

SUMMARY
Vascular morphogenesis requires a delicate gradient of Notch signaling controlled, in part, by the distribution of ligands (Dll4 and Jagged1). How Jagged1 (JAG1) expression is compartmentalized in the vascular plexus remains unclear. Here, we show that Jag1 mRNA is a direct target of zinc-finger protein 36 (ZFP36), an RNA-binding protein involved in mRNA decay that we find robustly induced by vascular endothelial growth factor (VEGF). Endothelial cells lacking ZFP36 display high levels of JAG1 and increase angiogenic sprouting in vitro. Furthermore, mice lacking Zfp36 in endothelial cells display mispatterned and increased levels of JAG1 in the developing retinal vascular plexus. Abnormal levels of JAG1 at the sprouting front alters NOTCH1 signaling, increasing the number of tip cells, a phenotype that is rescued by imposing haploinsufficiency of Jag1. Our findings reveal an important feedforward loop whereby VEGF stimulates ZFP36, consequently suppressing Jag1 to enable adequate levels of Notch signaling during sprouting angiogenesis.
Graphical Abstract
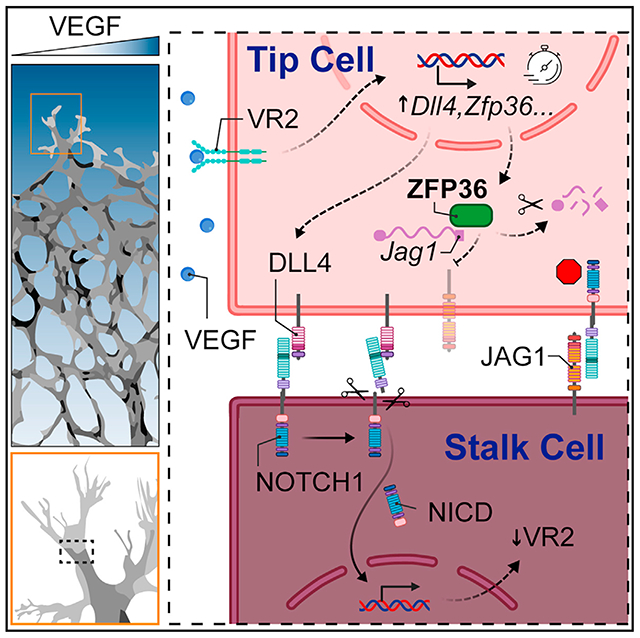
In brief
How are Notch ligand gradients generated in an otherwise homogeneous vascular plexus? Sunshine et al. show that the RNA decay protein ZFP36 prevents expression of Jagged1 where levels of VEGF are high. In this manner, Notch signaling at the angiogenic front specifies tip cells and promotes stalk cell differentiation and emergence of vascular sprouts.
INTRODUCTION
The formation of a hierarchical vascular tree requires timely and dose-specific activation of multiple signaling pathways. At the top of this list is vascular endothelial growth factor (VEGF), a critical regulator of sprouting angiogenesis.1 Through classic activation of VEGF receptor 2 tyrosine kinase activity, VEGF initiates a cascade of downstream events that promote endothelial cell (EC) proliferation and fosters EC specification into tip and stalk cells under the instruction of the Notch signaling pathway.2–4 In fact, VEGF raises canonical Notch ligand Delta-like 4 (DLL4) levels, creating a boundary between ligand-expressing (tip cells) and receptor-expressing cells (stalk cells).5–8 The balance between tip and stalk cells is indispensable to the morphogenesis of a functional vascular tree. Genetic or pharmacological inactivation of Notch signaling, through deletion of Notch1 or Dll4, results in an overwhelming number of tip cells.5,6,9–11 The excess of tip cells halts angiogenesis as alone, tip cells are unable to form trunks for vascular elongation, resulting in an underdeveloped vascular system.
In addition to DLL4, Jagged1 (JAG1) is a second important ligand for Notch in the vasculature. This transmembrane protein is expressed by ECs, pericytes, and smooth muscle cells,12 and it is critical in orchestrating the recruitment of mural cells (pericytes and smooth muscle cells) and promoting the formation of arteries.13,14 JAG1 is also essential in specifying smooth muscle cells and preventing them from adopting a default chondrocyte fate,12,15,16 and it is critical for maintaining this fate in the adult.17 In the immature vascular plexus, the leading front with tip and stalk cells is largely devoid of JAG1 expression.18 The reason for this heterogeneous pattern is unclear but has been shown to hold functional consequences because JAG1 in the endothelium appears to antagonize trans-activation through DLL4.19
Cells are endowed with multiple intrinsic mechanisms that regulate the levels and compartmentalization of protein expression. One such process involves post-transcriptional regulation through targeting mRNA transcripts for degradation.20 Tristetraprolin, also known as zinc-finger protein 36 homolog (ZFP36), is a potent mRNA decay protein.21 ZFP36 and the related family members butyrate response factors 1 and 2 (ZFP36L1 and ZFP36L2, respectively), recognize and bind to cis-acting adenylate and uridylate (AU)-rich elements (AREs) within the 3′ untranslated regions (3′ UTRs) of transcripts.22 Sub-sequent interactions with additional trans-acting mRNA decay machinery promote the targeted mRNA for deadenylation.23,24
ZFP36 levels are usually low or undetectable in most cell types under quiescent conditions. However, robust induction can occur in response to growth factors.25,26 Many reported ZFP36 mRNA targets are cytokines and growth factors involved in inflammation, cell cycle, and carcinogenesis.27 Of note, previous experiments from our laboratory on EC regeneration revealed robust expression of ZFP36 in the proliferative zone of injured arteries.28,29 These initial findings prompted interest in the potential physiological function of ZFP36 in the vasculature and its role at critical times when VEGF is highly expressed during angiogenesis.
Here, we uncovered robust but temporally restricted, EC induction of ZFP36 by VEGF. We demonstrate that Jag1 is a direct target of ZFP36 and found that ZFP36 titrates Notch signaling to facilitate proper tip and stalk cell dynamics during sprouting angiogenesis. Our findings imply that post-transcriptional ZFP36 regulation is an additional intermediary step in VEGF-Notch crosstalk via negative feedforward regulation of JAG1 expression.
RESULTS
VEGF exposure upregulates the RNA decay protein ZFP36
Given the high induction of ZFP36 during regeneration of arteries29 and in the context of its upregulation by multiple growth factors,26 we asked whether ZFP36 could be induced by VEGF. Bulk RNA sequencing of human umbilical vein ECs (HUVECs) stimulated with VEGF for 1 h revealed that ZFP36 was within the top 20 induced transcripts (Figures 1A and 1B). Time-course experiments confirmed the RNA sequencing (RNA-seq) findings at the protein level (Figure 1C) and further revealed that protein induction was short lived, peaking at 1 h and retaining high levels for an additional 1.5 h. Importantly, other members of the ZFP36 family, ZFP36L1 and ZFP36L2,27 were also expressed by ECs but were not induced by VEGF (Figure 1C). The response of ZFP36 to VEGF exposure was transcriptional, as shown by a clear peak of ZFP36 mRNA at 30 min that preceded protein induction (Figure 1D). Moreover, the transient nature of this induction and the presence of additional molecular weight forms recognized by the antibody implied additional post-transcriptional regulation and post-translational modifications, such as autoregulation of mRNA and/or control of protein stability through phosphorylation.30,31 The rapid VEGF sensitivity was reproduced in multiple HUVEC biological replicates and was noted to extend to human aortic and human dermal microvascular ECs (HAECs and HDMECs, respectively) (Figures S1A-S1D). Interestingly, while ZFP36 induction patterns were similar across different vascular beds, relative levels of ZFP36L1 and ZFP36L2 were variable. Both were highly expressed in HAECs; however, ZFP36L2 was higher in HUVECs and ZFP36L1 was higher in HDMECs. Induction of ZFP36 by VEGF was also confirmed by immunocytochemistry on endothelial monolayers, showing presence of ZFP36 both in the nucleus and cytoplasm (Figures 1E and 1F). Combined, the rapid changes of ZFP36 levels in response to VEGF and its known function as an mRNA decay factor implied a role in transient post-transcriptional regulation of selected targets downstream VEGF signaling.
Figure 1. VEGF triggers a robust induction of ZFP36 in ECs.
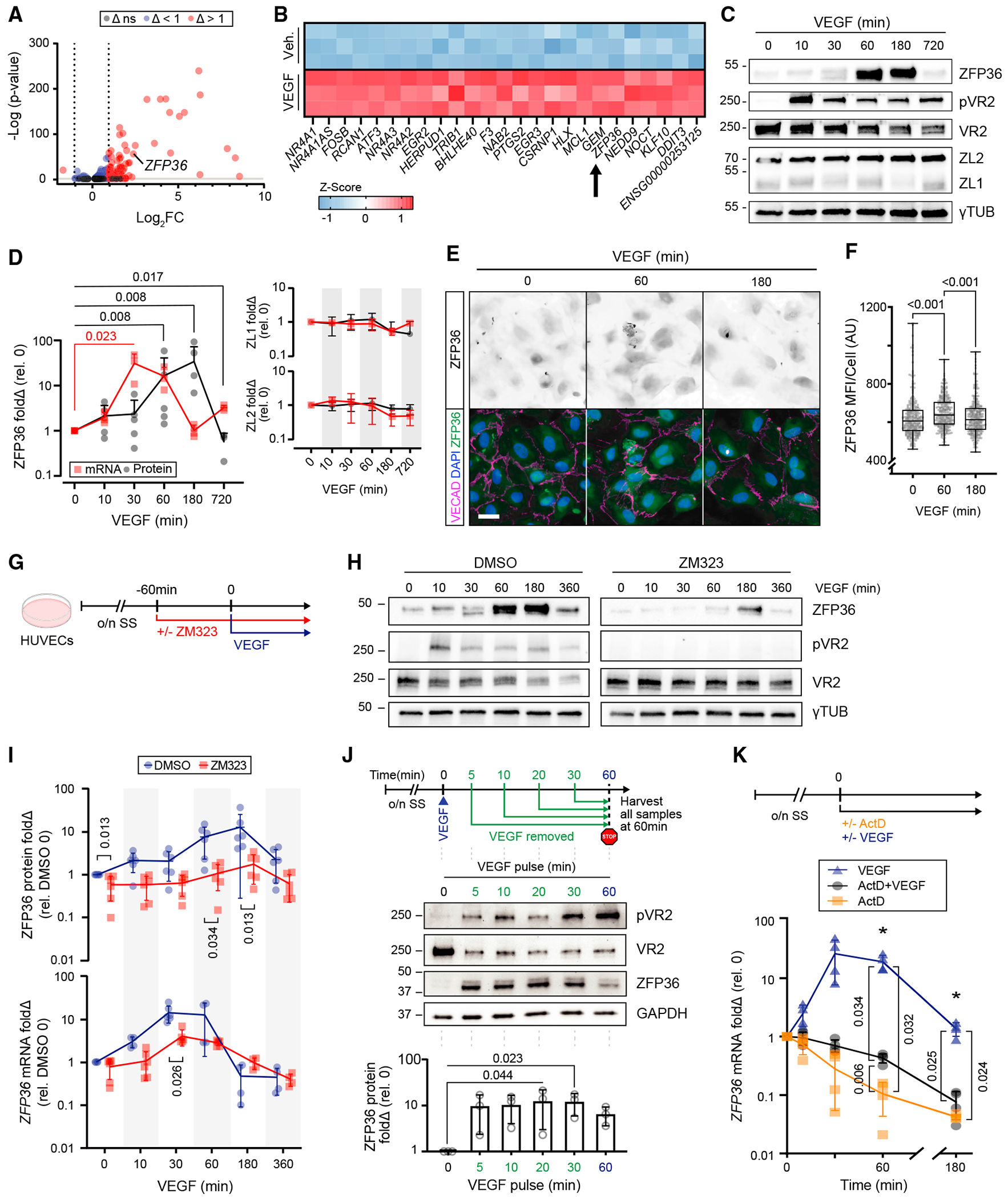
(A) Volcano plot of RNA-seq differential expression log2fold change (Δ) of HUVECs with or without VEGF for 1 h. Non-significant (ns) cutoff was set to −ILog(p) > 2.
(B) Z score heatmap of the top 25 differentially expressed genes after exposure to vehicle (control [Ctrl]) or VEGF165 (100 ng/mL) for 1 h (n = 3 biological replicates).
(C) Representative immunoblots of ZFP36, phospho-VEGFR2 (pVR2), VEGFR2 (VR2), ZFP36L1 (ZL1), ZFP36L2 (ZL2), and γ-tubulin (γTUB) from HUVEC lysates after VEGF stimulation for the indicated times.
(D) Immunoblot quantification of fold change (Δ) relative to control (n = 5 biological replicates; except t = 720, n = 3 biological replicates) and RT-qPCR transcripts of ZFP36, ZL1, and ZL2 (n = 3 biological replicates). Data for ZFP36 are individual replicates with a line representing mean. Data for ZL1 and ZL2 are represented with mean ± SD. Statistical analysis by Mann-Whitney test.
(E) Representative immunofluorescence of ZFP36, VE-cadherin (VECAD), and DAPI on HUVECs treated with or without VEGF at the indicated times (scale bar, 25 μm).
(F) Quantification of ZFP36 mean fluorescence intensity (MFI) per cell (n = 300 cells). Data are presented as individual replicates, with the overlaid box extending from the 25th to 75th percentiles with whiskers showing minimum and maximum values. Statistics: Kruskal-Wallis with post-hoc Dunn’s multiple-comparisons test.
(G) Experimental design. Confluent HUVEC monolayers were exposed to the VR2 inhibitor ZM323881 (ZM323) or vehicle for 1 h following overnight serum starvation (o/n SS), followed by VEGF165 stimulation.
(H) Representative immunoblots of ZFP36, pVR2, VR2, and γTUB from HUVECs treated with VEGF at the indicated times in the presence of DMSO (vehicle control) or ZM323 (VEGFR2 inhibitor).
(I) Graph representing protein levels (n = 5 biological replicates) and RT-qPCR transcripts (n = 4–6, combination of technical replicates with a minimum of 3 biological replicates) for ZFP36 after VEGF time course in the presence or absence of ZM323. Data presented are mean from biological replicates ± SD. Statistics: multiple Mann-Whitney tests with Holm-Šídák method to adjust for multiple comparisons.
(J) Experimental design of pulse-chase VEGF treatment with associated representative WB and quantification. WB quantification is presented with individual replicates and mean ± SD relative to control (n = 3 biological replicates). Statistics: Freidman’s test with post-hoc Dunn’s multiple-comparisons test.
(K) Experimental design of actinomycin D (ActD) treatment and qPCR quantification of mRNA normalized to hprt (n = 4 biological replicates) mean fold change ± SD relative to control. Kruskal-Wallis with post-hoc Dunn’s multiple-comparisons test.
Next, to determine whether ZFP36 induction used canonical VEGF signaling, pretreatment with a selective small-molecule VEGFR2 (VR2) inhibitor (ZM323881)32 was utilized to block receptor activation (Figures 1G and 1H). In the presence of VR2 inhibition, ZFP36 induction was drastically reduced at both protein and transcript levels (Figures 1H and 1I). Interestingly, only a short pulse of VEGF (5 min) was sufficient to trigger potent upregulation of ZFP36 by 1 h (Figure 1J). Finally, we confirmed that VEGF-mediated induction of ZFP36 required transcription by co-treatment with actinomycin D (ActD) (Figure 1K). Together, these results indicate a functional role of ZFP36-mediated mRNA decay in ECs downstream of VEGF signaling.
ZFP36 binds to the 3′ UTR of Jag1
To investigate the potential function of ZFP36 in the endothelium, we used enhanced UV crosslinking and immunoprecipitation (eCLIP-seq) to unbiasedly identify putative ZFP36 target mRNAs at the transcriptome-wide level.26 This approach preserves RNA-protein complexes by promoting covalent interactions, thus facilitating immunoprecipitation of ZFP36 and its associated transcripts. Subsequent sequencing of libraries generated from the pull-down templates allows the identification of binding sites with high precision.33 Recognizing the potential overlapping roles of ZFP36L1 and ZFP36L2 in transcript binding, libraries were generated using immortalized embryonic fibroblasts from mice with concurrent flox alleles in Zfp36, Zfp36l1, and Zfp36l2 loci (Figure 2A). RNA libraries were generated from serum-stimulated parental and from Cre recombinase-in-fected cells (Figure 2B). The approach was replicated with high reproducibility and identified many of the previously published targets.26,34–36 Importantly, the archetypal RNA binding feature that preferentially targets AREs was fully reproduced, as indicated by a nearly log-fold enrichment of the UAUUUAUU motif relative to competing binding sites.26 Among previously unreported ZFP36 targets with direct relation to endothelial biological context was JAG1. Using Integrative Genomics Viewer software, we found that ZFP36 bound to the 3′ UTR from wild-type cells, but the peaks were absent from triple-floxed knockout cells (TKO) in independent replicates (Figure 2C). The absence of peaks in the controls (TKO) further confirmed that the signal corresponded to ZFP36 binding to the Jag1 3′ UTR and did not originate from off-target affinities of the ZFP36 antibody. Importantly, the major peak showed several ARE motifs in the 3′ UTR of Jag1, consistent with its known affinity for this sequence and indicative of robust binding in this region (Figure 2C, boxed sequence and asterisk; Figure S2A, full ms-JAG1 3′ UTR sequence). The CLIP results were further confirmed using CLIP-qPCR and compared with the previously identified ZFP36-binding target Ptgs2 (Figure S2B).
