

Key Points
Question
Can rapid trio genome sequencing (rtGS) be deployed in a national public health care setting?
Findings
In this cohort study that included all neonatal intensive care units in Israel, rtGS in 130 neonates suspected of having a genetic disorder revealed a diagnosis in 50% (12 chromosomal and 52 monogenic disorders and 1 uniparental disomy). Immediate precision medicine was offered for 9% of diagnosed participants, and the mean turnaround time for rapid report was 7 days.
Meaning
These findings suggest that clinical rtGS can be implemented in the neonatal acute care setting in a national public health care system.
This cohort study evaluates the feasibility, diagnostic efficacy, and clinical utility of rapid trio gene sequencing in neonatal intensive care units in Israel.
Abstract
Importance
National implementation of rapid trio genome sequencing (rtGS) in a clinical acute setting is essential to ensure advanced and equitable care for ill neonates.
Objective
To evaluate the feasibility, diagnostic efficacy, and clinical utility of rtGS in neonatal intensive care units (NICUs) throughout Israel.
Design, Setting, and Participants
This prospective, public health care–based, multicenter cohort study was conducted from October 2021 to December 2022 with the Community Genetics Department of the Israeli Ministry of Health and all Israeli medical genetics institutes (n = 18) and NICUs (n = 25). Critically ill neonates suspected of having a genetic etiology were offered rtGS. All sequencing, analysis, and interpretation of data were performed in a central genomics center at Tel-Aviv Sourasky Medical Center. Rapid results were expected within 10 days. A secondary analysis report, issued within 60 days, focused mainly on cases with negative rapid results and actionable secondary findings. Pathogenic, likely pathogenic, and highly suspected variants of unknown significance (VUS) were reported.
Main Outcomes and Measures
Diagnostic rate, including highly suspected disease-causing VUS, and turnaround time for rapid results. Clinical utility was assessed via questionnaires circulated to treating neonatologists.
Results
A total of 130 neonates across Israel (70 [54%] male; 60 [46%] female) met inclusion criteria and were recruited. Mean (SD) age at enrollment was 12 (13) days. Mean (SD) turnaround time for rapid report was 7 (3) days. Diagnostic efficacy was 50% (65 of 130) for disease-causing variants, 11% (14 of 130) for VUS suspected to be causative, and 1 novel gene candidate (1%). Disease-causing variants included 12 chromosomal and 52 monogenic disorders as well as 1 neonate with uniparental disomy. Overall, the response rate for clinical utility questionnaires was 82% (107 of 130). Among respondents, genomic testing led to a change in medical management for 24 neonates (22%). Results led to immediate precision medicine for 6 of 65 diagnosed infants (9%), an additional 2 (3%) received palliative care, and 2 (3%) were transferred to nursing homes.
Conclusions and Relevance
In this national cohort study, rtGS in critically ill neonates was feasible and diagnostically beneficial in a public health care setting. This study is a prerequisite for implementation of rtGS for ill neonates into routine care and may aid in design of similar studies in other public health care systems.
Introduction
Genetic disorders and birth defects account for 30% of morbidity and 40% of mortality in neonatal intensive care units (NICUs).1,2,3,4,5 Overlapping clinical features in this age group make reaching a diagnosis by standard-of-care testing challenging. It is hypothesized that early etiologic diagnosis, facilitated by next-generation sequencing, has the potential to revolutionize clinical care, improve prognosis, and offer precision life-saving therapy or aid in palliative care decisions in critically ill neonates.6 Next-generation sequencing has been proven superior to standard-of-care testing in providing an accurate diagnosis.7,8,9
A recent retrospective analysis10 of 60 diagnosed infants from 5 centers in the Netherlands demonstrated the clinical utility of rapid exome sequencing (rES) for critically ill neonates as defined by increased diagnostic yield, shorter time to diagnosis, and net health care savings. The authors recommended widespread implementation of rES as a first-tier genetic test in critically ill neonates with suspected genetic disorders.10
Earlier studies, including NSIGHT1 and NICUseq, which compared genome sequencing (GS) with standard-of-care testing, have revealed GS to have a higher diagnostic yield even when compared with ES.9,11,12,13 An additional advantage of GS over ES, shown in the NSIGHT2 study, is the possibility for rapid and ultrarapid turnaround times (TATs) of as soon as 3 days.7 Taken together with the ability to detect diverse genomic variants in a single test, including chromosomal copy-number abnormalities, single-nucleotide variants (SNVs), triplet repeat expansions, uniparental disomy (UPD), and variants in noncoding regions, GS is the preferred comprehensive bedside genomic testing in the critically ill, for whom rapid clinical decisions are needed.14 Furthermore, rapid GS (rGS) was shown to be economically beneficial mainly through significantly shorter lengths of hospital stay in infants with a diagnostic GS test compared with undiagnosed children.15
Prospective studies utilizing next-generation sequencing in NICUs have shown a mean diagnostic rate of approximately 36% in different cohorts. Common indications for ES in early studies included mainly neonates and young infants with congenital anomalies or neurologic phenotypes, while recent studies broadened the indications to include any critically ill child suspected of having a genetic disorder.11,13,14,15,16,17
Diagnostic results affected clinical management decisions in 25% to 65% of patients across cohorts and enabled reproductive planning for 27% of families of diagnosed patients.15,18,19 Although an ultrarapid sequencing and analysis system with scalable diagnosis in less than a day was recently published,20 it has been suggested that a 2- to 3-week diagnostic pipeline is sufficient to impact most clinical decision making.18
Most published studies have been performed in a single center or in a restricted region, such as the rGS Baby Bear and NICUseq projects, each including up to 5 medical centers.11,15 Diagnostic efficacy and change-of-management assessments were comparable in these studies.
In public health care systems, limited resources challenge the widespread implementation of advanced sequencing technologies into routine inpatient clinical practice. Israel has a universal health care system. To our knowledge, we have conducted the first prospective national pilot of trio rGS (rtGS) as a single test for genomic diagnosis in critically ill neonates. Our primary objective was to assess the feasibility and diagnostic efficacy of rtGS for critically ill neonates in the Israeli national health care system. A secondary objective was to assess the clinical utility defined as the outcomes of rtGS for precision medicine and recurrence risk reduction in families.
Methods
Study Design and Objectives
We performed a prospective national pilot of rtGS in critically ill neonates. The project was a collaboration between the Community Genetics Department in the Israeli Ministry of Health (MOH), the Israeli Association of Medical Genetics, and the Israeli Neonatal Society. All medical genetics institutes (n = 18) and NICUs (n = 25) belonging to the Israeli national health care system participated in the project. The study was approved by the MOH Medical Research Ethics Committee. Written informed consent for GS was obtained from parents (eMethods 1 in Supplement 1). The report follows the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cohort studies.
