 96% ethanol (Sigma).
Stopping of the reaction, purification of the ethylated probe and binding assays were as described above. Separated free and bound probes were cleaved at modified phosphates by incubating recovered DNA in 50 μl of phosphate buffer (50 mM Na2HPO4, 5 mM EDTA [pH 7.0]) at 90°C for 15 min. Subsequently, 2 μl of 1 M NaOH was added to the mixture, and incubation was continued for another 30 min at 90°C. Cleaved DNA was ethanol precipitated, washed twice with 70% ethanol, and analyzed on a sequencing gel as described above. Maxam-Gilbert sequencing reactions of the same fragment were run in parallel with the probes. In all these studies, the protein concentration used was calibrated by a mobility shift assay to yield an approximately 1:1 ratio of bound to free probe.
Mutagenesis of the conserved leucines.
Oligonucleotide-directed mutagenesis was used to generate mutants with site-specific mutations in the NirA dimerization domain.
The nirA cDNA fragment containing the dimerization region was cloned into pBluescriptII KS(+) in the negative orientation, and thus the mutated priming oligodeoxynucleotides correspond to the coding strand of the nirA gene. In the oligonucleotides shown below, numbers in parentheses refer to nucleotide positions in the nirA locus (5) and boldface characters indicate mutated bases:
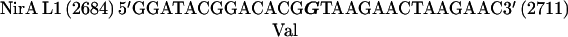 |
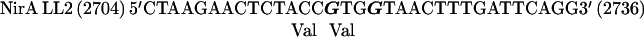 |
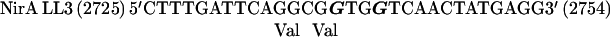 |
Mutagenesis was carried out as previously described (20). Five picomoles of each oligonucleotide, phosphorylated with T4 DNA kinase, was hybridized to 0.2 pmol of uracil-containing single-stranded DNA. The single-stranded DNA template was prepared after transformation of CJ236, an E. coli strain which contains the dut ung double mutation, with the pBluescript derivative plasmid and subsequent infection with the M13KO7 helper phage. The complementary strand of DNA was synthesized in the presence of T4 DNA polymerase and T4 DNA ligase. The double-stranded DNA was transformed into competent DH5α cells (Bethesda Research Laboratories), and clones were screened directly by DNA sequencing.
Phage immunity test.
Bacterial cells transformed with plasmids expressing different λ repressor fusion proteins were tested for sensitivity to λ phages. Phages of different virulent phenotype were assayed by spot tests, at concentrations varying from 102 to 104 phages per spot, on lawns of transformed bacteria. The λ phage used as a wild-type control is that of Bailone and Galimbert (2), and λvir is λ668, an ultravirulent phage carrying several mutations affecting both lambda operators (2).
RESULTS
Binding of NirA protein to wild-type and mutant probes.
There are four NirA binding sites in the intergenic region between niiA and niaD. All these sites are involved in the induction of either or both genes (28). The four sites correspond to the consensus sequence CTCCGHGG. We shall number the bases 5′ to 3′ from 1 to 8 in the top strand and from 1* to 8* in the bottom strand (Fig. 2).
FIG. 2.

Summary of protection and interference footprinting studies on all four consensus NirA binding sites in the niiA-niaD intergenic region. For the sake of consistency, the orientations of sites 2 and 3 have been inverted with respect to their orientations in the intergenic region. Hence, upper strands in sites 1 and 4 are niaD coding strands whereas upper strands in sites 2 and 3 are niiA coding strands. Regions protected from DNase I digestion are shown as brackets above and under each strand (28). Dots represent guanine contacts identified by methylation protection. Triangles between bases symbolize phosphate contacts identified by ethylation interference. Purine contacts (squares) or pyrimidine contacts (diamonds) were identified by missing-base interference, and symbols above the line between the two strands denote contacts on the upper strand whereas symbols under this line denote contacts on the lower strand. In all cases, solid and open symbols represent strong and weak protection or interference, respectively. Both depurination and depyrimidination interference experiments were carried out for sites 2 and 3; only depurination interference was carried out for sites 1 and 4. Footprint experiments for sites 1, 3, and 4 were carried out with the GST-NirA(1–125) fusion protein, while all techniques used for site 2 were carried out both with the GST-NirA(1–125) fusion protein and with the NirA(1–125) peptide with virtually identical results (see Fig. 3 through 5). See Materials and Methods for descriptions of probes and footprinting techniques used. Numbers indicate the position of the first nucleotide shown for each sequence in the niiA-niaD intergenic region as previously described (28). Note that the boundaries of DNase I protection do not always correspond exactly to those published previously (28). Careful densitometry was carried out on the gels used to construct this figure, and while exact boundaries could be somewhat subjective, we believe the present ones to be accurate.