Figure 2. JAG1 is a direct target for ZFP36 binding.
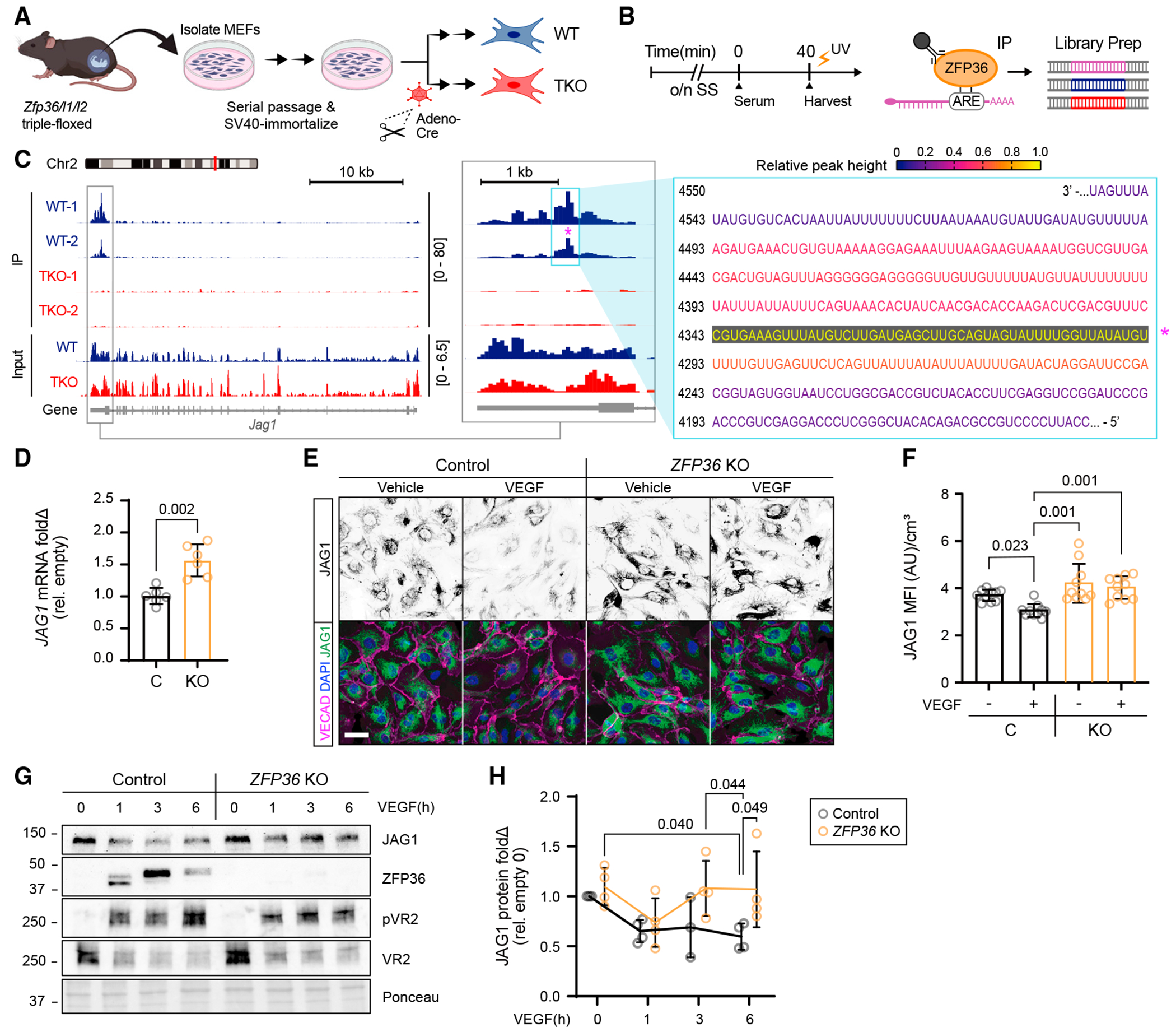
(A) Schematic for generation of Zfp36/l1/l2 triple-floxed MEF cells and adenovirus-based approach for in vitro Cre recombinase delivery (adeno-Cre) or GFP control (adeno-GFP) to derive Zfp36/l1/l2 triple-floxed wild-type (WT) and triple-floxed knockout (TKO) cells from an isogenic cell population.
(B) Schematic of the experimental design for eCLIP.
(C) Integrative Genomics Viewer-generated eCLIP-seq data showing the ZFP36 binding site on Jag1 mRNA within the 3′ UTR. Track height scale is denoted in brackets. Relative peak height from CLIP data was used to color code corresponding mRNA nucleotides associated with the highest binding peak (*).
(D) Quantification of JAG1 mRNA mean fold change (Δ) ± SD by RT-qPCR from CRISPR control and ZFP36 KO HUVECs normalized to HPRT (n = 3 biological replicates, 2 technical replicates each). Statistics: Kruskal-Wallis with post hoc Dunn’s multiple-comparisons test.
(E) Representative immunofluorescence of JAG1, VECAD, and DAPI in CRISPR control and ZFP36 KO HUVECs treated with or without VEGF for 1 h (scale bar, 50 μm).
(F) Quantification of Jag1 MFI ± SD normalized to cell volume per field of view (n = 10 fields). Statistics: Kruskal-Wallis with post hoc Dunn’s multiple-comparisons test.
(G) Representative immunoblots of JAG1, ZFP36, pVR2, and VR2 protein expression from CRISPR control and ZFP36 KO HUVECs stimulated with VEGF at the indicated times. The included corresponding Ponceau stain is used as a loading control reference.
(H) Quantification of protein fold change (Δ) relative to empty control and normalized to total protein (n = 3 biological replicates, with an additional technical replicate in all but control 3 h). Data represent mean ± SD. Statistical analysis: mixed-effects analysis with post hoc Tukey’s multiple-comparisons test.
MEF, mouse embryonic fibroblast; C, CRISPR Ctrl; KO, ZFP36 KO.
Based on sequence and ARE motif similarities between mouse and human Jag1, we suspected conserved ZFP36 JAG1-binding affinity across species and cell types (Figure S2C). To test the effect of ZFP36 on human ECs, we generated CRISPR control and KO cells. Because levels of ZFP36 are difficult to detect without VEGF stimulation, cells were exposed to VEGF for 1 h to verify that HUVEC KO for ZFP36 (ZFP36 KO) failed to be induced (Figure S3A). We also observed that the increased baseline levels of JAG1 in ZFP36 KO cells were stable even after 1 h of VEGF exposure (Figures S3A and S3B). These increases, at first glance, did not appear to phenotypically change the cells because ZFP36 KO did not affect key behaviors of migration or proliferation, and ZFP36 KO cells appeared otherwise normal in cultured monolayers (Figures S3C–S3F). Consistent with changes in protein abundance, we also found that JAG1 transcripts were elevated by approximately 50% in the absence of ZFP36 (Figure 2D). Increases in JAG1 in the absence of ZFP36 were also validated by immunocytochemistry (Figures 2E and 2F). The corresponding relationship between ZFP36 induction by VEGF and JAG1 levels was also tested through a time course of VEGF stimulation in control and KO cells. Consistent with the notion that ZFP36 regulates JAG1 levels, ECs exposed to VEGF, which upregulates ZFP36, showed a sustained reduction in JAG1 protein (1–6 h). However, the same was not observed in ECs where ZFP36 was inactivated (Figures 2G and 2H).
To further scrutinize the specificity of ZFP36 in human-derived ECs, we compared the 3′ UTRs of mouse (m) and human (h) Jag1 sequences and generated reporter constructs: a control construct with the full-length (FL) mJag1-3′ UTR and a second construct where the region corresponding to the major CLIP peak was deleted (Δ) (Figures 3A and 3B). Wild-type ECs infected with the resulting lentiviruses confirmed the relevance of this region in the Jag1 mRNA for regulation by endogenous ZFP36. The ARE-deleted construct exhibited higher mean nuclear enhanced green fluorescent protein (GFP, reporter) than the FL construct (Figures 3C and 3D). VEGF stimulation also resulted in reduced fluorescent signal in mΔ-transfected cells, however, to the same relative degree as the vehicle-treated group. This was surprising, as we expected that VEGF treatment would increase mRNA decay activity by induction of ZFP36 and, therefore, only reduce fluorescent signal in mFL-transfected cells. There are several possible interpretations for the outcome. While the mΔ construct no longer has the peak binding sequence, still retains other ARE motifs that can potentially bind to Zfp36, albeit with reduced affinity. This idea is supported by the fact that the VEGF-mediated reductions seen in mFL cells was still significantly lower than in mΔ-transfected cells. Alternatively, the ZFP family members ZFP36L1 and ZFP36L2 could also have potential redundancy in transcript binding. Though we did not perform a transcript binding analysis with the other family members, L1 and L2, because they are not sensitive to VEGF induction (Figure 1C), we predict that, in this specific context, they may not contribute to Jag1 transcript stability. Moreover, the relatively stable half-life (~26 h) of GFP might not be ideal to assess decay dynamics of a relatively rapid post-transcriptional mechanism. Thus, similar experiments were also performed with luciferase reporters using the human 3′ UTR sequence (Figures 3E–3G). An additional benefit of using a dual-luciferase assay is the differential readout (more quantitative) in relation to a co-transfected independent control vector to normalize for transfection efficiency. In control cells, deletion of the homologous human peak binding region (hΔ) increased the level of luciferase by 2-fold, an effect that was achieved at baseline in ZFP36 null cells with the FL construct (hFL) (Figure 3F). When transfections were done in the presence or absence of VEGF (to modulate ZFP36 levels), differences were exacerbated according to the trending difference in FL JAG1-3′ UTR (FL) in control cells (Figure 3G). These effects were no longer observed when either the binding region was removed (′-transfected cells) or in the context of ZFP36 KO cells.
Figure 3. Zfp36 regulates reporter expression levels depending on Jag1 3’ UTR domains.
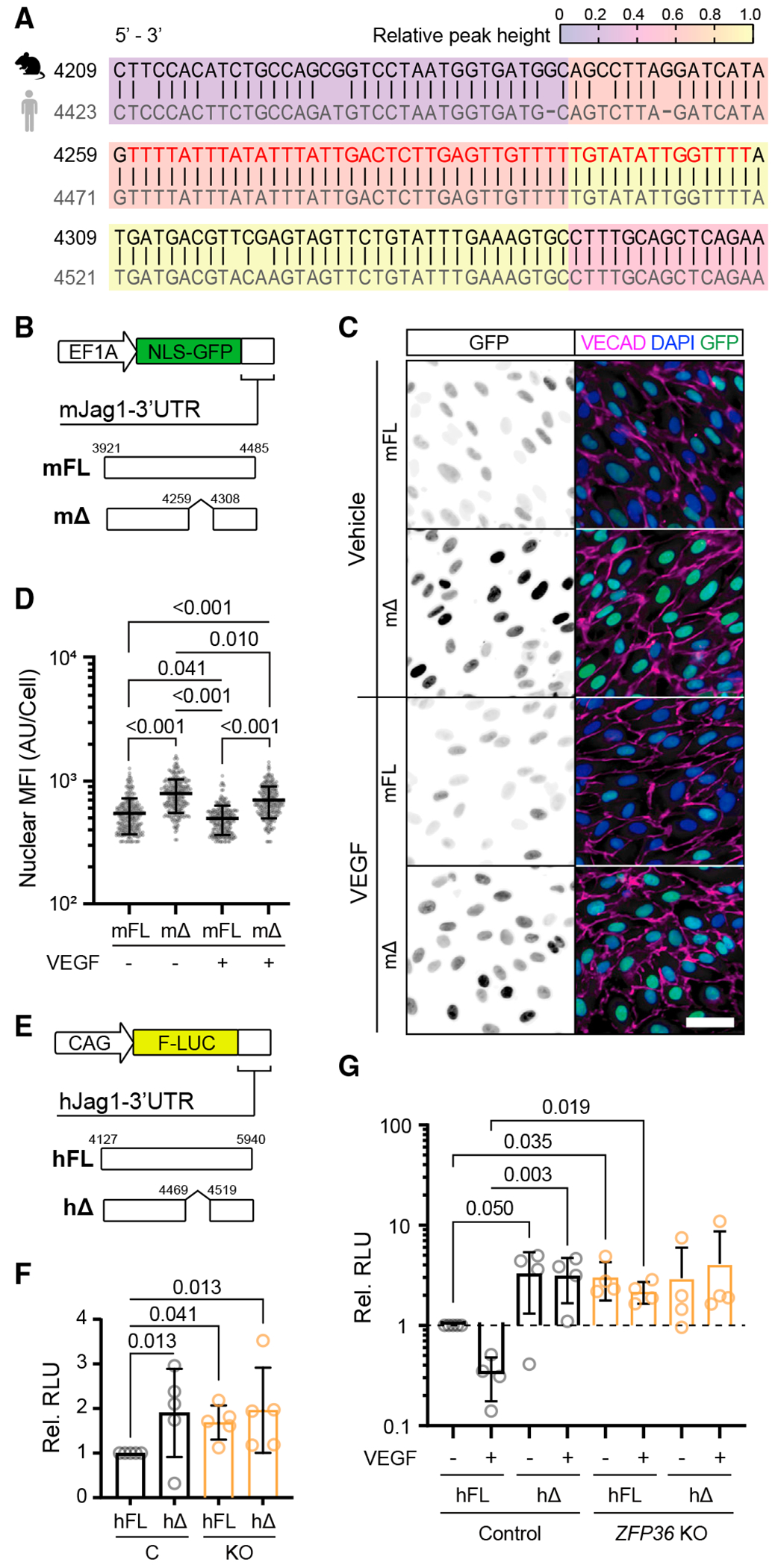
(A) BLASTN sequence alignment (5′ to 3′) of mouse Jag1 (NM_013882.5) and human JAG1 (NM_000214.3) mRNA associated with the peak ZFP36 binding domain identified in eCLIP-seq experiments (red lettering).
(B) Jag1 lentivirus reporter with nuclear localization signal (NLS)-GFP mJag1 3′ UTR with and without the peak binding sequence (4,260–4,307).
(C) Representative immunofluorescence of VECAD and DAPI on Jag1-GFP-UTR-infected HUVECs stimulated with and without VEGF for 1 h (scale bar, 50 μm). The first column shows monochromatic GFP to facilitate visualization of levels.
(D) Quantification of GFP nuclear MFI per cell ± SD (n > 300 cells). Statistics: Kruskal-Wallis with post hoc Dunn’s multiple-comparisons test.
(E) Luciferase constructs fused to the human JAG1 3′ UTR with and without the putative peak binding sequence (4,470–4,518).
(F) Mean luciferase activity ± SD in CRISPR control and ZFP36 KO HUVECs cotransfected with JAG1 3′ UTR either full length (FL) or without the peak binding sequence (Δ) and Renilla luciferase. Data are normalized to Renilla luciferase and presented relative to control HUVECs transfected with FL (n = 3 biological replicates with an additional technical replicate). Statistics: Kruskal-Wallis with post hoc uncorrected Dunn’s test.
(G) Same as (F) but with or without VEGF treatment for 1 h. Statistics: Kruskal-Wallis with post hoc uncorrected Dunn’s test.
