

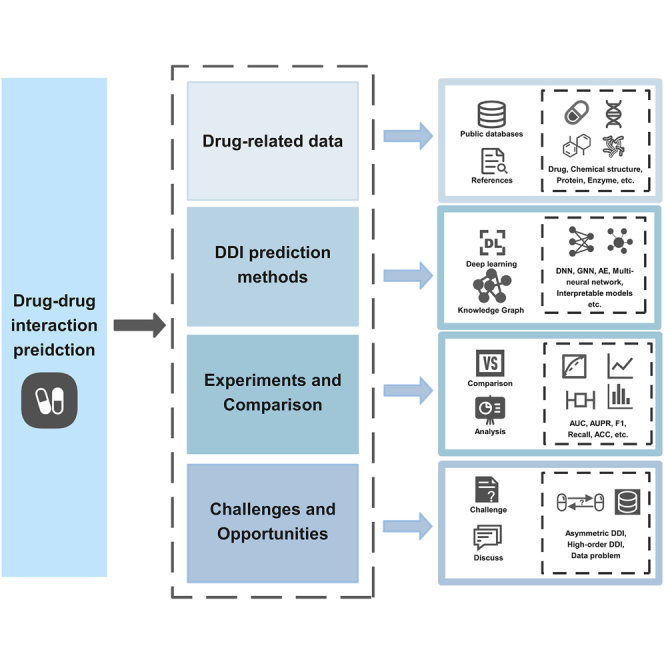
Summary
Drug-drug interactions (DDIs) can produce unpredictable pharmacological effects and lead to adverse events that have the potential to cause irreversible damage to the organism. Traditional methods to detect DDIs through biological or pharmacological analysis are time-consuming and expensive, therefore, there is an urgent need to develop computational methods to effectively predict drug-drug interactions. Currently, deep learning and knowledge graph techniques which can effectively extract features of entities have been widely utilized to develop DDI prediction methods. In this research, we aim to systematically review DDI prediction researches applying deep learning and graph knowledge. The available biomedical data and public databases related to drugs are firstly summarized in this review. Then, we discuss the existing drug-drug interactions prediction methods which have utilized deep learning and knowledge graph techniques and group them into three main classes: deep learning-based methods, knowledge graph-based methods, and methods that combine deep learning with knowledge graph. We comprehensively analyze the commonly used drug related data and various DDI prediction methods, and compare these prediction methods on benchmark datasets. Finally, we briefly discuss the challenges related to drug-drug interactions prediction, including asymmetric DDIs prediction and high-order DDI prediction.
Subject areas: Health sciences, Medicine, Medical specialty, Health informatics, Biological sciences, Bioinformatics, Pharmacoinformatics
Graphical abstract
Health sciences; Medicine; Medical specialty; Health informatics; Biological sciences; Bioinformatics; Pharmacoinformatics
Introduction
The use of drug combinations is common and necessary to treat patients with complex diseases.1,2 However, when drugs are concomitantly administered to a patient, the effects of the drugs may be enhanced or weakened, which may also cause side effects, these kinds of interactions are called drug-drug interactions (DDIs).3 For example, the serum concentration of dofetilide decreases when it is taken with dabrafenib together, whereas its serum concentration increases when taken with dalfopristin.4 Better knowledge of the incidence of DDIs and the drugs most frequently involved, can be helpful in a more accurate assessment of their overall clinical importance.5 Although a large number of DDIs are found during the clinical trials, there are still many DDIs on the market.6 Unknown DDIs can lead to unsafe treatments and even medication errors in those patients who are receiving polypharmacy.7 According to the US Centers for Disease Control and Prevention, more than 10% of people take five or more drugs at the same time, and even worse, 20% of older adults take at least 10 drugs,8 this phenomenon makes DDIs more likely to occur. Moreover, DDIs were estimated to be responsible for 4.8% of hospitalization in the elderly, an 8.4-fold increase compared to the general population.9 According to relevant statistics, DDIs cause a large number of deaths every year, and cause DDI-related costs of 177 billion US dollars.10 Therefore, it is very important to discover more potential DDIs.
Predicting potential DDI helps reduce unanticipated drug interactions and drug development costs and optimizes the drug design process.11 However, traditional experimental methods for DDIs identification and prediction, such as testing cytochrome P450 or transporter-related interaction, face challenges such as high cost and long duration.4 In some cases, researchers may suffer from limitations.12 Therefore, machine learning13 and deep learning14 based computational methods are proposed to solve these problems in traditional DDIs identification. The workflow of DDIs prediction using computational methods is shown in Figure 1. First, researchers need to collect available data from publicly available biomedical data sources such as databases or relevant literatures, including DDIs, targets, genes, proteins, etc. These data can provide valuable information for identifying potential DDIs. In the second step, advanced models utilizing machine learning and deep learning techniques are developed to identify DDIs. Then, in order to evaluate and validate the predictive performance of these proposed methods, it is necessary to compare them with state-of-the-art methods, perform prediction tasks on different gold standard datasets, and analyze them. Finally, these predicted interactions are validated in vitro and in vivo. Most methods generally classified DDI prediction tasks into binary classification task, multi-class task and multi-label task. The binary-class task is to predict whether there is an interaction between two drugs, multi-class task is to predict the type of DDI between drug pairs, and multi-label task is to predict the interaction types when there are two or more interactions between drug pairs.
Figure 1.

The workflow of drug-drug interactions prediction
Machine learning is used to teach machines how to handle data more effectively and relies on various algorithms to address data-related problems.15 Machine learning methods, including support vector machine (SVM), random forest, decision tree, naive Bayes, and K-nearest neighbors, have been widely applied in various fields such as computer vision, prediction, semantic analysis, natural language processing, and information retrieval.16 Traditional machine learning-based methods have made significant contributions to the field of DDIs prediction. Cheng et al.17 calculated four types of similarities of drugs as the features of drug pairs, including drug phenotypic similarity, therapeutic similarity, chemical structure similarity and genomic similarity, and finally applied naive Bayes, decision tree, K-nearest neighbors, logistic regression and support vector machine to predict DDIs. Li et al.18 have developed a probability ensemble approach (PEA) to build DDIs prediction model using drug molecules and pharmacological characteristics. Mei et al.19 developed a logistic regression model with L2 regularization, which used a simple f drug target profile representation to depict drugs and drug pairs to predict DDIs. Song et al.20 integrated the 2D molecular structure similarity, 3D pharmacophoric similarity, interaction profile fingerprint (IPF) similarity, target similarity and adverse drug effect (ADE) similarity to obtain the feature representation of drugs, and utilized SVM to predict candidate DDIs.
Deep learning techniques have been widely developed and applied due to their excellent performance on large-scale and high-dimensional datasets.21 Deep learning allows computational models that are composed of multiple processing layers to learn representations of data with multiple levels of abstraction,22 which can effectively find complex structures in high-dimensional data, and is more flexible than traditional methods based on Bayesian, random walk, support vector machine, and so on. Therefore, deep learning has been well applied in image recognition,23 computer vision,24 NLP,25 and speech recognition.26 Furthermore, deep learning has also gained extensive usage in the field of drug discovery including drug molecular activity prediction,27 molecular property prediction,28 target identification29 and DTI (drug-target interaction) prediction.30
In addition, there has been increasing interest in extending deep learning methods to graph data,31 the use of graph has also brought new breakthroughs in deep learning. Knowledge graph (KG) has been widely used in various business and scientific fields.32 Knowledge graph is a multi-relationship graph containing multiple types of entities and edges, in which nodes correspond to entities, and edges correspond to relations between the two connected entities.33 KG is mainly composed of triples, generally represented as , where denotes the set of entities, denotes the relations, denotes the set of edges, the edge denotes the existence of a relation between entities to , where , is the head entity and is the tail entity. The rich information provided by KG, containing structured and unstructured knowledge, can be input into deep learning models to find hidden connections between entities.
The use of drug related biomedical data is critical for developing high-performance DDI prediction models. Although many public databases have provided various drug-related information, there is still lacking of comprehensive summary of drug related entities and their interactions in current popular data sources, and the required data cannot be accurately and quickly obtained when developing computational prediction methods. In addition, many effective DDI prediction methods have been proposed, but there are still many issues that need to be considered. Certain models are unable to effectively utilize DDIs data, and the imbalance between positive and negative samples also needs to be resolved. Finally, the datasets and experiments used to analyze and evaluate the proposed prediction methods are generally not unified in many studies.
The advances in DDI prediction have been reviewed in detail from different aspects in recent years.8,34,35,36,37,38 For example, Zhang et al.35 reviewed deep learning-based methods for extracting DDIs from biomedical literatures. Han et al.8 reviewed DDI prediction models based on machine learning and organized into: traditional similarity, traditional classification, network diffusion, matrix factorization, ensemble-based approach and based on literature approach. This review can provide useful guidance for interested researchers to further promote bioinformatics algorithms to predict DDI. Compared with previous reviews, our study provides a more comprehensive and integrative analysis of deep learning-based and knowledge graph-based prediction methods and related biomedical data used in DDI prediction. Firstly, we summarize various sources of biomedical data used in DDI prediction methods. Secondly, the popular DDI prediction methods based on deep learning and knowledge graph are analyzed. Then, these computational methods are compared on the same datasets briefly. Finally, we discuss the limitations and challenges for developing DDI prediction methods.
In the recent review by Lin et al.,37 chemical structure based, network based, NLP based and hybrid methods for DDI prediction have been summarized, and provided an updated and accessible guide to the broad researchers and development community with different domain knowledge. In our review, we focus on analyzing and classifying existing computational methods for potential DDI prediction using deep learning and knowledge graph, respectively. Although the methods covered in our survey have certain overlaps with Lin et al.,37 we complement it with computational methods based on KG, which have recently demonstrated attractive prediction precision enhancement in DDI prediction. We further discuss the advantages and disadvantages of various prediction models. Furthermore, we have organized some benchmark datasets from the literatures that commonly used in DDI prediction tasks. The common validation strategies and evaluation metrics used in DDI prediction studies are also included to guide researchers to efficiently evaluate and verify the predictive ability of their developed methods in future studies.
Drug-related data
The explosive growth of large-scale genomic and phenotypic data, as well as data of small molecular compounds with granted regulatory approval,39 has enabled new developments for DDIs prediction. In addition to DDIs information, drug features are often utilized in DDI prediction, including chemical substructures, targets, enzymes, pathways, genes, transporters, side effects, indications, etc. The effective use of these drug features enables the model to learn comprehensively and improve performance. This study briefly summarizes the common public available databases and datasets involved in current researches on DDI prediction.
Drug-related public databases
Predicting DDIs often requires the use of multiple characteristics of drugs as well as known DDIs. The most commonly used databases include DrugBank,40 Drug Repurposing Knowledge Graph(DRKG),41 Kyoto Encyclopedia of Genes and Genomes(KEGG),42 Bio2RDF,43 TWOSIDES,44 SIDER,45 PubChem46 and DrugCentral.47 A detailed description of these public databases has been presented in Table 1. These public databases can be classified based on the content and function into drug omics data, drug adverse effects and drug knowledge graph databases.
Table 1.
The available databases
| Category | Database | Available data |
URL | API | ||
|---|---|---|---|---|---|---|
| Entities | Drug properties | Drug-related interactions | ||||
| Drug omics data | DrugBank40 | Drug, Target, Enzyme, Transporter, Protein, Disease, Gene, Carrier, Metabolite, Pathway, Compound, ATC, Adverse response |
Type, Chemical structure, Category, Approval status, Chemical identifiers, Indication, Function, Action | Drug-drug interaction, Drug-food interaction, Drug-metabolite interaction, Drug-protein interaction, Drug-transcript interaction, Drug-target interaction | https://go.drugbank.com/ | https://docs.drugbank.com/v1/ |
| KEGG42 | Pathway, Gene, Genome, Protein, Compound, Glycan, Enzyme, Variant, Disease, Drug, ATC, Target, Metabolism | Structure, Drug class, Chemical reaction, Chemical structure similarity | Drug-drug interaction, Drug-gene interaction, Drug-disease interaction | https://www.kegg.jp/ | https://www.kegg.jp/kegg/rest/keggapi.html | |
| PubChem46 | Taxonomy, Compound, Protein, Gene, Pathway, Cell line, Substance, Side effect, Bioactivity, Target | Structure, Indication | Chemical-chemical interaction, Chemical-gene interaction, Chemical-disease interaction | https://pubchem.ncbi.nlm.nih.gov/ | https://pubchem.ncbi.nlm.nih.gov/rest/pug | |
| DrugCentral47 | Drug, Target, Disease, Protein | Substructure, Adverse event, Similarity, Active ingredient, Indication, Drug mode of action, Pharmacologic action, Pharmacokinetic properties, Bioactivity | Drug-drug interaction, Drug-disease interaction, Drug-target interaction | https://drugcentral.org/ | https://drugcentral.org/OpenAPI | |
| Drug knowledge graph | DRKG41 | Anatomy, Biological process, Cellular component, Compound, Disease, Gene, Molecular function, Pathway, Pharmacologic class, Side effect, Symptom, ATC, Tax | SMILES | Compound-Compound interaction, Compound-side effect interaction, Compound-ATC classification interaction, Compound-pharmacologic class interaction, Compound-disease interaction, Compound-gene interaction | https://github.com/gnn4dr/DRKG | – |
| Bio2RDF43 | Drug, Gene, Protein, Compound, Disease, Cell | Chemical structure, Pharmacological property | Drug-adverse event interaction | http://bio2rdf.org/ | https://github.com/bio2rdf/bio2rdf-api | |
| Drug adverse effects | TWOSIDES44 | Drug, Side effect | Indication | Drug-side effect interaction | https://www.tatonettilab.org/resources/tatonetti-stm.html | – |
| SIDER45 | Drug, Side effect, ATC, Target | Chemical structure, Indication | Drug-side effect interaction | http://sideeffects.embl.de | – | |
Note: ’-’ indicates that the database does not provide API.
