

Abstract
The use of microbial inoculants in agriculture as biofertilisers and/or biopesticides is an appealing alternative to replace or reduce the practice of agrochemicals. Plant microbiota studies are revealing the different bacterial groups which are populating plant microbiomes re‐energising the plant probiotic bacteria (PPB) translational research sector. Some single‐microbial strain bioinoculants have proven valid in agriculture (e.g., based on Trichoderma, mycorrhiza or rhizobia); however, it is now recommended to consider multistrain consortia since plant‐beneficial effects are often a result of community‐level interactions in plant microbiomes. A limiting step is the selection of a fitting combination of microbial strains in order to accomplish the best beneficial effect upon plant inoculation. In this study, we have used a subset of 23 previously identified and characterised rice‐beneficial bacterial colonisers to design and test a series of associated experiments aimed to identify potential PPB consortia which are able to co‐colonise and induce plant growth promotion. Bacterial strains were co‐inoculated in vitro and in planta using several different methods and their co‐colonisation and co‐persistence monitored. Results include the identification of two 5‐strain and one 2‐strain consortia which displayed plant growth‐promoting features. Future practical applications of microbiome research must include experiments aimed at identifying consortia of bacteria which can be most effective as crop amendments.
Microbial inoculants in agriculture offer an appealing alternative to agrochemicals, with plant microbiota studies re‐energizing research on plant probiotic bacteria (PPB). While single‐microbial strain bioinoculants have shown validity, it is now recommended to consider multistrain consortia for better plant‐beneficial effects due to community‐level interactions in plant microbiomes. In this study, we identified two 5‐strain and one 2‐strain consortia displaying plant growth‐promoting features, emphasizing the importance of identifying effective bacterial consortia for future crop amendments.
INTRODUCTION
In the last decade, there has been an increase in the use of chemical fertilisers in agriculture in every area of the world (FAOSTAT) reaching alarming levels in terms of sustainability and environmental impact (Gouda & Saranga, 2018). It is now therefore critical to introduce alternative sustainable practices which will result in the elimination or reduction in the use of agrochemicals. The use of plant probiotic bacteria (PPB) as biofertilisers and/or biopesticides represents an appealing alternative (Menendez & Garcia‐Fraile, 2017) and the recent plant microbiome‐related technologies has re‐energised this translational research sector (Menendez & Garcia‐Fraile, 2017). PPB can have several plant‐beneficial properties such as biological nitrogen fixation, solubilising phosphate and potassium and biosynthesizing phytohormones (Lugtenberg & Kamilova, 2009). PPB can also have biocontrol properties by acting as protective agents against pathogens via the production of antimicrobial compounds, by providing a protective barrier and via induction of plant immunity.
Plants harbour a rich and complex microbiome in its compartments like the rhizosphere, endosphere, phyllosphere and seed and this intimate relationship between microbes and plants is becoming an important research field. The rhizosphere, which is the nearest soil (up to 2 mm) area to the roots, hosts a rich microbial plant community that is strongly influenced by root exudates which in return provides a series of beneficial outcomes related to plant growth (Mendes et al., 2013). A small percentage of the microbes which colonise the rhizosphere also enter the plant colonising the root endosphere thus becoming endophytes and some are then able to move to other plant organs (Compant et al., 2021). Plant microbiomes are dominated by bacteria which mainly belong to four bacterial phyla, the Proteobacteria, Bacteroidetes, Actinobacteria and Firmicutes (Lundberg et al., 2012). For many years, PPB have been selected in the laboratory on the basis of expressing the plant‐growth‐promoting phenotypes listed above such as nitrogen fixation, pathogen suppression and nutrient mineralisation (Compant et al., 2019). Applications of PPB as products in agriculture however have often proved unstable since they fail to colonise in a natural microbiome context at the density which is needed to express the plant‐beneficial traits (Hu et al., 2021). In addition, introducing PPB in the rhizosphere plant compartment could result in the alteration of the rhizosphere and endosphere microbiome composition and functioning (Delgado‐Baquerizo et al., 2016; Hu et al., 2021; Mueller & Sachs, 2015).