Inactivation of ZFP36 in ECs alters the distribution and levels of Jag1
Animal models with global deletion in Zfp36 develop inflammatory phenotypes. In fact, many identified mRNA binding targets are involved in regulating inflammatory transcripts, particularly tumor necrosis factor alpha (TNF-α) in macrophages.37 To clarify the role of ZFP36 in the endothelium and under physiological conditions, we generated tamoxifen-inducible EC-specific KO mice. Induction of endothelial Zfp36 deletion with tamoxifen shortly post birth did not result in drastic alterations in the phenotype of the mice, or reveal spontaneous inflammatory phenotypes despite high induction efficiency. To test the consequence of Zfp36 deletion to Jag1 levels, we first isolated liver ECs from Cre-negative and Cre-positive Zfp36f/f mice that were exposed to tamoxifen shortly post birth (Figure 4A). The efficiency of the isolation and relative endothelial purity of the cultures was assessed by evaluation of the tdTomato reporter by fluorescence microscopy and flow cytometry (Figures 4B and 4C). Using these cells, we evaluated levels of Jag1 by flow cytometry and western blots (Figures 4D and 4E). In both cases, absence of Zfp36 yielded a significant increase in both cell surface and total Jag1 protein. We also verified levels of Jag1 protein in vivo. Because aortic ECs from adult mice normally express Jag1, we predicted that this expression would be elevated in the absence of endothelial Zfp36. Accordingly, animals pulsed with tamoxifen as neonates showed that reporter-positive cells in adults (Figures 4F–4H, tomato, arrowheads) had higher levels of JAG1 than adjacent non-deleted cells (due to incomplete efficiency) (Figures 4F–4H, lack of tomato reporter, arrows). Similar experiments were also performed in endothelial-specific triple deletion (Zfp36, Zfp36l1, and Zfp36l2) animals (Z36T), which yielded the same outcome (Figures 4G and 4I). Specifically, reporter-positive ECs (Figures 4G and 4I, tomato positive, arrowheads) showed higher levels of JAG1 than adjacent reporter-negative ECs (Figures 4G and 4I, tomato-negative, arrows). These results were more robust than for single Zfp36 deletion (Figures 4G and 4I), suggesting a potential overlapping effect by ZFP36L1 and ZFP36L2 family members.
Figure 4. Deletion of zfp36 in the endothelium increases Jag1 expression in vivo.
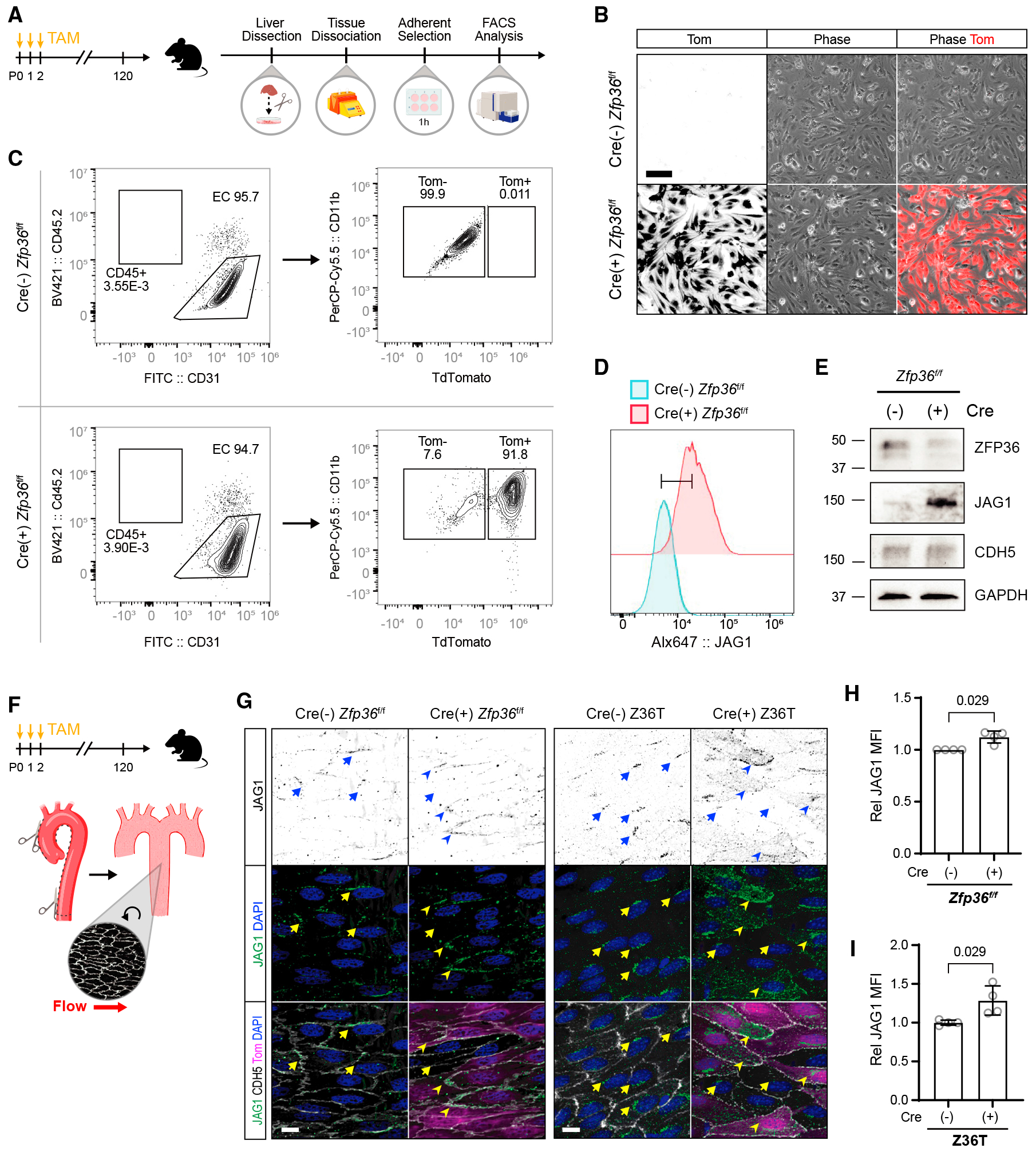
(A) Experimental design for isolation of ECs from tamoxifen-fed inducible Cdh5-Cre Z fp36f/f TdTomato (Tom) reporter mice.
(B) Representative Tom fluorescence and phase images from selected liver ECs in culture (scale bar, 100 μm). The first column shows the monochromatic conversion of the last column.
(C) Flow cytometry gating strategy and analysis of liver ECs.
(D) Flow cytometry analysis of surface Jag1 expression from Cre(−),Tom(−) ECs and Cre(+),Tom(+) ECs gated from the experiment shown in (C). The y axis represents normalized (relative to mode) cell numbers.
(E) Immunoblot of Zfp36, Jag1, VECAD, and GAPDH from EC lysates.
(F) Schematic of aortic dissection for en face immunohistochemistry.
(G) Representative immunohistochemistry of JAG1, VECAD, and DAPI from tamoxifen (TAM)-fed Cdh5-Cre zfp36f/f Tom mice and from Cdh5-Cre triple zfp36f/f/1f/f/2f/f (Z36T) Tom mice, as indicated (scale bars, 10 μm). A Tom signal indicates recombination of the reporter in the respective cells. Several areas of positive fluorescent signal are marked on Tom(−) (arrows) and Tom(+) (arrowheads) cells. The first row shows monochromatic view of the green (JAG1) channel.
(H) Quantification of Jag1 MFI ± SD from TAM-fed Cdh5-Crezfp36f/f Tom mice, averaged from a minimum of 2 fields of view per animal (n = 4). For Cre(+) animals, only Tom(+) cell areas were measured. Statistics: Mann-Whitney test.
(I) Quantification of JAG1 MFI ± SD from TAM-fed Cdh5-Cre triple zfp36f/fl1f/fl2f/f (Z36T) Tom mice, averaged from a minimum of 2 fields of view per animal (n = 4). For Cre(+) animals, only Tom(+) cell areas were measured. Statistics: Mann-Whitney test.
ZFP36 affects Notch signaling and sprouting angiogenesis by altering Jag1 expression
Notch signaling is initiated by binding to transmembrane ligands that interact with Notch receptors in trans. For this, the expression of the receptor (Notch) and the ligand (Dll4, Jag1, and others) should be in adjacent cells. Conversely, when receptor and ligand are both expressed by the same cell, pairing of receptor-ligand at the cell surface in cis leads to suppression of the pathway.38 To test the effects of ZFP36 deletion and the resulting elevated levels of JAG1 on Notch signaling, we examined nuclear Notch (ICD) in ZFP36 KO and control cells. These experiments were conducted in confluent cultures according to the integrity of continuous vascular endothelial (VE)-cadherin, to ensure trans-activation of the Notch pathway. Control and ZFP36 KO cultures were either exposed to vehicle or VEGF for 1 h, a time consistent with induction of ZFP36. We found that VEGF-treated cells had lower levels of Notch ICD in the nucleus, reflecting lower signaling. Importantly, in the case of ZFP36 KO cells, both control and VEGF-treated cells showed a noticeable reduction in nuclear Notch (Figures 5A and 5B). The findings indicate that excess Jagged1, due to the absence of ZFP36 regulation, is sufficient to tilt the delicate balance of cis versus trans ligand-receptor interactions and alter the levels of Notch signaling. When presenting control and ZFP36 KO HUVECs with exogenous ligand for 24 h, Notch signaling is no longer reduced, and the cells appear to signal within control levels. This indicates that heterotypic interactions mitigate the effects of potential cis inhibition (Figures S4A–S4D). The functional implications of JAG1 excess were further explored using sprouting bead assays in vitro with control and ZFP36 KO HUVECs (Figures 5C–5G). As anticipated, we found that absence of ZFP36 yielded a significant increase in the number and length of the sprouts that was not driven by differences in cell number (Figure 5G), a finding consistent with reduced Notch signaling.
Figure 5. Zfp36KO cells display increased sprouting angiogenesis.
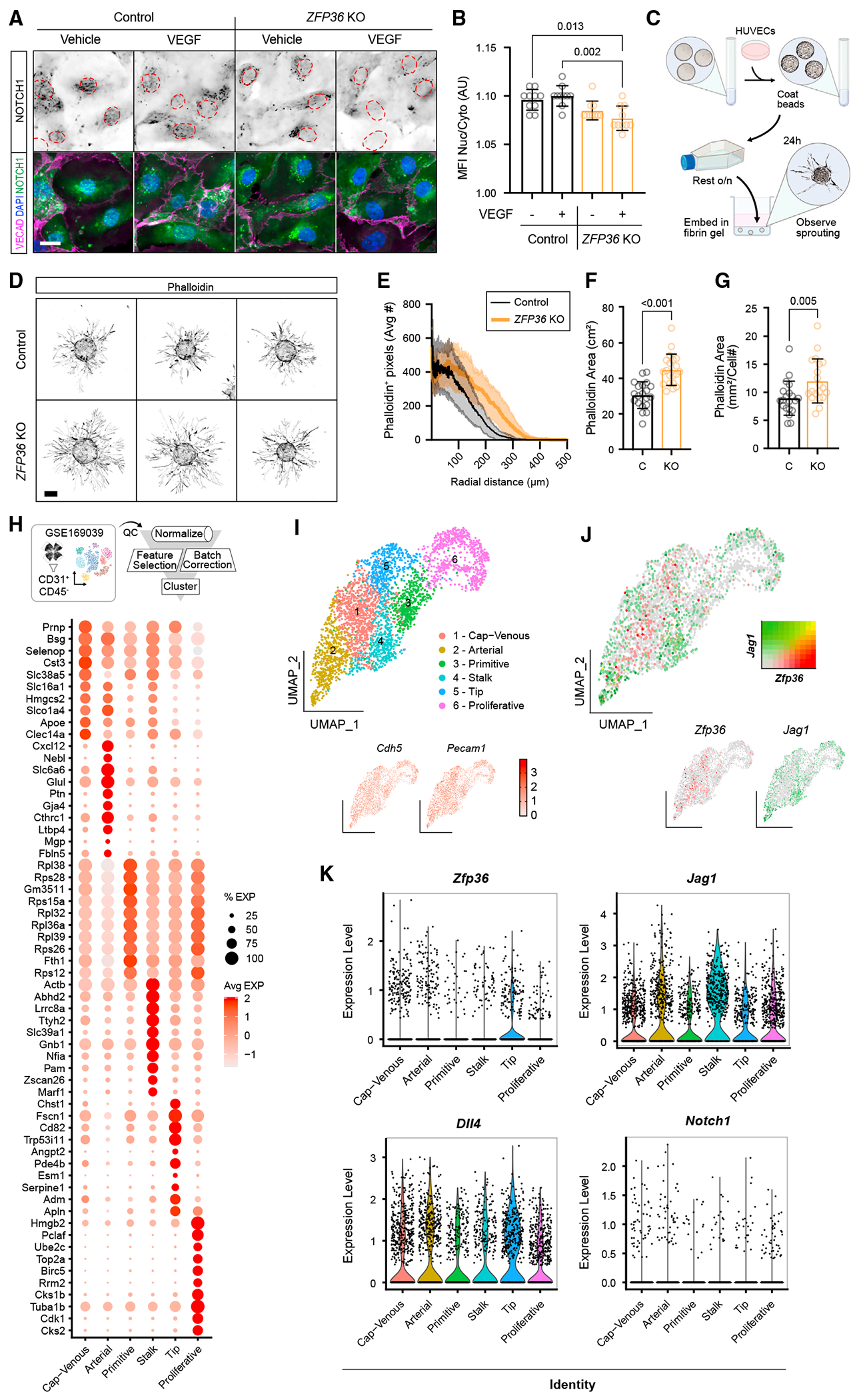
(A) Representative immunofluorescence of NOTCH1, VECAD, and DAPI in CRISPR control and ZFP36 KO HUVECs with or without VEGF for 1 h (scale bar, 25 μm). Dashed outlines indicate individual nuclei. The first row shows monochromatic NOTCH1 to facilitate visualization.
(B) Ratio of nuclear to cytosolic NOTCH1 MFI ± SD per field of view (n = 10 fields). Statistics: Kruskal-Wallis with post hoc Dunn’s multiple-comparisons test.
(C) Schematic of the microcarrier bead angiogenesis assay.
(D) Representative images of the angiogenesis assay phalloidin staining results from CRISPR empty and ZFP36 KO HUVECs post 24 h (scale bar, 100 μm). (E–G) CRISPR control and ZFP36 KO HUVEC microcarrier bead assay quantification of phalloidin distance relative to bead border (E), overall phalloidin area (sans bead area) (F), and overall phalloidin area normalized to cell number (G). Data bars and error lines indicate mean ± SD (n = 20 technical replicates). Statistics: Mann-Whitney test.
(H) Schematic of data processing from publicly available scRNA-seq data from CD31-enriched P6 and P10 WT retina (GSE169039) with corresponding dot plot for identification of endothelial clusters.
(I) UMAP cluster analysis of EC identity and the endothelial markers Cdh5 and Pecam1.
(J) Relative individual Zfp36 (red) and Jag1 (green) expression in endothelial clusters with corresponding feature-blended UMAP.
(K) Violin plots showing Zfp36, Jag1, Dll4, and Notch1 transcripts in the respective cell clusters.
The mouse retina is a classic site where to visualize vascular morphogenesis in a planar manner. By P6, the angiogenic front can be noted at the edge, with progressive differentiation toward the center of the retina.39 Using this platform, we first sought to determine whether Zfp36 was expressed in the retinal endothelium at the relevant time points. For this, we took advantage of available single-cell RNA-seq (scRNA-seq) datasets published by two independent groups.40,41 Critically, these results indicated that transcripts for Zfp36 (and Jag1) were present in developing retina ECs (Figures 5H–5K). This information was imperative because none of the commercially available antibodies tested were able to faithfully recognize mouse Zfp36 by immunocytochemistry (using null mice as controls) in our hands. Distribution of Zfp36- and Jag1-expressing cells in the uniform manifold approximation and projection (UMAP) in relation to the subpopulations of ECs in the retina (Figures 5I and 5J, red dots for Zfp36 and green dots for Jag1) revealed no overlap between the transcripts (yellow dots), supporting the concept that Zfp36 decreases levels of Jag1 transcripts. Importantly, examining specific cluster expression levels demonstrated that Zfp36 was higher in tip cells, the region of greatest exposure to VEGF and where Jag1 was canonically low (Figure 5K). As anticipated, Dll4 was also highest in the tip cell population, while Jagged was highest in the stalk cell population. In contrast, Notch1 receptor levels were very low and distributed evenly across all clusters.
Next, we evaluated the distribution and levels of Jag1 in the retinal vasculature of Zfp36f/f Cdh5 Cre-negative and Cre-positive mice. As anticipated, endothelial deletion of Zfp36 yields higher levels of overall Jag1 within CD31-positive vasculature (Figures 6A–6C). The increase in Jag1 was equivalent at the angiogenic front and in the inner primary plexus (Figure 6D). We acknowledge, however, that by using immunocytochemistry (IHC), it is difficult to account for the cellular source of Jag1 expression. The inner plexus, for instance, includes pericytes and smooth muscle cells, which also express Jag1. When we performed IHC analysis on a known Zfp36 target and tip cell marker, uPAR,42 we observed significant upregulation primarily at the angiogenic front (Figures S5A and S5B). The angiogenic front is unique in that it is generally only composed of sprouting tip ECs, which express high levels of Zfp36 based on scRNA-seq analysis. Therefore, we expect that the cells responsible for protein increases in this area are endothelial. Higher levels of uPAR and Jag1 were even more pronounced when all three ZFP36 family members were inactivated in the endothelium (Figures S5C, S5D, 6E, and 6F). Jag1 expression was increased by nearly 3-fold at the angiogenic front and in the inner plexus (Figures 6E and 6F). Combined, these findings validate that Jag1 is indeed an important target of Zfp36 in vivo and further highlight that expression of Zfp36 in the tip cells prevents expression of Jag1 at the angiogenic front.