All sequencing and data analyses were performed at the Tel-Aviv Sourasky Medical Center (TASMC) Genomics Center. Each NICU was assigned a corresponding genetics institute, either in the same center (n = 18) or from an adjacent medical facility (n = 7). Results were reported to the medical genetics team caring for the infant. The medical genetics team was responsible for disclosing results to the NICU staff and families. Follow-up genetic counselling was planned at the discretion of the medical genetics teams. Clinical utility (Hebrew-translated Clinician-Reported Genetic Testing Utility Index [C-GUIDE]21) and outcome questionnaires were completed by the NICU staff within 14 days of receiving the secondary report (Figure 1A; eMethods 1 in Supplement 1).
Figure 1. Study Design, Enrollment, Timeline, and Results.
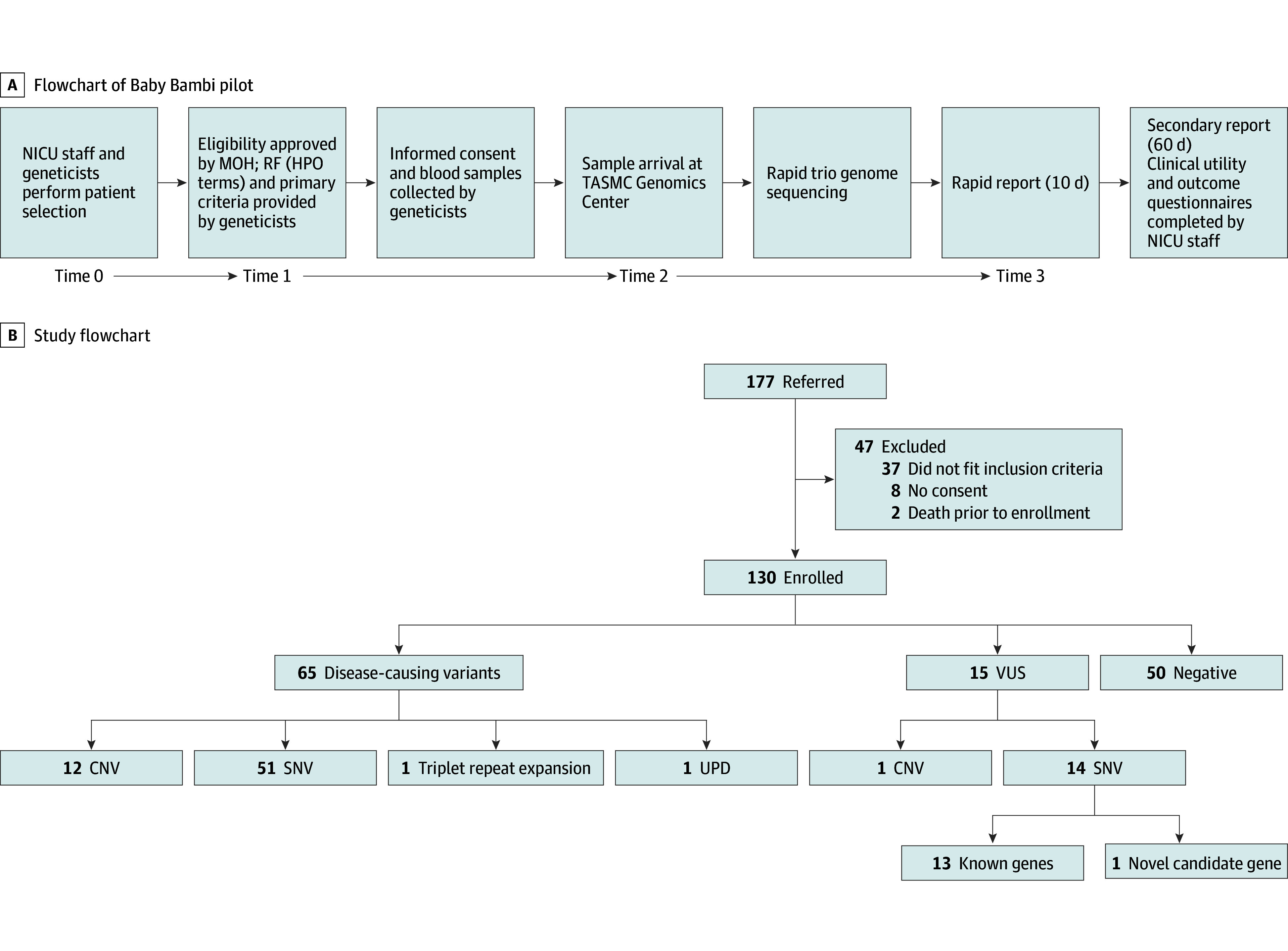
A, Flowchart of the Baby Bambi pilot. Timelines are indicated are as follows: Time 0 to time 1: up to 1 day; time 1 to time 2: 1 to 5 working days; time 2 to time 3: 10 days. B, Flowchart depicting pilot referral, enrollment, and results. Of the 47 excluded patients, 8 could not provide biparental consent on account of religious beliefs. CNV indicates copy-number variation; MOH, Ministry of Health; NICU, neonatal intensive care unit; RF, requisition form; SNV, single-nucleotide variant; TASMC, Tel Aviv Sourasky Medical Center; UPD, uniparental disomy; VUS, variant of unknown significance.
Study Cohort
Critically ill neonates (n = 130) were selected by the practicing neonatologist. A requisition form (eMethods 1 in Supplement 1) listing inclusion criteria (eTable 1 in Supplement 1) and a medical summary report were reviewed for eligibility by the MOH Community Genetics Department. Only 1 primary inclusion criterion could be selected for each patient, with unlimited secondary category criteria (eTable 1 and eMethods 1 in Supplement 1). Upon approval, written parental informed consent was obtained and trio samples were delivered to the TASMC Genomics Center (Figure 1A). Ethnicity was self-reported and subcategorized as Jewish, non-Jewish, or mixed. Mixed ethnicity was defined as both Jewish and non-Jewish background. Consanguinity was self-reported and defined by a shared common ancestor.
GS and Analysis
All samples were subjected to the Illumina DNA PCR-Free Library Prep and sequenced on the NovaSeq 6000 (Illumina) using S1/S2 reagent kit version 1.5, 150 × paired-end. Variant analysis was performed using a 2-step approach. Step 1 consisted of primary rapid analysis, which was performed on the TruSight Software Suite (TSS [Illumina]), relying on phenotype-driven variant prioritization using Human Phenotype Ontology (HPO) terms provided by the referring medical geneticist. Variants prioritized as related to the phenotype were manually reviewed by the bioinformatics and pediatric genetics team and classified according to the American College of Medical Genetics and Genomics (ACMG) criteria.22 Inconclusive or suspected candidate results were discussed on a case-by-case basis with the referring team at each site. Pathogenic and likely pathogenic (P/LP) variants and variants of unknown significance (VUS) highly suspected to be causative were reported to the referring geneticist via a rapid report. Step 2 included secondary analysis performed on Franklin data analysis software (Genoox). This step involved reanalysis for undiagnosed cases and analysis for ACMG actionable secondary findings (SFs),23 unless parents opted out. These were reported back to the referring geneticist via a comprehensive final report. Rapid and secondary analyses were compared for compatibility.