Previous work carried out with a glutathione S-transferase (GST)–NirA(1–125) fusion protein suggested strongly that NirA binds to asymmetrical, nonrepeated sequences. We have now carried out similar experiments with a NirA protein not carrying any GST sequences in order to eliminate any artifacts due to GST dimerization sequences, artifacts known to occur for the AlcR-DNA complex (22). We have extended the analysis to mutations in all nucleotides of the consensus sequence and have estimated quantitatively the effect of each mutation. The results are shown in Table 1. All bases of the CTCCGHGG consensus sequence are essential for binding. C1 cannot be replaced by an A, and this shows that the palindromic sequences TCCGCGGA, CCGCGG, and CC2XGG, which are found within one or more NirA binding sites, are not recognized by NirA. Table 1 shows that a G3 is not acceptable, while previous data showed that a T3 is not acceptable (28). It can be proposed that the consensus sequence is an imperfect variant of the palindromic sequence CTCCGGAG. This is shown not to be the case for probe 6/7GA (Table 1). The base in position 6 can be A, T, or C but not G (Fig. 2), which would be the base symmetrical with the C in position 3. It can be further proposed that the consensus sequence is an imperfect variant of a longer, 10-bp symmetrical sequence, CTCCGCGGAG (probe HS). Table 1 shows that this longer, symmetrical sequence has no higher affinity for NirA than the asymmetrical, physiological sequence it contains. This was confirmed by gel shift experiments where the concentration of protein varied from that resulting in 10% binding of the probe to saturating concentrations. The kinetics of binding of the wild type and the 10-bp symmetrical sequence (HS) are, within the limits of this technique, identical (results not shown). Figure 3 gives an example of a gel retardation experiment of the type used to obtain the data shown in Table 1. We show, in this figure, the effect of mutating the crucial first C to A.
TABLE 1.
Summary of the gel shift analysis of 10 double-stranded DNA oligonucleotides containing mutations in the core binding sequencea
| Probeb | Sequence | % of binding relative to the WT probec |
|---|---|---|
| WT2 | 5′GCCCAA CTCCGCGG AAATTCA3′ | 100 |
| 1A | 5′GCCCAA ATCCGCGG AAATTCA3′ | <5 |
| 2G | 5′GCCCAA CGCCGCGG AAATTCA3′ | <5 |
| 3G | 5′GCCCAA CTGCGCGG AAATTCA3′ | <5 |
| 4G | 5′GCCCAA CTCGGCGG AAATTCA3′ | 7 |
| 5A | 5′GCCCAA CTCCACGG AAATTCA3′ | <5 |
| 6G | 5′GCCCAA CTCCGGGG AAATTCA3′ | 6 |
| 7T | 5′GCCCAA CTCCGCTG AAATTCA3′ | <5 |
| 8T | 5′GCCCAA CTCCGCGT AAATTCA3′ | 8 |
| HS | 5′GCCCAA CTCCGCGG AGATTCA3′ | 79 |
| WT4 | 5′CTGTTT CTCCGAGG CTGAAAG3′ | 100 |
| 6/7GA | 5′CTGTTT CTCCGGAG CTGAAAG3′ | 8 |
Band shift analysis of probes containing wild-type or mutated NirA binding sites. Double stranded 20- to 30-mer oligonucleotides containing different derivatives of binding site 2 or binding site 4 were tested for their affinity to bind NirA(1–125) in mobility shift assays. Only relevant sequences of one strand containing the consensus binding site in the 5′ to 3′ direction are shown. Probes with nonmutated target sequences corresponding to site 2 (WT2) or site 4 (WT4) in the niiA-niaD intergenic region were used as references.
Mutated probes are named according to the position of base substitutions in the 5′CTCCGHGG3′ consensus sequence: 1A, C to A at position 1; 2G, C to G at position 2; 3G, C to G at position 3; 4G, C to G at position 4; 5A, G to A at position 5; 6G, C to G at position 6; 7T, G to T at position 7; 8T, G to T at position 8; HS, A to G at position +2 3′ of the consensus sequence, thereby creating a symmetric inverted repeat (5′CTCCG3′); 6/7GA, A to G and G to A at positions 6 and 7, respectively, thereby creating a symmetric inverted repeat (5′CTCC3′). The mutated bases are shown in boldface type in the sequences.
The percentage of probe shifted was obtained by dividing the disintegrations per minute of each shifted probe by the disintegrations per minute of the corresponding free probe. The ratio obtained for the wild-type probes WT2 and WT4 was set to 100%, and ratios for mutant probes are relative to those of the wild-type probes. The relative retardations of WT2 and WT4 are identical within the limits of this assay.
FIG. 3.

Gel retardation experiment with the wild type and a mutant probe. In this experiment, we give a semiquantitative example of the effect of a point mutation on binding of the NirA(1–125) peptide. The probes used are WT2 (right) and 1A (left). Lanes: 1, free probe; 2 through 4, probes incubated with 360, 240, and 120 ng of protein, respectively. The results shown in Table 1 were obtained with probes incubated with 500 ng of protein.
Footprint analysis of the NirA-DNA binding.
We have investigated the binding of a GST-NirA(1–125) fusion protein by DNase I protection, missing-base (both purines and pyrimidines) interference, and methylation protection and/or interference. When both methylation techniques were used (site 2), the results were very similar. We have investigated the symmetry of binding for all natural sites by determining the phosphate contacts by ethylation interference. For one site (site 2), the one with the most important role in vivo, we have repeated all footprinting techniques (except DNase I protection) with the NirA(1–125) peptide cleaved from the GST protein. The collated results obtained with the fusion protein are shown in Fig. 2. The results obtained with the GST-NirA(1–125) fusion protein and with the NirA(1–125) peptide are virtually identical for all the techniques (but see below), validating the results shown in Fig. 2. For one of the techniques, depurination interference, we show comparatively the results with the GST-NirA(1–125) fusion protein and the NirA(1–125) peptide (Fig. 4). The results of depyrimidination interference and ethylation interference of site 2 carried out with the NirA(1–125) peptide are shown in Fig. 5. Densitometric analysis of missing-base interference, methylation interference, and protection and ethylation interference results obtained with the NirA(1–125) peptide and site 2 are shown in Fig. 6. The results of densitometric analysis of methylation protection of all three other sites complexed with the GST-NirA(1–125) fusion protein are virtually identical to that shown in Fig. 6B for site 2, including the quantitative differences in the extent of protection of the different G’s (data not shown).