Figure 6. Deletion of Zfp36 in the endothelium increases Jag1 expression in the developing retina vascular plexus.
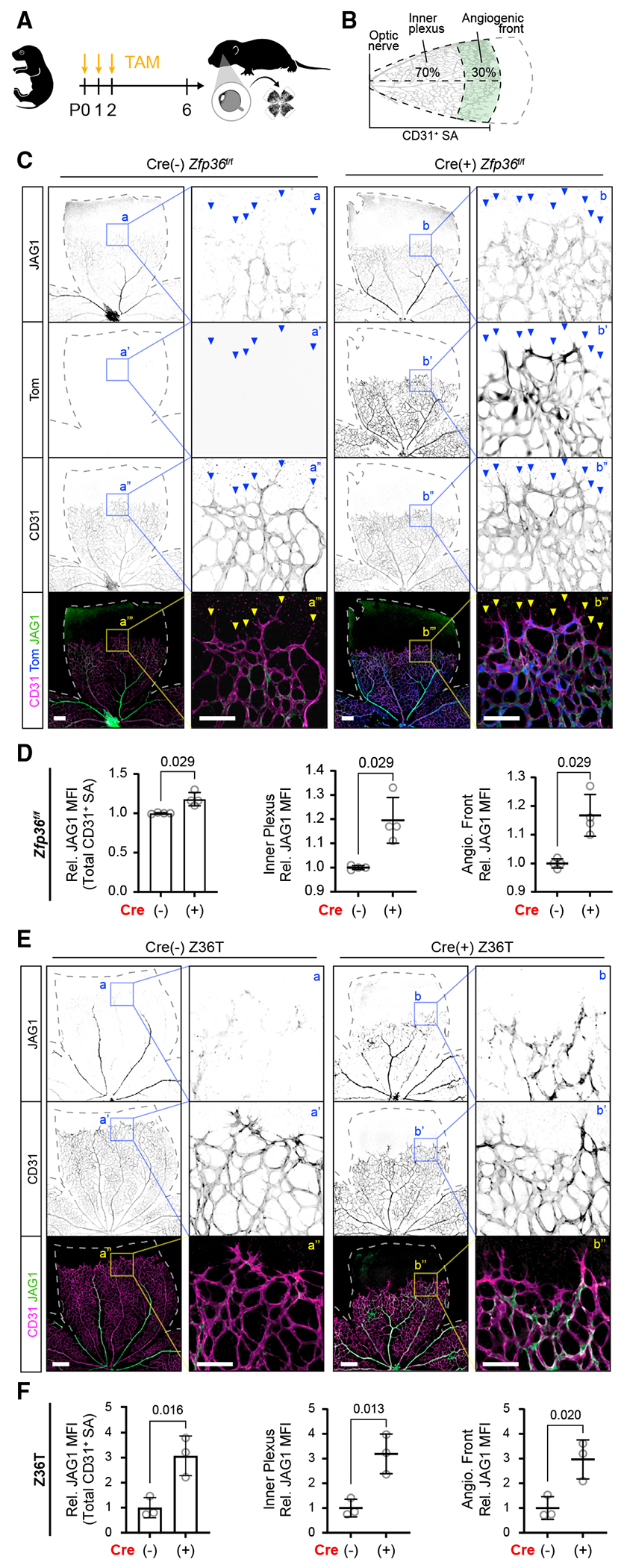
(A) Schematic of TAM feeding and post-natal retina harvest.
(B) Schematic of quantification of JAG1 MFI using the CD31+ surface area (SA) mask. This area was further subdivided into inner plexus and angiogenic front regions based on CD31+ SA convex hull area percentages as indicated.
(C) Representative JAG1, Tom, and CD31 immunohistochemistry of TAM-fed inducible Cdh5-Cre Zfp36f/f Tom reporter mice. Dashed lines outline the retina area, and arrows indicate angiogenic sprouts (low-magnification scale bar, 200 μm; higher-magnification scale bar, 100 μm).
(D) Quantification of JAG1 MFI ± SD within total CD31+ SA, inner plexus, and angiogenic front of TAM-fed inducible Cdh5-Cre Zfp36f/f mice relative to respective littermate cre(–) controls (n = 4 replicates each, comparisons from 3 independent litters). Statistics: Mann-Whitney test.
(E) Representative Jag1 and CD31 immunohistochemistry of TAM-fed inducible triple CDH5-Cre Zfp36f/fl1f/fl2f/f (Z36T) mice. Dashed lines outline the retina area (low-magnification scale bar, 300 μm; higher-magnification scale bar, 100 μm).
(F) Quantification of Jag1 MFI ± SD within CD31+ area inner plexus and angiogenic front from experimental and control littermates (n = 3 animals each). Statistics: unpaired two-tailed t test.
We then explored whether the balance of tip to stalk cell identity was compromised upon Zfp36 deletion. Using mice with endothelial-specific deletion of Zfp36 and Cre-negative controls, we found that outgrowth was impaired and the number of Esm1+Erg+ cells (tip cells) was increased in Zfp36ECKO retinas in comparison with littermate controls (Figures 7A–7D). Again, these effects were more pronounced in triple KO animals when all three Zfp36 family members were deleted in the endothelium (Figures S5E–S5H). Although in this model, attributing the delayed outgrowth solely to changes in tip and stalk cell dynamics is potentially confounded by the additional reduction in proliferation at the angiogenic front (Figure S5I and S5J). Changes in proliferation were not observed in the single-Zfp36 endothelial-null mice (Figures S5K and S5L).
Figure 7. Increased tip cells and delayed retinal angiogenesis from endothelial Zfp36 KO is rescued by Jag1 haploinsufficiency.
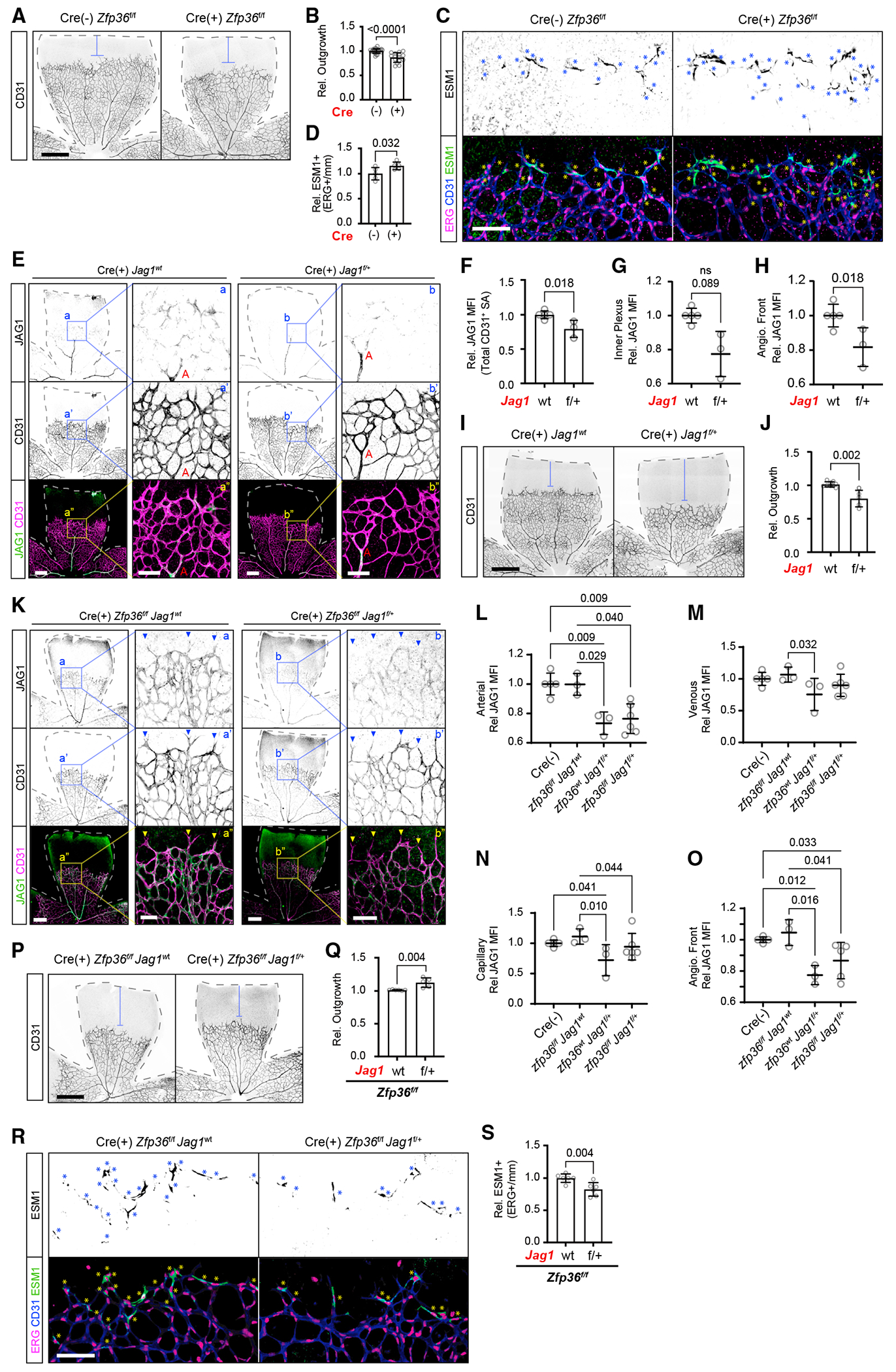
(A) Representative CD31 immunohistochemistry of TAM-fed inducible Cdh5-Cre Zfp36f/f mice. Dashed lines outline the retina area, and blue brackets indicate angiogenic outgrowth based on remaining retina area (scale bar, 500 μm).
(B) Quantification of outgrowth (CD31+ area/total area) averaged per animal of TAM-fed inducible Cdh5-Cre Zfp36f/f mouse retina relative to average littermate control (mean ± SD; n = 22 Cre(–) Zfp36f/f and 15 Cre(+) Zfp36f/f, derived from >3 independent litters). Statistics: Mann-Whitney test.
(C) Representative CD31, Esm1, and Erg immunohistochemistry of TAM-fed inducible Cdh5-Cre Zfp36f/f mice. Asterisks indicate Esm1+ Erg (scale bar, 100 μm).
(D) Quantification of Esm1+ Erg normalized to the width of the angiogenic front (mm) of TAM-fed inducible Cdh5-Cre Zfp36f/f mice relative to average littermate control (mean ± SD; n = 5 replicates each derived from 3 independent litters). Statistics: Mann-Whitney test.
(E) Representative JAG1 and CD31 immunohistochemistry of TAM-fed inducible CDH5-Cre(+) Jag1wt and Jag1f/+ mice. Dashed lines outline the retina area with the artery (A) labeled (low-magnification scale bar, 300 μm; higher-magnification scale bar, 100 μm).
(F–H) Quantification of JAG1 MFI ± SD within total CD31+ SA (F), inner plexus (G), and angiogenic front (H) in TAM-fed inducible CDH5-Cre(+) Jag1wt and Jag1f/+ mice (mean ± SD, n = 5 and 3, respectively). Statistics: Mann-Whitney test.
(I) Representative CD31 immunohistochemistry of TAM-fed inducible CDH5-Cre(+) Jag1wt and Jag1f/+ mice. Dashed lines outline the retina area, and blue brackets indicate angiogenic outgrowth based on remaining retina area (scale bar, 300 μm).
(J) Quantification of outgrowth (CD31+ area/total area) per retina of TAM-fed inducible CDH5-Cre(+) Jag1wt and Jag1f/+ mice (mean ± SD, n = 5 and 3, respectively). Statistics: Mann-Whitney test.
(K) Representative JAG1 and CD31 IHC of TAM-fed inducible CDH5-Cre(+) Zfp36f/f mice with either Jag1wt or Jag1f/+ alleles. Dashed lines outline the retina area (low-magnification scale bar, 300 μm; higher-magnification scale bar, 100 μm).
(L–O) Quantification of JAG1 MFI ± SD within CD31+ SA, subdivided by specific arterial (L), venous (M), or capillary (N) regions from the inner plexus in addition to the angiogenic front (O) in TAM-fed inducible CDH5-Cre(+) Zfp36f/fJag1wt, Zfp36wtJag1f/+, and Zfp36f/fJag1f/+ mice relative to littermate Cre(–) controls (n = 3, 3, 5, and 5 animals, respectively, derived from 4 independent litters). Statistics: Mann-Whitney test and Kruskal-Wallis with post hoc uncorrected Dunn’s test.
(P) Representative CD31 immunohistochemistry of TAM-fed inducible Cdh5-Cre(+) Zfp36f/f mice with either Jag1wt or Jag1f/+ alleles. Dashed lines outline the retina area, and blue brackets indicate angiogenic outgrowth based on remaining retina area (scale bar, 500 μm).
(Q) Quantification of outgrowth (CD31+ area/total area) averaged per animal of TAM-fed inducible Cdh5-Cre Zfp36f/f mice with either Jag1wt or Jag1f/+ retina relative to average littermate control (mean ± SD; n = 6 Jag1wt and 5 Jag1f/+; derived from 4 independent litters). Statistics: Mann-Whitney test.
(R) Representative CD31, Esm1, and Erg immunohistochemistry of TAM-fed inducible Cdh5-Cre(+) Zfp36f/f mice with either Jag1wt or Jag1f/+ alleles. Asterisks indicate Esm1+ Erg (scale bars, 100 μm).
(S) Quantification of Esm1+ Erg of TAM-fed inducible Cdh5-Cre Zfp36f/f mouse retina with either Jag1wt or Jag1f/+ alleles normalized to the width of the angiogenic front (mm) relative to average littermate control (mean ± SD; n = 7 replicates each, derived from 4 independent litters). Statistics: Mann-Whitney test.
Overall, our findings in vitro and in vivo were consistent with a role for ZFP36 in the regulation of Notch signaling by altering JAG1 levels and distribution. Then again, ZFP36 controls multiple other genes, including uPAR, which could likely participate in promoting the invading angiogenic front. Thus, additional experimentation was needed to determine to what degree the effect of Zfp36 was specifically due to JAG1 regulation instead of other genes, like uPAR. Along these lines, we considered a potential rescue experiment where genetic reduction in Jag1, in the context of Zfp36 deficiency, might return baseline levels of Jag1 and normalize biological readouts. These experiments could help confirm or deny direct causation. To achieve this, we first examined how removal of one Jag1 allele affected protein levels and vascular growth. Endothelial-specific heterozygous mice for Jag1 showed a reduction in Jag1 protein by about 20% in comparison to wild-type controls (Figures 7E–7H) and delayed angiogenic outgrowth (Figures 7I and 7J). Crosses between Cdh5-Cre Zfp36f/f and Jag1f/f animals eventually allowed us to obtain triple-transgenic mice with either one or two copies of Jag1 in the same litter. When compared to Jag1 wild type, lower levels of Jag1 protein were detected in Jag1f/+/Zfp36f/f/Cre+ mice in arteries, veins, capillaries, and the angiogenic front. In this context, inactivation of Zfp36 in Jag1 heterozygous mice resulted in an elevation in Jag1 protein, albeit not sufficiently high to return to baseline Jag1 (wild-type) levels (Figure 7K–7O). Despite this, the additional Jag1 protein was sufficient to rescue the biological phenotype of Zfp36ECKO. Specifically, both the increase in tip cells and the reduction in angiogenic outgrowth returned to baseline levels by Jag1 hap-loinsufficiency (Figures 7P–7S). Combined, these experiments corroborated that regulation of Jag1 by Zfp36 is biologically relevant. Moreover, as a supplemental merit, both compared experimental groups in Jag1 haploinsufficiency experiments were Cre positive and exposed to tamoxifen, providing an important control for concurrent presence of Cre and tamoxifen.43
DISCUSSION
In this study, we established that VEGF signaling induces ZFP36 in a transcriptionally dependent manner, resulting in a burst of protein expression that subsides relatively quickly. In turn, we identified Jag1 transcript as a target of ZFP36 and validated a role of this RNA binding protein in post-transcriptional stability of JAG1 in vitro and in vivo. We showed that ECs lacking ZFP36 exhibit constitutively higher levels of Jagged protein, reduced Notch1 signaling, and increased sprouting angiogenesis. In mice, endothelial-specific deletion of ZFP36 mirrored the effects found in vitro, with increased levels of JAG1 and increased number of Esm1-expressing tip cells in the immature vascular plexus that delays maturation and growth. This phenotype was mitigated by genetic reduction of Jag1 dosage through heterozygosity. From these observations, we present a critical functional role for ZFP36 as a post-transcriptional regulator of endothelial Notch signaling downstream of VEGF.