Dual diagnosis was defined as cases with more than 1 diagnosis related to the phenotype. Suspected dual diagnosis was defined as cases with 1 P/LP variant and an additional VUS deemed related to the phenotype. Samples were not analyzed for carrier state of autosomal recessive (AR) disorders.
Feasibility was assessed by calculating TAT, defined as the time from sample arrival at the sequencing laboratory to rapid report finalization (Figure 1A). Diagnostic efficacy was calculated for the proportion of diagnostic and negative results for the entire cohort and for the various indications for testing. rtGS results and inheritance patterns were further compared between Jewish and non-Jewish ethnicities. Cases with VUS in a gene associated with the patient’s phenotype, potentially expanding the phenotype or in a novel candidate gene, were categorized as highly suspected to be causative (possibly diagnosed).
Statistical Analysis
All statistical analyses were conducted using R version 4.1.1 (R Project for Statistical Computing). Fisher exact test was used for categorical comparisons due to the presence of small sample sizes in certain categories. For continuous variables, the nonparametric Kruskal-Wallis test was used.24 Statistical significance was set at P < .05, and all tests were 2-tailed.
A binary logistic regression (LR) model was built to analyze associations between variables and diagnosis status, chosen for its robustness and suitability for categorical outcomes (eMethods 2 in Supplement 1).
Results
Patient and Demographic Characteristics
A total of 130 patients were enrolled during a 15-month period (October 2021 to December 2022) (Figure 1B). There were 60 female neonates (46%) and 70 male neonates (54%). Mean (SD) age at referral was 12 (13) days (eFigure 1 in Supplement 1). There were 46 (35%) preterm neonates and 53 (41%) full-term neonates, among the 99 with data available. Ethnic backgrounds included Jewish (67 [51%]), non-Jewish (58 [45%]), and mixed ethnicity (5 [4%]) (Table 1; eFigure 2A-C in Supplement 1). The mortality rate within 84 days of enrollment was 27% (29 of 107 for whom data were available). The genetic diagnosis explained death in all diagnosed cases, but one infant died of sepsis. The outcome (deceased or alive) did not significantly differ across the diagnosed, possibly diagnosed, and undiagnosed subgroups (Table 1).
Table 1. Baby Bambi Cohort Demographic Characteristics and Rapid Trio Genome Sequencing Results.
| Characteristic | Total (N = 130) | Diagnosed (n = 65)a | Possible diagnosis (n = 15)b | Undiagnosed (n = 50)c | P value |
|---|---|---|---|---|---|
| Sex | |||||
| Male | 70 (54) | 34 (52) | 9 (60) | 27 (54) | >.99 |
| Female | 60 (46) | 31 (48) | 6 (40) | 23 (46) | |
| Gestational age | |||||
| Full term | 53 (41) | 29 (45) | 8 (53) | 16 (32) | .26 |
| Preterm | 46 (35) | 21 (32) | 4 (27) | 21 (42) | |
| Unknown | 31 (24) | 15 (23) | 3 (20) | 13 (26) | |
| Ethnicityd | |||||
| Jewish | 67 (51) | 28 (43) | 7 (47) | 32 (64) | .04 |
| Non-Jewish | 58 (45) | 36 (55) | 7 (47) | 15 (30) | |
| Mixed | 5 (4) | 1 (2) | 1 (6) | 3 (6) | |
| Outcome | |||||
| Deceased | 29 (22) | 14 (22) | 3 (20) | 12 (24) | .87 |
| Alive | 78 (60) | 40 (61) | 10 (67) | 28 (56) | |
| No data | 23 (18) | 11 (17) | 2 (13) | 10 (20) | |
| Primary category inclusion criteria | |||||
| Primary neurologic phenotypee | 35 (27) | 18 (28) | 4 (27) | 13 (26) | .55 |
| 2 Separate major anomalies | 47 (36) | 23 (36) | 6 (40) | 18 (36) | |
| Single otherwise rare abnormality | 14 (11) | 9 (14) | 1 (6) | 4 (8) | |
| Structural brain malformation | 14 (11) | 8 (13) | 0 | 6 (12) | |
| Unstable metabolic or endocrine abnormality | 12 (9) | 5 (8) | 4 (27) | 3 (6) | |
| Congenital heart failure or cardiomyopathy | 6 (5) | 2 (1) | 0 | 4 (8) | |
| Undefinedf | 2 (1) | 0 | 0 | 2 (4) | |
| Genomic variants (n = 80) | |||||
| Chromosomal abnormalities | 13 (10) | 12 (18) | 1 (7)f | 0 | NA |
| Common aneuploidyg | 3 (2) | 3 (4) | 0 | 0 | |
| Copy-number abnormalities | 8 (6) | 7 (11) | 1 (7) | 0 | |
| Unbalanced translocation | 2 (2) | 2 (3) | 0 | 0 | |
| Single-nucleotide variants | 65 (50) | 51 (78) | 14 (93) | 0 | |
| Triplet repeat expansion | 1 (1) | 1 (2) | 0 | 0 | |
| Uniparental heterodisomyh | 1 (1) | 1 (2) | 0 | 0 | |
| Inheritance | |||||
| Autosomal recessive | 28 (21) | 22 (34) | 6 (40)i | 0 | NA |
| De novo, sporadic | 24 (18) | 23 (36) | 1 (7) | 0 | |
| Autosomal dominant familial | 10 (8) | 5 (8) | 5 (32) | 0 | |
| X-linked recessive | 2 (2) | 1 (2) | 1 (7) | 0 | |
| X-linked dominant | 2 (2) | 1 (2) | 1 (7) | 0 | |
| Dual diagnosis | 1 (1) | 1 (2) | 0 | 0 | NA |
| Suspected dual diagnosis | 13 (10) | NA | NA | 0 | NA |
| Secondary findings (n = 112)j | 7 (63) | 4 (7) | 1 (8) | 2 (4) | .74 |
| TAT, mean (SD) [range], d | |||||
| Rapid report | 7.4 (2.7) [2-18] | 6.5 (2.3) [2-15] | 8.5 (3.3) [4-16] | 8.2 (2.8) [2-18] | .003 |
| Final report | 67.6 (26.1) [23-212] | 69.6 (28.9) [24-212] | 57.3 (22.4) [23-98] | 68.2 (23.0) [23-111] | .22 |
Abbreviation: TAT, turnaround time.
Disease-causing variant identified.
Variants of unknown significance in gene highly suspected to fully or partially explain phenotype identified.
Negative genome sequencing test.
Overall, 23 of 113 parents (58 of 113 were diagnosed; 55 of 113 had variants of unknown significance or were undiagnosed) were consanguineous; 17 parents did not have consanguinity ruled out or confirmed. By ethnicity, there was 1 consanguineous couple with Jewish ethnicity (negative rapid trio genome sequencing results), 22 consanguineous couples with non-Jewish ethnicity (14 with disease-causing variants identified, 3 with variants of unknown significance, and 5 with negative rapid trio genome sequencing results), and 0 consanguineous couples with mixed ethnicity. The difference among groups was not significant (P = .11).