Drug omics databases mainly include DrugBank,40 KEGG,42 PubChem46 and DrugCentral.47
-
(1)
DrugBank40 is an open-access DB, which collects data from various sources, including journal articles, electronic databases, and textbooks.48 DrugBank (version 5.1.10) contains 16,565 drug entries including 2,761 approved small molecule drugs, 1,610 approved biologics, 135 nutraceuticals and over 6,723 experimental (discovery-phase) drugs. Moreover, 5,302 non-redundant protein (i.e., drug target, enzyme, transporter and carrier) sequences are linked to these drug entries. The data have been validated by DrugBank curatorial staff, multiple automated data consistency checks have also been performed to ensure a uniformly high level of data integrity.49 It can provide rich and high-quality data that enables significant advances in the bioinformatics field. Moreover, DrugBank team has constantly optimized the interface of the database and enriched the search functions to make it more convenient for researchers to access and use.
-
(2)
KEGG42 is a daily updated, free database resource to help understand the high-level functions and utilities of the biological system, which contains fifteen manually curated databases and a computationally generated database,42 and the PATHWAY database is the most important component. KEGG has powerful graphical functions to provide a comprehensive understanding of the interaction networks of genes, proteins and compounds, based on graphical representation of biological objects and graphical computation technologies.50
-
(3)
PubChem46 is key open chemical information resource at the US National Institutes of Health (NIH), which has collected information of drugs from hundreds of data sources, including university labs, patent documents, government agencies, pharmaceutical companies, chemical vendors, publishers and a number of chemical biology resources. PubChem contains small molecules, chemical structures, identifiers, chemical and physical properties, and many others. In addition, it also offers rich query and analysis functions that allows chemical compounds to be searched by name and structural formula.
-
(4)
DrugCentral47 is a well-rounded drug information resource that integrates a wide range of drugs, chemical substructures and indications information. The majority of the data are collected and aggregated from online public resources, combined with manual curation of literature and drug label information.51 DrugCentral is updated every 2–3 years, and its team regularly monitors new drug approval from FDA, EMA, and PMDA to provide accurate and high-quality data for related researches.
DRKG41 and Bio2RDF43 are two comprehensive drug knowledge graph databases.
-
(1)
DRKG41 is a comprehensive biological knowledge graph, which contains genes, compounds, diseases and side effects, ATC, biological process, etc. It integrates data from six existing biological databases and publications to ensure integrity and usefulness of data, which includes 97,238 entities belonging to 13 entity-types, and 5,874,261 triplets belonging to 107 edge-types.
-
(2)
Bio2RDF43 is an open knowledge graph that uses Semantic Web technologies to build and provide the largest network of Linked Data for the Life Sciences, which contains ∼11 billion triples across 35 biomedical databases. Bio2RDF can create RDFs compatible Linked Data from a diverse set of heterogeneously formatted sources to support complex biomedical researches.
Both TWOSIDES44 and SIDER45 are drug adverse effects databases.
-
(1)
TWOSIDES44 is a multi-drug side effect resource that contains significant associations between drugs and adverse events, this database contains 868,221 significant associations between 59,220 pairs of drugs and 1,301 adverse events. But these associations are limited to only those that cannot be clearly attributed to either drug alone.44
-
(2)
SIDER45 (version 4.1) contains 1430 drugs, 5,868 side effects (SEs) and 139,756 drug-SE pairs, the information about marketed drugs and their recorded side effects is extracted from public documents and package inserts. The available information also includes frequency of side effect and side effect classifications, etc., can provide high-quality and valuable data for related studies. Moreover, the high quality of the extracted entities has also been ensured by manually annotating names, adding synonymous names and using an additional Natural Language Processing step.45
Standard datasets collected from the literatures
With the development of DDIs prediction research, the existing studies have provided many benchmark datasets to facilitate the evaluation of prediction methods. Tables 2 and 3 show some benchmark datasets collected from the existing literatures, which are applied on three different tasks.
Table 2.
Benchmark datasets collected from existing literatures for binary classification prediction task
| Dataset name | Drugs | DDIs | Drug-related information | Data resource |
|---|---|---|---|---|
| Db_152 | – | 1,178,210 | SMILES | DRKG |
| Db_253 | 548 | 48,584 | Substructure: 881 Target: 780 Enzyme: 129 Transporter: 78 Pathway: 253 Indication: 4,897 Side effect: 4,897 Off side effect: 9,496 |
TWOSIDES, SIDER, OFFSIDES, PubChem, DrugBank, KEGG |
| Db_354 | 1,537 | 34,282 | SMILES | DrugBank |
| Db_455 | 2,578 | 612,388 | – | DrugBank |
| Db_556 | – | 48,548 | SMILES | DrugBank,53 |
| Db_657 | 1,752 | 504,468 | Morgan fingerprint | DrugBank |
| Db_758 | 1,562 | 180,576 | Chemical structures, ATC, DBP (899 drug targets and 222 non-target proteins) | DrugBank,59,60 |
| Db_858 | 1,934 | 230,887 | ||
| Db_961 | 2,367 | 209,494 | Target: 2,411 Enzyme: 285 Pathway: 314 Substructure: 699 |
DrugBank, KEGG, PubChem |
| Db_1055 | 1,925 | 56,983 | – | KEGG |
| Db_1162 | 613 | 80,702 | Enzyme: 454 Pathway: 533 Side effect: 4,859 Substructure: 811 Target: 2,670 Node2vec: 613 PRL: 978 |
DrugBank, CTD,63 KEGG, SIDER, LINCS64,65,66,67 |
| Db_1268 | 841 | 82,620 | Target: 1,333 Enzyme: 214 Pathway: 307 Substructure: 619 |
DrugBank, KEGG, PubChem |
| Db_1369 | 1,850 | 443,046 | SMILES | Drugbank |
| Db_1469 | 1,322 | 83,040 | BIOSNAP70 | |
| Db_1571 | – | 2,898,937 | – | DrugBank, KEGG, TWOSIDES, MEDLINE,72 OFFSIDES, PharmGKB53,73,74,75,76 |
| Db_1677 | 10,533 | 1,195,972 | SMILES | DrugBank, OGB-biokg78 |
| Db_1777 | 1,925 | 56,983 | DrugBank, KEGG |
Note: ‘Drugs’ represents the number of drugs in the dataset, ‘DDIs’ represents the numbers of drug-drug interactions, ‘Drug-related information’ represents other drug-related features in the dataset, ‘Data resource’ represents the source of the data, ‘-’ represents no clear explanation in the original literature.
Table 3.
Benchmark datasets collected from existing literatures for DDI events prediction tasks
| Task | Dataset name | Drugs | DDIs | Types | Drug-related information | Data resource |
|---|---|---|---|---|---|---|
| Multi-class | Dm_c179 | 572 | 74,528 | 65 | Chemical substructure, Target, Pathway, Enzyme | DrugBank, KEGG |
| Dm_c252 | – | 172,426 | 81 | SMILES | Ryu et al.80 | |
| Dm_c381 | 1,697 | 190,728 | 86 | Chemical structure | Ryu et al.80 | |
| Dm_c482 | 1,704 | 191,400 | 86 | SMILES | DrugBank | |
| Dm_c583 | 1,258 | 323,539 | 100 | Substructure, Target, Enzyme | DrugBank | |
| Dm_c684 | 1,935 | 589,827 | 2 | Chemical structure | DrugBank | |
| Dm_c781 | 1,013 | 114,204 | 71 | Chemical structure | Ryu et al.80, Lin et al.83 | |
| Multi-label | Dm_l152 | – | 99,002 | 200 | SMILES | TWOSIDES |
| Dm_l285 | 10,533 | 1,195,972 | 39 | – | OGB-biokg78 | |
| Dm_l381 | 751 | 53,888 | 200 | Chemical structure | TWOSIDES,83 | |
| Dm_l480 | 1,861 | 192,284 | 86 | SMILES | DrugBank | |
| Dm_l585 | 3,797 | 1,236,361 | 2 | – | DrugBank | |
| Dm_l685 | 1,925 | 56,983 | 2 | – | KEGG | |
| Dm_l786 | 1,918 | 30,979 | 100 | – | TWOSIDES | |
| Dm_l887 | 1,597 | 188,258 | 106 | SMILES, Gene: 22,032 GO terms: 29,692 |
DrugBank, BioGrid88,89,90 | |
| Dm_l981 | 1,314 | 103,938 | 200 | Chemical structure | TWOSIDES | |
| Dm_l1091 | 1,317 | 198,697 | 86 | Protein, Substructure Fingerprint |
DrugBank, PubChem |
Note: ‘Task’ represents prediction task, ‘Drugs’ represents the number of drugs in the dataset, ‘DDIs’ represents the numbers of drug-drug interactions, and ‘Types’ indicates the number of DDI types, ‘Drug-related information’ represents other types of data, ‘Data resource’ represents the source of the data, ‘-’ represents no clear explanation in the original literature. There are a total of 2,322 drugs in 52.
In this review, we evaluate the performance of the models using different datasets in Tables 2 and 3. For the binary classification prediction task, six benchmark datasets including Dm_l4,80 Db_2,53 Db_3,54 Db_4,55 Db_556 and Db_657 are chosen for model performance evaluation. The multi-class prediction task is performed on three benchmark datasets including Dm_c1,79 Dm_c252 and Dm_c3.81 For the multi-label prediction task, three benchmark datasets including Dm_l1,52 Dm_l285 and Dm_l381 are selected for model evaluation.
DDIs prediction methods
We group the existing DDI prediction methods into three classes: methods based on deep learning, methods based on knowledge graph, and methods that combine deep learning with knowledge graph. Table 4 lists the methods corresponding to each class. The graphical summary of the overall methods is shown in Figure 2.
Table 4.