FIG. 4.

Purine missing-base interference footprints of GST-NirA(1–125) fusion protein (A) and NirA(1–125) peptide (B) on site 2 of the niiA-niaD intergenic region. “upper” and “lower” refer to strands as in Fig. 2. fp, free probe; b, bound probe. Only the sequence of the limited region bracketing the NirA binding sequence is shown. Solid squares, strongly interfering purines; open squares, partially interfering purines. The probe for site 2 is as described above. Footprint experiments have been carried out at least twice with identical results.
FIG. 5.

Pyrimidine missing-base (A) and ethylation (B) interference footprints of NirA(1–125) peptide on site 2 of the niiA-niaD intergenic region. “upper” and “lower” refer to strands as in Fig. 2. fp, free probe; b, bound probe; Py, Maxam-Gilbert pyrimidine reaction used as the sequence standard; filled symbols, strongly interfering bases or phosphates; open symbols, partially interfering bases or phosphates. squares (A), pyrimidines; triangles (B), phosphates. The probe is as described above. Footprint experiments have been carried out at least twice with identical results.
FIG. 6.

Densitometric scanning of footprints of binding site 2. (A) Methylation interference analysis, showing reduction in relative binding as a consequence of methylation of guanines. (B) Methylation protection analysis, showing reduction in relative methylation of guanines in the protein-DNA complex. (C) Purine and pyrimidine missing-base analysis, showing reduction in relative binding as a consequence of loss of each nucleoside. (D) Reduction in relative binding as a consequence of ethylation of each phosphate. Differences in the labelling of the lanes were normalized by equating the absorbance of bands clearly outside the interfering area for the free and bound lanes. All the experiments were carried out with the NirA(1–125) peptide; the probe is as in Fig. 2 through 4.
The conclusions of this study are straightforward. Loss of any of the eight bases of the consensus sequence in either strand interferes strongly with NirA binding. Loss of the variable base in position 6 (and of its complement, 3*) interferes strongly, irrespective of its nature. Missing-base interference (Fig. 6C) does not shed any light on the issue of symmetry of binding, except by the fact that all bases in an asymmetric sequence interfere without any suggestion of “internal symmetry” within the consensus. Methylation protection and, when studied (site 2), interference patterns are imperfectly symmetric. An obvious asymmetry is the strong interference found for G7.
While missing-base interference and methylation protection patterns are very similar in the four sites, phosphate contacts, as revealed by ethylation interference, show some differences between sites. Sites 2 and 3 show very similar patterns of phosphate contacts. It should be noticed that sites 2 and 3 have a C · G pair in the variable position, while sites 1 and 4 have T · A and A · T, respectively. Flanking sequences may also have some influence in the pattern of phosphate contacts (see Discussion). The phosphate contacts are not compatible with a symmetric pattern of binding for any of the four sites (Fig. 5 and 6). The interference by ethylation of the 2-3 bond, found in all four sites, has no equivalent in the 2*-3* bond. The strong interference by ethylation of the 4*-5* and 5*-6* bonds at sites 2 and 3 does not have a symmetrical counterpart in the 4-5 and 5-6 bonds. When phosphate contacts are seen in flanking sequences, these are also arranged completely (sites 2 and 3) or partially (sites 1 and 4) asymmetrically.
There are some minor differences between the ethylation interference results shown in Fig. 2 and 6D for site 2. The weak interferences seen in Fig. 6 (e.g., between positions 5 and 6 and positions 2* and 3*) are probably not genuine differences between the NirA(1–125) peptide and the GST-NirA(1–125) fusion protein but a result of the sensitivity of the densitometric methods used to calculate the data in Fig. 6. However, the phosphate contact 5′ of position 1 (Fig. 2) is not seen in the experiments carried out with the NirA(1–125) peptide and constitutes the only genuine qualitative difference observed between the GST-NirA(1–125) fusion protein and the NirA(1–125) peptide. This difference does not affect the pattern of asymmetry of phosphate contacts seen for site 2 with the GST-NirA(1–125) fusion protein and the NirA(1–125) peptide as discussed above.
NirA binds DNA as a dimer.
Two NirA partial peptides of different lengths were synthesized either separately or together from a vector containing a T7 promoter, by using an in vitro transcription-translation coupled system. The formation of the heterodimer was evident in gel shifts with a 239-bp 32P-labelled restriction fragment containing binding site 2 as a probe (Fig. 7). Heterodimers were never obtained when analogous His-tagged proteins expressed in E. coli were purified and mixed (data not shown), in either the native or denatured form (followed by renaturation). A similar situation has been described for Gal4p (6).
FIG. 7.

NirA binds DNA as a dimer. A coupled in vitro transcription-translation system was used to synthesize NirA(1–121) and NirA(1–222) either singly or jointly. A 239-bp ClaI-BanI fragment containing the NirA binding site 2 was used as a probe. The position of the heterodimer is indicated by an arrow.
Note that the NirA(1–121) protein used in this and previous experiments contains only the first, amino-terminal dimerization domain proposed by Schjerling and Holmberg (30), while the NirA(1–222) protein contains both domains. Therefore, the amino-terminal domain is sufficient for dimerization. The crucial importance of this domain is demonstrated by in vitro mutagenesis (see below). However, the carboxy-terminal dimerization domain does play a role. The difference in the affinity for the probe between the NirA(1–121) and NirA(1–222) proteins (Fig. 7) is genuine and has been observed in several independent experiments.
Mapping the NirA dimerization domain.