Primary neurologic phenotype included encephalopathy, seizures, and severe hypotonia.
See eTable 2 in Supplement 2.
Trisomy 21 and trisomy 18 (eTable 2 in Supplement 2).
Prader-Willi syndrome detected by methylation-specific multiplex ligation-dependent probe amplification; see Discussion and eTable 2 in Supplement 2.
Including novel candidate gene.
Data regarding secondary findings are valid for 112 of 130 patients (18 opted out on consent form: 11 in the diagnosed group; 2 in the variants of unknown significance group; 5 with negative results).
rtGS Results
Abnormal results were found in 80 neonates (62%), including 65 P/LP variants (diagnosed group), 14 VUS highly suspected as causative, and 1 patient with a candidate novel gene. Negative rtGS results were reported in 50 neonates (38%) (Table 1 and Figure 2A). SNVs were the most common type of disease-causing variants (51 of 65 [78%]), followed by chromosomal abnormalities (12 [18%]), 1 patient with a triplet repeat expansion (in DMPK), and 1 patient with UPD (Figure 2B). SNVs were all exonic, except for 1 case with 2 intronic VUSs (eTable 2 in Supplement 2). The distribution of abnormal variants did not differ significantly between full-term and preterm infants or between males and females. AR (22 [34%]) and de novo (23 [36%]) were the leading inheritance patterns among the diagnosed group, whereas dominant familial disorders were more likely classified as VUS (Table 1).
Figure 2. Rapid Trio Genome Sequencing (rtGS) Results.
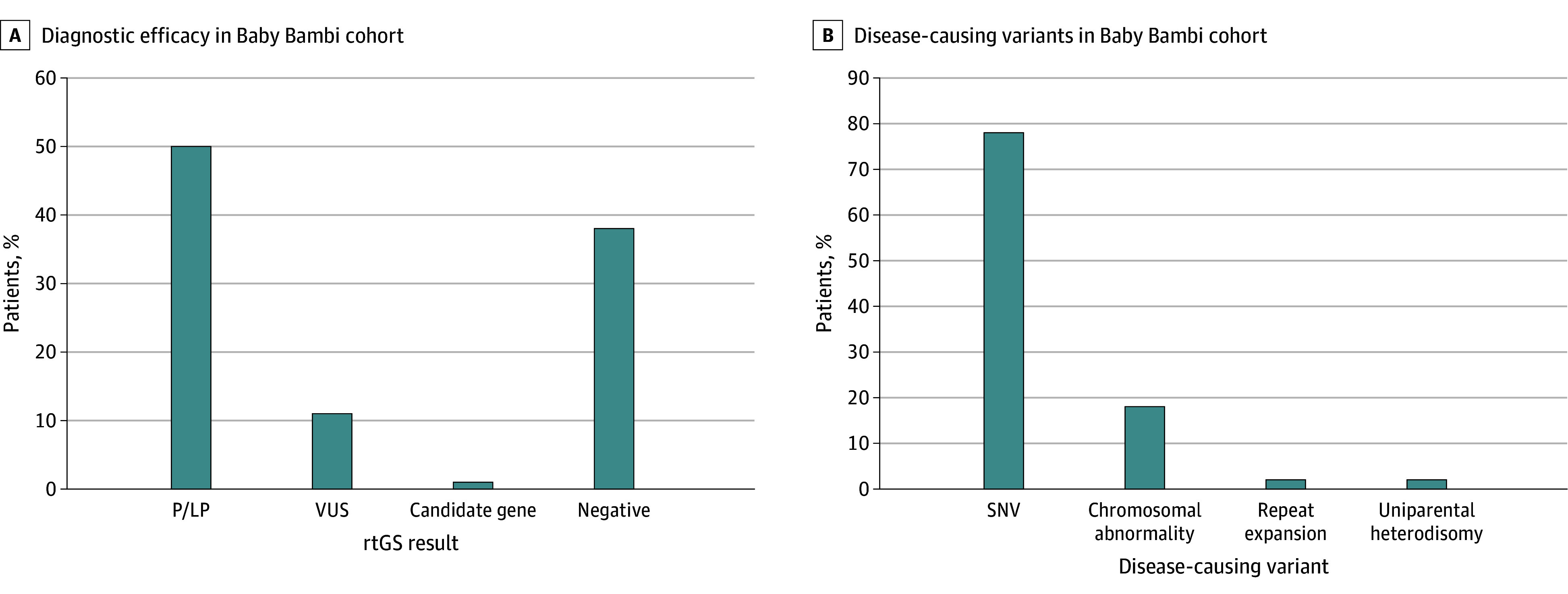
A, Diagnostic efficacy of rtGS in full Baby Bambi cohort (130 neonates). B, Types and proportions of disease-causing variants identified in the Baby Bambi cohort (65 neonates). LP indicates likely pathogenic; P, pathogenic; SNV, single-nucleotide variant; VUS, variant of unknown significance.
There was a minor significant difference in the distribution of diagnosed, undiagnosed, and VUS categories among Jewish and non-Jewish infants (Table 1). A total of 28 of 65 neonates with a diagnosis (43%) were Jewish and 36 (55%) were non-Jewish. Of 15 neonates with a possible diagnosis (ie, VUS), 7 (47%) were Jewish and 7 were non-Jewish (47%). Of 50 neonates with no diagnosis, 32 (64%) were Jewish and 15 (30%) were non-Jewish (P = .04), indicating a slightly higher diagnostic rate in the non-Jewish group. Dominant de novo disorders predominated in Jewish neonates (16 of 28 [57%] vs 7 of 36 [19%]), in contrast to AR disorders in non-Jewish neonates (19 of 36 [53%] vs 3 of 28 [11%]) (P < .001) eFigure 2D in Supplement 1). A nonsignificant higher diagnostic yield was seen in neonates of consanguineous couples (14 of 23 [60%]) compared with those of nonconsanguineous couples (44 of 90 [49%]) (P = .11). None of the detected diagnostic variants or VUS were known founder mutations in the Israeli population.
Definitive dual diagnosis was confirmed in 1 patient manifesting 2 de novo conditions, Treacher-Collins and Cleidocranial Dysplasia syndromes (eTable 2 in Supplement 2) and was suspected in 13 additional cases (20%). Follow-up data were unavailable to confirm causality of additional VUSs.
Feasibility
Mean (SD) TATs of rapid and secondary analysis reports were 7.4 (2.7) and 67.6 (26.1) days, respectively. Rapid reports of causative variants (mean [SD] TAT, 6.5 [2.3] days) had significantly shorter TATs than negative cases (mean [SD] TAT, 8.5 [3.3] days) (P = .003) (Table 1).
Compatibility of Rapid and Secondary Analyses
All variants detected by TSS were also detected by Franklin. In 8 cases, rapid analysis was negative, but secondary analysis revealed 2 pathogenic variants, increasing the diagnostic rate by 2%. Six additional VUSs were also found (eTable 2 in Supplement 2).