Methods related to deep learning and knowledge graph
| Technology | Method | Descriptions | Advantages | Limitations | Validate | Links |
|---|---|---|---|---|---|---|
| DNN | DeepDDI80 | The model predicts potential DDIs using only the drug name and structure, and can also be utilized to predict drug-food interactions. | It breaks the limitation of not being able to obtain detailed information about drugs. | DNN used in the model needs to be upgraded based on the training with more data on drug pair interactions. | N | https://bitbucket.org/kaistsystemsbiology/deepddi |
| DDIMDL79 | Four sub-models are constructed by using each drug feature and a joint DNN framework is used to combine the sub-models to learn cross-modality representations of drug-drug pairs. | DDIMDL has taken advantage of deep learning and diverse drug-related features to predict DDI events. | Ignoring the problem of unbalanced datasets, and the fewer number of some event interactions may lead to underfitting of the model. | N | https://github.com/YifanDengWHU/DDIMDL | |
| DANN-DDI68 | An attention neural network is designed to learn the representations of drug-drug pairs, which considers the different contributions of different features and their dimensions. | DANN-DDI can combine multiple drug information to predict novel drug-drug interactions and DDI-associated events. | The imbalanced data and the noise bring challenges, and for some events without detailed descriptions and proved interactions, the model need the interpretability. | Y | https://github.com/naodandandan/DANN-DDI | |
| BioChemDDI92 | A computational method that integrates multi-level information by applying the self-attention mechanism to efficiently integrates biochemical and network features. | Graph collapse is innovatively introduced to extract network structure, and biochemical information is utilized during the pre-training process. | The more reasonable way of selecting negative samples needs to be considered to reduce the noise from the imbalance in the original dataset. | N | http://120.77.11.78/BioChemDDI/ | |
| GNN | BI-GNN93 | The model treats the data as a bi-level graph, which the highest level represents the interactions between biological entities, and each entity itself is further expanded to its intrinsic graph representation. | The transductive setting of drug repurposing is considered and can also be extended to other biological link prediction tasks with different interaction biological entities. | The introduction of extra features may improve the performance of this model. | N | – |
| SSI-DDI82 | The task of DDI prediction between two drugs is decomposed into identifying pairwise interactions between their respective substructures, and directly operating on the raw molecular graph representations of the drugs. | The model can learn substructures directly from drug molecular graphs, and can improve the ability of both expert and non-expert users to interpret the results of the prediction. | When the order of drugs of DDIs are changed, the performance is affected even during the training phase; there is some noisy information leaked in during the substructure extraction phase, which might have affected performance in inductive setting. | N | https://github.com/kanz76/SSI-DDI | |
| DSN-DDI94 | The model iteratively learns modules using local and global representations, while learning drug substructures from intra-view and inter-view. | DSN-DDI shows the usefulness for real-world DDI applications and can also serve as a generalized framework in the drug discovery field. | The model’s ability to generalize to new drugs in the inductive learning setting needs to be further improved. | Y | https://github.com/microsoft/Drug-Interaction-Research/tree/DSN-DDI-for-DDI-Prediction | |
| Chen et al.95 | Only the compressed structural information extracted from molecular graphs is utilized to predict the DDI. | It achieves higher performance on both small-scale and large-scale datasets, and is more robust to the extremely low pairwise similarity information. | The graph convolution operator can only operate on flat 2D molecular graphs, which may lose some vital information conveyed by 3D structure. | N | – | |
| GCNMK62 | Two DDI graph kernels are used for the graph convolutional layers, namely, increased DDI graph consisting of ‘increased’-related DDIs and a decreased DDI graph consisting of ‘decreased’-related DDIs. | Benefiting from the two graph kernels, GCNMK model can be used to predict DDIs effectively. | This model can’t identify DDIs among isolated drugs. | Y | – | |
| RS-GCN86 | It is a new relation-dependent sampling model. The core of this approach is to assign a learnable probability to each relation type and update it. | RS-GCN specifically provides an advantage in scalability. It is also shown that learning edge type probabilities is indeed beneficial. | There is still room for further improvement in predicting on graph with complex relationships and less dense. | N | – | |
| MIRACLE96 | A novel unsupervised contrastive learning component is proposed to balance and integrate multi-view information. It can capture inter-view molecule structure and intra-view interactions between molecules simultaneously. | It is superior on small-scale, medium-scale, and large-scale datasets. | The performance of the model becomes gradually worse as the size of the dataset gets larger and larger. | N | https://github.com/isjakewong/MIRACLE | |
| MFFGNN97 | A new feature extraction module is proposed to capture the global features for the molecular graph and the local features for each atom of the molecular graph. | MFFGNN can effectively fuse the topological information in molecular graphs, the interaction information between drugs and the local chemical context in SMILES sequences. | The model cannot be extended to multi-type DDI prediction tasks. | N | https://github.com/kaola111/mff | |
| AE | Purkayastha et al.98 | The model incorporates different combinations of feature embeddings from the drug-target interaction network and chemical structure. | The model uses a GAE to effectively predict missing DDI links. | Lack of the exploration of other representations of drugs, such as textual description of the drugs and side effect-based interaction network representation of the drugs. | N | – |
| Lee et al.87 | Target similarity profiles (TSP), Gene Ontology (GO) term similarity profiles (GSP), as well as structural similarity profiles (SSP) are constructed and combined. | GSP and TSP increase the prediction accuracy when using SSP alone, and the proposed model identified a number of novel DDIs that are supported by medical databases or existing researches. | The DDIs predicted by this model and their clinical consequences are mostly unvalidated in DrugBank, and the experimental results can be changed for different settings including different dataset version or experimental environment. | N | – | |
| DDI-MDAE61 | A drug representation learning method, which can learn unified drug representations from multiple drug feature networks simultaneously. | DDI-MDAE can predict potential interactions for drugs with incomplete features even faced with large-scale, noisy and sparse data. | Only considering the structural topologies of drug feature networks is not enough to learn the more comprehensive drug representations. | N | – | |
| Ensemble | NMDADNN99 | NMDADNN extracts the unified drug mapping features by integrating five drug-related heterogeneous information sources. | Five drug-related heterogeneous information sources are effectively integrated. | The quality of drug similarity matrix may be improved by utilizing more drug-related sources and suitable similarity measures, this is one of its limitations. | Y | – |
| DPDDI58 | GCN is utilized to extract the network structure features of drugs from DDI network, and DNN as a predictor. | This is an effective and robust method proposed to predict potential DDIs by utilizing the DDI network information without considering the drug properties. | Interactions with the new drugs cannot be predicted. | N | https://github.com/NWPU-903PR/DPDDI | |
| -DDI100 | The framework encodes drugs and relationship embeddings, and builds the relation-aware refined features. | It significantly improves the DDI prediction performance over multiple real-world datasets and settings, and shows better generalization ability. | This model ignores the atom level of the pair interaction between drugs, the modeling of relation and the relation-aware module is relatively simple, the imbalanced data issue is also not solved. | Y | https://github.com/linjc16/R2-DDI | |
| MDF-SA-DDI83 | The model combines two drugs in four different ways and can predict unobserved interactions between new drugs. | Multi-source drug fusion is used to obtain better prediction of DDIs. | There is room for further improvement in the prediction of interactions between new drugs. | N | https://github.com/ShenggengLin/MDF-SA-DDI | |
| AMDE54 | AMDE encodes drug features from multiple dimensions, including information from both Simplified Molecular-Input Line-Entry System sequence and atomic graph of the drug. | AMDE integrates drug features from multiple dimensions to enhance the effectiveness of downstream prediction tasks. | The model can be further optimized to incorporate more drug features. | Y | https://github.com/wan-Ying-Z/AMDE-master | |
| SGRL-DDI84 | SGRL-DDI model captures the task-joint information by integrating relation graph convolutional networks with Balance and Status patterns and predicts directed DDIs. | It utilizes the Balance theory and Status theory to reveal pharmacological interaction patterns in the directed DDI network. | Only enhancive/depressive DDIs and directed DDIs can be predicted, and there is limitation in predicting specific events. | Y | https://github.com/NWPU-903PR/SGRL-DDI | |
| Interpretable | CASTER69 | A sequential pattern mining module is developed by using labeled and unlabeled chemical structure data. | The model improves generalizability and interpretable prediction. | The model can be further improved by extending it to chemical sub-graph embedding and incorporating metric learning. | N | https://github.com/kexinhuang12345/CASTER |
| STNN-DDI101 | Mapping drugs into SSI space based on a list of predefined substructures with specific chemical meanings, which allows STNN-DDI to perform multiple types of DDI predictions in both transductive and inductive scenarios in a unified form with an explicable manner. | The interpretability of the model is improved, and it can also predict interactions between new drugs. | The introduction of more features of drugs may be helpful in improving the performance. | N | https://github.com/zsy-9/STNN-DDI | |
| DeSIDE-DDI102 | The model uses drug-induced gene expression signatures followed by gating and translating embedding. | It can increase DDI prediction accuracy and provide model interpretability. | The datasets are very sparse in terms of side effect type, and the identification of the side effect mechanism remains challenging. | Y | https://github.com/GIST-CSBL/DeSIDE-DDI | |
| DSIL-DDI103 | Treat the substructure interactions as domain-invariant representations of DDIs. Moreover, a pluggable substructure interaction module and a practical loss function are proposed. | DSIL-DDI improves the generalization and interpretability of DDI prediction models. | Effective predictions between new drugs need to be considered. | N | – | |
| GGI-DDI104 | A method that employs granular computing to identify key substructures, drugs are granulated into a set of coarser granules. | It enhances the interpretability of DDI predictions, and offers a consistent framework for DDI prediction in both transductive and inductive scenarios. | The rich knowledge contained in the different biomedical entities associated with drugs (e.g., proteins, genes, and targets) is ignored. | N | https://codeocean.com/ | |
| KG | Celebi et al.11 | A simple disjoint cross-validation scheme is proposed to evaluate DDI predictions in the absence of known DDIs for drugs. | Different KGE methods for DDIs prediction are compared. | Because the embedding predictors are constructed using the black-box model, they are unable to provide the mechanistic explanations for predicted potential DDIs. | N | https://github.com/rcelebi/GraphEmbedding4DDI/ |
| DDI-BLKG105 | A method for discovering potential DDIs through the generation of the KG from disease-specific literatures. | It uses semantic relations connecting different drugs in the literatures, as features of DDI. | A qualitative analysis of the resulting KG should be conducted, to gain insights on errors generated by the automatic extraction tools. | N | https://github.com/kbogas/DDIBLKG | |
| BERTKG-DDI106 | BERTKG-DDI combines drug embeddings obtained from DDIs and other biomedical entities with an RC architecture based on domain-specific BioBERT embeddings. | This method is in line with the new direction of research of fusing various information to DDI prediction. | It is also essential to explore other external drug representation such as chemical structure, textual description in predicting DDI from textual corpus. | N | – | |
| DL and KG | Conv-LSTM71 | Using KG integrates 12,000 drug features from DrugBank, PharmaGKB, and KEGG drugs, embedding the nodes in the graph using various embedding approaches. | It can integrate multiple sources of drugs and related data for comprehensive information. | There is no interpretation for the predicted DDIs. | N | – |
| MDNN107 | A two-pathway framework including DKG-based pathway and heterogeneous feature (HF)-based pathway, which is designed to obtain the multimodal representations of drugs. | MDNN learns the representations from multimodal data and mines the inter-modality similarities from multiple sources, also exploits the topological structure information and semantic relations with DKG. | The dataset imbalance problem is ignored. | N | – | |
| KGNN55 | KGNN is an end-to-end framework that explores drugs’ topological structures in knowledge graph. | KGNN aggregates all topological neighborhood information received locally to extract both high-order structures and semantic relations. | The introduction of other drug features might be helpful to improve the prediction performance. | N | https://github.com/xzenglab/KGNN | |
| RANEDDI91 | This is a relation-aware network embedding model, which can embed multi-relational graph. | By considering the multi-relational information and relation-aware network structure information together, RANEDDI can learn the more representative entity embeddings. | It cannot predict DDIs effectively when training sets are very sparse or the drug has no neighbors, and performs poorly on some low-frequency DDI types. | N | https://github.com/DongWenMin/RANEDDI | |
| AAE-FOR-KG108 | A new knowledge graph embedding framework is proposed by introducing adversarial autoencoder (AAE) based on Wasserstein distances and Gumbel-Softmax relaxation. | AAE can generate high-quality negative samples, the Gumbel-Softmax relaxation and the Wasserstein distance help train the embedding model steadily. | Compared to small dataset, it is not suitable for large dataset which contains complex relationship types. | N | https://github.com/dyf0631/AAE_FOR_KG | |
| KG2ECapsule85 | This model integrates a probability-based negative sampling strategy to generate high-quality negative samples, and also utilizes a capsule network for non-linear transformation to enrich the representations of entities under specified relational space. | High-quality negative samples are generated, which refrains from the danger of introducing false negative. | Multi-hop is not considered, so the receptive field of each entity cannot be globally determine for accurate learning. This is one of its limitations. | N | https://github.com/Blair1213/KG2ECapsule | |
| MUFFIN52 | A bi-level cross strategy is proposed that includes cross- and scalar-level components to fuse multi-modal features well. | MUFFIN can jointly learn the drug representations based on both the drug-self structure information and the KG with rich bio-medical information. | The model lacks interpretability and the problem of data redundancy is also to be resolved. | N | https://github.com/xzenglab/MUFFIN | |
| DDKG77 | DDKG fully utilizes the information of biomedical KGs, and can learn the initial embeddings of drug nodes from their attributes in the KG, while considering both neighboring node embeddings and triple facts. | Drug attributes are integrated into the representation learning process to improve performance of DDI prediction. | It is difficult for DDKG to obtain the global optimal solution. | N | https://github.com/Blair1213/DDKG | |
| BioDKG-DDI109 | BioDKG-DDI integrates multi-feature with biochemical information, and predicts potential DDIs through an attention machine with superior performance. | It is a robust, yet simple method and can be used as a benefic supplement to the experimental process. | Transportability can be further improved by changing the way of extracting the feature of drug functional similarity. | N | – | |
| DeepLGF110 | DeepLGF is an inductive model that predicts DDIs through aggregating local-global multi-information based on the BKG. | This model is proposed to fully exploit biomedical knowledge graph (BKG) fusing local-global information, which improves the performance of DDIs prediction. | Random selection of negative samples can bring certain noise. Moreover, drug sequences as CS information are too simple and may only provide limited one-dimensional information. | N | https://github.com/MrPhil/DeepLGF | |
| 3WDDI56 | A method based on three-way decision, which combines knowledge graph embedding as supplementary features to enhance DDI prediction. | Delay decision is made for objects in the boundary region by integrating KG embedding feature, and improves the accuracy of decision-making. | Not considering diverse drug features and multi-omics data may introduce some limitations to the model. | N | – | |
| DGAT-DDI57 | A directed graph attention network to predict asymmetric DDIs, which learns embeddings of the source roles, the target roles and the self-roles of drugs. | DGAT-DDI is the first approach for predicting asymmetric interactions among drugs. | It cannot accommodate multitype asymmetric interactions, which is one of its limitations. | Y | https://github.com/F-windyy/DGATDDI | |
| MCFF-MTDDI81 | This model integrates the extra label information into KG-based multi-typed DDI prediction, and innovatively proposes a novel KG feature learning method and a State Encoder. | By using multi-channel feature fusion, biomedical KG-based features, extra label information and drug chemical structures are fused more effectively. | Extremely unbalanced data may lead to bad prediction outcomes. | N | https://github.com/ChendiHan111/MCFF-MTDDI |
Note: ‘Validate’ represents whether the method has been externally validated on independent dataset or in real clinical settings, ‘Y’ represents yes, and ‘N’ represents no, ‘-’ represents no clear explanation in the original literature.
Figure 2.

The computational methods for predicting DDIs are classified into three main groups: deep learning-based models, KG-based models, models which combine deep learning and KG
In particular, DL-based methods are categorized based on their underlying deep learning models.
DL and KG techniques
Deep learning uses large amounts of unsupervised data to automatically extract complex representations.111 It has been demonstrated that that deep learning is effective in discovering complex structures in high-dimensional data and is therefore applicable to many areas of science, business, and government.22 In this section, we review the various deep learning and KG techniques.