The ZPF36 family, which also includes ZFP36L1 and ZFP36L2, consists of RNA binding proteins involved in sequestration and/or decay of ARE-containing mRNAs.21,22 Through recognition and selective mRNA binding, they regulate metabolic pathways, inflammation, and immunity. 21–27 Furthermore, a role of ZFP36 in facilitating quick adaptive switches, particularly in inflammatory settings, has been highlighted in several pathological conditions, partially due to its rapid and robust induction by several cytokines.26,31,33–37,42,44 The findings presented here indicate that ZFP36 can also modulate developmental processes, such as angiogenesis, downstream of VEGF signaling. Importantly, we showed that induction of ZFP36 by VEGF is temporally restricted and presumably associated with the recycling of VEGFR2. Although we did not experimentally demonstrate the mechanisms responsible for ZFP36 reduction, it has been recognized that its activity, localization, and stability are regulated by phosphorylation.31 Intriguingly, some of the kinases involved in ZFP36 post-translational modifications are also downstream of VEGF, indicating a feedback loop of robust production and degradation. We suspect that these specific pulses of induction impose oscillatory cycles of regulation that refine and optimize the angiogenic process. In the case of Jag1, we found that its reduction titrates Notch signaling and fine-tunes the number of tip cells during angiogenic expansion.
The identification of an additional regulatory mechanism for Jag1 is far reaching, given the impact of the Notch signaling pathway in multiple biological and pathological settings. Within the context of a growing vascular plexus, these results fill a knowledge gap and contribute to explain the heterogeneous distribution of JAG1 in the developing vascular plexus (abundant in mature arteries but absent from the sprouting front).18 JAG1 is predominantly, though nonexclusively, expressed in adult arterial endothelium.45 In mature vessels, JAG1 mediates trans-Notch activation but mostly through heterotypic interactions with smooth muscle cells.46–48 Maturation of the vascular plexus requires acquisition of mural cells (pericytes and smooth muscle cells). It is in this context that high Jag1 expression is important. Endothelial JAG1 trans-activates Notch1 and Notch3 in smooth muscle cells, promoting mural cell investment and arterial fate.
Specific ligand enrichment is not unique to the mature vascular plexus; DLL4 is the predominant ligand for Notch in capillaries and in sprouting tip cells. In this manner, while JAG1 marks mature arterioles, DLL4 is a well-accepted marker for tip cells along with ESM1.49 At the angiogenic front, the expansion of a vascular plexus relies on the careful specification of tip and stalk cells. Tip cells at the edge of the plexus expand the network, while the adjacent stalk cells communicate to the rest of the network through the organization of tubular structures. VEGF and Notch coordinate this process, whereby VEGF drives Dll4 expression in tip cells that bind to adjacent Notch-expressing stalk cells, establishing their differential fates.50–52 Additionally, Dll4 is maintained through a Notch-dependent positive feedback loop.53 This relationship provides a feedforward mechanism by which Notch signaling can be propagated between adjacent ECs with very limited amount of ligand. In fact, this inter-endothelial continuous DLL4/Notch signaling is critical for endothelial quiescence and vascular stability.5,54–56 Thus, DLL4 is distributed throughout the plexus, regulating multiple aspects necessary for establishing and maintaining vascular networks. In addition to intrinsic Notch autoregulatory mechanisms, interactions with transcription factors, such as ERG, have been noted to reinforce Dll4 activation and repress Jag1.57 This mechanism, however, presents a paradox in that ERG expression is relatively uniform across the vascular plexus despite varying ligand expression. Therefore, the confined endothelial distribution of JAG1 has been acknowledged but not mechanistically understood.58 Critically, Dll4 transcripts have very few weak ARE motifs only noted to interact with ZFP36L1,59 which we found not to be induced by VEGF. Thus, our results depict a targeted auxiliary model for Jag1 suppression by ZFP36 that spatially matches its known expression and interaction profiles, hence satisfying a potential mechanism for ligand segregation.
The combinatorial contribution of Notch ligands in the endothelium remains poorly understood. Notably, EC-specific inactivation of each ligand yields distinct phenotypes, highlighting nonredundant ligand contributions to vascular morphogenesis. Deletion of Dll4 leads to excess of sprouts and absence of a differentiated vascular network, a phenotype that is replicated by inactivation of Notch1 in ECs.6,10,60 In contrast, EC-specific deletion of Jag1 yields a considerably reduced vascular plexus with a paucity of tip cells.19 These findings support the necessary interplay between ligands and a model where JAG1 functions antagonistically to canonical trans-EC DLL4 ligand binding, titrating signaling potential. In this context, we propose that ZFP36 acts as a rheostat for this balance by imposing pulses of transient Jag1 suppression that provide feedforward regulation and adequate vascular morphogenesis.
Limitations of the study
ZFP36L1 and ZFP36L2 have similar affinities for ARE motifs; thus, there is potential for functional redundancy in transcript regulation.61 Indeed, we observed more drastic vascular defects in endothelial-specific ZFP triple KO animals, supporting such potential redundancy. However, such a phenotype could also emerge from alternative targets because others have noted changes in metabolism or the cell cycle in response to the genetic silencing of these other family members.25,26,62–64 In addition, and in relation to ZFP36 specifically, we also acknowledge the confounding probability of additional ZFP36 RNA-binding targets and addressed this caveat through two key experiments. First, we examined ZFP36 expression in the developing vascular plexus of the retina and found, on a single-cell basis, that Jag1 and Zfp36 were, for the most part, mutually exclusive. Next, we attempted to genetically rescue the increased Jag1 imposed by the absence of ZFP36 through inactivating one Jag1 allele. Indeed, using a Jag1 heterozygous background, absence of ZFP36 did not increase the number of tip cells; instead, they approach wild-type levels. This genetic rescue corroborates that the observed phenotype was indeed due to overexpression of Jag1.
The link between VEGF and ZFP36 induction uncovers another facet in our understanding of the molecular crosstalk that drives angiogenesis. Importantly, while we mainly utilized developmental models in this study, an analogous crosstalk is most likely present in pathological angiogenesis, where a similar paradigm of tip-talk cells is at play.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, M. Luisa Iruela-Arispe, Ph.D. (arispe@northwestern.edu).
Materials availability
Materials used in this study are commercially available.
Data and code availability
RNA-seq data generated in this study has been deposited at GEO (GSE235462). This paper also analyzes existing, publicly available data. The accession numbers for these datasets are listed in the key resources table.
Other original data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-BRF1/2 | Cell Signaling Tech | Cat#2119; RRID: AB_10695874 |
| Rat monoclonal anti- CD11b, PerCP-Cy5.5 conjugated | Biolegend | Cat#101228; RRID: AB_893232 |
| Rat monoclonal anti-CD31 | BD Biosciences | Cat#557355; RRID: AB_396660 |
| Rat monoclonal anti-CD31, FITC conjugated, Clone MEC 13.3 | BD Biosciences | Cat#553372; RRID: AB_394818 |
| Hamster monoclonal anti-CD31 (clone 2H8) | Bogen65 | N/A |
| Rat monoclonal anti-CD45, Brilliant Violet 421 conjugated | Biolegend | Cat#109831; RRID: AB_10900256 |
| Goat polyclonal anti-CDH5 | R&D Systems | Cat#AF938; RRID: AB_355726 |
| Rabbit monoclonal anti-CDH5 | Cell Signaling Tech | Cat#2500; RRID: AB_10839118 |
| Hamster monoclonal anti-CDH5 (clone Hec1) | Ali et al.66 | N/A |
| Rabbit monoclonal anti-ERG | Abcam | Cat#Ab115555; RRID: AB_10898854 |
| Rabbit monoclonal anti-ERG- Alexa Fluor 647 | Abcam | Cat#Ab196149 |
| Rabbit monoclonal anti-ERG- Alexa Fluor 488 | Abcam | Cat#Ab196374 |
| Goat polyclonal anti-ESM1 | R&D Systems | Cat#AF1999; RRID: AB_2101810 |
| Rabbit polyclonal anti-gamma-Tubulin | Abcam | Cat#11321; RRID: AB_297926 |
| Mouse monoclonal anti-GAPDH | Millipore Sigma | Cat#MAB374; RRID: AB_2107445 |
| Rabbit polyclonal anti-phospho-Histone H3 (Ser10) | Cell Signaling Tech | Cat#9701; RRID:AB_331535 |
| Rabbit monoclonal anti-JAG1 | Cell Signaling Tech | Cat#2620; RRID: AB_10693295 |
| Goat polyclonal anti-JAG1 | Sigma-Aldrich | Cat#J4127; RRID: AB_260348 |
| Mouse monoclonal anti-JAG1 (E–12) | Santa Cruz | Cat#Sc-390177; RRID: AB_2892141 |
| Rabbit monoclonal anti-NICD (Val1744) | Cell Signaling Tech | Cat#4147; RRID: AB_2153348 |
| Rabbit monoclonal anti-Notch1 | Cell Signaling Tech | Cat#3608; RRID: AB_2153354 |
| Rabbit monoclonal anti-phospho-VR2 (Tyr1175) | Cell Signaling Tech | Cat#2478; RRID: AB_31377 |
| Goat polyclonal anti-uPAR | R&D Systems | Cat#AF534; RRID: AB_2165351 |
| Rabbit monoclonal anti-VR2 | Cell Signaling Tech | Cat#2479; RRID: AB_2212507 |
| Rabbit monoclonal anti-ZFP36 | Cell Signaling Tech | Cat#71632; RRID: AB_2799806 |
| Rabbit polyclonal anti-ZFP36 | Millipore Sigma | Cat#ABE285; RRID: AB_11205589 |
| Bacterial and virus strains | ||
| Ad-Cre-GFP | Vector Biolabs | Cat#1700 |
| Ad-GFP | Vector Biolabs | Cat#1060 |
| lentiCRISPR v2 | Sanjana et al.67 | Cat#52961; RRID:Addgene_52961 |
| Biological samples | ||
| Human umbilical vein endothelial cells | Lonza | Cat# C2517A; Lot# 18TL072772, 18TL072771, 18TL061650, 21TL169354, 21TL195719, 20TL293905, 0000632996, 0000296747 |
| Human umbilical vein endothelial cells, pooled | Lonza | Cat#C2519A; Lot#0000460587 |
| Human aortic endothelial cells | University of California, Los Angeles | N/A |
| Human Dermal Microvascular Endothelial Cells | PromoCell | Cat#C-12212 |
| Chemicals, peptides, and recombinant proteins | ||
| ZM323881 hydrochloride | Tocris | Cat#2475/1 |
| Actinomycin D | Invitrogen | Cat#A7592 |
| VEGFA165 | Peprotech | Cat#100-20 |
| Recombinant human Jagged1 Fc Chimera | R&D systems | Cat#1277 |
| Human IgG, Fc fragment | Sigma-Aldrich | Cat#AG714 |
| eBioscience 1xRBC lysis buffer | Invitrogen | Cat#00-4333-57 |
| Methalcholine chloride, 100.4% | MP Biomedicals | Cat#0219023105 |
| Lipofectamine 2000 | Thermo Fisher | Cat#11668019 |
| Restore Western Blot Stripping Buffer | Thermo Fisher | Cat#21059 |
| Dynabeads Protein A | Thermo Fisher | Cat#10001D |
| ProLong Gold Antifade Mountant | Thermo Fisher | Cat#P36930 |
| Puromycin | Invitrogen | Cat#ANTPR1 |
| Polybrene | Millipore Sigma | Cat#TR-1003-G |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11873580001 |
| Recombinant Human Jagged1 Fc Chimera Protein, CF | R&D Systems | Cat#1277-JG-050 |
| Cytodex 3 microcarriers | Cytiva | Cat#17048501 |
| Fibronogen | Sigma-Aldrich | Cat#F-8630 |
| Aprotinin | Sigma-Aldrich | Cat#A-1153 |
| Thrombin | Sigma-Aldrich | Cat#T-3399 |
| 2.5% Trypsin, 10x | Corning | Cat#MT25054CI |
| Paraformaldehyde (PFA) 4%, in PBS | Thermo Fisher | Cat#AAJ61899AP |
| Triton X-100 | Thermo Fisher | Cat#BP151500 |
| Tween 20 | Sigma-Aldrich | Cat#P9416 |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat#017-000-121 |
| Phalloidin-AF488 | Thermo Fisher | Cat#A12379 |
| Hoechst 33342 | Enzo | Cat#ENZ-52401 |
| Critical commercial assays | ||
| RNeasy Plus Micro Kit | Qiagen | Cat#74034 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| TruSeq Total RNA library prep kit | Illumina | Cat#20020594 |
| Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit | Bio-Rad | Cat#1704271 |
| Thermo Scientific Pierce Detergent Compatible Bradford Assay | Fisher Scientific | Cat#PI23246 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat#23227 |
| 4–20% Mini-PROTEAN TGX Precast Protein Gels | Bio-Rad | Cat#4561095, 4561094 |
| 4–20% Criterion TGX Stain-Free Protein Gel | Bio-Rad | Cat#5678093 |
| Liver dissociation kit, mouse | Miltenyi | Cat#130-105-807 |
| Superscript III First-Strand Synthesis System | Invitrogen | Cat#18080051 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat#1725274 |
| Dual-Glo Luciferase Assay System | Promega | Cat#E2940 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher | Cat#L3000015 |
| Deposited data | ||
| HUVEC RNAseq | This paper | GSE235462 |
| Retinal single-cell mRNA profiles of WT P6 mice (GSM5350878) | Zarkada et al.40 | GSE175895 |
| eCLIP-seq | Cicchetto et al.26 | PRJNA943291 |
| Experimental models: Cell lines | ||
| Lenti-X 293T | Takara | Cat#632180 |
| Experimental models: Organisms/strains | ||
| Mouse: Tg(Cdh5-cre/ERT2)1Rha | (Sorensen et al.68 | N/A |
| Mouse: Zfp36f/f | Qiu et al.69 | N/A |
| Mouse: Gt(ROSA)26Sortm14(CAG tdTomato)Hze | Jackson Laboratory | RRID: IMSR_JAX:007914 |
| Mouse: Jag1f/f | Mancini et al.70 | N/A |
| Mouse: Zfp36f/fZl1f/fZl2f/f | This paper | N/A |
| Oligonucleotides | ||
| qPCR primers (Table S1) | See Table S1 | N/A |
| gRNA ZFP36 Forward: CACCGTGCCCGTGCCATCCGACCA | This paper | N/A |
| gRNA ZFP36 Reverse: AAACTGGTCGGATGGCACGGGCAC | This paper | N/A |
| Recombinant DNA | ||
| pLV[Exp]-Puro-EF1A>NLS-EGFP: {mJag1_3′ UTR_565bp} | This Paper - Vector Builder custom order | Cat#VB220720-1510tzf |
| pLV[Exp]-Puro-EF1A>NLS-EGFP: {mJag1_3′ UTR_517bp(del 48bp)} | This Paper - Vector Builder custom order | Cat#VB220720-1515agk |
| pRP[Exp]-Hygro-CAG-Luciferase&{hJAG1_3UTR_1814bp} | This Paper – Vector Builder custom order | Cat#VB230730-1401fzh |
| pRP[Exp]-Hygro-CAG-Luciferase&{hJAG1_3UTR’(del 331bp-429bp)} | This Paper – Vector Builder custom order | Cat#VB230807-1714sjb |
| psPAX2 | Addgene | Cat# 12260; RRID: Addgene_12260 |
| pMD2.G | Addgene | Cat# 12259; RRID: Addgene_12259 |
| pCMV-GFP | Matsuda et al.71 | Cat#11153; RRID: Addgene_11153 |
| Software and algorithms | ||
| FIJI | Schindelin et al.72 | RRID:SCR_002285 |
| Imaris (v9.9.0) | Bitplane | RRID:SCR_007370 |
| Seurat (v4.1.1) | Hao et al.73 | RRID:SCR_016341 |
| NIS Elements | Nikon | RRID:SCR_014329 |
| Image Lab Software | BioRad | RRID:SCR_014210 |
| CFX Manager (v3.1) | BioRad | RRID:SCR_017251 |
| STAR (v2.7.3) | Dobin et al.74 | RRID:SCR_004463 |
| FlowJo | BD Biosciences | RRID:SCR_008520 |
| BioRender | BioRender | RRID:SCR_018361 |
| Adobe Illustrator | Adobe | RRID:SCR_010279 |
| Prism 9 | Graphpad | RRID:SCR_002798 |
| UMI-tools | GitHub | RRID:SCR_017048 |
| PureCLIP | GitHub | https://github.com/skrakau/PureCLIP |
| DESeq2 | GitHub | RRID:SCR_015687 |
| FastQC | GitHub | RRID:SCR_014583 |
| AREsite2 | Gruber et al.75 | http://nibiru.tbi.univie.ac.at/AREsite2/welcome |
| BioTek Gen5 | Agilent | RRID:SCR_017317 |
| Wound_healing_size_tool | Suarez-Arnedo et al.76 | https://github.com/AlejandraArnedo/Wound-healing-size-tool/wiki |
| Other | ||
| HiSeq3000 | Illumina | Cat#SY-401-3001 |
| IncuCyte S3 Live Cell Analysis System | Sartorius | Cat#4647; RRID:SCR_023147 |
| Glass bottom well-plates | Cell Vis | Cat# P06-1.5H-N, P12-1.5H-N, P24-1.5H-N |
| BioTek Synergy H1 Microplate Reader | Agilent | Cat#SH1M2-SN; RRID:SCR_019748 |
| Incucyte Wound Maker 96-Tool | Sartorius | Cat# 4563 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
Endothelial cells (HUVECs, HAECs, and HDMECs) were cultured with 10% fetal bovine serum (FBS) (Omega Scientific #FB-11) in either MCDB-131 (VEC Technologies; MCDB131-WOFBS) or EBM-2 Basal Medium (Lonza; CC-3156) supplemented with 1% penicillin/streptomycin and EGM-2 supplements (Lonza; CC-4176) sans kit FBS. HEK293T and MEF cell lines were cultured in DMEM containing 1 mM pyruvate and 4 mM glutamine supplemented with 10% FBS and 1% penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2 and atmospheric oxygen. MEF isolation, immortalization and adenoviral treatments were performed as previously described.26
Mouse models
All animal procedures were approved and performed in accordance with Northwestern University Institutional Animal Care and Use Committee. All mouse strains were maintained on a C57BL/6J background, both female and male mice were used for tissue analysis. For inducible endothelial deletion of Zfp36, Tg(Cdh5-Cre/ERT2)1Rha mice77 were crossed with Zfp36f/f mice or with Zfp36f/fl1f/fl2f/f mice.69,78 These lines were then further crossed with R26RTd Cre reporter line (Gt(ROSA)26Sortm14(CAG tdTomato)Hze).79 For developmental post-natal comparisons at P6, internal littermate controls were achieved by crossing Cre(−) with Cre(+) parents. For Jag1 dosage experiments, mice Cdh5-Cre(+) and Zfp36f/f were further crossed with Jag1f/f mice80 and backcrossed in order to obtain Cdh5-Cre(+) litters containing Zfp36f/fJag1wt and Zfp36f/fJag1f/+ for comparison. Three consecutive days of tamoxifen administration was given by oral gavage (5uL at 20 mg/mL) starting on day of birth. Adult studies were conducted on age mice at 120 days.