Due to the richness in microbial diversity of the plant microbiome, scientists are beginning to be more interested in PPB inoculants based on a consortium of microbial species which have multiple plant‐beneficial features and are well represented in the microbiome (Trivedi et al., 2020). The possible applications of microbial consortia have been evidenced with strains belonging to the genera of Azospirillum, Arthrobacter and Agrobacterium were inoculated in barley showing improved PGP (plant growth promotion) potential (Lin et al., 2019). Similar consortium studies with beneficial outcomes were also performed in wheat combining bacterial strains and a mycorrhizal fungus (Singh & Kapoor, 1999). Studies using microbial consortia are also of interest in relation to abiotic stress as well as nutrient acquisition (Vurukonda et al., 2016; Wang et al., 2012). Microbial consortia can also be used for biocontrol purposes by protecting host plants against pathogens resulting in the alleviation of disease symptoms and increase in biomass (reviewed by Minchev et al., 2021).
Multi‐strain PPB consortia can potentially perform better than single‐strain inoculants due to ecologically and functional complementarity which can arise in a microbiome context. Initial omics studies are evidencing that increasing the probiotic consortium richness can result in better colonisation and consequently improved plant growth and protection from pathogens (Hu et al., 2016, 2021). In addition, a PPB consortium can also potentially drive/shift the residential microbiome towards an improvement in plant‐growth‐promoting effects. A major limiting step is how to select for the most appropriate combination of microbial strains in order to achieve the best effect upon plant inoculation. In this study, we present an approach on how to identify the best consortium from a collection of 23 bacterial strains which were previously identified and characterised plant‐beneficial bacteria isolated from sterilised rice samples. Most plant‐beneficial inoculation studies involve the use of single microbial strains; the use of a large consortium in order to identify beneficial consortia of a few strains is not common and is the aim of this work. The 23 strains have been selected from a large culture collection of over 1300 rice bacterial endophytes for their ability to colonise the rhizosphere and endosphere displaying several plant‐beneficial properties (Bertani et al., 2016). These 23 rice‐beneficial bacterial strains were used here to design and test a series of connected experiments aimed to identify potential PPB consortia which are able to synergistically co‐colonise and induce plant growth promotion.
EXPERIMENTAL PROCEDURES
23 bacterial isolates used in this study and growth conditions
The 23 isolates used for this study have been carefully selected from a large collection of rice endophytic bacteria (Bertani et al., 2016). Different criteria were used, these were (i) plant compartments (root −34%, stem −52% or leaf −13%), in which the strains were isolated, (ii) vegetative states (tillering—34%, booting −13%, or milky maturation −52%), (iii) members of the most numerous groups, at phyla and taxa level (Actinobacteria −26%, Firmicutes 9%, Bacteroidetes 9%, α‐Proteobacteria 13%, β‐Proteobacteria 9%‐, γ‐Proteobacteria 34%), (iv) selection using data obtained from in vitro and in planta PGP features. The selection was performed in order to have the most heterogeneous and representative group of strains as a starting collection to use for the design of the best PPB consortia.
The set of bacterial rice endophytic strains was routinely grown at 28°C in Luria–Bertani (LB) broth medium (Miller, 1972) or tryptic soy (TS) medium (Sigma‐Aldrich).
Experimental design for the identification of PPB consortia and co‐growth tests of the bacterial endophytic strains
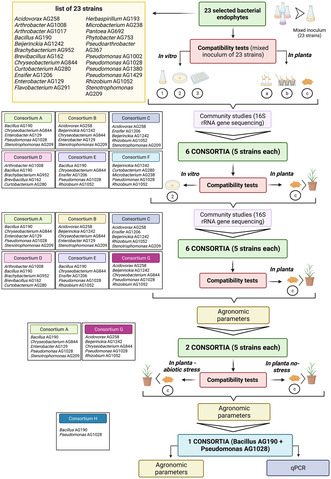
A set of associated experiments was designed aimed at identifying a potential consortium (or consortia) of bacteria which can be used as a plant‐beneficial inoculum. The summary of the rationale and strategy is presented in Figure 1. A first set of compatibility growth experiments was performed using all the 23 strains grown together in several different ways both in vitro (in liquid and solid media) and in the in planta (in rice using three different inoculation methods, i.e. root dipping, furrow and seed inoculation; as described below) in the root compartment. In all these conditions, the 23 bacterial‐strain community was monitored and assayed using 16S rRNA amplicon sequencing and analysis (as described below). Based on the results of this first set of compatibility growth experiments, a set of six consortia having 5‐bacterial strains each was then designed with the aim to create the most heterogenous groups of bacterial strains having metabolic and plant‐beneficial abilities complementary and being able to co‐exist without competing in the conditions tested here. All the six designed consortia were tested again using an in vitro co‐growth in plate media and in planta co‐growth using the seed‐inoculation method (these two methods resulted to be the most informative and easy to optimise according to the first compatibility test) and the bacterial community composition again determined using 16S rRNA amplicon sequencing and analysis. Some 5‐strain consortia were further studied for in planta growth promotion as described below.