DNN
DNN can learn more complex and abstract high-level features than shallow neural networks.112 Given an input sample fixed at the input layer, the other units of the network compute their values based on the activity of the units that they are connected to in the next layer.113 Furthermore, whether it is a linear or nonlinear relationship, DNN has the ability to discover suitable parameters to convert inputs into corresponding outputs. In the DDI prediction field, DNNs are widely used to develop prediction frameworks. Figure 3 provides a specific example of DDI prediction method using DNN. DNN can be processed through two main phases: Forward Propagation (FP) and Backward Propagation (BP).
-
(1)
FP: The input data are propagated from the input layer to the output layer. Each layer utilizes the outputs of the previous layer as inputs, and the predicted outputs for given inputs are obtained by fully connected between layers.
-
(2)
BP: Backward propagation is the process of calculating the gradients of the loss function with respect to the weights and biases of the neural network. It allows the network to learn and adjust its parameters during the training process. BP also involves Weight Gradient Calculation (WG) and Weight Update (WU). WG refers to the computation of the gradients of the loss function with respect to the weights of the neural network and WU focuses on adjusting the weights of the neural network based on the calculated weight gradient.
Figure 3.

The process of using DNN for DDI prediction
The feature vectors of drug a and b are first combined and then fed into the DNN to predict DDI.
GNN
Graphical representation is a useful tool to represent potential relationships among entities in the field of science and engineering, such as computer vision, pattern recognition, data mining.114,115,116,117,118 Graph Neural Networks (GNNs) are deep learning-based methods that operate on graphs domain, due to its convincing performance, GNNs have been widely used in recent years.119 Figure 4 reviews popular GNN models include GCN, GAT. For DDI prediction, drugs are regarded as nodes in the graph, these nodes are connected to form a network, an edge denotes an interaction between drugs. GNN can effectively capture the relationships among nodes and edges in DDI graphs, thereby enhancing the accuracy of DDI prediction through comprehensive training and modeling.
Figure 4.

The widely used GNN frameworks
(A) GNN; (B) GCN; (C) GAT.
GCN
Graph convolutional neural network (GCN) is one of the most commonly used methods in GNN. GCN can effectively leverage the graph structure and aggregate node information from neighboring nodes in a convolutional manner. It has significant expressive power to learn graph representations and achieve superior performance in various tasks and applications.120 Figure 4B) shows a simple example of GCN. After constructing a DDI graph, the node representations are updated by aggregating the features of neighbor nodes, and the commonly used aggregation methods include Sum and Average. Then, the obtained node representations are input into the activation function for nonlinear transformation, which can help the model learn more complex feature representations. The new node representations finally obtained are utilized as the inputs of the next layer, the local and global information of nodes are gradually fused through several iterations.
GAT
Graph Attention Network (GAT) introduces an attention mechanism into the process of computing weights between nodes. By stacking layers in which nodes are able to attend over their neighborhoods’ features, GAT enables (implicitly) specifying different weights to different nodes in a neighborhood, without requiring any kind of computationally intensive matrix operation (such as inversion) or depending on knowing the graph structure upfront.121 Figure 4C) shows a single graph attentional layer, which inputs a set of node features and applies a weight matrix to every node, then uses a shared self-attention for each node to compute attentional coefficients. The softmax function is utilized to normalize the neighbor nodes and to compute linear combination of node features.
AE
AE122 is a nonlinear unsupervised neural network model, which consists of encoder and decoder. The encoder maps the input to the representation space, while the decoder reconstructs the original input from the representation.123 Due to its advantages in data dimensionality reduction and feature extraction,124 autoencoder has been applied to DDI prediction with favorable results. By setting appropriate dimensions for the encoder, the most important features can be learned. Figure 5 demonstrates the reconstruction of the input data using an AE model. There are two phases in the training process of the autoencoder: the encoder phase and the decoder phase.
-
(1)
Encoder phase: The Encoder consists of a series of hidden layers that receive the input data and gradually compress the data. The parameters of encoder are learned by minimizing the reconstruction error, which ensures to capture the main features of the input data.
-
(2)
Decoder phase: In this phase, the representations of hidden layers are mapped back to the original input space through the decoder. The parameters of decoder are also learned by minimizing the reconstruction error to recover the original data as possible.
Figure 5.

Reconstructing SMILES of a drug using AE
AE involves two processes: encoder and decoder. The encoder compresses the input high-dimensional data into low-dimensional data, while the decoder returns the low-dimensional data into high-dimensional data, and the goal is sample reconstruction.
KG
Knowledge graph (KG) is a multi-relational semantic network, which exhibits robust expressive capabilities and modeling flexibility. Figure 6 illustrates an example of a node embedding and its updating process on knowledge graph, which obtains the node’s multi-hop neighborhood features by propagating and aggregating information, and utilizes these features to update the node’s embedding vector.
Figure 6.

Updating embedding of node D1 in KG
After initializing the embedding of D1, the first-order neighborhood information is recursively propagated and aggregated, the same operation to aggregate the second-order neighborhood information, and then to update the embedding of node D1.
However, when using KGs to calculate semantic relations between entities, it is often necessary to design special graph algorithms to achieve this, but such graph algorithms have high computational complexity and poor scalability.125 Therefore, some knowledge graph embedding methods have been proposed. KGE models ingest graph data in triplets form where they learn global graph low-rank latent features, which preserve the graph’s coherent structure, the common KGE models include TransE, DistMult, HolE, etc.126
DL-based methods
DNN-based methods
In the real world, it is generally not possible to obtain all the detailed drug information. In order to ease these limitations, Ryu et al.80 proposed DeepDDI which only used drug names and structure information as inputs. DeepDDI generated a structure similarity profile (SSP) for each drug, obtained the structurel features of the drug, and then merged two SSPs of a drug pair, input them to DNN for prediction. DeepDDI could provide important information about drug prescriptions and even dietary recommendations when taking certain drugs, as well as guidelines during drug development. Although these studies have made significant contributions to DDI prediction, considering more features is necessary for comprehensive study and better performances.
To effectively integrate different features of drugs, Deng et al.79 proposed a multi-modal deep learning framework named DDIMDL. Four drug features including chemical substructure, target, enzyme and pathway, were effectively utilized and each type of these features was fed into a sub-model with a multilayer neural network, and then were combined using a joint DNN framework to learn cross-modal representations of drug pairs for DDI events prediction. Liu et al.68 proposed DANN-DDI that utilized SDNE for learning drug nodes embeddings from five drug-related feature networks, connected the learned drug embeddings and neural network to represent drug pairs, and then used DNN to predict DDI. However, it is difficult for these methods to preserve higher-order structure, and they might tend to obtain local optimal solutions. Moreover, integrating drug features without the attention machine may lead to limited prediction performance. Attention mechanism can be used as a resource allocation scheme, which is the main means to solve the problem of information overload.127 Ren et al.92 used attention mechanism to integrate drug features, and proposed BioChemDDI computational method to construct highly representative integrated feature descriptors, which were input to the DNN for DDI prediction. The experimental results showed the effectiveness of the attention mechanism and also utilized the deep network structure information.
GNN-based methods
To overcome the current limitations of using single drug compound structures or using only DDI data, Bai et al.93 proposed BI-GNN which constructed a bi-level graph based on DDIs, in which the highest level contained the interaction graph of DDIs and the representation graphs of entities. The lower level representation graph neural network generated vector representations for each representation graph. The higher level interaction graph neural network further propagated information from the lower level graph embeddings to neighboring nodes in the interaction graph, then provided the final graph representations to a fully connected network to obtain a final link prediction score. This design of features captured the whole molecular structure of drugs. In fact, DDI usually only depend on a few substructures. Nyamabo et al.82 have proposed a DDI prediction method named SSI-DDI based on the assumption that DDI is actually caused by chemical substructure-substructure interactions. SSI-DDI used the GAT layer with shared weights to extract the raw molecular graph representations of drugs and extract substructure information. The task of DDI prediction was decomposed into substructure-substructure interactions prediction. Li et al.94 proposed DSN-DDI based on GNN, which iteratively learned drug substructures from the single drug(intra-view) and the drug pair(inter-view). Each GNN had its own unique receptive field, each one aggregated information from neighboring nodes, which resulted in updating the nodes and extracting the substructures.
Chen et al.95 utilized GCN to convert the molecular data with irregular structure into the corresponding molecular data in low dimensional vector spaces. Then, the learned embedding vectors were used to predict whether there has DDI between two drugs or DDI type. Wang et al.62 proposed a multi-kernel graph convolutional network (GCNMK). GCNMK used two graph cores as the graph convolution layers, in which an increased DDI graph and a decreased DDI graph were constructed from the ‘increase’-related and ‘decrease’-related DDIs. The two graphs and the drug feature matrices were fed into the first GCN layer composed of two GCN blocks, then the low-dimensional representation vectors of drugs were generated using the second layer composed of one GCN block. Finally, the two drug feature vectors were concatenated to form a DDI vector, which were then inputted into a block with three fully connected layers to predict potential DDIs. Feeney et al.86 proposed RS-GCN to model the importance of neighborhood sampling relationship types in the network, in which each relationship type was assigned a learning probability and updated through a reinforcing-based method. The results showed that this model can learn the right balance: relation-type probabilities that reflect both frequency and importance, and also offered some kind of explanation. However, these graph-based studies only focus on a single view of the drug, the combination of multi-view information inside the DDI network is often ignored.
GCN is also a promising way to handle multiple views, Wang et al.96 proposed MIRACLE to simultaneously capture the relationships between molecular structures across views and the molecular-molecular interactions within views. MIRACLE regarded the DDI network as a multi-view graph in which each node represented an instance of a drug molecule graph. In the learning stage, GCN was utilized to encode DDIs, while the key aware attention information propagation method was used to capture the drug molecular structure information. Although the models mentioned above have taken into account the structure, sequence or interaction information of the drugs, while they neglect the synergistic effects between them. He et al.97 proposed MFFGNN to efficiently integrate topological information in molecular graphs, DDIs information and local chemical contexts in SMILES sequences. In MFFGNN, the bi-directional gate recurrent unit extracted local chemical context information from SMILES sequences, while the graph interactions network with graph wrap unit extracted the topological structure features of the drugs from the given molecular graphs. Finally, GCN fused the intra-drug features and external DDI features to update the drug representations.
AE-based methods
Purkayastha et al.98 proposed an effective method to fuse multiple drug features to predict DDIs. The proposed model leveraged the drug-target interaction (DTI) network to learn drug embeddings. In order to obtain drug representations from rich chemical structures, a variational autoencoder was also constructed. Finally, the obtained different combinations of feature embeddings of the drugs were incorporated and input into a Graph Auto Encoder to predict missing DDI links in the network. Lee et al.87 proposed a DDI prediction method based on constructed Target Similarity Profile (TSP), Gene Ontology (GO), Term Similarity Profile (GSP) as well as the SSP. An autoencoder was trained to minimize the difference between inputs and outputs, while training it to minimize the prediction error of DDI labels. However, the relationships between these biomedical events are usually non-linear across all types of drug features. Additionally, some datasets may lack labels or contain noise, which can potentially have adverse effects on prediction models.
In order to predict DDIs from large-scale, noisy and sparse data, Zhang et al.61 proposed a multi-modal deep AE-based method named DDI-MDAE to learn drug representations. This computational method first utilized an AE to learn uniform drug representations from multiple drug feature networks simultaneously. Next, four operators were applied to the learned drug embeddings to represent drug-drug pairs. Finally, a random forest model was trained using the representations to predict DDIs.
Multi-neural network ensemble-based methods
The above proposed prediction methods have achieved excellent performance when using one type of neural network alone. To utilize the advantages of various deep learning models, researchers have begun to combine different neural networks to predict potential DDIs, thus developing many high-performance and high-robust computing methods. Figure 7 shows an example of using both GCN and DNN, the GCN extracts the features from the DDI network and inputs them into the DNN network for prediction.
Figure 7.

An example of a model that utilizes both GCN and DNN
Yan et al.99 proposed NMDADNN, which constructed similarity networks by integrating five heterogeneous sources of drug-related information, after unifying these similarities with AE, a DNN was constructed to predict DDI. Feng et al.58 proposed DPDDI to obtain network structure features of drugs. In DPDDI, GCN was utilized to capture the topological relationships among drugs in the DDI network and extract their network structural features. DNN acted as a predictor to associate the potential feature vectors with the feature vectors of the corresponding drug pairs to predict DDIs. Lin et al.100 proposed a novel DDI prediction framework(-DDI). After encoding drugs through DeeperGCN,128 the feature refinement module was constructed to obtain mutually aware drugs. Furthermore, the relation embeddings could be updated by incorporating the drug information. In this way, the DDI prediction can be improved with more informative and distinguishable features. Finally, these refined features for drugs and relation were utilized to train and predict the possibility of DDI. However, the feature fusion methods utilized in these models are simple, and more effective fusion methods can be designed to improve the performance.