METHOD DETAILS
Cell culture treatments
VEGF and serum stimulation
Stock recombinant Human VEGFA165 (VEGF) (Peprotech) was prepared in sterile molecular grade water in single-use aliquots. After washing cells once with serum-free media cells were serum starved overnight followed by direct application of VEGF at a final concentration of 100 ng/mL for indicated times. In the case of 10% FBS treatments (MEF experiments), FBS was re-introduced as in standard culture conditions (10% final concentration) post-overnight serum starvation.
VEGF burst experiment
Cultured HUVECs were washed once in serum-free media and serum-starved overnight before adding VEGF directly to culture media as described above. At indicated times cells were washed twice with serum-free media to remove VEGF. Leaving on the second wash, all cultures were harvested for protein after total time of 1 h to examine ZFP36 induction.
Inhibitor treatments
ZM323881 hydrochloride (ZM323) (Tocris) and Actinomycin D (ActD) (Invitrogen) stock solutions were prepared according to manufacturer’s instructions. After overnight serum starvation described above, ZM323 or ActD was added directly to culture media 1 h prior to VEGF time course experiments at a final concentration of 1 μM and 10 μg/mL respectively.
Lentivirus production and infection
Lentivirus particles were produced in 293T cells by co-transfecting with a construct of interest and second-generation packaging plasmids psPAX2 and pMD2.G (Addgene; gift from Didier Trono). At 24 and 48 h, lentivirus-rich medium was collected and pooled. After centrifuging 1250 rpm for 5 min, supernatant was filtered (0.22 μm) and aliquot for single use and frozen at −80°C for long term storage. For transduction on HUVECs, aliquots were applied directly to cells overnight in the presence of 8 μg/mL polybrene. Infected cells were cultured in regular growth medium for 48 h prior to beginning puromycin (1 mg/mL) antibiotic selection where applicable.
Migration assay
Cells were seeded in 96-well image lock plates (Essen Bioscience) to confluence. After waiting for adherence (4-6h) scratches were made in each well using Incucyte woundmaker tool according to manufacturer instructions. Cells were washed twice with culture media to remove cell debris. Automated time course imaging was performed using Incucyte S3 (Sartorius) in humidified incubator at 37°C with 5% CO2 set to capture each well every hour until wound closure. Analysis was performed using ImageJ software plugin for wound healing analysis.81
Proliferation assay
Control or ZFP36 KO HUVECs infected with lentivirus CMV-GFP were seeded in 96-well plates at 5,000 cells per well. Automated image capture was performed using Incucyte S3 (Sartorius) in humidified incubator at 37°C with 5% CO2 and set to capture both phase and GFP images every hour until confluence was reached. Media was changed every other day as necessary. Confluence analysis was performed using ImageJ. Briefly, GFP images were binarized to calculate Area Fraction of endothelial coverage in the field of view over time.
JAG1-UTR construct experiments and staining
Lentivirus constructs were manufactured through VectorBuilder custom lentivirus gene expression packaging service. HUVECs were infected according to manufacturer’s instructions using MOI 4 with 8 μg/mL final concentration polybrene. HUVEC infection and puromycin selection were otherwise performed as described above. After VEGF stimulation, Cells were fixed with 4% (w/v) paraformaldehyde diluted in 1xPBS (PFA) for 10 min at room temp. After three 5 min washes in 1xPBS cells were directly stained for 30 min with Alx647 conjugated-VE-CAD clone Hec1 antibody (graciously provided by Dr. William Muller - Northwestern University, Chicago) and DAPI. After three additional 5 min washes in 1xPBS cells were directly imaged.
Luciferase reporter assay
Plasmid constructs were manufactured through VectorBuilder custom design vector services. Control or ZFP36 KO HUVECs were trypsinzed and transfected in suspension using Lipofectamine 3000 (ThermoFisher) according to manufacturer’s instructions and seeded to opaque-white 96-well clear-bottom culture plates. For each L3000 reaction, a Renilla Luciferase reporter was co-transfected 10:1 (Firefly to Renilla) for normalization of transfection efficiency. After 24h, plates were media changed and cells were allowed to recover post-transfection. Luciferase activity was measured the following day using DualGlo Luciferase Assay System (Promega) according to the manufacturer’s instructions. For VEGF stimulation, serum starvation was performed overnight after the 24h media change and VEGF was added the following day at indicated times before measurement.
Jag1-Fc coated culture plates
Recombinant human Jagged1 Fc Chimera (R&D systems) was reconstituted according to manufacturer’s instructions (200 κg/mL in sterile 1xPBS) and stored frozen at −80°C in single use aliquots. This stock or control human IgG, Fc fragment (Sigma-Aldrich) was diluted to final concentration of 10 μg/mL and incubated rocking overnight at 4°C in respective wells of 12-well culture plate. Wells were washed once with sterile 1xPBS and cells were seeded in wells to confluence. After 24 h, cell lysates were harvested for immunoblotting or quantitative RT-PCR.
Bulk RNA-seq analysis
Total RNA was extracted and purified using RNeasy mini kit (Qiagen) according to manufacturer’s instructions. RNA libraries were prepared using the Illumina TruSeq Total RNA library prep kit according to manufacturer’s instructions. Following barcoding, 18 samples per lane were sequenced on a HiSeq3000 using 50 bp single-end protocol. Reads were QC’d using FastQC in batch mode and mapped to the mouse genome (mm10) using STAR aligner version 2.3.1. The count data were normalized using DESeq2′s median of ratios method.82 Differential expression analysis was performed using DEseq2 with statistically significant genes called using adjusted p value cutoffs of less than 0.1.83
Cell lysis and immunoblotting
Cells were lysed in modified RIPA buffer (50 mM Tris pH8, 150 mM NaCl, 0.5% w/v Na-Desoxycholate, 1% w/v Triton X-100, 0.1% w/v SDS, 200 μM Na3VO4, 1x protease inhibitor cocktail) after washing once with cold 1xPBS. Proteins were separated by SDS-PAGE gradient (4–20%) gel (Bio-Rad) and transferred using 20 min semi-dry transfer using Trans-blot Turbo (Bio-Rad) onto nitrocellulose membranes and incubated overnight at 4°C with primary antibodies (see key resources table). HRP-conjugated secondary antibodies (1:10,000) were applied in species dependent manner at room temperature for 1 h. Immuno-complexes were detected by enhanced chemiluminescence with SuperSignal West Pico PLUS or Femto Maximum Sensitivity Substrate (ThermoFisher Scientific; PI34580 & PI34095) using ChemiDoc Imaging system (Bio-Rad Laboratories). Quantification of bands by densitometry analysis was performed using ImageLab Software (Bio-Rad Laboratories).
Quantitative RT-PCR
Total RNA from cell culture was extracted and purified using RNeasy mini kit (Qiagen) according to manufacturer’s instructions. Complementary DNA synthesis was performed with Superscript III reverse transcription First-Strand synthesis kit (Invitrogen). qPCR was performed for each sample in duplicate and gene expression was normalized with the housekeeping gene (HPRT) and relative expression calculated using the ΔΔCt method. Primer sets were synthesized by Integrated DNA Technologies, Inc (See Table S1 for primer sequences).
Immunocytochemistry
Staining and imaging
Cells cultured on glass bottom 6 or 12-well plates (Cell Vis) were fixed with 2% PFA for 15 min followed by permeabilization with blocking buffer (3% v/v normal donkey serum, 0.3% v/v Triton X-100, and 0.05% v/v Tween 20 diluted in 1xPBS) for 1 h at room temperature. Primary antibodies (listed in key resources table) were incubated overnight at 4°C diluted in blocking buffer. After washing 3 × 5 min with 1xPBS, fluorescent-tagged secondary antibodies were applied and incubated for 1 h at room temperature. After washing another 3 × 5min with 1xPBS, imaging was performed using A1R HD25 confocal microscope (Nikon) using ×20 objective. z stack scan feature was used to capture cell volume. For figure images, Denoise.AI (Nikon) was employed to remove Poisson shot noise.
Image analysis
Cell mean fluorescence intensity (MFI) measurements were quantified on non-denoised images using Imaris software (Imaris 9.9.0, Bitplane) ‘Cells’ feature. Where applicable, VECAD was used for cell borders and DAPI for nuclear area. MFI was calculated per cell or as an average of cells within field of view. In the case of Notch1, MFI was calculated as a fraction of nuclear/cytosolic as a proxy for pathway activation.
eCLIP-seq processing and analysis
This paper analyzes existing, publicly available data.26 Accession numbers for the datasets are listed in the key resources table. Reads were processed and aligned as previously described. Briefly, reads were aligned to the mouse mm10 genome. Then using a combination of umi_tools,84 cutadapt,85 and STAR74 sequences were aligned and de-duplicated. Peaks were called with pureclip,86 using an input control for each CLIP library. To identify ZFP36-specific peaks, peaks were identified in both the Zfp36/l1/l2 wildtype (WT) and triple knockout (TKO) MEF conditions for each library; peaks identified in TFKO.1 libraries were excluded from all downstream analyses.
AREsite alignment
Adenosine-uridine rich element (ARE) motifs in mouse Jag1 and human JAG1 3′ UTR were identified using publicly available database AREsite2 using all available default motifs.75 Bed files were extracted and aligned to genomes (mm10 and hg38 respectively) using Integrative Genomics Viewer. Overlapping sequences were collapsed for final presentation and aligned to available eCLIP binding peaks.
CLIP-qPCR
CLIP-qPCR validation of sequencing was performed as previously described.26 Briefly, Zfp36/l1/l2 wildtype (TFWT) or triple knockout (TFKO.1) MEFs were serum deprived overnight, stimulated for 40 min with 10% FBS, UV-irradiated, snap frozen, and stored at −80°C as described for eCLIP-seq experiments.26 At the time of lysis, DNase digestion was performed for 5 min at 37°C. Importantly, RNase inhibitor was added to the lysates and no RNase digestion step was performed to ensure recovery of full-length transcripts in complex with ZFP36. Protein quantification using a BCA assay was performed to ensure equal amounts of protein from TFWT or TFKO.1 MEF conditions were used for subsequent immunoprecipitation (IP). For each IP, 100 μL Protein A Dynabeads pre-conjugated with 25 μg anti-ZFP36 antibody (Millipore ABE285) was incubated rocking for 1 h at 4°C. On-bead Proteinase K (NEB) digestion was performed to release RNA, which was then purified with acid-phenol:chloroform, pH 4.5 (with IAA, 125:24:1) and concentrated to 20 μL final volume (Zymo). Isolated RNA was used for cDNA synthesis (Biorad iScript), then diluted 5-fold in nuclease-free water in preparation for qPCR using QuantStudio5 (Applied Biosystems). Relative ZFP36 binding enrichment to target mRNAs over background was calculated according to previously described methods deriving ΔΔCt with Rplpo as the reference gene for IP samples.87 Tuba1b was used as a negative control, Ptgs2 served as a positive control. Data are presented as ZFP36 target binding enrichment fold change relative to TFKO.1 cells; signal from TFKO.1 conditions is independent of ZFP36. (Primer sequences listed in key resources table).
Microcarrier bead angiogenesis assay
Microcarrier bead cell coating
Microbead angiogenesis assay was performed as previously described.77 Briefly, trypsinized HUVECs were coated on dextrancoated microcarrier beads at a ratio of approximately 400:1. Coated beads were rested overnight in culture media in humidified incubator at 37°C with 5% CO2 and atmospheric oxygen. The next day beads were washed and resuspended at 500 beads/mL in PBS solution containing 10 mg/mL fibrinogen & 15 U/mL aprotinin. This mixture was then carefully mixed inside 24-well glass bottom well containing a droplet of 10 U/mL thrombin to form and embed the HUVEC coated beads in fibrin gel. After polymerization, sprouting was allowed to occur for 24 h.
Staining and imaging
After 24 h, wells were fixed with 4% PFA followed by permeabilization with blocking buffer (3% v/v normal donkey serum, 0.3% v/v Triton X-100, and 0.05% v/v Tween 20 diluted in 1xPBS)for 1 h at room temperature. Phalloidin and Hoechst diluted in blocking buffer were incubated overnight at 4°C. Imaging was performed using a CSU-W1 confocal microscope (Nikon) with ×20 objective. z stack scan feature was used to capture entire bead and sprouting volume. For figure images, Denoise.AI (Nikon) was employed to remove Poisson shot noise.
Image analysis
Non-denoised z stack images were imported to ImageJ for analysis. From maximum intensity projections, bead area was manually masked for exclusion and individual channels were threshold (Li algorithm) to obtain overall measured phalloidin area and nuclei counts. To assess outgrowth distance, Euclidean distance maps (binary with 10 iterations) were generated from each bead mask. Binary phalloidin area was used to generate a selection area. This area was then restored on distance maps to generate a histogram of positive pixels over radial distance. Histogram data was compiled, and pixel distance converted to microns.
Liver endothelial isolation and flow cytometry
Aged tamoxifen-treated Cre(−) Zfp36f/f and Cre(+) Zfp36f/f tdTom reporter mice were sacrificed and perfused through the left ventricle with 10mL DMEM. Liver lobes were collected and washed once with HBSS and returned to fresh DMEM. Tissue was roughly minced with surgical scissors. Liver pieces were then digested using liver dissociation kit (Miltenyi) according to manufacturer’s instructions. Cell pellets were resuspended in 1 mL RBC lysis buffer and incubated for 4 min at RT with frequent vortexing. This reaction was quenched using 12 mL HBSS +10% FBS. Cells were pelleted at 300xg for 10 min. If the pellet was still red, RBC lysis was repeated however all subsequent incubations were only 1 min without vortexing. When the pellet was clean, it was resuspended in DMEM and cells counted with trypan blue. Cells were then plated at a concentration of 1.5×106 cells/well of 6-well plate. Cells were then placed in humidified incubator at 37°C with 5% CO2 for 1 h in MCDB-131 + 10% FBS to for adherent endothelial enrichment. After washing away non-adherent cells, adherent cells were trypsinzed and collected, pelleted, and stained in FACS buffer (0.5% bovine serum albumin, 1 mM EDTA and 0.05% sodium azide in PBS) with indicated antibodies for 30 min on ice. When secondaries were necessary, cells were washed twice with FACs buffer before incubating with secondary for 30 min on ice. For direct flow analysis, cells were washed and then fixed with 1% PFA before analysis using Cytek Northern Lights flow cytometer. Data were analyzed with FlowJo (BD Biosciences).