FIGURE 1.
Experimental rationale followed in this study in order to identify beneficial bacterial consortia from a set of 23 beneficial bacterial strains isolated from rice sterilised roots. See text for details.
The bacterial strains were independently grown in liquid media (LB or TSB), washed twice and resuspended in phosphate‐buffered‐saline (PBS) solution. An equal amount of each strain (aliquot of 100 μL OD600 = 1 in PBS solution) was then pooled in a solution in PBS. This mixed solution containing all strains in a comparable amount was used as inoculum for all in vitro and in planta compatibility tests as described here below.
In vitro co‐growth in plate media
An aliquot of 100 μL of the strain mixed inoculum in PBS was spotted in the centre of TSA plates and samples were then collected at times 0, 43 and 80 h and DNA was extracted and used for independent 16S rRNA amplicon library preparation (see below).
In vitro co‐growth in liquid media
An aliquot of 100 μL of the strain‐mixed inoculum in PBS was transferred to flasks with 25 mL of TSB and incubated at 28°C and 150 rpm. Samples were collected at times 0, 22 and 43 h and DNA was extracted and used for independent 16S rRNA amplicon library preparation (see below).
In vitro co‐growth in static biofilm microtitre plates and on seeds: an aliquot of 200 μL of the strain mixed inoculum in PBS was transferred to biofilm‐microtiter plates and to 50 mL falcon tube flask containing 100 rice surface sterilised seeds immersed in 5 mL of TSB. Samples were incubated at 28°C with no agitation and biofilms from microtitre plates or seeds were collected at times 0, and 144 h and DNA was extracted and used for independent 16S rRNA amplicon library preparation (see below).
In planta co‐growth: different plant inoculation methods were used to assay for root colonisation (rhizosphere and endosphere). Seeds were surfaced sterilised and plants were grown in a growth chamber (photoperiod 16 h light\8 h dark and humidity at 70%). The soil substrate was composed of gardening soil (Ecoter TS Terriccio universale, autoclaved at 121°C for 40 min). For the seed inoculation, seeds were immersed in the strain‐mixed inoculum in PBS for 15 min and transferred to growth tubes. For the root dipping inoculation, plants were grown in sterile pure water until the development of roots (7 days). The roots were immersed in the strain‐mixed inoculum in PBS for 5 min and transferred to growth tubes. For the furrow inoculation, an aliquot of 1 mL of the strain mixed inoculum in PBS was inoculated directly in the growth tube (a small cavity was created on the soil), followed by transferring of the 7‐day‐old seedlings. Plant samples were collected from washed macerated roots at days 10, 15 and 20 (root dipping and furrow inoculation) and at days 20, 25 and 30 (seed inoculation).
For each time‐point five biological replicates (for in vitro co‐growth tests or in planta) were sampled and DNA was extracted and used for independent 16S rRNA amplicon library preparation (see below).
DNA extraction, amplicon library preparation and sequencing
The DNA from all the in vitro co‐growth experiments was extracted using the Bacterial Genomic DNA isolation kit (Norgen Bioyek Corp.) whereas from the in planta co‐growth experiments with the Power Soil DNA isolation kit (Qiagen) following the manufacturer's instructions. The DNA quality and quantity were determined by using a NanoDrop device (Thermo Scientific). The DNA extracted was used to amplify the V3 and V4 hypervariable region of the 16S rRNA gene using barcoded primers and PCR conditions following Illumina Inc.'s protocol (Illumina Inc.). Briefly, individual barcoded libraries were directly prepared by PCR using long primers (16S_Amplicon_PCR_Fw: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG; 16S_Amplicon_PCR_Rv:GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC) (Klindworth et al., 2013) incorporating the Illumina adapter sequences, which allow pooling multiple samples into one run of sequencing. Following the first amplification, a cleaning step was performed using the AMPure XP bead clean‐up (A63880l; Beckman Coulter Inc.). A second PCR reaction was then performed to attach dual index and Illumina sequencing adapters using the Nextera XT Index Kit; followed by a final AMPure XP bead clean‐up. Amplicons size, integrity, and purity were checked using the Bioanalyzer equipment (Agilent Inc.) and the library concentration was measured by fluorimetric quantification using Qubit 2 (Invitrogen Inc.). Finally, libraries sequencing was performed using 2 × 250 bp MiSeq.