Secondary and Incidental Findings
Actionable secondary findings23 were detected in 7 families (5%), including known founder variants in BRCA1/2 (eTable 2 in Supplement 2). Incidental findings were reported in 3 families (2%), and familial segregation was offered: a maternally inherited COL4A5 variant led to subsequent diagnosis of familial Alport syndrome; a paternally inherited pre-alteration expanded DMPK allele; and a homozygous VWF variant for which preventive measures were offered.
rtGS Result Variables
The distribution of the various primary and secondary inclusion criteria categories among the 3 groups (diagnosed, possibly diagnosed, and undiagnosed) was analyzed. For the primary category, a neurologic phenotype and multiple congenital anomalies were the most common (82 [63%]) (Table 1). No significant differences were found in the distribution of diagnostic variants or VUSs across primary criteria (Table 1). Abnormality of prenatal development or birth (HP:0001197, 36 [28%]) and brain imaging abnormality (HP:0410263, 34 [26%]) were the leading secondary category indications for testing, although they did not characterize a positive or negative rtGS result (eTable 3 in Supplement 1).
The LR model identified 3 secondary category inclusion criteria with significant association with a positive rtGS result: hepatic failure (HP:0001399), with a coefficient of 2.84 (P = .05); seizures (HP:0001250), with a coefficient of 2.20 (P = .03); and generalized hypotonia (HP:0001290), with a coefficient of 1.53 (P = .04). Conversely, abnormal kidney morphology (HP:0012210) with a coefficient of −3.43 and abnormality of the endocrine system (HP:0000818) with a coefficient of −1.81, each showed a significant correlation with a negative rtGS result (P = .01 and P = .04, respectively) (eAppendix and eFigure 3 in Supplement 1).
Pathogenicity Factors of VUS
Possible diagnosis was suggested in 15 cases with VUS suspected of explaining the phenotype. We applied the model trained on the diagnosed cases to assess causality of these VUS. By this model, a calculated probability score greater than 0.5 was assigned a score of 1 and was considered supportive of the clinical association between a reported VUS and the patient’s phenotype. A calculated probability score of less than 0.5 was assigned a score of 0, suggesting a negative association (eAppendix in Supplement 1). This approach supported causality of detected VUSs in nearly two-thirds of the possibly diagnosed group (Table 2; eAppendix in Supplement 1). It is essential to note that these variables should be interpreted cautiously.
Table 2. Logistic Regression Model Score of 15 Possibly Diagnostic Variants of Unknown Significance.
| ID No. | Sex assigned at birth | Primary category inclusion criteriaa | Secondary category inclusion criteria, HPO terms | HPO terms submitted in analysis pipeline | Gene | NM_number | Variant | Phase | Syndrome or disorder | MIM No. | Inheritance | Probabilityb | Model scoreb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Female | 2 | HP:0001627, HP:0000924, HP:0000818 | Abnormal heart morphology, HP:0001627; femoral bowing, HP:0002980; tibial bowing, HP:0002982; talipes equinovarus, HP:0001762; kyphoscoliosis, HP:0002751; respiratory distress, HP:0002098; abnormal facial shape, HP:0001999; macrocephaly, HP:0000256; ventricular septal defect, HP:0001629; cryptorchidism, HP:0000028; rocker bottom foot, HP:0001838 | PHEX | NM_000444.6 | c.751A>G, p.Lys251Glu | Heterozygote | Hypophosphatemic rickets | 307800 | XLD, maternal | 0.644 | 1 |
| 2 | Male | 2 | HP:0001197 | Bifid scrotum, HP:0000048; cleft lip, HP:0410030; cleft palate, HP:0000175 | GLI3 | NM_000168.6 | c.613C>T, p.Arg205Cys | Heterozygote | Pallister Hall syndrome vs Greig cephalopolysyndactyly syndrome | 146510 vs 175700 | Paternal | 0.784 | 1 |
| ARHGAP29 | NM_004815.4 | c.2127_2128dupTG, p.Gly710fs | Heterozygote | Cleft lip with or without cleft palate | MONDO:0016034, ORPHA:199306 | De novo | |||||||
| 3 | Male | 5 | HP:0001627, HP:0000818 | Abnormal prolactin level, HP:0040086; abnormal pulmonary artery morphology, HP:0030966; hypertelorism, HP:0000316; brachyturricephaly, HP:0000244; atrial septal defect, HP:0001631; central hypothyroidism, HP:0011787; hyperinsulinemia, HP:0000842; wide nasal bridge, HP:0000431 | NA | NA | Chromosome 6:110 747 678-117 730 196 (approximately 6.983 Mb) dup | NA | Nonrecurrent copy number abnormality | NA | De novo | 0.294 | 0 |
| 4 | Female | 2 | HP:0001627, HP:0011024 | Situs inversus totalis, HP:0001696; biliary atresia, HP:0005912 | New candidate gene | NA | NA | Compound heterozygote | NA | NA | NA | 0.657 | 1 |
| 5 | Female | 2 | HP:0000951, HP:0011024 | Anal atresia, HP:0002023; preaxial hand polydactyly, HP:0001177; ventricular septal defect, HP:0001629; supernumerary ribs, HP:0005815 | GLI3 | NM_000168.5 | c.1984T>G p.Ser662Ala | Heterozygote | Pallister-Hall syndrome | 146510 | Maternal | 0.209 | 0 |
| 6 | Female | 3 | HP:0002086 | Ventilator dependence with inability to wean, HP:0005946; pulmonary arterial hypertension, HP:0002092; abnormal pulmonary interstitial morphology, HP:0006530; abnormal respiratory system physiology, HP:0002795 | SLC18A3 | NM_003055.3 | c.680T>C, p.Val227Ala | Homozygote | Myasthenic syndrome, congenital, 21, presynaptic | 617239 | AR | 0.233 | 0 |
| MYPN | NM_032578.3 | c.83G>C, p.Gly28Ala | Homozygote | Nemaline myopathy 11 | 617336 | AR | |||||||
| DMD | NM_004006.2 | c.3506T>C, p.Met1169Thr | Hemizygote | Becker muscular dystrophy | 300376 | XLR | |||||||
| 7 | Female | 5 | HP:0000818, HP:0001250 | Hypoglycemia, HP:0001943; hyperinsulinemic hypoglycemia, HP:0000825; convulsive status epilepticus, HP:0032660; congestive heart failure, HP:0001635; patent ductus arteriosus, HP:0001643; mitral regurgitation, HP:0001653; tricuspid regurgitation, HP:0005180; patent foramen ovale, HP:0001655; microcephaly, HP:0000252 | HNF4A | NM_001287184.1 | c.5C>T; p.Ser2Leu | Heterozygote | Fanconi renotubular syndrome 4, with maturity-onset diabetes of the young vs maturity-onset diabetes only | 616026 vs 125850 | Paternal | 0.559 | 1 |
| 8 | Male | 1 | HP:0001197, HP:0410263 | Hypoplasia of the corpus callosum, HP:0002079; abnormal facial shape, HP:0001999; postaxial polydactyly, HP:0100259; talipes calcaneovalgus, HP:0001884 | EVC2 | NM_147127.3 | c.1421T>C, p.Met474Thr | Homozygote | Ellis-van Creveld syndrome | 225500 | AR | 0.691 | 1 |