Latent feature fusion via Siamese network is a useful method, Lin et al.83 proposed MDF-SA-DDI computational method, which based on multi-source drug fusion and multi-source feature fusion with self-attentive mechanism in order to predict interaction events between two drugs. After combining two drugs in four different ways, the combined drug feature representations were fed into four different drug fusion networks (Siamese network, convolutional neural network, and two autoencoders) to obtain the potential feature vectors of the drug pairs, then fused with potential features using a transformation block with self-attentive mechanism. The multi-source drug fusion can provide diverse information from different views to deep learning models, and accurately predict DDI events. In addition to this method of encoding drug features, there have also been some models that directly encode SMILES of drugs. Pang et al.54 proposed attention-based multidimensional feature encoder method (AMDE) that effectively utilized this information to enhance the accuracy of DDI prediction. AMDE utilized the MPAN model as the graphic encoder to process the atomic graphs of drugs and generate 2D atomic graph feature vectors. Meanwhile, a sequence encoder was used to process the sequence data generated by the SMILES to generate 1D sequence feature vectors. Finally, all feature vectors were fed into multidimensional decoder to predict whether there is an interaction between two drugs.
Although these methods have achieved inspiring results, they neglected the pharmacological changes that DDI could induce enhancement and inhibition, as well as the different pharmacological roles of two drugs in an interaction. Therefore, Feng et al.84 proposed a new graph representation learning model SGRL-DDI. This model leveraged Balance theory and Status theory from social networks to characterize pharmacological patterns of DDI, and organized DDI entries into a signed and directed network that reflects the relational semantics between drugs. Two-layer embedding and an extra enhancer based on social theory were utilized to represent drugs in DDIs network, which each embedding layer was constructed by a multi-relation GNN and a two-layer MLP. Finally, the concatenation vectors of two drug latent features are fed into two dense DNNs to achieve two tasks of predicting enhancive/depressive DDIs and predicting the directed DDIs.
Deep learning models with interpretability
Although DL models have shown good performance for DDIs prediction, they usually need a large number of parameters, which are difficult to interpret.129 DL prediction models with good interpretability can not only help researchers understand the trigger mechanism of DDIs, but also ensure safer medication, so it is especially crucial to improve the interpretability of deep learning models. In recent years, more and more models have begun to focus on this issue. In this section, we review models that consider the problem of non-interpretability of deep learning.
Huang et al.69 proposed a computational method named CASTER which utilized a dictionary learning module to help researchers to understand how this model makes prediction and identify which sub-structures can possibly lead to the interaction. After generating frequent sub-structures for the input drug SMILES sequences, CASTER generated a functional representation for each frequent sub-structure, then exploited the encoder to generate a matrix of latent feature vectors, and projected the resulting potential vector into the subspace defined by the matrix. The basis vector in the matrix was associated with a frequent molecular sub-graph, and its corresponding projection coefficient revealed the statistical importance of having the sub-graph in the molecular graphs of the drug pairs so that they would interact, thus explaining the rationality behind CASTER’s predictions. Yu et al.101 proposed a novel substructure-aware tensor neural network model STNN-DDI, which mapped drugs into an SSI space based on a list of predefined substructures with specific chemical meanings, thereby improving the interpretability of the model. STNN-DDI characterized the SSI space by using the learned substructure × substructure × interaction tensor (ST), which was a 3-D tensor and expanded by a series of rank-one tensors. It represented the occurring probability of drug-drug pair as the sum of the occurring probability of the substructures included in this drug pair, and the types of SSI were defined as the same as the corresponding types of DDI. Potential DDIs were obtained by learning the probability of each SSI under a list of predefined chemical substructures. This form improved interpretability of the model, because both known drugs and new drugs are mapped into a common SSI space no matter whether a drug has an interaction or not. Kim et al.102 proposed DeSIDE-DDI, which can provide the interpretable expression level for DDI prediction with drug-induced gene expression signatures. Specifically speaking, the model engineered dynamic drug features using a gating mechanism to mimic drug co-administration effects by imposing attention to important genes. The concept of translating embeddings was also introduced, which considered a side effect as a relationship between two drugs and applied the margin-based loss function, implying positive pairs of drug combinations are positioned closely on the given side effect space. Via using drug-induced gene expression signatures followed by gating and translating embedding can provide model interpretability and increase the accuracy of the model.
Tang et al.103 proposed DSIL-DDI, a pluggable substructure interaction module, which not only considered the fine-grained properties of substructures, but also introduced the attention mechanism to recognize which substructure interaction representations contributed more to the DDI representation. The substructure representations of the input drugs extracted by GNN were regarded as the property spectrum of the substructure, which represented the extent to which chemical substructure responds to different properties. In the process of using Hadamard product to model the fine-grained attribute interactions, attention weights were introduced to help observe which substructure interaction representations contributes more to the DDI representation. Furthermore, experiments were also performed to demonstrate the interpretability of DSIL-DDI. Yu et al.104 proposed GGI-DDI, which utilized granular computing to extract key subgraphs between drug pairs and enhance the interpretability of DDI prediction models, the key chemical substructures were defined as coarser granules. In detail, after aggregating the node information in the atomic graphs of drugs using GINE, the attention mechanism and bond information were fused to obtain the embedding of nodes and subgraphs for determining the importance of nodes. Then, a local and global score function were introduced to evaluate the local significance and long-range dependencies the embedding of each subgraph, respectively, to help identify the key subgraphs related to drugs and were reconstructed to create new graphs. Coarser particles (key chemical substructures) were identified via multiple granulations on graphs of drug pairs, and a cross-attention mechanism was employed to compute the attention score for each coarser granule, thereby obtaining their final representation. This interpretability provided by GGI-DDI can guide the advancement of novel drug development and poly-drug therapy strategies.
KG-based methods
In recent years, biological data and knowledge bases have been increasingly built on Semantic Web technologies.130 Many bioinformatics databases have started to use Semantic Web-related technologies and use these data as Linked Open Data (LOD).131 By using LOD data, Celebi et al.11 proposed a model to evaluate DDI in the absence of a known DDI. Different knowledge graph embedding methods were used to extract drug feature vectors from KG, such as RDF2Vec, TransE, and TransD. Finally, some common classifiers were utilized to predict DDIs. Bougiatiotis et al.105 proposed DDI-BLKG, predicted DDIs by generating disease-specific KG from biomedical publications and manually curated databases. The human-curated drug database was utilized to train a classifier that identifies patterns of interactions between drug pairs. The predictive potential and usefulness of the method were demonstrated through a small-scale qualitative evaluation. Based on representation learning, Mondal et al.106 proposed a DDI prediction method named BERTKG-DDI, which has used a knowledge graph that contains target-target, drug-drug, drug-disease, disease-target, disease-target interactions, and enhanced entity representations to train the relational classification model.
Combination of DL with KG
Recent studies have demonstrated the effectiveness of deep learning models in extracting node features from KG and achieved remarkable results. When using deep learning for DDI prediction, it is common to make predictions based on the features of drug nodes. On the other hand, KG focuses on prediction based on features of drug nodes, neighbor nodes and relations. As a result, mixing these approaches can compliment each other, and offer richer information. Additionally, the labels or feature information provided by KG can also enhance the performance of deep learning models. Thus, the integration of DL and KG offers opportunities for improvement in DDI prediction tasks.
Karim et al.71 have proposed a novel DDI prediction method, Conv-LSTM, which based on a KG including drug-related information information from KEGG, DrugBank, TWOSIDES and the literatures. The method utilized RDF2Vec, SimpleIE, TransE, KGloVe, CrossE and PBG for the KG embeddings. Then input these embeddings into CNN-LSTM layer, where the CNN extracted local relationship values in the drug features, and the LSTM captured the overall relationship from the features extracted by the CNN. Nevertheless, Conv-LSTM has limitations on obtaining the rich features of drugs from structural information and semantic relations. Furthermore, it has not considered the drug multimodal data coherence and complementarity together. To effectively assist the joint representation learning of multi-modal data related to DDI events, Lyu et al.107 proposed a multi-modal deep neural network (MDNN) for DDI events prediction. A dual-path framework containing a drug knowledge graph (DKG)-based pathway and a heterogeneous feature (HF)-based pathway was proposed to extract structural information and semantic relations from the DKG to learn drug representations. Moreover, a multi-modal fusion neural layer was utilized to explore the complementarity among multi-modal representations of drugs.
But one neglected deficiency is that model DDI as an independent data sample and do not consider their multiple related correlations in knowledge graph. Moreover, directly learning the latent embedding of nodes in KG could also bring some limitations. Therefore, Lin et al.55 proposed KGNN to obtain rich neighborhood information for each entity in KG. The central idea of KGNN was to consider both high-order structures and semantic relations, by using GNN to encode the drugs and topological neighborhood information to distributed representations, which facilitated the prediction of DDI events. Nevertheless, Yu et al.91 found that both the multi-relational information and network structure information can affect the learning of entity embeddings, thereby proposed a relation-aware network embedding model (RANEDDI) to predict potential DDIs. After embedding entities and relations into the vector space using RotatE, a relation-aware information propagation mechanism was utilized to extract the relation-aware network structure information of the entities by propagating their neighbor’s information under different relations. Finally, DNN acted as the predictor to predict the probability of the certain interaction between drug pairs. Experiments have also demonstrated its robust performance even in the case of a scarcity of labeled DDIs.
Several deep learning approaches have been proposed to embed Drug Knowledge Graphs (DKGs) for predicting unknown DDIs. Training a KG embedding models requires negative samples, but most embedding models have been generating negative triplets via a uniform negative sampling strategy, and the obtained samples are too simple to train the model effectively. Thus Dai et al.108 proposed AAE-FOR-KG, a knowledge graph embedding framework. AAE-FOR-KG utilized adversarial autoencoder to generate high-quality negative samples. Based on positive and negative triples, discriminator could learn drugs and interaction embeddings effectively. Compared to other traditional knowledge graph embedding methods, it has better performance. Su et al.85 also proposed KG2ECapsule, which generated high-quality negative samples based on a probability negative sampling strategy. KG2ECapsule constructed a Graph-to-Embedding Layer, which could recursively propagate embeddings from the neighbors of the entity as well as the relations between them. In addition, a two-layer capsule network has been innovatively integrated to obtain entity representations in a non-linear form under specified relational space. Finally, these entity representations were utilized to predict the DDI. Experiments also demonstrated the effectiveness of probability-based sampling strategy and non-linear transformations.
It has been proved that the performance of DDI prediction can be improved by considering KG with rich bio-medical information and drug molecular structure information or SMILES sequence simultaneously. Chen et al.52 proposed a multi-scale feature fusion deep learning model named MUFFIN. MUFFIN utilized MPNN to extract molecular structure features from SMILES, and TransE to extract semantic features from KG. Then, crossed these features to learn local and global features using CNN and flattening operations. Meanwhile, the fine-grained interaction features between two different features were obtained using the element product method, the obtained features were stitched together for prediction. Su et al.77 proposed DDKG, which learned the initial embeddings of drugs from the corresponding attributes of the nodes in KG using an encoder-decoder layer. To learn accurate global representations of drug nodes, the model recursively propagated and aggregated the first-order neighborhood information along the top-ranked network paths which determined by the embeddings of neighbor nodes and triples. Finally, estimated the interaction probability for pairwise drugs with their respective representations.
Considering some unavailable information can lead to inaccuracy of drug features extraction, Ren et al.109 proposed BioDKG-DDI, which used Mol2Context-vec to extract molecular features, combined ComplEx-DURA with DKG to obtain drug global features, and incorporated drug functional similarity features as supplementary information. Such approach enabled the combination of drug molecular features, drug global association information and drug functional similarity features, and then the features were integrated and input to DNN for prediction. Moreover, Ren et al.110 proposed DeepLGF, which utilized the BFGNN model to construct a heterogeneous network of drugs to obtain the bio-local information, learned the global feature information of BKG by the KGE method of ComplEx, finally obtained a stable and effective model to predict potential DDI after integration. Nevertheless, this model relies on obtaining detailed and complete information about the drug at once. In fact, only the SMILES sequences of the drugs are easy to obtain, while others information needed further experiments to discover. The uncertainty and incompleteness brought about by drug information might hinder the accuracy of DDI prediction. Three-way decision-making can solve this problem. Thus Hao et al.56 proposed 3WDDI based on the computational method of three-way decision making.132 After embedding KG using ComplEx, CNN was utilized as the decision function to classify the drug pairs into positive region, negative region and boundary region based on the chemical structure features of the drugs using SCNN. By combining the knowledge graph embedding features, the robustness of the decision could be improved by delay decision for objects in the boundary region.
Many biological experiments have proved pharmacological asymmetry between DDIs, but most of the above models did not consider this situation. Feng et al.57 designed DGAT-DDI model for asymmetric DDIs prediction by learning the embeddings of source-role, target-role, and self-role of drugs. The method viewed the knowledge graph as directed graph and used GAT to aggregate information of entities and neighboring nodes to generate source and target embeddings, while using MLP to obtain self-role embeddings. After aligning these embeddings, asymmetric DDIs were predicted. However, extra label information has not yet been applied to the task of multi-label DDI prediction. To overcome the problem, Han et al.81 proposed a multi-channel feature fusion model MCFF-MTDDI, which utilized drug chemical structure features, drug pairs’ extra label features, and KG features of drugs. For the target drug pair, after extracting the structural feature vector, the three types of KG representations and the extra label vector, MCFF-MTDDI utilized a state encoder to fuse these three KG representations vectors and obtain two KG fusion representations. In addition, a multi-channel feature fusion framework based on GRU was established, which comprehensively integrated the information of two KG feature channels, structural feature channels, and additional label feature channels. Finally, multi-class prediction and multi-label DDIs prediction were performed respectively.