We assumed that a coiled-coil dimerization element is present in NirA in a position similar to that of other proteins of this group. Since the sequences bound by NirA are identical to those bound by the N. crassa protein NIT4, we assumed that the essential residues of linker and dimerization elements had to be conserved. Interestingly, while the Zn cluster and putative linker domains are almost identical between the two organisms, the sequence of the putative dimerization domain is not so highly conserved. However, hydrophobic amino acids are present at crucial positions of both proteins. Two coiled coils, comprising residues 98 to 111 and 115 to 139, have been predicted within the region following the zinc cluster of NirA (30). We have tested the dimerization activity of a peptide encompassing the whole putative dimerization element, from residues 78 to 148 of the NirA protein (Fig. 1). The nirA fragment coding for this peptide was introduced into a vector containing a sequence coding for the λ cI DNA binding domain without its dimerization sequences. Since the cI repressor functions as a dimer, substituting its dimerization domain with a heterologous protein region leads to a functional repressor only if the heterologous region is able to dimerize (3, 15). The results are shown in Fig. 8. The putative NirA dimerization domain is able to confer DNA binding activity to the cI DNA binding domain. This is seen by the immunity to wild-type λ phage (but not to an ultravirulent λ phage, multiply mutated in both oR and oL) conferred by the appropriate plasmid to E. coli cells.
FIG. 8.

Phage immunity test. λ phages of wild-type (λ) and ultravirulent (λvir) phenotypes (2) were assayed on lawns of bacteria by spot tests at increasing phage titers (from left to right, 102, 103, and 104 PFU per spot). Bacteria were transformed with plasmids containing the λ repressor fused to the actual or putative dimerization elements indicated below. “none,” CSH50 E. coli cells transformed with pcI-NH2 (expressing the NH2-terminal domain of λ repressor without any putative dimerization element); NirA, the same cells transformed with pcI-NDD, containing the putative NirA dimerization domain; Rop, a positive control transformed with pC132, expressing a cI-Rop fusion (the Rop protein efficiently dimerizes to form a bundle of four antiparallel helices); Rop*, transformed with pC135 expressing a cI-Rop* fusion with a mutated Rop protein unable to dimerize (7).
Mutagenesis of the NirA dimerization domain.
To determine whether dimerization was a prerequisite for binding, we mutagenized the leucines included in a putative coiled-coil domain of the NirA(1–121) protein. The first hydrophobic residues of heptad repeats predicted to form a coiled coil are Leu98 and Leu105 or, alternatively, Leu99 and Leu106 (30). Leu91 is 7 residues upstream of Leu98 and thus could also be included in a potential coiled coil. We mutagenized Leu91, Leu98 and Leu99 simultaneously, or Leu105 and Leu106 simultaneously. Some combinations of these mutations were also analyzed. All the mutations were conservative changes from leucine to valine. These changes should prevent the formation of a coiled-coil dimerization element while not altering the overall hydrophobicity or the α-helical structure of this peptidic region (1). The mutant proteins were synthesized in vitro and examined for DNA binding activity in gel shift assays with a DNA fragment containing NirA binding site 2 as a probe. The results are shown in Fig. 9. Mutation of Leu91Val (L1) does not affect binding or dimerization, since the complex formed has the same mobility as the wild-type complex. Simultaneous mutations at Leu98 and Leu99 (LL2) do not seem to affect binding (but see below), while a double substitution at Leu105 and Leu106 (LL3) affects binding drastically without completely abolishing it. Simultaneous mutation of these four leucine residues has a very marked effect on binding. It is clear that a dimer is formed, since the complex moves with a mobility even lower than that of the wild-type complex. This effect is more marked when Leu98, Leu99, Leu105, and Leu106 are all mutagenized. Thus, Leu98 or Leu99 must contribute to dimerization, even if this is not obvious when they are mutated on their own. This is supported by the fact that a double substitution at Leu98 and Leu99 decreases the mobility of the complex very slightly. This effect is genuine, since it has been observed in several independent experiments. The conclusion that can be drawn is that Leu98 or Leu99 and Leu105 or Leu106 are involved in the formation of a coiled coil. When they are mutated, the putative coiled coil can still form, even when all the leucines are mutated to valine, but it has a less rigid structure, which could account nicely for the decrease in the mobility of the complex. It is quite clear that proper dimerization is necessary for efficient binding.
FIG. 9.

DNA binding activities of NirA proteins mutated in the dimerization element. Wild type, NirA(1–121) peptide as described in the legend to Fig. 7. Mutants: L1, Leu91Val; LL2, Leu98Val and Leu99Val; LL3, Leu105Val and Leu106Val. A plus sign between the symbols for each mutation denotes the presence of two or three mutations in the dimerization element. Equal amounts of in vitro-synthesized protein substitutions were incubated with the probe described in the legend to Fig. 6, containing NirA binding site 2. The NirA protein present in the transcription-translation reactions was quantified by Western blot analysis with polyclonal antibodies against the NirA(1–125) peptide followed by densitometry.
It is worth noticing that either Leu105 or Leu106, whose simultaneous mutation has the most drastic effect on the binding and mobility of the DNA-protein complex, is the first hydrophobic residue in the second heptad of the first short coiled coil predicted by Schjerling and Holmberg (30) (see above). However Leu91, whose mutation does not affect binding, is not included in either of the two predicted coiled coils.