| ASH1L | NM_018489.1 | c.6929G>C; p.Gly2310Ala | Heterozygote | Intellectual developmental disorder, autosomal dominant 52 | 617796 | De novo | |||||||
| ADAMTSL1c | NM_001040272.6 | c.1414G>A, p.Gly472Arg | Homozygote | Microcephaly, facial dysmorphism, ocular anomalies, multiple congenital anomalies syndrome | ORPHA:521445 | Unknown | |||||||
| 9 | Male | 1 | HP:0001250, HP:0001298 | NA | CHD2 c | NM_001271.4 | c.295-125A>G | Heterozygote | Developmental and epileptic encephalopathy 94 | 615369 | De novo | 0.678 | 1 |
| CACNA1A | NM_001127222.2 | c.294-16973C>T | Heterozygote | Developmental and epileptic encephalopathy 42 | 617106 | De novo | |||||||
| 10 | Female | 5 | HP:0001939, HP:0011024, HP:0001290 | Thin upper lip vermilion, HP:0000219, chin with horizontal crease, HP:0011823, micrognathia, HP:0000347, jaundice, HP:0000952, conjugated hyperbilirubinemia, HP:0002908, infantile axial hypotonia, HP:0009062, cholestasis, HP:0001396, acholic stools, HP:0011985, long philtrum, HP:0000343, nasogastric tube feeding, HP:0040288 | GLIS3 c | NM_001042413.2 | c.2538G>A (p.Pro846Pro) maternal; c.1787C>T (p.Pro596Leu) paternal | Compound heterozygote | Neonatal diabetes with congenital hypothyroidism | 610199 | AR | 0.457 | 0 |
| 11 | Female | 1 | HP:0001250 | Dystonia, HP:0001332; feeding difficulties in infancy, HP:0008872; spasticity, HP:0001257; abnormal thalamic MRI signal intensity, HP:0012696; abnormal basal ganglia MRI signal intensity, HP:0012751 | RYR3 c | NM_001036.6 | c.1623C>A (p.Asn541Lys) maternal; c.9218G>A (p.Arg3073His) paternal | Compound heterozygote | Congenital myopathy 20 | 620310 | AR | 0.885 | 1 |
| 12 | Female | 1 | HP:0001250 | Myoclonic seizure, HP:0032794; abnormal isoelectric focusing of serum transferrin, HP:0003160 | GRIN1 | NM_007327.4 | c.1630C>T, p.Arg544Cys | Homozygote | Developmental and epileptic encephalopathy 101 | 619814 | AR | 0.889 | 1 |
| 13 | Female | 5 | HP:0000818 | Conjugated hyperbilirubinemia, HP:0002908, intrauterine growth retardation, HP:0001511, hyperinsulinemic hypoglycemia, HP:00008250, hypoglycemia, HP:0001943 | KCNJ11 | NM_000525.3 | c.635G>A, p.Ser212AsnS | Heterozygote | Hyperinsulinemic hypoglycemia, familial, 2 | 601820 | Maternal | 0.176 | 0 |
| GLUD1 | NM_005271.5 | c.1582T>A, p.Tyr528Asn | Heterozygote | Hyperinsulinism-hyperammonemia syndrome | 606762 | Paternal | |||||||
| IGFR1 | NM_000875.5 | c.1633G>A; p.Gly545Ser | Heterozygote | Insulin-like growth factor I, resistance to | 606762 | Paternal | |||||||
| 14 | Male | 2 | HP:0001197, HP:0000924, HP:0410263 | Bilateral cleft lip and palate, HP:0002744; foot oligodactyly, HP:0001849; agenesis of corpus callosum, HP:0001274 | FGFR1 c | NM_023110.3 | c.748C>G, p.Arg250Gly | Heterozygote | Holoprosencephaly, hypogonadotropic hypogonadism 2 with or without anosmia | 147950 | AD (paternal) | 0.942 | 1 |
| CHD6 | NM_032221.5 | c.176A>G, p.Lys59Arg | Heterozygote | Unknown | 616114 (gene) | AD (maternal) | |||||||
| 15 | Female | 2 | HP:0012210 | Pneumothorax, HP:0004876; polycystic kidney diseases, HP:0000113; macrosomia, HP:0001520 | FOXP3 c | NM_014009.4 | c.188C>T, p.Pro63Leu | Hemizygote | Immune dysregulation-polyendocrinopathy-enteropathy-X-linked syndrome | 304790 | XLR (maternal) | 0.061 | 0 |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; HPO, human phenotype ontology; ID, identification; MIM, Mendelian Inheritance in Man; MRI, magnetic resonance imaging; NA, not applicable; XLD, X-linked dominant; XLR, X-linked recessive.
For primary category inclusion criteria see eTable 1 in Supplement 1.
For LR model (LR) see Methods section and eMethods 2 in Supplement 1.
Variant detected in secondary analysis.
Clinical Utility
The outcomes of rtGS for clinical management were evaluated via questionnaires completed by the NICU staff. The response rate was 82% (107 cases). Among respondents, change of management was reported for 24 cases (22%), all in the diagnosed group, although in most cases the type of change was not disclosed. rtGS affected decisions regarding invasive procedures in 10 cases (9%). Among diagnosed patients, results led to early tailored medication in 6 of 65 (9%), transfer to nursing care facilities in 2 (3%), and supportive care in 2 (3%) (Table 3).
Table 3. Short-Term Tailored Medical Management in 10 Neonates in the Baby Bambi Cohort .
| Gene | Transcript | Variant | Phase | Inheritance | Syndrome | MIM No. | Precision management |
|---|---|---|---|---|---|---|---|
| Tailored precision drugs | |||||||
| ODC1 | NM_002539.3 | c.1307_1311 delinsT, p.Thr436IlefsX11 | Heterozygote | De novo | Bachmann-Bupp syndrome | 619075 | Eflornithine (research drug) |
| PNPO | NM_018129.3 | c.284G>A p.Arg95His | Homozygote | AR | Pyridoxamine 5-prime-phosphate oxidase deficiency | 610090 | Pyridoxal phosphate supplement |
| COL1A1 | NM_000088.4 | c.1066G>T, p.Gly356Cys | Heterozygote | De novo | Osteogenesis imperfecta 3 | 166200 | Bisphosphonates |
| KCNQ1 | NM_000218.3 | c.421G>A, p.Val141Met | Heterozygote | De novo | Short QT syndrome, 2 | 192500 | Channel-specific blockade of IK1 |
| COL1A1 | NM_000088.4 | c.3488G>A, p.Gly1163Glu | Heterozygote | De novo | Osteogenesis imperfecta 2-3 | 166200 | Bisphosphonates |
| MVK | NM_000431.4 | c.1129G>A, p.Val377Ile (maternal); c.1039G>A, p.Gly347Arg (paternal) | Compound heterozygote | AR | Mevalonate kinase deficiency | 260920 | Interleukin 1 receptor antagonist (anakinra), bone marrow transplant |
| Palliative care | |||||||
| SETBP1 | NM_015559.3 | c.2608G>A, p.Gly870Ser | Heterozygote | De novo | Schinzel-Giedion midface retraction syndrome | 269150 | Transferred to nursing home |
| STXBP1 | NM_003165.3 | c.1095_1096del, p.Cys366ProfsTer13 | Heterozygote | De novo | Developmental and epileptic encephalopathy 4 | 612164 | Transferred to nursing home |
| CHD2 | NM_001271.4 | c.295-125A>G | Heterozygote | De novo | Developmental and epileptic encephalopathy 94 | 615369 | Supportive care |
| CACNA1A | NM_001127222.2 | c.294-16973C>T | Heterozygote | De novo | Developmental and epileptic encephalopathy 42 | 617106 | |
| Trisomy 18 | NA | NA | NA | NA | NA | NA | Supportive care |
Abbreviations: AR, autosomal recessive; MIM, Mendelian Inheritance in Man; NA, not applicable.