Aorta en face preparation
Mice were intraperitoneally injected with 100 μL methalcholine (10 mg/mL in 1xPBS) before sacrifice. 2% (w/v) PFA was perfused through the left ventricle of the heart; the aorta’s length was then measured before removal. Under a dissecting microscope, small branching vessels and adventitia were removed before the vessel was cut open longitudinally. Fileted tissue pieces were then pinned down in a 35 mm silicon-coated dish before proceeding with additional 2% (w/v) PFA fixation for 20 min at room temperature.
Aorta en face immunohistochemistry
Pinned aortae in silicon-coated dishes were washed 3 × 5 min with Hank’s balanced salt solution (HBSS) before incubating in blocking/permeabilization buffer (0.3% Triton X-100, 0.05% Tween 20, 3% Normal Donkey Serum in HBSS) for 1 h at room temperature. Aortae were incubated in primary antibody overnight at 4°C, washed 3 × 5 min with 1xHBSS, and then incubated in secondary antibody for 1 h at room temperature. To mount, aortae were stretched and pinned to its in situ length on a silicon-coated dish. A glass coverslip was then placed under the pinned aorta, ProLong Gold mounting reagent was used to cover the tissue, and then a second glass coverslip was placed over the tissue to seal. After curing overnight at room temperature, the glass-tissue-glass sandwich was transferred onto a glass microscope slide and imaged.
Retina immunohistochemistry
At P6, post-enucleation, whole eyes were fixed directly in 4% PFA for 15min, followed by retinal dissection in 2% PFA and total fixation time of 1 h in 2% PFA. The retinas were washed 3 × 5 min in 1xPBS before blocking for 1 h at room temperature. Primary antibodies were incubated overnight at 4 °C in blocking buffer. The following day retinas were washed 3 × 5 min in 1xPBS before secondary antibodies applied for 1 h at room temperature. Retinas were again washed 3 × 5 min in 1xPBS and flat-mounted on slides in ProLong Gold Antifade Mountant (Fisher Scientific #P36930).
IHC confocal imaging
Imaging was performed using A1R HD25 confocal microscope (Nikon). z stack and tile scan features were used to image the entire retinal surface and superficial plexus. Tiles were stitched into a single large image (NIS-Elements, Nikon). For figure images, Denoise.AI (Nikon) was employed to remove Poisson shot noise. Images were acquired using ×20 objective.
Image quantification and analysis
Aorta single field Z-stacks (non-denoised) were analyzed in ImageJ for JAG1 mean fluorescent intensity for each field of view. Manual selections were made in some instances in Cre(+) animals to only include Tom(+) cells. For outgrowth calculations, Denoise.ai (Nikon) processed retina images were analyzed with ImageJ (FIJI). Imported images were threshold to create binary images for total area and CD31 area. A median of 0.5 pixels was applied to remove noise. Convex hulls were generated from CD31 area and used to calculate percent outgrowth from total area. Percent outgrowth was then normalized relative to littermate controls. Using CD31 (BD) binary images created as described above, selection masks were generated to measure vasculature specific mean fluorescent intensity (MFI) of uPAR (R&D Systems) or JAG1 (Sigma) on corresponding un-denoised images. For vascular plexus subdivision, previously generated CD31 convex hulls were rescaled centered to 70%. This new scaled selection was used to divide CD31 binary into inner plexus and angiogenic front specific surfaces. Mean fluorescent intensity measurements were normalized by subtracting mean background fluorescent intensity and made relative to littermate controls. Counting ESM1+ and pHH3+ ECs was performed manually using Imaris spots feature. Positive counts were made for ERG that co-localized with the respective marker. Counts outside CD31+ area were excluded.
scRNA-seq processing and analysis
The expression matrices for the scRNA-seq samples were downloaded from GEO accession: GSE175895. All six samples were merged, and only the WT samples were used to make the plots. The R package Seurat73 (v4.1.1) was used to cluster the cells in the merged matrix. Cells with less than 100 genes or 500 transcripts or more than 10,000 transcripts or 15% of mitochondrial expression were first filtered out as low-quality cells. The NormalizeData function was used to normalize the expression level for each cell with default parameters. The FindVariableFeatures function was used to select variable genes with default parameters. The ScaleData function was used to scale and center the counts in the dataset. Principal component analysis (PCA) was performed on the variable genes. The RunHarmony function from the Harmony package was applied to remove potential batch effect among samples processed in different batches. Uniform Manifold Approximation and Projection (UMAP) dimensional reduction was performed using the RunUMAP function. The clusters were obtained using the FindNeighbors and FindClusters functions with the resolution set to 0.5. The cluster marker genes were found using the FindAllMarkers function. Pecam1 and Cdh5 expression were used to identify the endothelial cluster. Sub-clustering on ECs was performed with the same quality controls and Seurat steps described above. Heatmaps, violin plots and gene expression plots were generated by DoHeatmap, VlnPlot and featurePlot functions, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters were calculated using Prism 8 (Graphpad) and are specified in figure legends. Unless otherwise stated, we calculated p values for time course datasets using non-parametric Kruskal-Wallis with post-hoc Dunn’s multiple comparison test. For single comparisons we used Mann-Whitney tests. All significant results, defined as having a p value <0.05, are specified for each figure.
Supplementary Material
Highlights.
VEGF signaling induces expression of ZFP36
ZFP36, an RNA-binding protein, binds to the 3′ UTR Jagged1 mRNA
Endothelial inactivation of ZFP36 shows mislocalized and increased Jagged1
Inactivation of ZFP36 attenuates Notch1 signaling rescued by Jag1 haploinsufficiency
ACKNOWLEDGMENTS
We thank the Broad Stem Cell Research Center (BSCRC) and Jonsson Comprehensive Cancer Center (JCCC) at UCLA for sequencing libraries, the Feinberg School of Medicine, and the Robert H. Lurie Comprehensive Cancer Center (RHLCCC) and associated core facilities at Northwestern University (supported by Cancer Center Support grant NCI CA060553): the Mouse Histology and Phenotyping Core, RHLCCC Flow Cytometry Facility, and Center for Advanced Microscopy. A special thanks goes to Michelle Steel and Snezana Mirkov for assistance with husbandry and animal experiments. This work was supported by the NIH (R35HL140014 to M.L.I.-A.) and Ruth L. Kirschstein National Research Service Awards (GM007185 and T32HL069766) and a Whitcome Fellowship (UCLA) (to H.L.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113627.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, and Betsholtz C (2003). VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol 161, 1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawson ND, Vogel AM, and Weinstein BM (2002). Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127–136. [DOI] [PubMed] [Google Scholar]
- 3.Sainson RCA, Aoto J, Nakatsu MN, Holderfield M, Conn E, Koller E, and Hughes CCW (2005). Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 19, 1027–1029. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z-J, Shirakawa T, Li Y, Soma A, Oka M, Dotto GP, Fairman RM, Velazquez OC, and Herlyn M (2003). Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol. Cell Biol 23, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leslie JD, Ariza-McNaughton L, Bermange AL, McAdow R, Johnson SL, and Lewis J (2007). Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 134, 839–844. [DOI] [PubMed] [Google Scholar]
- 6.Hellström M, Phng L-K, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson A-K, Karlsson L, Gaiano N, et al. (2007). Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–780. [DOI] [PubMed] [Google Scholar]
- 7.Siekmann AF, and Lawson ND (2007). Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445, 781–784. [DOI] [PubMed] [Google Scholar]
- 8.Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, et al. (2010). Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol 12, 943–953. [DOI] [PubMed] [Google Scholar]
- 9.Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, and Wiegand SJ (2007). Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 104, 3219–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suchting S, Freitas C, le Noble F, Benedito R, Bréant C, Duarte A, and Eichmann A (2007). The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 104, 3225–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, and Thurston G (2006). Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444, 1032–1037. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Kennard S, and Lilly B (2009). NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res 104, 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, and Epstein JA (2008). Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 105, 1955–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedrosa A-R, Trindade A, Fernandes A-C, Carvalho C, Gigante J, Tavares AT, Diéguez-Hurtado R, Yagita H, Adams RH, and Duarte A (2015). Endothelial Jagged1 antagonizes Dll4 regulation of endothelial branching and promotes vascular maturation downstream of Dll4/Notch1. Arterioscler. Thromb. Vasc. Biol 35, 1134–1146. [DOI] [PubMed] [Google Scholar]
- 15.Lindsell CE, Shawber CJ, Boulter J, and Weinmaster G (1995). Jagged: a mammalian ligand that activates Notch1. Cell 80, 909–917. [DOI] [PubMed] [Google Scholar]
- 16.Briot A, Jaroszewicz A, Warren CM, Lu J, Touma M, Rudat C, Hofmann JJ, Airik R, Weinmaster G, Lyons K, et al. (2014). Repression of Sox9 by Jag1 is continuously required to suppress the default chondrogenic fate of vascular smooth muscle cells. Dev. Cell 31, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breikaa RM, Denman K, Ueyama Y, McCallinhart PE, Khan AQ, Agarwal G, Trask AJ, Garg V, and Lilly B (2022). Loss of Jagged1 in mature endothelial cells causes vascular dysfunction with alterations in smooth muscle phenotypes. Vascul. Pharmacol 145, 107087. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann JJ, and Luisa Iruela-Arispe M (2007). Notch expression patterns in the retina: An eye on receptor-ligand distribution during angiogenesis. Gene Expr. Patterns 7, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, and Adams RH (2009). The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124–1135. [DOI] [PubMed] [Google Scholar]
- 20.Garneau NL, Wilusz J, and Wilusz CJ (2007). The highways and by-ways of mRNA decay. Nat. Rev. Mol. Cell Biol 8, 113–126. [DOI] [PubMed] [Google Scholar]
- 21.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, and Blackshear PJ (1996). A Pathogenetic Role for TNFα in the Syndrome of Cachexia, Arthritis, and Autoimmunity Resulting from Tristetraprolin (TTP) Deficiency. Immunity 4, 445–454. [DOI] [PubMed] [Google Scholar]
- 22.Lai WS, Kennington EA, and Blackshear PJ (2002). Interactions of CCCH zinc finger proteins with mRNA: non-binding tristetraprolin mutants exert an inhibitory effect on degradation of AU-rich element-containing mRNAs. J. Biol. Chem 277, 9606–9613. [DOI] [PubMed] [Google Scholar]
- 23.Lykke-Andersen J, and Wagner E (2005). Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 19, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabian MR, Frank F, Rouya C, Siddiqui N, Lai WS, Karetnikov A, Blackshear PJ, Nagar B, and Sonenberg N (2013). Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nat. Struct. Mol. Biol 20, 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newton R, Shah S, Altonsy MO, and Gerber AN (2017). Glucocorticoid and cytokine crosstalk: Feedback, feedforward, and co-regulatory interactions determine repression or resistance. J. Biol. Chem 292, 7163–7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cicchetto AC, Jacobson EC, Sunshine H, Wilde BR, Krall AS, Jarrett KE, Sedgeman L, Turner M, Plath K, Iruela-Arispe ML, et al. (2023). ZFP36-mediated mRNA decay regulates metabolism. Cell Rep. 42, 112411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makita S, Takatori H, and Nakajima H (2021). Post-Transcriptional Regulation of Immune Responses and Inflammatory Diseases by RNA-Binding ZFP36 Family Proteins. Front. Immunol 12, 711633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald AI, Shirali AS, Aragón R, Ma F, Hernandez G, Vaughn DA, Mack JJ, Lim TY, Sunshine H, Zhao P, et al. (2018). Endothelial Regeneration of Large Vessels Is a Biphasic Process Driven by Local Cells with Distinct Proliferative Capacities. Cell Stem Cell 23, 210–225.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirali AS, Romay MC, McDonald AI, Su T, Steel ME, and Iruela-Arispe ML (2018). A multi-step transcriptional cascade underlies vascular regeneration in vivo. Sci. Rep 8, 5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks SA, Connolly JE, and Rigby WFC (2004). The role of mRNA turnover in the regulation of tristetraprolin expression: evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol 172, 7263–7271. [DOI] [PubMed] [Google Scholar]
- 31.Rezcallah MC, Al-Mazi T, and Ammit AJ (2021). Cataloguing the phosphorylation sites of tristetraprolin (TTP): Functional implications for inflammatory diseases. Cell. Signal 78, 109868. [DOI] [PubMed] [Google Scholar]
- 32.Whittles CE, Pocock TM, Wedge SR, Kendrew J, Hennequin LF, Harper SJ, and Bates DO (2002). ZM323881, a novel inhibitor of vascular endothelial growth factor-receptor-2 tyrosine kinase activity. Microcirculation 9, 513–522. [DOI] [PubMed] [Google Scholar]
- 33.Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukherjee N, Jacobs NC, Hafner M, Kennington EA, Nusbaum JD, Tuschl T, Blackshear PJ, and Ohler U (2014). Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 15, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore MJ, Blachere NE, Fak JJ, Park CY, Sawicka K, Parveen S, Zucker-Scharff I, Moltedo B, Rudensky AY, and Darnell RB (2018). ZFP36 RNA-binding proteins restrain T cell activation and anti-viral immunity. Elife 7, e33057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedlyarov V, Fallmann J, Ebner F, Huemer J, Sneezum L, Ivin M, Kreiner K, Tanzer A, Vogl C, Hofacker I, and Kovarik P (2016). Tristetraprolin binding site atlas in the macrophage transcriptome reveals a switch for inflammation resolution. Mol. Syst. Biol 12, 868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rappl P, Brüne B, and Schmid T (2021). Role of tristetraprolin in the resolution of inflammation. Biology 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.del Álamo D, Rouault H, and Schweisguth F (2011). Mechanism and significance of cis-inhibition in Notch signalling. Curr. Biol 21, R40–R47. [DOI] [PubMed] [Google Scholar]