DISCUSSION
The NirA protein mediates the induction by nitrate and nitrite of the genes involved in nitrate assimilation in A. nidulans. It is extremely similar in a number of domains to the NIT4 protein of N. crassa; the DNA binding domains are almost identical (5). The latter protein was reported to bind the perfect palindromic sequence TCCGCGGA. Note that this sequence is contained in site 2 of the niiA-niaD intergenic region of A. nidulans. However, both sites reported for N. crassa contain a C (C1 in our consensus) before the “first” T (T2 of our consensus) and one of them contains a C in place of the “last” A (base in position 9 after our consensus) (12). If we consider the composite 9-bp sequence CTCCGHGGA, we observe that the first C is conserved among all the N. crassa and A. nidulans sites and the last A (position 9) is present only in site 2 of A. nidulans and one of the sites of N. crassa. Thus, comparative evidence indicates that both proteins bind to an asymmetrical site of the form CTCCGHGG. For the A. nidulans protein, the conclusive evidence is provided by the mutagenesis of the C in position 1 (see Results).
We have investigated the asymmetry of binding by all possible interference reactions. Removal of any of the bases within the consensus sequence interferes strongly with binding. There is some generally partial, variable asymmetrical interference outside the consensus sequence; this will be discussed below. Methylation interference (and protection) is consistent only with asymmetrical binding. While methylation of G5, G5*, G8, and G8* interferes with binding (but not always to the same extent), interference by methylation of G7 has no symmetrical counterpart, since there is no G in 7*. The pattern of methylation interference clearly differs from that found in proteins recognizing the CGG motif. For both Lac9p and UaY, the only base where methylation interferes with binding is the second G (13, 32). This is the only G whose N7 accepts a proton from the ɛ-NH2 group of a conserved lysine in the structures of the Gal4p and Ppr1p protein-DNA complexes (24, 25). The phosphate contacts as revealed by ethylation interference are compatible only with asymmetrical binding of NirA.
Missing purine and methylation interference patterns of the N. crassa NIT4 protein are strikingly similar to what is reported in this article. These studies were conducted with a β-galactosidase fusion protein containing amino acids 1 to 202 of NIT4, and it is reassuring that three different proteins, the NirA(1–125) peptide, the GST-NirA(1–125) fusion protein, and the NIT4(1–202)–β-galactosidase fusion protein, gave such consistent results. The comparison can be extended to the flanking sequences of different sites in both organisms. In N. crassa site 1, the sequence 5′AAAT is found 5′ of base 1*. The three A’s are revealed by methylation interference. In A. nidulans site 2, the sequence in the same position is 5′ATTT. The 3 T’s are revealed by missing-base interference (Fig. 2). When methylation protection of this site was carried out under conditions where the N3 sites of the A’s are methylated, a striking hypersensitivity of the A’s in positions 9 and 10 and a strong protection of the two A’s 5′ to position 1 were apparent (data not shown). Site 2 of N. crassa has 3 A’s following position 8*. These three A’s are revealed both by missing-base interference and methylation interference. We found exactly the same sequence in site 4 of A. nidulans (Fig. 2). Two of the A’s immediately 3′ to 8* are revealed by missing-base interference, and there are clear phosphate contacts in the corresponding phosphodiester bonds (Fig. 2). When the methylation protection of these region was studied as above, again the same two A’s showed clear methylation protection (data not shown). The role of external AT sequences is not identical in different sites. We have investigated the effect of distamycin, a drug that binds in the minor groove of AT-rich regions (16). Distamycin (30 μM) completely inhibits the binding of the GST-NirA(1–125) fusion protein to site 4 but not to sites 2 and 3. Interestingly, binding to site 1, which is preceded by a long run of AT sequences, is also strongly inhibited by distamycin (31). These results are again consistent with an overall asymmetry of binding. The two sites where binding is inhibited by distamycin have AT-rich sequences on the 5′ site of position 1 (site 4 actually on both 5′ and 3′ sites), site 3 has no AT-rich sequences, and site 2 has an AT-rich sequence 3′ of position 8.
An obvious way in which a protein can recognize a nonrepeated, asymmetrical sequence is if the sequence is bound by a monomer. Other work from this laboratory has already shown that the zinc binuclear cluster AlcR protein can bind as a monomer (8, 22) and that the single sites play physiological roles in vivo (27). Residues of a monomer located in different places along the DNA binding domain could recognize the two half-sites of the NirA binding sequence. It should be noted that, besides the cluster of basic residues in the first loop, common to all the proteins of the Zn binuclear cluster class, NirA and NIT4 contain two further clusters of basic residues, one amino terminal to the Zn motif and the second in the putative linker element. Results shown above establish unequivocally that NirA binds as a dimer. Cross-linking studies with a truncated NIT4 protein (residues 48 to 179) suggests that the unbound protein also exists as a dimer in solution (12).
Mutation of putative crucial residues in the dimerization element results in drastically diminished binding. Since we are detecting the protein by the binding assay, it is impossible to determine whether this is due to a reduced affinity of the dimer, to the defective formation of the dimer, or to a combination of the two. However, the altered mobility of the complex shows that the mutation of the dimerization element changes the overall structure of the dimer-DNA complex and presumably that of the free dimeric protein, if the latter forms at all.
Inverted and everted repeats of the same sequences can be recognized by proteins differing in their dimerization and/or linker elements in such a way that the crucial base recognition residues have opposite orientations. This, however, maintains an overall symmetry of the binding domains in relation to each other (14). To account for the recognition of direct repeats by dimeric proteins, it has been proposed that the dimer must have an asymmetric head-to-tail orientation (36). Interestingly, it has recently been shown for Cyp1p (Hap1p) that such head-to-tail orientation is not determined by the linker or dimerization element but by the DNA binding domains (37). In fact, as the Cyp1p protein is a monomer in solution (37), the asymmetric pattern of dimerization is determined by the DNA binding domain/DNA-specific contacts. The relevance of this observation will be made clear below.