Increased recurrence risk in future offspring could be confirmed for all inherited disorders in the diagnosed group (30 [46%]), including 22 families with AR, 5 with autosomal dominant, 2 with X-linked disorders, and 1 family with confirmed parental balanced reciprocal chromosomal translocation (Table 1). A parental balanced reciprocal translocation was highly suspected in an additional case (eTable 2 in Supplement 2), but parental studies were unavailable.
Discussion
To our knowledge, this is the first national study evaluating the feasibility of rtGS in a public health care setting. Israel has a unique universal health care system, an annual birth rate of approximately 180 000, and an estimated 12% admission rate to NICUs.25 In this collaborative study, we evaluated the feasibility, diagnostic efficacy, and clinical utility of rtGS in the Israeli national public health care system, including 130 critically ill neonates enrolled by NICUs and geneticists throughout the country. Rapid mean TAT of 7 days proved feasibility, with a substantial 2 days shorter TAT in the diagnosed group. This is compatible with the immediate report of a diagnosis once a definitive P/LP variant was detected and the tendency to reevaluate VUS and negative cases. We found that rapid return of diagnostic rtGS results is possible on a national scale and within the capacities of a public health care system in an adequate timeframe for change-of-management decisions without compromising prognosis.11,19
Diagnostic efficacy in our cohort was relatively high at 50% (considering only P/LP variants), compared with the average 30% to 40% published previously. Several earlier studies have reported high diagnostic rates of 57% to 73%,26,27 but these were conducted in small homogeneous cohorts. Indeed, a recent review of 21 prospective studies encompassing 1654 infants19 indicated a negative correlation between diagnostic rate and cohort size, noting rates of less than 30% in cohorts of 100 or more participants. Our high diagnostic yield may be explained by the unique Israeli population, still showing high rates of consanguinity and endogamy as reflected by a high risk of AR disorders and significantly higher diagnostic rates in the non-Jewish participants (eFigure 2D in Supplement 1). Interestingly, we did not detect any known Israeli founder variants; thus, national preconception carrier screening programs would have detected none of the carrier parents.
Most studies limit their reports to P/LP variants alone, although 21 VUSs in 184 participants (11%) were reported in Project Baby Bear.15 In our cohort, VUSs have also been suspected as either fully or partially related to the child’s phenotype in 12% (15 of 130) (Table 3). Using an LR model developed on the definitive diagnoses, we were able to support causality in 64% (9 of 15) of VUSs. Long-term follow up, including continued phenotyping and familial segregation studies, will shed light on these associations. Application of this novel approach in a larger cohort may aid in the interpretation of VUSs and raise diagnostic rates.
We used a rather narrow definition of clinical utility, focusing on precision medical treatment and family planning possibilities, critical for families with a severely affected or deceased child (46% of diagnosed patients) (eTable 2 in Supplement 2). The possibility of offering early tailored medical therapy is expected to substantially impact prognosis. Tailored management could be offered in 9% of our diagnosed patients (6 of 65), including prompt inclusion of 1 patient in a clinical trial (Table 3). Overall, 22% of utility questionnaire respondents reported a change of management for their patients. This rate is somewhat lower than the 25% to 65% reported in the literature; however, the definition of change of management is not uniform across studies, affecting its rate.
A causative genetic variant was not detected in 38% to 50% of our patients. Some of these infants’ phenotypes do not have a genetic etiology, while others may be identified in the future by clinical refinement, periodic reanalysis, new research findings, and/or advanced -omics technologies.
Limitations
This study has limitations, including possible bias due to lack of systematic evaluation of all newborns eligible for the study. Participation in the pilot was not mandatory; thus, eligible neonates could have been tested by other means. Referral bias could potentially affect diagnostic rates, eg, centers serving population with high rates of consanguinity. Despite this possible limitation, diagnostic rate did not significantly differ among consanguineous (14 of 65) and nonconsanguineous (44 of 90), families (P = .11) but was slightly higher in non-Jewish vs Jewish infants. In addition, clinical utility may be underestimated, as it was assessed short term. Long-term follow-up data regarding outcomes in terms of survival, growth and development, medical procedures, and more will provide additional evidence of clinical utility of rtGS in ill neonates. Furthermore, there are still limitations to the GS bioinformatics tools, as exhibited in a hypotonic infant with Prader-Willi syndrome (eTable 2 in Supplement 2). Neither TSS nor Franklin detected the maternal UPD, which was subsequently observed on methylation-specific multiplex ligation-dependent probe amplification ordered by the treating geneticist. Retrospective reanalysis revealed a complex mixed maternal UPD spanning approximately 81 megabase on chr15q11.2-q26.3. Following this experience, Franklin improved their algorithm to detect complex heterodisomy. Furthermore, sound clinical judgement is still required, even in the era of artificial intelligence. A neonate was found homozygous for a GBE variant (eTable 2 in Supplement 2) classified as VUS according to ACMG guidelines22; however, her phenotype and family history were highly compatible with the severe type of glycogen storage disease type IV, and the variant was reclassified as disease causing. The latter 2 cases highlight the importance of the ongoing crosstalk between bioinformaticians and treating physicians.