Sequence data processing and bioinformatic analyses
The .fastq files were imported into qiime2 (Bolyen et al., 2019), the clustering of reads into Amplicon Sequence Variants ASVs was done using the DADA2 plugin (Callahan et al., 2016) and taxonomic assignment was done based on a customised dataset formed by the 16S rRNA gene sequences of the 23 isolates used in this study. For clustering and comparing the ASVs based on sequence similarity, the cd‐hit program was used, setting the parameter of sequence similarity equal to 98% for two sequences to be considered part of the same cluster. All the ASVs not matching with any of the 16S rRNA gene sequences of the customised dataset were removed from the analysis. The obtained dataset was imported in R using the package qiime2R (Bolyen et al., 2019), and the subsequent analyses and plots were drawn using either phyloseq, microbiome or reshape2 R‐packages (McMurdie & Holmes, 2013). DESeq2 (Love et al., 2014) and MaAsLin2 (Mallick et al., 2021) were used for differential abundance analysis when required.
In planta plant growth promotion assays
The six 5‐strain consortia designed were tested for plant‐growth‐promoting activity in rice in a plant growth chamber; conditions were photoperiod 16 h light\8 h dark; humidity at 70%; and temperature 28°C. The consortia were seed inoculated in the following way; seeds were surface‐sterilised for 50 min in 50% commercial bleach and washed six times in sterile ultrapure water. Seeds were immersed in the corresponding inoculum and incubated overnight at room temperature under gentle agitation (approximately 40 rpm). The growth substrate for plants was gardening soil (Ecoter TS Terriccio universale), autoclaved at 121°C for 40 min. The experiment was run for 40 days and agronomic parameters analysed were shoot and root dry weight in two time‐points—20 and 40 days after inoculation/planting. Each treatment contained 25 technical replicates, being 10 replicates for each time‐point of plant growth analysis and five replicates for root microbiome analysis. Irrigation was performed weekly with Hoagland solution and two times per week with tap water.
The 5‐strain consortia were tested for plant‐growth‐promoting activity in rice under salt and nitrogen stress. The growth conditions were the same as described above with the variation that for the irrigation for salt stress assay, stressed control and inoculated treatments were irrigated with 80 mM NaCl, three times per week (first 2 weeks) and with 130 mM NaCl, three times per week (last 2 weeks). Whereas for the nitrogen stress assay, stressed control and inoculated treatments were irrigated with modified Hoagland solution, in which N content was reduced from 15 to 1 mM.
The in planta experiment with the 2‐strain consortia of strain AG1028 and AG190 was performed using the same conditions above with no stress applied with the only variation that the soil was not autoclaved.
Statistical analysis
The results were analysed in Prism 8.1.2 by average comparison, using one‐way ANOVA, two‐way analysis of variance with Tukey's post hoc multiple comparison test was used for multigroup comparison analysis, and Tukey test at 95% significance (p < 0.05).