Experiments and comparison
Evaluation metrics
In general, the performance of model is usually evaluated through experiments, therefore, the learning ability of the model needs to be tested by using a test set. Cross-validation (CV) is commonly utilized to train models, and the basic idea is to split the original data into the train set and the test set. The train set for training the model and the test set for evaluating the performance of the model. The most popular method is the K-CV, which the original data is divided into K subsets, one of them is used as the test set while K-1 subsets as the train set. The process is then repeated K times, with each subset being used as the test set once. The final performance score of the model is obtained by taking the average of the evaluation scores obtained during these iterations. Normally, the values of K are set to 5 and 10.
Evaluating the performance of a model requires not only feasible experimental methods of evaluation, but also evaluation metrics to measure the generalization ability of the model. In the classification task, the samples can be grouped into true positive (TP), false positive (FP), true negative (TN) and false negative (FN) based on the true and prediction labels, which are referred to as ‘confusion matrix’, as shown in Table 5. Accuracy, precision, recall and F1 are commonly used evaluation metrics in classification problems. Accuracy means the number of correctly classified samples as a proportion of the total number of samples, and precision means the proportion of samples classified as TP among all positive samples. The calculation formulas are as follows.
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
Table 5.
Confusion matrix
| Real | Predict |
|
|---|---|---|
| Positive | Negative | |
| Positive | TP | FN |
| Negative | FP | TN |
Receiver operating characteristic (ROC), by setting out several different continuous values of the continuous variables, thereby calculating a series of true positive rate and false positive case rate, and plotted as curves for the vertical and horizontal axes respectively, the larger the area under the curve, the higher the accuracy rate is indicated, and the area under the curve is also called AUC. Precision-recall curve (PR), with Recall on the horizontal axis and Precision on the vertical axis, shows model performance at different classification thresholds, AUPR is the area under PR curve. The values of AUC and AUPR range from 0 to 1, and larger values indicate better model performance.
Experiment results
We compare the models in binary classification prediction, multi-class prediction, and multi-label prediction tasks, respectively. The experiment results are shown in Figures 8, 9, and 10.
Figure 8.

Performance evaluation under binary classification task
Figure 9.

Performance evaluation under multi-class task
Figure 10.

Performance evaluation under multi-label task
Figure 8 shows the comparison results of different models under binary classification task on six benchmark datasets. The performance of DDIs prediction achieved by these models were all measured in terms of ACC, AUC, AUPR and F1. For Dm_l4, we can observe that GNN-based method SSI-DDI achieves the highest AUC of 96.14. MFFGNN (AUPR = 96.81, F1 = 92.54), which also belongs to GNN-based model, achieves the highest AUPR and F1, respectively. This may be attributed to the fact that the features from drug sequences and molecular graphs are integrated and comprehensively learned, while some DNN-based models (e.g., DeepDDI and DDIMDL) only consider the structural information and features of drugs. Interestingly, for Db_3, AMDE, which based on the integration of multi-neural networks shows similar AUC and ACC as KGNN that belongs to the models of combine DL and KG. KGNN and 3WDDI, which belong to the combined DL and KG models perform well and obtain the best AUPR and AUC on three different datasets. KGNN obtains AUC of 99.12 and AUPR of 98.92 on Db_4, also obtains AUC of 99.1 and AUPR of 98.9 on Db_6. For Db_5, the AUC and AUPR result of 3WDDI is 95.82 and 96.14, respectively.
For multi-class prediction task, we can observe from Figure 9 that MUFFIN (F1 = 94.95, ACC = 96.48, Precision = 95.68, Recall = 94.82) and MCFF-MTDDI (AUPR = 97.57, AUC = 99.81, ACC = 97.74), the models combining DL and KG, achieve the best performance on Dm_c2 and Dm_c3, respectively. The achievement of high performance means that the combination of KG-based features and drug features or fusion with additional information is feasible. For Dm_c1, the AUPR and AUC of MDF-SA-DDI that based on multi-neural network ensemble achieve the best performance, 97.37 and 99.89, respectively. The success of MDF-SA-DDI may be attributed to the utilization of a novel approach to fuse the information of drug pairs, i.e., four different drug fusion networks are utilized to obtain latent feature vectors of the drug pairs. Furthermore, we discover that the DNN-based model DeepDDI has the same AUC as the AE-based model Lee et al.87 on Dm_c1, but the latter has the better performance in general.
Actually, multi-label prediction is more difficult than binary and multi-class prediction. From Figure 10, we can observe that MUFFIN, KG2ECapsule and MCFF-MTDDI, which belong to the combination of DL and KG methods achieve the best performance on three datasets, respectively. For Dm_l2, the AUC and AUPR of KG2ECapsule respectively increased by 23.03% and 31.16% compared to KGNN, which is a surprising improvement. The success of KG2ECapsule may be attributed to modeling the triplets and integrating the relations of edges into embedding. On Dm_l3, the AUC of MCFF-MTDDI is 93.15, and AUPR is 74.36, which demonstrate the importance of using extra label information of drug pairs. It is worth mentioning that DL and KG methods are more often used in multi-label prediction task with better results.
Challenges and opportunities
Although excellent results have been achieved using deep learning and knowledge graph for DDI prediction, there are still some issues that need to be resolved, which are summarized as follows.
-
(1)
Asymmetric drug-drug interactions prediction. In the field of DDI prediction, relevant computational methods are well developed, and it is difficult to further improve. In most computational methods related to DDIs prediction, such as the model using DKG as input, the relationships in DKG are undirected, and the drugs in the triples are symmetric, but that doesn’t fit the real world. When Wicha et al.133 evaluated their model using a dataset of 200 combination experiments in Saccharomyces cerevisiae, they found that 67% of the interactions were monodirectional. In addition, asymmetric DDIs prediction can also help patients determine the order of medication, which greatly affects the effectiveness of medication. Bible et al.134 demonstrated that for paclitaxel, cytarabine, topotecan, doxorubicin and etoposide, synergy was more pronounced when the agents were administered before flavopiridol rather than concomitant with or following flavopiridol. Therefore, in future researches, we can turn our attention to asymmetric DDIs prediction.
-
(2)
High-order drug-drug interactions prediction. The vast majority of current methods have been developed to predict DDI, and few methods predict interactions among multiple drugs. Ning et al.135 proposed a purely data-driven fashion for representing, discovering, quantifying, and visualizing high-order DDIs. Peng et al.136 proposed a model for predicting high-order DDI based on deep learning techniques. There is still much room for development in predicting high-order DDIs, which should receive more attention in future research.
-
(3)
Datasets. While utilizing a large amount of complex data can provide the model with rich drug features, it can also introduce a significant level of noise. In addition, many datasets only contain known drug pairs with interactions, lacking validated drug pairs without known interactions. These issues create excessive deviation between experimental results and actual values, which significantly reduces the accuracy of the experiment. Therefore, constructing high-quality and available negative samples can further improve the accuracy of the DDI prediction methods.
Discussion
It’s important to discuss the clinical relevance and practical applications of DDI prediction methods. How to translate these predictions into actionable insights for healthcare providers and patients is a very important task. Therefore, we can do the following.
-
(1)
The computational model is applied to various drug-drug pairs to predict whether and what kind of interactions exist between these drug-drug pairs, to generate sentences describing relevant interactions, which suggest specific pharmacological effects (e.g., “the decreased therapeutic efficacy” and “the increased anticoagulant activities”) in addition to “the increased risk or severity of bleeding”.
-
(2)
To validate the accuracy of the model’s predictions, outputs need to be compared with the consistent descriptions on the DDIs present in the Drugs.com (https://www.drugs.com/), which provides additional information regarding DDIs.
-
(3)
Except the drug pairs examined above by comparing with the information presented in the Drugs.com, there may be drug pairs for which ADEs possible causal mechanisms are not available elsewhere. Therefore, the output sentences describing additional DDI types for the drug pairs with the reported ADEs can serve as the likely causal mechanisms of DDIs for further validation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China [grant number 61972134, 61802113, 61802114]; and the Science and Technology Development Plan Project of Henan Province [grant number 222102210238, 212102210091].
Declaration of interests
No competing interest is declared.
References
- 1.Lu D.-Y., Lu T.R., Yarla N.S., Wu H.Y., Xu B., Ding J., Zhu H. Drug combination in clinical cancer treatments. Rev. Recent Clin. Trials. 2017;12:202–211. doi: 10.2174/1574887112666170803145955. [DOI] [PubMed] [Google Scholar]
- 2.Fisusi F.A., Akala E.O. Drug combinations in breast cancer therapy. Pharm. Nanotechnol. 2019;7:3–23. doi: 10.2174/2211738507666190122111224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asada M., Miwa M., Sasaki Y. Enhancing drug-drug interaction extraction from texts by molecular structure information. arXiv. 2018 doi: 10.48550/arXiv.1805.05593. Preprint at. [DOI] [Google Scholar]
- 4.Yu H., Mao K.T., Shi J.Y., Huang H., Chen Z., Dong K., Yiu S.M. Predicting and understanding comprehensive drug-drug interactions via semi-nonnegative matrix factorization. BMC Syst. Biol. 2018;12:101–110. doi: 10.1186/s12918-018-0532-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker M.L., Kallewaard M., Caspers P.W.J., Visser L.E., Leufkens H.G.M., Stricker B.H.C. Hospitalisations and emergency department visits due to drug–drug interactions: a literature review. Pharmacoepidemiol. Drug Saf. 2007;16:641–651. doi: 10.1002/pds.1351. [DOI] [PubMed] [Google Scholar]
- 6.Yan Z., Zhao L., Wei X., Zhang Q. In: Heled J., Yuan A., editors. Vol. 173. EDP Sciences; 2018. Improved label propagation model to predict drug-drug interactions. (MATEC Web of Conferences). [Google Scholar]
- 7.Karbownik A., Szałek E., Sobańska K., Grabowski T., Wolc A., Grześkowiak E. Pharmacokinetic drug-drug interaction between erlotinib and paracetamol: a potential risk for clinical practice. Eur. J. Pharm. Sci. 2017;102:55–62. doi: 10.1016/j.ejps.2017.02.028. [DOI] [PubMed] [Google Scholar]
- 8.Han K., Cao P., Wang Y., Xie F., Ma J., Yu M., Wang J., Xu Y., Zhang Y., Wan J. A review of approaches for predicting drug–drug interactions based on machine learning. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.814858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu Q. US Department of Health and Human Services, Centers for Disease Control and Prevention; 2010. Prescription Drug Use Continues to Increase: US Prescription Drug Data for 2007-2008. [Google Scholar]
- 10.Giacomini K.M., Krauss R.M., Roden D.M., Eichelbaum M., Hayden M.R., Nakamura Y. When good drugs go bad. Nature. 2007;446:975–977. doi: 10.1038/446975a. [DOI] [PubMed] [Google Scholar]
- 11.Celebi R., Uyar H., Yasar E., Gumus O., Dikenelli O., Dumontier M. Evaluation of knowledge graph embedding approaches for drug-drug interaction prediction in realistic settings. BMC Bioinf. 2019;20:726. doi: 10.1186/s12859-019-3284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safdari R., Ferdousi R., Aziziheris K., Niakan-Kalhori S.R., Omidi Y. Computerized techniques pave the way for drug-drug interaction prediction and interpretation. Bioimpacts. 2016;6:71–78. doi: 10.15171/bi.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jordan M.I., Mitchell T.M. Machine learning: Trends, perspectives, and prospects. Science. 2015;349:255–260. doi: 10.1126/science.aaa8415. [DOI] [PubMed] [Google Scholar]