- 39.Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, Seaward MR, Willett KL, Aderman CM, Guerin KI, et al. (2010). The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci 51, 2813–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zarkada G, Howard JP, Xiao X, Park H, Bizou M, Leclerc S, Künzel SE, Boisseau B, Li J, Cagnone G, et al. (2021). Specialized endothelial tip cells guide neuroretina vascularization and blood-retina-barrier formation. Dev. Cell 56, 2237–2251.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chavkin NW, Genet G, Poulet M, Jeffery ED, Marziano C, Genet N, Vasavada H, Nelson EA, Acharya BR, Kour A, et al. (2022). Endothelial cell cycle state determines propensity for arterial-venous fate. Nat. Commun 13, 5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Souhibani N,Al-Ahmadi W, Hesketh JE, Blackshear PJ, and Khabar KSA (2010). The RNA-binding zinc-finger protein tristetraprolin regulates AU-rich mRNAs involved in breast cancer-related processes. Oncogene 29, 4205–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brash JT, Bolton RL, Rashbrook VS, Denti L, Kubota Y, and Ruhrberg C (2020). Tamoxifen-Activated CreERT Impairs Retinal Angiogenesis Independently of Gene Deletion. Circ. Res 127, 849–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiedje C, Diaz-Muñoz MD,Trulley P, Ahlfors H, Laaß K, Blackshear PJ, Turner M, and Gaestel M (2016). The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 44, 7418–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Briot A, Civelek M, Seki A, Hoi K, Mack JJ, Lee SD, Kim J, Hong C, Yu J, Fishbein GA, et al. (2015). Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J. Exp. Med 212, 2147–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, and Epstein JA (2008). Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 105, 1955–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manderfield LJ, High FA, Engleka KA, Liu F, Li L, Rentschler S, and Epstein JA (2012). Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation 125, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breikaa RM, Denman K, Ueyama Y, McCallinhart PE, Khan AQ, Agarwal G, Trask AJ, Garg V, and Lilly B (2022). Loss of Jagged1 in mature endothelial cells causes vascular dysfunction with alterations in smooth muscle phenotypes. Vascul. Pharmacol 145, 107087. [DOI] [PubMed] [Google Scholar]
- 49.Rocha SF, Schiller M, Jing D, Li H, Butz S, Vestweber D, Biljes D, Drexler HCA, Nieminen-Kelhä M, Vajkoczy P, et al. (2014). Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ. Res 115, 581–590. [DOI] [PubMed] [Google Scholar]
- 50.Tung JJ, Tattersall IW, and Kitajewski J (2012). Tips, stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb. Perspect. Med 2, a006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benedito R, and Hellström M (2013). Notch as a hub for signaling in angiogenesis. Exp. Cell Res 319, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 52.Mack JJ, and Iruela-Arispe ML (2018). NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol 25, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caolo V, van den Akker NMS, Verbruggen S, Donners MMPC, Swennen G, Schulten H, Waltenberger J, Post MJ, and Molin DGM (2010). Feed-forward signaling by membrane-bound ligand receptor circuit: the case of NOTCH DELTA-like 4 ligand in endothelial cells. J. Biol. Chem 285, 40681–40689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dou G-R, Wang Y-C, Hu X-B, Hou L-H, Wang C-M, Xu J-F, Wang Y-S, Liang Y-M, Yao L-B, Yang A-G, and Han H (2008). RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J. 22, 1606–1617. [DOI] [PubMed] [Google Scholar]
- 55.Phng L-K, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, Ondr JK, Rao S, Lang RA, Thurston G, and Gerhardt H (2009). Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev. Cell 16, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mack JJ, Mosqueiro TS, Archer BJ, Jones WM, Sunshine H, Faas GC, Briot A, Aragón RL, Su T, Romay MC, et al. (2017). NOTCH1 is a mechanosensor in adult arteries. Nat. Commun 8, 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shah AV, Birdsey GM, Peghaire C, Pitulescu ME, Dufton NP, Yang Y, Weinberg I, Osuna Almagro L, Payne L, Mason JC, et al. (2017). The endothelial transcription factor ERG mediates Angiopoietin-1-dependent control of Notch signalling and vascular stability. Nat. Commun 8, 16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofmann JJ, and Iruela-Arispe ML (2007). Notch signaling in blood vessels: who is talking to whom about what? Circ. Res 100, 1556–1568. [DOI] [PubMed] [Google Scholar]
- 59.Desroches-Castan A, Cherradi N, Feige J-J, and Ciais D (2011). A novel function of Tis11b/BRF1 as a regulator of Dll4 mRNA 3’-end processing. Mol. Biol. Cell 22, 3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, et al. (2017). Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat. Cell Biol 19, 915–927. [DOI] [PubMed] [Google Scholar]
- 61.Ciais D, Cherradi N, and Feige J-J (2013). Multiple functions of triste-traprolin/TIS11 RNA-binding proteins in the regulation of mRNA biogenesis and degradation. Cell. Mol. Life Sci 70, 2031–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vogel KU, Bell LS, Galloway A, Ahlfors H, and Turner M (2016). The RNA-Binding Proteins Zfp36l1 and Zfp36l2 Enforce the Thymic β-Selection Checkpoint by Limiting DNA Damage Response Signaling and Cell Cycle Progression. J. Immunol 197, 2673–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuura Y, Noguchi A, Sakai S, Yokota N, and Kawahara H (2020). Nuclear accumulation of ZFP36L1 is cell cycle-dependent and determined by a C-terminal serine-rich cluster. J. Biochem 168, 477–489. [DOI] [PubMed] [Google Scholar]
- 64.Wu F, Kaczynski T, Matheson LS, Liu T, Wang J, Turner M, and Mu X (2020). Zfp36l1 and Zfp36l2 balances proliferation and differentiation in the developing retina. Preprint at bioRxiv. [Google Scholar]
- 65.Bogen S, Pak J, Garifallou M, Deng X, and Muller WA (1994). Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J. Exp. Med 179, 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ali J, Liao F, Martens E, and Muller WA (1997). Vascular endothelial cadherin (VE-cadherin): cloning and role in endothelial cell-cell adhesion. Microcirculation 4, 267–277. [DOI] [PubMed] [Google Scholar]
- 67.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sörensen I, Adams RH, and Gossler A (2009). DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113, 5680–5688. [DOI] [PubMed] [Google Scholar]
- 69.Qiu L-Q, Stumpo DJ, and Blackshear PJ (2012). Myeloid-specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. J. Immunol 188, 5150–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mancini SJC, Mantei N, Dumortier A, Suter U, MacDonald HR, and Radtke F (2005). Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood 105, 2340–2342. [DOI] [PubMed] [Google Scholar]
- 71.Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gruber AR, Fallmann J, Kratochvill F, Kovarik P, and Hofacker IL (2011). AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 39, D66–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suarez-Arnedo A, Torres Figueroa F, Clavijo C, Arbeláez P, Cruz JC, and Muñoz-Camargo C (2020). An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS One 15, e0232565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kempers L, van der Biji I, van Stalborch A-MD, Ponsioen B, and Margadant C (2021). Fast in vitro protocol for the visualization and quantitative high-throughput analysis of sprouting angiogenesis by confocal microscopy. STAR Protoc. 2, 100690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sörensen I, Adams RH, and Gossler A (2009). DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113, 5680–5688. [DOI] [PubMed] [Google Scholar]
- 79.Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, Pannell R, Siebel CW, MacDonald HR, De Keersmaecker K, et al. (2010). Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat. Immunol 11, 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lizama CO, Hawkins JS, Schmitt CE, Bos FL, Zape JP, Cautivo KM, Borges Pinto H, Rhyner AM, Yu H, Donohoe ME, et al. (2015). Repression of arterial genes in hemogenic endothelium is sufficient for haematopoietic fate acquisition. Nat. Commun 6, 7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suarez-Arnedo A, Torres Figueroa F, Clavijo C, Arbeláez P, Cruz JC, and Muñoz-Camargo C (2020). An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS One 15, e0232565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10. [Google Scholar]
- 86.Krakau S, Richard H, and Marsico A (2017). PureCLIP: capturing target-specific protein-RNA interaction footprints from single-nucleotide CLIP-seq data. Genome Biol. 18, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martindale JL, Gorospe M, and Idda ML (2020). Ribonucleoprotein immunoprecipitation (RIP) analysis. Bio. Protoc 10, e3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated in this study has been deposited at GEO (GSE235462). This paper also analyzes existing, publicly available data. The accession numbers for these datasets are listed in the key resources table.
Other original data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-BRF1/2 | Cell Signaling Tech | Cat#2119; RRID: AB_10695874 |
| Rat monoclonal anti- CD11b, PerCP-Cy5.5 conjugated | Biolegend | Cat#101228; RRID: AB_893232 |
| Rat monoclonal anti-CD31 | BD Biosciences | Cat#557355; RRID: AB_396660 |
| Rat monoclonal anti-CD31, FITC conjugated, Clone MEC 13.3 | BD Biosciences | Cat#553372; RRID: AB_394818 |
| Hamster monoclonal anti-CD31 (clone 2H8) | Bogen65 | N/A |
| Rat monoclonal anti-CD45, Brilliant Violet 421 conjugated | Biolegend | Cat#109831; RRID: AB_10900256 |
| Goat polyclonal anti-CDH5 | R&D Systems | Cat#AF938; RRID: AB_355726 |
| Rabbit monoclonal anti-CDH5 | Cell Signaling Tech | Cat#2500; RRID: AB_10839118 |
| Hamster monoclonal anti-CDH5 (clone Hec1) | Ali et al.66 | N/A |
| Rabbit monoclonal anti-ERG | Abcam | Cat#Ab115555; RRID: AB_10898854 |
| Rabbit monoclonal anti-ERG- Alexa Fluor 647 | Abcam | Cat#Ab196149 |
| Rabbit monoclonal anti-ERG- Alexa Fluor 488 | Abcam | Cat#Ab196374 |
| Goat polyclonal anti-ESM1 | R&D Systems | Cat#AF1999; RRID: AB_2101810 |
| Rabbit polyclonal anti-gamma-Tubulin | Abcam | Cat#11321; RRID: AB_297926 |
| Mouse monoclonal anti-GAPDH | Millipore Sigma | Cat#MAB374; RRID: AB_2107445 |
| Rabbit polyclonal anti-phospho-Histone H3 (Ser10) | Cell Signaling Tech | Cat#9701; RRID:AB_331535 |
| Rabbit monoclonal anti-JAG1 | Cell Signaling Tech | Cat#2620; RRID: AB_10693295 |
| Goat polyclonal anti-JAG1 | Sigma-Aldrich | Cat#J4127; RRID: AB_260348 |
| Mouse monoclonal anti-JAG1 (E–12) | Santa Cruz | Cat#Sc-390177; RRID: AB_2892141 |
| Rabbit monoclonal anti-NICD (Val1744) | Cell Signaling Tech | Cat#4147; RRID: AB_2153348 |
| Rabbit monoclonal anti-Notch1 | Cell Signaling Tech | Cat#3608; RRID: AB_2153354 |
| Rabbit monoclonal anti-phospho-VR2 (Tyr1175) | Cell Signaling Tech | Cat#2478; RRID: AB_31377 |
| Goat polyclonal anti-uPAR | R&D Systems | Cat#AF534; RRID: AB_2165351 |
| Rabbit monoclonal anti-VR2 | Cell Signaling Tech | Cat#2479; RRID: AB_2212507 |
| Rabbit monoclonal anti-ZFP36 | Cell Signaling Tech | Cat#71632; RRID: AB_2799806 |
| Rabbit polyclonal anti-ZFP36 | Millipore Sigma | Cat#ABE285; RRID: AB_11205589 |
| Bacterial and virus strains | ||
| Ad-Cre-GFP | Vector Biolabs | Cat#1700 |
| Ad-GFP | Vector Biolabs | Cat#1060 |
| lentiCRISPR v2 | Sanjana et al.67 | Cat#52961; RRID:Addgene_52961 |
| Biological samples | ||
| Human umbilical vein endothelial cells | Lonza | Cat# C2517A; Lot# 18TL072772, 18TL072771, 18TL061650, 21TL169354, 21TL195719, 20TL293905, 0000632996, 0000296747 |
| Human umbilical vein endothelial cells, pooled | Lonza | Cat#C2519A; Lot#0000460587 |
| Human aortic endothelial cells | University of California, Los Angeles | N/A |
| Human Dermal Microvascular Endothelial Cells | PromoCell | Cat#C-12212 |
| Chemicals, peptides, and recombinant proteins | ||
| ZM323881 hydrochloride | Tocris | Cat#2475/1 |
| Actinomycin D | Invitrogen | Cat#A7592 |
| VEGFA165 | Peprotech | Cat#100-20 |
| Recombinant human Jagged1 Fc Chimera | R&D systems | Cat#1277 |
| Human IgG, Fc fragment | Sigma-Aldrich | Cat#AG714 |
| eBioscience 1xRBC lysis buffer | Invitrogen | Cat#00-4333-57 |
| Methalcholine chloride, 100.4% | MP Biomedicals | Cat#0219023105 |
| Lipofectamine 2000 | Thermo Fisher | Cat#11668019 |
| Restore Western Blot Stripping Buffer | Thermo Fisher | Cat#21059 |
| Dynabeads Protein A | Thermo Fisher | Cat#10001D |
| ProLong Gold Antifade Mountant | Thermo Fisher | Cat#P36930 |
| Puromycin | Invitrogen | Cat#ANTPR1 |
| Polybrene | Millipore Sigma | Cat#TR-1003-G |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11873580001 |
| Recombinant Human Jagged1 Fc Chimera Protein, CF | R&D Systems | Cat#1277-JG-050 |
| Cytodex 3 microcarriers | Cytiva | Cat#17048501 |
| Fibronogen | Sigma-Aldrich | Cat#F-8630 |
| Aprotinin | Sigma-Aldrich | Cat#A-1153 |
| Thrombin | Sigma-Aldrich | Cat#T-3399 |
| 2.5% Trypsin, 10x | Corning | Cat#MT25054CI |
| Paraformaldehyde (PFA) 4%, in PBS | Thermo Fisher | Cat#AAJ61899AP |
| Triton X-100 | Thermo Fisher | Cat#BP151500 |
| Tween 20 | Sigma-Aldrich | Cat#P9416 |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat#017-000-121 |
| Phalloidin-AF488 | Thermo Fisher | Cat#A12379 |
| Hoechst 33342 | Enzo | Cat#ENZ-52401 |
| Critical commercial assays | ||
| RNeasy Plus Micro Kit | Qiagen | Cat#74034 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| TruSeq Total RNA library prep kit | Illumina | Cat#20020594 |
| Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit | Bio-Rad | Cat#1704271 |
| Thermo Scientific Pierce Detergent Compatible Bradford Assay | Fisher Scientific | Cat#PI23246 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat#23227 |
| 4–20% Mini-PROTEAN TGX Precast Protein Gels | Bio-Rad | Cat#4561095, 4561094 |
| 4–20% Criterion TGX Stain-Free Protein Gel | Bio-Rad | Cat#5678093 |
| Liver dissociation kit, mouse | Miltenyi | Cat#130-105-807 |
| Superscript III First-Strand Synthesis System | Invitrogen | Cat#18080051 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat#1725274 |
| Dual-Glo Luciferase Assay System | Promega | Cat#E2940 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher | Cat#L3000015 |
| Deposited data | ||
| HUVEC RNAseq | This paper | GSE235462 |
| Retinal single-cell mRNA profiles of WT P6 mice (GSM5350878) | Zarkada et al.40 | GSE175895 |
| eCLIP-seq | Cicchetto et al.26 | PRJNA943291 |
| Experimental models: Cell lines | ||
| Lenti-X 293T | Takara | Cat#632180 |
| Experimental models: Organisms/strains | ||
| Mouse: Tg(Cdh5-cre/ERT2)1Rha | (Sorensen et al.68 | N/A |
| Mouse: Zfp36f/f | Qiu et al.69 | N/A |
| Mouse: Gt(ROSA)26Sortm14(CAG tdTomato)Hze | Jackson Laboratory | RRID: IMSR_JAX:007914 |
| Mouse: Jag1f/f | Mancini et al.70 | N/A |
| Mouse: Zfp36f/fZl1f/fZl2f/f | This paper | N/A |
| Oligonucleotides | ||
| qPCR primers (Table S1) | See Table S1 | N/A |
| gRNA ZFP36 Forward: CACCGTGCCCGTGCCATCCGACCA | This paper | N/A |
| gRNA ZFP36 Reverse: AAACTGGTCGGATGGCACGGGCAC | This paper | N/A |
| Recombinant DNA | ||
| pLV[Exp]-Puro-EF1A>NLS-EGFP: {mJag1_3′ UTR_565bp} | This Paper - Vector Builder custom order | Cat#VB220720-1510tzf |
| pLV[Exp]-Puro-EF1A>NLS-EGFP: {mJag1_3′ UTR_517bp(del 48bp)} | This Paper - Vector Builder custom order | Cat#VB220720-1515agk |
| pRP[Exp]-Hygro-CAG-Luciferase&{hJAG1_3UTR_1814bp} | This Paper – Vector Builder custom order | Cat#VB230730-1401fzh |
| pRP[Exp]-Hygro-CAG-Luciferase&{hJAG1_3UTR’(del 331bp-429bp)} | This Paper – Vector Builder custom order | Cat#VB230807-1714sjb |
| psPAX2 | Addgene | Cat# 12260; RRID: Addgene_12260 |
| pMD2.G | Addgene | Cat# 12259; RRID: Addgene_12259 |
| pCMV-GFP | Matsuda et al.71 | Cat#11153; RRID: Addgene_11153 |
| Software and algorithms | ||
| FIJI | Schindelin et al.72 | RRID:SCR_002285 |
| Imaris (v9.9.0) | Bitplane | RRID:SCR_007370 |
| Seurat (v4.1.1) | Hao et al.73 | RRID:SCR_016341 |
| NIS Elements | Nikon | RRID:SCR_014329 |
| Image Lab Software | BioRad | RRID:SCR_014210 |
| CFX Manager (v3.1) | BioRad | RRID:SCR_017251 |
| STAR (v2.7.3) | Dobin et al.74 | RRID:SCR_004463 |
| FlowJo | BD Biosciences | RRID:SCR_008520 |
| BioRender | BioRender | RRID:SCR_018361 |
| Adobe Illustrator | Adobe | RRID:SCR_010279 |
| Prism 9 | Graphpad | RRID:SCR_002798 |
| UMI-tools | GitHub | RRID:SCR_017048 |
| PureCLIP | GitHub | https://github.com/skrakau/PureCLIP |
| DESeq2 | GitHub | RRID:SCR_015687 |
| FastQC | GitHub | RRID:SCR_014583 |
| AREsite2 | Gruber et al.75 | http://nibiru.tbi.univie.ac.at/AREsite2/welcome |
| BioTek Gen5 | Agilent | RRID:SCR_017317 |
| Wound_healing_size_tool | Suarez-Arnedo et al.76 | https://github.com/AlejandraArnedo/Wound-healing-size-tool/wiki |
| Other | ||
| HiSeq3000 | Illumina | Cat#SY-401-3001 |
| IncuCyte S3 Live Cell Analysis System | Sartorius | Cat#4647; RRID:SCR_023147 |
| Glass bottom well-plates | Cell Vis | Cat# P06-1.5H-N, P12-1.5H-N, P24-1.5H-N |
| BioTek Synergy H1 Microplate Reader | Agilent | Cat#SH1M2-SN; RRID:SCR_019748 |
| Incucyte Wound Maker 96-Tool | Sartorius | Cat# 4563 |