The results obtained for NirA contradict the picture of the class of Zn binuclear cluster proteins as a monotonous group, recognizing the same CGG sequence in different orientations as described above and/or separated by different sequence lengths (14, 29). If we assume that both Zn binuclear clusters are involved in recognizing the octet of base pairs that constitute the NirA and NIT4 binding sites, it follows that each half of the asymmetrical sequence must bind to different amino acids. Thus, either different amino acids within the first loop make different contacts in each monomer or one monomer may contact DNA by using amino acids outside the first loop. This is discussed below. For the models developed to account for the binding of the proteins recognizing CGG half-sites in ascomycetes, it is assumed, and shown in some cases, that the amino acid/base contacts are identical and that what varies is their orientation or separation along the DNA axis or both (14, 23–25, 29, 37). Any model able to account for the specificity of NirA binding must postulate that the specific base contacts and phosphate contacts made by the two zinc clusters are different.
This is, as far as we are aware, the only case hitherto described where each subunit of a dimer protein binds to such an asymmetrical, nonrepeated DNA sequence, not only within the class of the Zn binuclear cluster transcription factors but also among all DNA binding proteins. The closest precedent seems to be the E47 protein of Drosophila melanogaster. This protein belongs to a group of helix-loop-helix transcription factors which bind as dimers to the so-called E box, a CAXXTG sequence. While some proteins of this group bind to perfectly symmetrical CACGTG or CAGCTG sequences, E47 prefers a CACCTG sequence. The difference seems to depend on one residue of the DNA binding domain. The E47 has an hydrophobic valine at the junction of the DNA binding domain and helix 1. The proteins of this group which recognize symmetrical sequences have an arginine in this position (11).
It is clearly impossible to reduce the NirA binding sequence to a tandem repetition of two half-sites. If we try to reduce it to an inverted repeated sequence, we could write CzzCGzzG, where z denotes the residues where no symmetry can be seen. Most striking is the asymmetry in the 3 and 3* positions. C is canonical in position 3 (and cannot be mutated to a G), but C is the only base that cannot be accepted in position 3* (i.e., a G is not acceptable in position 6 [Table 1]). However, this skeleton of an inverted repeat could provide a basis to explain NirA binding. Only two lysines make specific base contacts in the Ppr1p and Gal4p/DNA complexes. These two lysines of the first loop are flanked by hydrophobic residues in both proteins. On the other hand, the first loop of NirA an NIT4 is CRRRKSKC, where all side chains of residues between the Zn-chelating cysteines, including that of serine, are able to form hydrogen bonds. It can be proposed that the two zinc binuclear clusters of a symmetrical dimer bind to the “skeleton sequence” in such a way that different residues are able to make base and phosphate contacts in each half-site. If half-sites are separated by less than a whole helix turn, the overall structure of the complex will necessarily be asymmetrical. In addition, asymmetrical binding could be generated by local deviations of DNA from its canonical structure. Independently of its origin, this asymmetry will be reflected in either distortion of the linker and/or dimerization elements, thus allowing symmetrical binding to the DNA half-sites, or, if suitable side chains are available, different residues may establish bases and backbone contacts to each half-site. The binding of UaY and Ppr1p to apparently perfectly symmetrical sequences (CGG-6X-CCG) show, after more detailed chemical and crystallographic analysis, respectively, obvious asymmetrical features (25, 32). A comparison with Cyp1p (Hap1p) binding is again relevant here. It has been proposed that the latter protein has a very “weak” dimerization element (37). Thus, the recognition of direct repeats, imposed in an unspecified manner by DNA-protein binding sequence interaction, would be permitted by the flexibility of the dimerization element. The opposite is true for NirA. Here the dimerization element is “strong” enough to act as a dimerization element in vivo for the cI repressor and dimerization is not completely abolished even after mutagenesis of several residues. Two tandemly arranged coiled-coil regions, potentially acting as dimerization elements, have been proposed for NirA (30), and this work has shown that while the amino-terminal domain in essential for NirA dimerization, the carboxy-terminal domain may participate in proper dimer assembly. Thus, we can expect the dimer structure to be quite rigid. The result of an asymmetry of base contacts imposed by the protein-DNA interaction would not be resolved here by a rotation around the axis of the dimerization element (as proposed for Cyp1p) (37), permitting the recognition of a direct repeat, but by a “sliding” along the first loop of the zinc cluster in such a way that different side chains make crucial contacts. The interplay of specific contacts by the first loop of the zinc cluster, the structure of the linker element, and the stability of the dimerization element indeed determine the specificity of binding of zinc binuclear cluster proteins but in a more varied and richer fashion than has been hitherto supposed. While this article was being completed, the crystal structure of the Put3p/DNA complex was published (33). The CGG-10X-CCG sequence, is as expected, recognized by a protein dimer. The complex is highly asymmetric, in spite of the symmetry of the DNA sequence. However, the asymmetry does not involve the two Zn binuclear clusters, as proposed here for NirA; indeed, the only base-specific contact made by a side chain is by a histidine, which occupies the same crucial position in the cluster as the lysine in Gal4p and Ppr1p. The asymmetry is due to contacts in the minor groove that are made by the linker and dimerization regions.
In spite of substantial work describing either minor (25, 32) or considerable (11, 33) asymmetry of binding by dimeric proteins, the structural and thermodynamic parameters which determine such asymmetries remain elusive. The NirA-DNA interaction provides the most striking example of such an asymmetrical complex.