- 14.Goodfellow I., Bengio Y., Courville A. MIT press; 2016. Deep Learning. [Google Scholar]
- 15.Mahesh B. Machine learning algorithms-a review. Int. J. Sci. Res. 2020;9:381–386. [Google Scholar]
- 16.Shinde P.P., Shah S. 2018 Fourth international conference on computing communication control and automation (ICCUBEA) IEEE; 2018. A Review of Machine Learning and Deep Learning Applications. [Google Scholar]
- 17.Cheng F., Zhao Z. Machine learning-based prediction of drug–drug interactions by integrating drug phenotypic, therapeutic, chemical, and genomic properties. J. Am. Med. Inform. Assoc. 2014;21:e278–e286. doi: 10.1136/amiajnl-2013-002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P., Huang C., Fu Y., Wang J., Wu Z., Ru J., Zheng C., Guo Z., Chen X., Zhou W., et al. Large-scale exploration and analysis of drug combinations. Bioinformatics. 2015;31:2007–2016. doi: 10.1093/bioinformatics/btv080. [DOI] [PubMed] [Google Scholar]
- 19.Mei S., Zhang K. A machine learning framework for predicting drug–drug interactions. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-97193-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song D., Chen Y., Min Q., Sun Q., Ye K., Zhou C., Yuan S., Sun Z., Liao J. Similarity-based machine learning support vector machine predictor of drug-drug interactions with improved accuracies. J. Clin. Pharm. Ther. 2019;44:268–275. doi: 10.1111/jcpt.12786. [DOI] [PubMed] [Google Scholar]
- 21.Ye F. Particle swarm optimization-based automatic parameter selection for deep neural networks and its applications in large-scale and high-dimensional data. PLoS One. 2017;12:e0188746. doi: 10.1371/journal.pone.0188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 23.Wu M., Chen L. 2015 Chinese automation congress (CAC) IEEE; 2015. Image recognition based on deep learning. [Google Scholar]
- 24.Esteva A., Chou K., Yeung S., Naik N., Madani A., Mottaghi A., Liu Y., Topol E., Dean J., Socher R. Deep learning-enabled medical computer vision. NPJ Digit. Med. 2021;4:5. doi: 10.1038/s41746-020-00376-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez M.M., Kalita J. Deep Learning applied to NLP. arXiv. 2017 doi: 10.48550/arXiv.1703.0309. Preprint at. [DOI] [Google Scholar]
- 26.Abdel-Hamid O., Mohamed A.r., Jiang H., Deng L., Penn G., Yu D. Convolutional neural networks for speech recognition. IEEE/ACM Trans. Audio Speech Lang. Process. 2014;22:1533–1545. [Google Scholar]
- 27.Kato Y., Hamada S., Goto H. 2016 international conference on advanced informatics: concepts, theory and application (ICAICTA) IEEE; 2016. Molecular activity prediction using deep learning software library. [Google Scholar]
- 28.Feinberg E.N., Sur D., Wu Z., Husic B.E., Mai H., Li Y., Sun S., Yang J., Ramsundar B., Pande V.S. PotentialNet for molecular property prediction. ACS Cent. Sci. 2018;4:1520–1530. doi: 10.1021/acscentsci.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng X., Zhu S., Lu W., Liu Z., Huang J., Zhou Y., Fang J., Huang Y., Guo H., Li L., et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020;11:1775–1797. doi: 10.1039/c9sc04336e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torng W., Altman R.B. Graph convolutional neural networks for predicting drug-target interactions. J. Chem. Inf. Model. 2019;59:4131–4149. doi: 10.1021/acs.jcim.9b00628. [DOI] [PubMed] [Google Scholar]
- 31.Liu Z., Han X. Deep learning in knowledge graph. Deep Learning in Natural Language Processing, Deng L. and Liu Y.(eds), Springer, Singapore. 2018. pp. 117–145. [Google Scholar]
- 32.Zou X. Journal of Physics: Conference Series. IOP Publishing; 2020. A survey on application of knowledge graph. [Google Scholar]
- 33.Park N., Kan A., Dong X.L., Zhao T., Faloutsos C. Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining. 2019. Estimating node importance in knowledge graphs using graph neural networks. [Google Scholar]
- 34.Qiu Y., Zhang Y., Deng Y., Liu S., Zhang W. A comprehensive review of computational methods for drug-drug interaction detection. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022;19:1968–1985. doi: 10.1109/TCBB.2021.3081268. [DOI] [PubMed] [Google Scholar]
- 35.Zhang T., Leng J., Liu Y. Deep learning for drug–drug interaction extraction from the literature: a review. Brief. Bioinform. 2020;21:1609–1627. doi: 10.1093/bib/bbz087. [DOI] [PubMed] [Google Scholar]
- 36.Zeng X., Tu X., Liu Y., Fu X., Su Y. Toward better drug discovery with knowledge graph. Curr. Opin. Struct. Biol. 2022;72:114–126. doi: 10.1016/j.sbi.2021.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Lin X., Dai L., Zhou Y., Yu Z.G., Zhang W., Shi J.Y., Cao D.S., Zeng L., Chen H., Song B., et al. Comprehensive evaluation of deep and graph learning on drug–drug interactions prediction. Brief. Bioinform. 2023;24:bbad235. doi: 10.1093/bib/bbad235. [DOI] [PubMed] [Google Scholar]
- 38.Vo T.H., Nguyen N.T.K., Kha Q.H., Le N.Q.K. On the road to explainable AI in drug-drug interactions prediction: A systematic review. Comput. Struct. Biotechnol. J. 2022;20:2112–2123. doi: 10.1016/j.csbj.2022.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J., Zheng S., Chen B., Butte A.J., Swamidass S.J., Lu Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2016;17:2–12. doi: 10.1093/bib/bbv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ioannidis, V.N., Song, X., Manchanda, S., Li, M., Pan, X., Zheng, D., Ning, X., Zeng, X., and Karypis, G. Drkg-drug repurposing knowledge graph for covid-19. https://github.com/gnn4dr/DRKG.2020. Accessed 01 May 2023.
- 42.Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Callahan A., Cruz-Toledo J., Ansell P., Dumontier M. The Semantic Web: Semantics and Big Data: 10th International Conference, ESWC 2013, Montpellier, France, May 26-30, 2013. Proceedings 10. Springer; 2013. Bio2RDF release 2: improved coverage, interoperability and provenance of life science linked data. [Google Scholar]
- 44.Tatonetti N.P., Ye P.P., Daneshjou R., Altman R.B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012;4:125ra31. doi: 10.1126/scitranslmed.3003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhn M., Letunic I., Jensen L.J., Bork P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016;44:D1075–D1079. doi: 10.1093/nar/gkv1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avram S., Bologa C.G., Holmes J., Bocci G., Wilson T.B., Nguyen D.T., Curpan R., Halip L., Bora A., Yang J.J., et al. DrugCentral 2021 supports drug discovery and repositioning. Nucleic Acids Res. 2021;49:D1160–D1169. doi: 10.1093/nar/gkaa997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wishart D.S., Knox C., Guo A.C., Shrivastava S., Hassanali M., Stothard P., Chang Z., Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wishart D.S., Knox C., Guo A.C., Cheng D., Shrivastava S., Tzur D., Gautam B., Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanehisa M. ‘In silico’simulation of biological processes: Novartis Foundation Symposium 247. Wiley Online Library; 2002. The KEGG database. [PubMed] [Google Scholar]
- 51.Ursu O., Holmes J., Knockel J., Bologa C.G., Yang J.J., Mathias S.L., Nelson S.J., Oprea T.I. DrugCentral: Online Drug Compendium. Nucleic acids research. 2016;45:gkw993. doi: 10.1093/nar/gkw993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y., Ma T., Yang X., Wang J., Song B., Zeng X. MUFFIN: multi-scale feature fusion for drug–drug interaction prediction. Bioinformatics. 2021;37:2651–2658. doi: 10.1093/bioinformatics/btab169. [DOI] [PubMed] [Google Scholar]
- 53.Zhang W., Chen Y., Liu F., Luo F., Tian G., Li X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinf. 2017;18:18. doi: 10.1186/s12859-016-1415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pang S., Zhang Y., Song T., Zhang X., Wang X., Rodriguez-Patón A. AMDE: a novel attention-mechanism-based multidimensional feature encoder for drug–drug interaction prediction. Brief. Bioinform. 2022;23:bbab545. doi: 10.1093/bib/bbab545. [DOI] [PubMed] [Google Scholar]
- 55.Lin X., Quan Z., Wang Z.-J., Ma T., Zeng X. IJCAI. 2020. KGNN: Knowledge Graph Neural Network for Drug-Drug Interaction Prediction. [Google Scholar]
- 56.Hao X., Chen Q., Pan H., Qiu J., Zhang Y., Yu Q., Han Z., Du X. Enhancing drug–drug interaction prediction by three-way decision and knowledge graph embedding. Granul. Comput. 2023;8:67–76. doi: 10.1007/s41066-022-00315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng Y.-Y., Yu H., Feng Y.H., Shi J.Y. Directed graph attention networks for predicting asymmetric drug–drug interactions. Brief. Bioinform. 2022;23:bbac151. doi: 10.1093/bib/bbac151. [DOI] [PubMed] [Google Scholar]
- 58.Feng Y.-H., Zhang S.-W., Shi J.-Y. DPDDI: a deep predictor for drug-drug interactions. BMC Bioinf. 2020;21:419. doi: 10.1186/s12859-020-03724-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Z., Guo F., Gu J., Wang Y., Li Y., Wang D., Lu L., Li D., He F. Similarity-based prediction for anatomical therapeutic chemical classification of drugs by integrating multiple data sources. Bioinformatics. 2015;31:1788–1795. doi: 10.1093/bioinformatics/btv055. [DOI] [PubMed] [Google Scholar]
- 60.Shi J.-Y., Mao K.T., Yu H., Yiu S.M. Detecting drug communities and predicting comprehensive drug–drug interactions via balance regularized semi-nonnegative matrix factorization. J. Cheminform. 2019;11:28. doi: 10.1186/s13321-019-0352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y., Qiu Y., Cui Y., Liu S., Zhang W. Predicting drug-drug interactions using multi-modal deep auto-encoders based network embedding and positive-unlabeled learning. Methods. 2020;179:37–46. doi: 10.1016/j.ymeth.2020.05.007. [DOI] [PubMed] [Google Scholar]
- 62.Wang F., Lei X., Liao B., Wu F.X. Predicting drug–drug interactions by graph convolutional network with multi-kernel. Brief. Bioinform. 2022;23:bbab511. doi: 10.1093/bib/bbab511. [DOI] [PubMed] [Google Scholar]
- 63.Davis A.P., Grondin C.J., Johnson R.J., Sciaky D., Wiegers J., Wiegers T.C., Mattingly C.J. Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res. 2021;49:D1138–D1143. doi: 10.1093/nar/gkaa891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iorio F., Bosotti R., Scacheri E., Belcastro V., Mithbaokar P., Ferriero R., Murino L., Tagliaferri R., Brunetti-Pierri N., Isacchi A., di Bernardo D. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA. 2010;107:14621–14626. doi: 10.1073/pnas.1000138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K., et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grover A., Leskovec J. node2vec: Scalable feature learning for networks. Proceedings of the 22nd ACM SIGKDD international conference on Knowledge discovery and data mining; ACM. 2016 doi: 10.1145/2939672.2939754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang K., Xiao C., Glass L.M., Zitnik M., Sun J. SkipGNN: predicting molecular interactions with skip-graph networks. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-77766-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu S., Zhang Y., Cui Y., Qiu Y., Deng Y., Zhang Z., Zhang W. Enhancing drug-drug interaction prediction using deep attention neural networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023;20:976–985. doi: 10.1109/TCBB.2022.3172421. [DOI] [PubMed] [Google Scholar]
- 69.Huang K., Xiao C., Hoang T., Glass L., Sun J. Vol. 34. 2020. Caster: Predicting drug interactions with chemical substructure representation; pp. 702–709. (Proceedings of the AAAI conference on artificial intelligence). [Google Scholar]
- 70.Zitnik M., Sosic R., Leskovec J. BioSNAP Datasets: Stanford Biomedical Network Dataset Collection. 2018. http://snap.stanford.edu/biodata
- 71.Karim M.R., Cochez M., Jares J., Uddin M., Beyan O., Decker S. Drug-drug interaction prediction based on knowledge graph embeddings and convolutional-LSTM network. Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics; ACM. 2019 [Google Scholar]
- 72.Herrero-Zazo M., Segura-Bedmar I., Martínez P., Declerck T. The DDI corpus: An annotated corpus with pharmacological substances and drug–drug interactions. J. Biomed. Inform. 2013;46:914–920. doi: 10.1016/j.jbi.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 73.Whirl-Carrillo M., McDonagh E.M., Hebert J.M., Gong L., Sangkuhl K., Thorn C.F., Altman R.B., Klein T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012;92:414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dhami D.S., Kunapuli G., Das M., Page D., Natarajan S. Drug-drug interaction discovery: kernel learning from heterogeneous similarities. Smart Health. 2018;9–10:88–100. doi: 10.1016/j.smhl.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sridhar D., Fakhraei S., Getoor L. A probabilistic approach for collective similarity-based drug–drug interaction prediction. Bioinformatics. 2016;32:3175–3182. doi: 10.1093/bioinformatics/btw342. [DOI] [PubMed] [Google Scholar]