Conclusions
In this study, we found that rapid TAT of approximately 7 days was feasible in a national public health care setting with a 50% to 62% diagnostic rate, engaging both NICU staff and medical geneticists in a collaborative model. Early precision medicine, made available via rtGS, holds promise for significant clinical utility in this patient group. Following the success of the Baby Bambi national pilot, rtGS in critically ill neonates has been implemented in routine clinical care.
eMethods 1. Supplemental Methods and Translated Copies of Informed Consent, Clinical Utility, Outcome, and Requisition Forms
eTable 1. Study Inclusion and Exclusion Criteria
eMethods 2. Statistical Analyses
eReferences.
eAppendix. Supplemental Results
eFigure 1. Neonatal Age at Referral for rtGS
eFigure 2. Ethnic Backgrounds of Baby Bambi Pilot Population
eTable 3. Distribution of Secondary Category Inclusion Criteria (HPO Terms) Among Diagnosed, Possibly Diagnosed, and Undiagnosed Patients
eFigure 3. Variables of Diagnostic vs Negative rtGS Results
eTable 2. Baby Bambi Cohort Results
Data Sharing Statement
References
- 1.Arth AC, Tinker SC, Simeone RM, Ailes EC, Cragan JD, Grosse SD. Inpatient hospitalization costs associated with birth defects among persons of all ages—United States, 2013. MMWR Morb Mortal Wkly Rep. 2017;66(2):41-46. doi: 10.15585/mmwr.mm6602a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baxter SK, King MC. A time to sequence. JAMA Pediatr. 2017;171(12):e173435. doi: 10.1001/jamapediatrics.2017.3435 [DOI] [PubMed] [Google Scholar]
- 3.Berry MA, Shah PS, Brouillette RT, Hellmann J. Predictors of mortality and length of stay for neonates admitted to children’s hospital neonatal intensive care units. J Perinatol. 2008;28(4):297-302. doi: 10.1038/sj.jp.7211904 [DOI] [PubMed] [Google Scholar]
- 4.Murphy SL, Xu J, Kochanek KD, Arias E. Mortality in the United States, 2017. NCHS Data Brief. 2018;(328):1-8. [PubMed] [Google Scholar]
- 5.Weiner J, Sharma J, Lantos J, Kilbride H. How infants die in the neonatal intensive care unit: trends from 1999 through 2008. Arch Pediatr Adolesc Med. 2011;165(7):630-634. doi: 10.1001/archpediatrics.2011.102 [DOI] [PubMed] [Google Scholar]
- 6.Marouane A, Olde Keizer RACM, Frederix GWJ, Vissers LELM, de Boode WP, van Zelst-Stams WAG. Congenital anomalies and genetic disorders in neonates and infants: a single-center observational cohort study. Eur J Pediatr. 2022;181(1):359-367. doi: 10.1007/s00431-021-04213-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kingsmore SF, Cakici JA, Clark MM, et al. ; RCIGM Investigators . A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am J Hum Genet. 2019;105(4):719-733. doi: 10.1016/j.ajhg.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imafidon ME, Sikkema-Raddatz B, Abbott KM, et al. Strategies in rapid genetic diagnostics of critically ill children: experiences from a Dutch university hospital. Front Pediatr. 2021;9:600556. doi: 10.3389/fped.2021.600556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med. 2018;3:6. doi: 10.1038/s41525-018-0045-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olde Keizer RACM, Marouane A, Kerstjens-Frederikse WS, et al. ; RADICON-NL consortium . Rapid exome sequencing as a first-tier test in neonates with suspected genetic disorder: results of a prospective multicenter clinical utility study in the Netherlands. Eur J Pediatr. 2023;182(6):2683-2692. doi: 10.1007/s00431-023-04909-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krantz ID, Medne L, Weatherly JM, et al. ; NICUSeq Study Group . Effect of whole-genome sequencing on the clinical management of acutely ill infants with suspected genetic disease: a randomized clinical trial. JAMA Pediatr. 2021;175(12):1218-1226. doi: 10.1001/jamapediatrics.2021.3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20(4):435-443. doi: 10.1038/gim.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu B, Kang W, Wang Y, et al. Application of full-spectrum rapid clinical genome sequencing improves diagnostic rate and clinical outcomes in critically ill infants in the China Neonatal Genomes Project. Crit Care Med. 2021;49(10):1674-1683. doi: 10.1097/CCM.0000000000005052 [DOI] [PubMed] [Google Scholar]
- 14.Stark Z, Ellard S. Rapid genomic testing for critically ill children: time to become standard of care? Eur J Hum Genet. 2022;30(2):142-149. doi: 10.1038/s41431-021-00990-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimmock D, Caylor S, Waldman B, et al. Project Baby Bear: rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet. 2021;108(7):1231-1238. doi: 10.1016/j.ajhg.2021.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mestek-Boukhibar L, Clement E, Jones WD, et al. Rapid Paediatric Sequencing (RaPS): comprehensive real-life workflow for rapid diagnosis of critically ill children. J Med Genet. 2018;55(11):721-728. doi: 10.1136/jmedgenet-2018-105396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Diemen CC, Kerstjens-Frederikse WS, Bergman KA, et al. Rapid targeted genomics in critically ill newborns. Pediatrics. 2017;140(4):e20162854. doi: 10.1542/peds.2016-2854 [DOI] [PubMed] [Google Scholar]
- 18.French CE, Delon I, Dolling H, et al. ; NIHR BioResource—Rare Disease; Next Generation Children Project . Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 2019;45(5):627-636. doi: 10.1007/s00134-019-05552-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Callahan KP, Mueller R, Flibotte J, Largent EA, Feudtner C. Measures of utility among studies of genomic medicine for critically ill infants: a systematic review. JAMA Netw Open. 2022;5(8):e2225980. doi: 10.1001/jamanetworkopen.2022.25980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owen MJ, Lefebvre S, Hansen C, et al. An automated 13.5-hour system for scalable diagnosis and acute management guidance for genetic diseases. Nat Commun. 2022;13(1):4057. doi: 10.1038/s41467-022-31446-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayeems RZ, Luca S, Ungar WJ, et al. The Clinician-Reported Genetic Testing Utility Index (C-GUIDE): preliminary evidence of validity and reliability. Genet Med. 2022;24(2):430-438. doi: 10.1016/j.gim.2021.10.005 [DOI] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller DT, Lee K, Chung WK, et al. ; ACMG Secondary Findings Working Group . ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(8):1381-1390. doi: 10.1038/s41436-021-01172-3 [DOI] [PubMed] [Google Scholar]
- 24.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47(260):583-621. doi: 10.1080/01621459.1952.10483441 [DOI] [Google Scholar]
- 25.Israel Ministry of Health . Recommendation for manpower and infrastructure management in departments for intensive and special treatment of newborns and premature babies (premature babies). Accessed June 21, 2023. https://www.gov.il/he/departments/policies/mr52-2013
- 26.Willig LK, Petrikin JE, Smith LD, et al. Whole-genome sequencing for identification of mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3(5):377-387. doi: 10.1016/S2213-2600(15)00139-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168. doi: 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods 1. Supplemental Methods and Translated Copies of Informed Consent, Clinical Utility, Outcome, and Requisition Forms
eTable 1. Study Inclusion and Exclusion Criteria
eMethods 2. Statistical Analyses
eReferences.
eAppendix. Supplemental Results
eFigure 1. Neonatal Age at Referral for rtGS
eFigure 2. Ethnic Backgrounds of Baby Bambi Pilot Population
eTable 3. Distribution of Secondary Category Inclusion Criteria (HPO Terms) Among Diagnosed, Possibly Diagnosed, and Undiagnosed Patients
eFigure 3. Variables of Diagnostic vs Negative rtGS Results
eTable 2. Baby Bambi Cohort Results
Data Sharing Statement