ACKNOWLEDGMENTS
Joseph Strauss and M. Isabel Muro-Pastor contributed equally to this work.
We thank F. Gigliani for CSH50 E. coli cells, plasmid pC132, and λ phages used in this work; G. Cesareni for plasmid pC135; and F. Lema for polyclonal antibodies against the NirA(1–125) protein. J.S. is grateful to C. P. Kubicek, Technical University, and to the Department of Medical Biochemistry, University of Vienna, for laboratory space. Wilhelm Guschlbauer is thanked for helpful discussion.
This work was supported by CEE grants SCI-CT92-0815 and BIO2-CT93-0147. The work at Vienna was supported by grant 6105 from the Jubilaeumsfonds der Oesterreichischen Nationalbank to J.S. J.S. was supported by a studentship of Austauschprogramm France-Austria and by a short-term EMBO fellowship (ASTF 7958). M.I.M.-P. has been the recipient of, successively, a postdoctoral fellowship from the Ministerio de Educación y Ciencia of the Spanish Government, CE fellowship BIO-CT-94-8102, and a fellowship from Fondation pour la Recherche Médicale.
REFERENCES
- 1.Alber T. Structure of the leucine zipper. Curr Opin Genet Dev. 1992;2:205–210. doi: 10.1016/s0959-437x(05)80275-8. [DOI] [PubMed] [Google Scholar]
- 2.Bailone A, Galimbert F. Nucleotide sequence of the operators of λ ultravirulent mutants. Nucleic Acids Res. 1980;8:2147–2164. doi: 10.1093/nar/8.10.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battaglia P A, Longo F, Ciotta C, Del Grosso M F, Ambrosini E, Gigliani F. Genetic tests to reveal Tat homodimer formation and select Tat homodimer inhibitor. Biochem Biophys Res Commun. 1994;201:701–708. doi: 10.1006/bbrc.1994.1757. [DOI] [PubMed] [Google Scholar]
- 4.Burger G, Tilburn J, Scazzocchio C. Molecular cloning and functional characterization of the pathway-specific regulatory gene nirA, which controls nitrate assimilation in Aspergillus nidulans. Mol Cell Biol. 1991;11:795–802. doi: 10.1128/mcb.11.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burger G, Strauss J, Scazzocchio C, Lang B F. nirA, the pathway-specific regulatory gene of nitrate assimilation in Aspergillus nidulans, encodes a putative GAL4-type zinc finger protein and contains four introns in highly conserved regions. Mol Cell Biol. 1991;11:5746–5755. doi: 10.1128/mcb.11.11.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carey M, Kakidani H, Leatherwood J, Mostashari F, Ptashne M. An amino terminal fragment of GAL4 binds DNA as a dimer. J Mol Biol. 1989;209:423–432. doi: 10.1016/0022-2836(89)90007-7. [DOI] [PubMed] [Google Scholar]
- 7.Castagnoli L, Vetriani C, Cesareni G. Linking an easily detectable phenotype to the folding of a common structural motif. Selection of rare turn mutations that prevent the folding of Rop. J Mol Biol. 1994;237:378–387. doi: 10.1006/jmbi.1994.1241. [DOI] [PubMed] [Google Scholar]
- 8.Cerdan R, Collin D, Lenouvel F, Felenbok B, Guittet E. The Aspergillus nidulans transcription factor AlcR forms a stable complex with its half-site DNA: a NMR study. FEBS Lett. 1997;408:235–240. doi: 10.1016/s0014-5793(97)00430-4. [DOI] [PubMed] [Google Scholar]
- 9.Cove D J, Pateman J A. Independently segregating genetic loci concerned with nitrate reductase activity in Aspergillus nidulans. Nature. 1963;198:262–263. doi: 10.1038/198262a0. [DOI] [PubMed] [Google Scholar]
- 10.Cubero B, Scazzocchio C. Two different adjacent and divergent zinc-finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J. 1994;13:407–415. doi: 10.1002/j.1460-2075.1994.tb06275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellenberger T, Fass D, Arnaud M, Harrison S C. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev. 1994;8:970–980. doi: 10.1101/gad.8.8.970. [DOI] [PubMed] [Google Scholar]
- 12.Fu Y H, Feng B, Evans S, Marzluf G A. Sequence-specific DNA binding by NIT4, the pathway-specific regulatory protein that mediates nitrate induction in Neurospora. Mol Microbiol. 1995;15:935–942. doi: 10.1111/j.1365-2958.1995.tb02362.x. [DOI] [PubMed] [Google Scholar]
- 13.Halvorsen Y D C, Nanadabalan K, Dickson R C. Identification of base and backbone contacts used for DNA sequence recognition and high-affinity binding by LAC9, a transcription activator containing a C6 zinc finger. Mol Cell Biol. 1991;11:1777–1784. doi: 10.1128/mcb.11.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellauer K, Rochon M H, Turcotte B. A novel DNA binding motif for yeast zinc cluster proteins: the Leu3p and Pdr3p transcriptional activators recognize everted repeats. Mol Cell Biol. 1996;16:6096–6102. doi: 10.1128/mcb.16.11.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J C, O’Shea E K, Kim P S, Sauer R T. Sequence requirements for coiled-coils: analysis with λ repressor-GCN4 leucine zipper fusions. Science. 1990;250:1400–1403. doi: 10.1126/science.2147779. [DOI] [PubMed] [Google Scholar]