- 76.Zhang P., Wang F., Hu J., Sorrentino R. Label propagation prediction of drug-drug interactions based on clinical side effects. Sci. Rep. 2015;5 doi: 10.1038/srep12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Su X., Hu L., You Z., Hu P., Zhao B. Attention-based knowledge graph representation learning for predicting drug-drug interactions. Brief. Bioinform. 2022;23:bbac140. doi: 10.1093/bib/bbac140. [DOI] [PubMed] [Google Scholar]
- 78.Hu W., Fey M., Zitnik M., Dong Y., Ren H., Liu B., Catasta M., Leskovec J. Open graph benchmark: Datasets for machine learning on graphs. Adv. Neural Inf. Process. Syst. 2020;33:22118–22133. [Google Scholar]
- 79.Deng Y., Xu X., Qiu Y., Xia J., Zhang W., Liu S. A multimodal deep learning framework for predicting drug–drug interaction events. Bioinformatics. 2020;36:4316–4322. doi: 10.1093/bioinformatics/btaa501. [DOI] [PubMed] [Google Scholar]
- 80.Ryu J.Y., Kim H.U., Lee S.Y. Deep learning improves prediction of drug–drug and drug–food interactions. Proc. Natl. Acad. Sci. USA. 2018;115:E4304–E4311. doi: 10.1073/pnas.1803294115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han C.-D., Wang C.C., Huang L., Chen X. MCFF-MTDDI: multi-channel feature fusion for multi-typed drug–drug interaction prediction. Brief. Bioinform. 2023;24:bbad215. doi: 10.1093/bib/bbad215. [DOI] [PubMed] [Google Scholar]
- 82.Nyamabo A.K., Yu H., Shi J.-Y. SSI–DDI: substructure–substructure interactions for drug–drug interaction prediction. Brief. Bioinform. 2021;22:bbab133. doi: 10.1093/bib/bbab133. [DOI] [PubMed] [Google Scholar]
- 83.Lin S., Wang Y., Zhang L., Chu Y., Liu Y., Fang Y., Jiang M., Wang Q., Zhao B., Xiong Y., Wei D.Q. MDF-SA-DDI: predicting drug–drug interaction events based on multi-source drug fusion, multi-source feature fusion and transformer self-attention mechanism. Brief. Bioinform. 2022;23:bbab421. doi: 10.1093/bib/bbab421. [DOI] [PubMed] [Google Scholar]
- 84.Feng Y.-H., Zhang S.W., Feng Y.Y., Zhang Q.Q., Shi M.H., Shi J.Y. A social theory-enhanced graph representation learning framework for multitask prediction of drug–drug interactions. Brief. Bioinform. 2023;24:bbac602. doi: 10.1093/bib/bbac602. [DOI] [PubMed] [Google Scholar]
- 85.Su X., You Z.-H., Huang D.S., Wang L. Biomedical knowledge graph embedding with capsule network for multi-label drug-drug interaction prediction. IEEE Trans. Knowl. Data Eng. 2022;35:5640–5651. [Google Scholar]
- 86.Feeney A., Gupta R., Thost V., Angell R., Chandu G., Adhikari Y., Ma T. Relation matters in sampling: a scalable multi-relational graph neural network for drug-drug interaction prediction. arXiv. 2021 doi: 10.48550/arXiv.2105.13975. Preprint at. [DOI] [Google Scholar]
- 87.Lee G., Park C., Ahn J. Novel deep learning model for more accurate prediction of drug-drug interaction effects. BMC Bioinf. 2019;20:415–418. doi: 10.1186/s12859-019-3013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O'Donnell L., Oster S., Theesfeld C., Sellam A., et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu H., Dong W., Shi J. RANEDDI: Relation-aware network embedding for drug-drug interaction prediction. Inf. Sci. 2022;582:167–180. [Google Scholar]
- 92.Ren Z.-H., Yu C.Q., Li L.P., You Z.H., Pan J., Guan Y.J., Guo L.X. BioChemDDI: Predicting Drug–Drug Interactions by Fusing Biochemical and Structural Information through a Self-Attention Mechanism. Biology. 2022;11:758. doi: 10.3390/biology11050758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bai Y., Gu K., Sun Y., Wang W. Bi-level graph neural networks for drug-drug interaction prediction. arXiv. 2020 doi: 10.48550/arXiv.2006.14002. Preprint at. [DOI] [Google Scholar]
- 94.Li Z., Zhu S., Shao B., Zeng X., Wang T., Liu T.Y. DSN-DDI: an accurate and generalized framework for drug–drug interaction prediction by dual-view representation learning. Brief. Bioinform. 2023;24:bbac597. doi: 10.1093/bib/bbac597. [DOI] [PubMed] [Google Scholar]
- 95.Chen X., Liu X., Wu J. 2019 IEEE International conference on bioinformatics and biomedicine (BIBM) IEEE; 2019. Drug-drug interaction prediction with graph representation learning. [Google Scholar]
- 96.Wang Y., Min Y., Chen X., Wu J. Multi-view graph contrastive representation learning for drug-drug interaction prediction. Proceedings of the Web Conference 2021; ACM. 2021 [Google Scholar]
- 97.He C., Liu Y., Li H., Zhang H., Mao Y., Qin X., Liu L., Zhang X. Multi-type feature fusion based on graph neural network for drug-drug interaction prediction. BMC Bioinf. 2022;23:224. doi: 10.1186/s12859-022-04763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Purkayastha S., Mondal I., Sarkar S., Goyal P., Pillai J.K. 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE) IEEE; 2019. Drug-drug interactions prediction based on drug embedding and graph auto-encoder. [Google Scholar]
- 99.Yan X.-Y., Yin P.W., Wu X.M., Han J.X. Prediction of the drug–drug interaction types with the unified embedding features from drug similarity networks. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.794205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin J., Wu L., Zhu J., Liang X., Xia Y., Xie S., Qin T., Liu T.Y. R2-DDI: relation-aware feature refinement for drug–drug interaction prediction. Brief. Bioinform. 2023;24:bbac576. doi: 10.1093/bib/bbac576. [DOI] [PubMed] [Google Scholar]
- 101.Yu H., Zhao S., Shi J. STNN-DDI: a substructure-aware tensor neural network to predict drug–drug interactions. Brief. Bioinform. 2022;23:bbac209. doi: 10.1093/bib/bbac209. [DOI] [PubMed] [Google Scholar]
- 102.Kim E., Nam H. DeSIDE-DDI: interpretable prediction of drug-drug interactions using drug-induced gene expressions. J. Cheminform. 2022;14:9–12. doi: 10.1186/s13321-022-00589-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang Z., Chen G., Yang H., Zhong W., Chen C.Y.C. DSIL-DDI: A Domain-Invariant Substructure Interaction Learning for Generalizable Drug–Drug Interaction Prediction. IEEE Trans. Neural Netw. Learn. Syst. 2023:1–9. doi: 10.1109/TNNLS.2023.3242656. [DOI] [PubMed] [Google Scholar]
- 104.Yu H., Wang J., Zhao S.Y., Silver O., Liu Z., Yao J., Shi J.Y. GGI-DDI: Identification for key molecular substructures by granule learning to interpret predicted drug-drug interactions. Expert Syst. Appl. 2024;240 [Google Scholar]
- 105.Bougiatiotis K., Aisopos F., Nentidis A., Krithara A., Paliouras G. Artificial Intelligence in Medicine: 18th International Conference on Artificial Intelligence in Medicine, AIME 2020, Minneapolis, MN, USA, August 25–28, 2020, Proceedings 18. Springer; 2020. Drug-drug interaction prediction on a biomedical literature knowledge graph. [Google Scholar]
- 106.Mondal I. Towards Incorporating Entity-specific Knowledge Graph Information in Predicting Drug-Drug Interactions. arXiv. 2020 doi: 10.48550/arXiv.2012.11142. Preprint at. [DOI] [Google Scholar]
- 107.Lyu T., Gao J., Tian L., Li Z., Zhang P., Zhang J. IJCAI. 2021. MDNN: A Multimodal Deep Neural Network for Predicting Drug-Drug Interaction Events. [Google Scholar]
- 108.Dai Y., Guo C., Guo W., Eickhoff C. Drug–drug interaction prediction with Wasserstein Adversarial Autoencoder-based knowledge graph embeddings. Brief. Bioinform. 2021;22:bbaa256. doi: 10.1093/bib/bbaa256. [DOI] [PubMed] [Google Scholar]
- 109.Ren Z.-H., Yu C.Q., Li L.P., You Z.H., Guan Y.J., Wang X.F., Pan J. BioDKG–DDI: predicting drug–drug interactions based on drug knowledge graph fusing biochemical information. Brief. Funct. Genomics. 2022;21:216–229. doi: 10.1093/bfgp/elac004. [DOI] [PubMed] [Google Scholar]
- 110.Ren Z.-H., You Z.H., Yu C.Q., Li L.P., Guan Y.J., Guo L.X., Pan J. A biomedical knowledge graph-based method for drug–drug interactions prediction through combining local and global features with deep neural networks. Brief. Bioinform. 2022;23:bbac363. doi: 10.1093/bib/bbac363. [DOI] [PubMed] [Google Scholar]
- 111.Najafabadi M.M., Villanustre F., Khoshgoftaar T.M., Seliya N., Wald R., Muharemagic E. Deep learning applications and challenges in big data analytics. J. Big Data. 2015;2:1–21. [Google Scholar]
- 112.Sze V., Chen Y.H., Yang T.J., Emer J.S. Efficient processing of deep neural networks: A tutorial and survey. Proc. IEEE. 2017;105:2295–2329. [Google Scholar]
- 113.Larochelle H., Bengio Y., Louradour J., Lamblin P. Exploring strategies for training deep neural networks. J. Mach. Learn. Res. 2009;10 [Google Scholar]
- 114.Washio T., Motoda H. State of the art of graph-based data mining. SIGKDD Explor. Newsl. 2003;5:59–68. [Google Scholar]
- 115.Bunke H., Riesen K. Recent advances in graph-based pattern recognition with applications in document analysis. Pattern Recogn. 2011;44:1057–1067. [Google Scholar]
- 116.Jiao L., Chen J., Liu F., Yang S., You C., Liu X., Li L., Hou B. Graph representation learning meets computer vision: A survey. IEEE Trans. Artif. Intell. 2023;4:2–22. [Google Scholar]
- 117.Zhang Y., Lei X., Fang Z., Pan Y. CircRNA-disease associations prediction based on metapath2vec++ and matrix factorization. Big Data Min. Anal. 2020;3:280–291. [Google Scholar]
- 118.Fan C., Lei X., Guo L., Zhang A. Predicting the associations between microbes and diseases by integrating multiple data sources and path-based HeteSim scores. Neurocomputing. 2019;323:76–85. [Google Scholar]
- 119.Zhou J., Cui G., Hu S., Zhang Z., Yang C., Liu Z., Wang L., Li C., Sun M. Graph neural networks: A review of methods and applications. AI Open. 2020;1:57–81. [Google Scholar]
- 120.Zhang S., Tong H., Xu J., Maciejewski R. Graph convolutional networks: a comprehensive review. Comput. Soc. Netw. 2019;6:1–23. doi: 10.1186/s40649-019-0069-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Velickovic P., Casanova A., Lio P., Cucurull G., Romero A., Bengio Y. Graph attention networks. stat. 2017;1050:10–48550. [Google Scholar]
- 122.Rumelhart D.E., Hinton G.E., Williams R.J. Institute for Cognitive Science, University of California, San Diego La; 1985. Learning Internal Representations by Error Propagation. [Google Scholar]
- 123.Tschannen M., Bachem O., Lucic M. Recent advances in autoencoder-based representation learning. arXiv. 2018 doi: 10.48550/arXiv.1812.05069. Preprint at. [DOI] [Google Scholar]
- 124.Kasun L.L.C., Yang Y., Huang G.B., Zhang Z. Dimension reduction with extreme learning machine. IEEE Trans. Image Process. 2016;25:3906–3918. doi: 10.1109/TIP.2016.2570569. [DOI] [PubMed] [Google Scholar]
- 125.Dai Y., Wang S., Xiong N.N., Guo W. A survey on knowledge graph embedding: Approaches, applications and benchmarks. Electronics. 2020;9:750. [Google Scholar]
- 126.Mohamed S.K., Nounu A., Nováček V. Biological applications of knowledge graph embedding models. Brief. Bioinform. 2021;22:1679–1693. doi: 10.1093/bib/bbaa012. [DOI] [PubMed] [Google Scholar]
- 127.Niu Z., Zhong G., Yu H. A review on the attention mechanism of deep learning. Neurocomputing. 2021;452:48–62. [Google Scholar]
- 128.Li G., Xiong C., Thabet A., Ghanem B. Deepergcn: All you need to train deeper gcns. arXiv. 2020 doi: 10.48550/arXiv.2006.07739. Preprint at. [DOI] [Google Scholar]
- 129.Gómez-Bombarelli R., Wei J.N., Duvenaud D., Hernández-Lobato J.M., Sánchez-Lengeling B., Sheberla D., Aguilera-Iparraguirre J., Hirzel T.D., Adams R.P., Aspuru-Guzik A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018;4:268–276. doi: 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Katayama T., Wilkinson M.D., Aoki-Kinoshita K.F., Kawashima S., Yamamoto Y., Yamaguchi A., Okamoto S., Kawano S., Kim J.D., Wang Y., et al. BioHackathon series in 2011 and 2012: penetration of ontology and linked data in life science domains. J. Biomed. Semantics. 2014;5:1–13. doi: 10.1186/2041-1480-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smith B., Ceusters W., Klagges B., Köhler J., Kumar A., Lomax J., Mungall C., Neuhaus F., Rector A.L., Rosse C. Relations in biomedical ontologies. Genome Biol. 2005;6:1–15. doi: 10.1186/gb-2005-6-5-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hu B.Q. Three-way decisions space and three-way decisions. Inf. Sci. 2014;281:21–52. [Google Scholar]
- 133.Wicha S.G., Chen C., Clewe O., Simonsson U.S.H. A general pharmacodynamic interaction model identifies perpetrators and victims in drug interactions. Nat. Commun. 2017;8:2129. doi: 10.1038/s41467-017-01929-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bible K.C., Kaufmann S.H. Cytotoxic synergy between flavopiridol (NSC 649890, L86-8275) and various antineoplastic agents: the importance of sequence of administration. Cancer Res. 1997;57:3375–3380. [PubMed] [Google Scholar]
- 135.Ning X., Schleyer T., Shen L., Li L. 2017 IEEE International Conference on Healthcare Informatics (ICHI) IEEE; 2017. Pattern discovery from directional high-order drug-drug interaction relations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peng B., Ning X. Deep learning for high-order drug-drug interaction prediction. Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics; ACM. 2019 [Google Scholar]