- 16.Kopka M L, Yoon C, Goodsell D, Pjura P, Dickerson R. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc Natl Acad Sci USA. 1985;82:1376–1380. doi: 10.1073/pnas.82.5.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulmburg P, Prangé T, Mathieu M, Sequeval D, Scazzocchio C, Felenbok B. Correct intron splicing generates a new type of a putative zinc-binding domain in a transcriptional activator of Aspergillus nidulans. FEBS Lett. 1991;280:11–16. doi: 10.1016/0014-5793(91)80193-7. [DOI] [PubMed] [Google Scholar]
- 18.Kulmburg P, Sequeval D, Lenouvel F, Mathieu M, Felenbok B. Identification of the promoter region involved in the autoregulation of the transcriptional activator ALCR in Aspergillus nidulans. Mol Cell Biol. 1992;12:1932–1939. doi: 10.1128/mcb.12.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulmburg P, Judewicz N, Mathieu M, Lenouvel F, Sequeval D, Felenbok B. Specific binding sites for the activator protein ALCR, in the alcA promoter of the ethanol regulon of Aspergillus nidulans. J Biol Chem. 1992;267:21146–21153. [PubMed] [Google Scholar]
- 20.Kunkel T A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Lenouvel F, Nikolaev I, Felenbok B. In vitro recognition of specific DNA targets by AlcR, a zinc binuclear cluster activator different from the other proteins of this class. J Biol Chem. 1997;272:15521–15526. doi: 10.1074/jbc.272.24.15521. [DOI] [PubMed] [Google Scholar]
- 23.Liang S D, Marmorstein R, Harrison S C, Ptashne M. DNA sequence preferences of GAL4 and PPR1: how a subset of Zn2Cys6 binuclear cluster proteins recognizes DNA. Mol Cell Biol. 1996;16:3773–3780. doi: 10.1128/mcb.16.7.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marmorstein R, Carey M, Ptashne M, Harrison S C. DNA recognition by GAL4: structure of a protein-DNA complex. Nature (London) 1992;356:408–415. doi: 10.1038/356408a0. [DOI] [PubMed] [Google Scholar]
- 25.Marmorstein R, Harrison S C. Crystal structure of a PPR1-DNA complex: DNA recognition by proteins containing a Zn2Cys6 binuclear cluster. Genes Dev. 1994;8:2504–2512. doi: 10.1101/gad.8.20.2504. [DOI] [PubMed] [Google Scholar]
- 26.Maxam A M, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:497–559. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 27.Panozzo C, Capuano V, Fillinger S, Felenbok B. The zinc binuclear cluster activator AlcR is able to bind to single sites, but requires multiple repeated sites for synergistic activation of the alcA gene in Aspergillus nidulans. J Biol Chem. 1997;272:22859–22865. doi: 10.1074/jbc.272.36.22859. [DOI] [PubMed] [Google Scholar]
- 28.Punt P J, Strauss J, Smit R, Kinghorn J R, van den Hondel C A M J J, Scazzocchio C. The intergenic region between the divergently transcribed niiA and niaD genes of Aspergillus nidulans contains multiple NirA binding sites which act bidirectionally. Mol Cell Biol. 1995;15:5688–5699. doi: 10.1128/mcb.15.10.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reece R, Ptashne M. Determinants of binding-site specificity among yeast C6 zinc cluster proteins. Science. 1993;261:909–911. doi: 10.1126/science.8346441. [DOI] [PubMed] [Google Scholar]
- 30.Schjerling P, Holmberg S. Comparative amino acid sequence analysis of the C6 zinc cluster family of transcriptional regulators. Nucleic Acids Res. 1996;24:4599–4607. doi: 10.1093/nar/24.23.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strauss J. Bases moléculaires de la régulation des gènes de la voie d’assimilation du nitrate chez Aspergillus nidulans. Ph.D. thesis. Centre d’Orsay, Orsay, France: Université Paris Sud; 1993. [Google Scholar]
- 32.Suárez T, Vieira de Queiroz M, Oestreicher N, Scazzocchio C. The sequence and binding specificity of UaY, the specific regulator of the purine utilization pathway in Aspergillus nidulans, suggest an evolutionary relationship with the PPR1 protein of Saccharomyces cerevisiae. EMBO J. 1995;14:1453–1467. doi: 10.1002/j.1460-2075.1995.tb07132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swaminathan K, Flynn P, Reece R J, Marmorstein R. Crystal structure of a PUT3-DNA complex reveals a novel mechanism for DNA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nat Struct Biol. 1997;4:751–759. doi: 10.1038/nsb0997-751. [DOI] [PubMed] [Google Scholar]
- 34.Unkles S E, Hawker K L, Grieve C, Campbell E I, Montague P, Kinghorn J R. crnA encodes a nitrate transporter in Aspergillus nidulans. Proc Natl Acad Sci USA. 1991;88:204–208. doi: 10.1073/pnas.88.1.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a.Wolschek, M., F. Narendja, J. Karlseder, B. Cubero, C. Scazzocchio, C. P. Kubicek, and J. Strauss. Unpublished data. [DOI] [PMC free article] [PubMed]
- 35.Yuan G F, Fu Y H, Marzluf G A. nit4, a pathway-specific regulatory gene of Neurospora crassa, encodes a protein with a putative binuclear zinc DNA-binding domain. Mol Cell Biol. 1991;11:5735–5745. doi: 10.1128/mcb.11.11.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L, Guarente L. The yeast activator HAP1—a GAL4 family member—binds DNA in a directly repeated orientation. Genes Dev. 1994;8:2110–2119. doi: 10.1101/gad.8.17.2110. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Guarente L. The C6 zinc cluster dictates asymmetric binding by HAP1. EMBO J. 1996;15:4676–4681. [PMC free article] [PubMed] [Google Scholar]