Genome sequences of the rice endophytic bacterial isolates
The majority of the bacterial isolates used in this study have been whole genome sequenced with the exception of Acidovorax AG258, Arthrobacter AG1008, Arthrobacter AG1017, Beijerinckia AG1242, Curtobacterium AG280, Herbaspirillum AG193, Pseudomonas AG1380 and Rhizobium AG1052. The complete genomes of 15 rice bacterial endophytes were sequenced with the Illumina MiSeq platform using 150 bp paired‐end reads. Each Whole Genome Shotgun project has been deposited at JGI IMG portal and is accessible through the Sequencing Project ID, as listed below. Bacillus AG190 ID Gp0255572; Brachybacterium AG952 ID Gp0255558; Brevibacillus AG162 ID Gp0255576; Chryseobacterium AG844 ID Gp0255568; Ensifer AG1206 ID Gp0255581; Enterobacter AG129 ID Gp0544153; Flavobacterium AG291 ID Gp0255570; Microbacterium AG238 ID Gp0255564; Pantoea AG692 ID Gp0592337; Phytobacter AG753 ID Gp0255595; Pseudoarthrobacter AG367 ID Gp0255557; Pseudomonas AG1002 ID Gp0255602; Pseudomonas AG1028 ID Gp0255604; Pseudomonas AG1429 ID Gp0255600; Stenotrophomonas AG209 ID Gp0255606.
Absolute quantification real‐time PCR for in planta detection and estimation of Pseudomonas fulva AG1028 and Bacillus megaterium AG190
Genomic DNA of bacterial strains AG1028 and AG190 was purified as previously reported (Better et al., 1983). In order to formulate standard curves to then use in absolute quantification real‐time PCR (qPCR) assays, for strain AG190 we selected the gene marR, which encodes for the MarR family transcriptional regulator and PCR amplifications were performed a 557 bp fragment was PCR amplified with primers 190V‐F/R (Table S1). For strain AG1028 on the other hand, we chose gene hp, which encodes for a hypothetical protein (sequence ID: WP_147177139.1) and a 549 bp fragment was PCR amplified with primers 1028V‐F/R (Table S1). Both amplified fragments were confirmed to be highly specific as determined via nucleotide searches in the NCBI GenBank BLAST and via PCR amplification among 10 different bacteria. These two fragments were obtained by conventional PCR using genomic DNA of the strains as template and cloned in the pGEM®‐T Easy Vector (Promega A1360, Promega Co.). The two plasmids harbouring the PCR fragments were purified from Escherichia coli DH5α using the E.Z.N.A.® Plasmid DNA Midi Kit (Omega Bio‐tek, Inc.) and the concentration quantified by a NanoDrop ND‐1000 spectrophotometer (NanoDrop Technologies, Thermo Scientific Instruments). The plasmids were linearised with SpeII and to calculate the number of construct gene copies, the following formula was used: concentration of pGEM::557marR or 549 hp (ng·μL)/[the molecular weight (660 Da·bp) × Avogadro's constant (6.022 × 1023 molecules/mol) × base pairs number of pGEM::590marR or 549 hp]. 1 μL of pGEM::557marR contained 2.37 × 109 gene copies and 1 μL of pGEM::549 hp contained 4.17 × 109 gene copies (Table S2). Serial decimal dilutions of linearised pGEM::557marR (2.37–2.37 × 109) and pGEM::549 hp (4.17–4.17 × 109) were done in double‐distilled water to prepare the standard curve for qPCR (Figure S1). The standard dilutions were then aliquoted and stored at −80°C until use. Four replicates of each dilution were added to each qPCR.
To detect and quantify the presence of B. megaterium AG190 and P. fulva AG1028 in inoculated rice roots at different growth time points, the forward q‐190‐F and reverse q‐190‐R primers using a TaqMan™ MGB Probe (Applied Biosystems) Probe‐190 (Table S1) were designed for strain AG190, amplifying a 115 bp fragment of marR, and the forward q‐1028‐F and reverse q‐1028‐R primers Probe‐1028 (Table S1) were designed for strain AG1028, amplifying a 121 bp fragment of hp. For qPCR, we used PowerUp SYBR green master mix (Applied Biosystems) and the amplification reactions were performed in Bio‐Rad CFX96 Real‐Time PCR Detection System. As negative controls, double‐distilled sterile water and the aliquots taken after DNase treatment during the DNA extraction procedure were used. The number of gene copies was defined as the average of quadruplicate reactions.
The amplification efficiencies of two strains reference genes of the standard curve were calculated, regression coefficient of the standard curve of B. megaterium AG190 was 0.9903 (Figure S1A) and regression coefficient of the standard curve of P. fulva AG1028 was 0.9991 (Figure S1B), based on the equation E = 10–1/slope−1, their amplification efficiencies were 100.81% and 108.64% respectively.
RESULTS
The 23 bacterial strain collection and design rationale for the isolation of a beneficial bacterial consortia
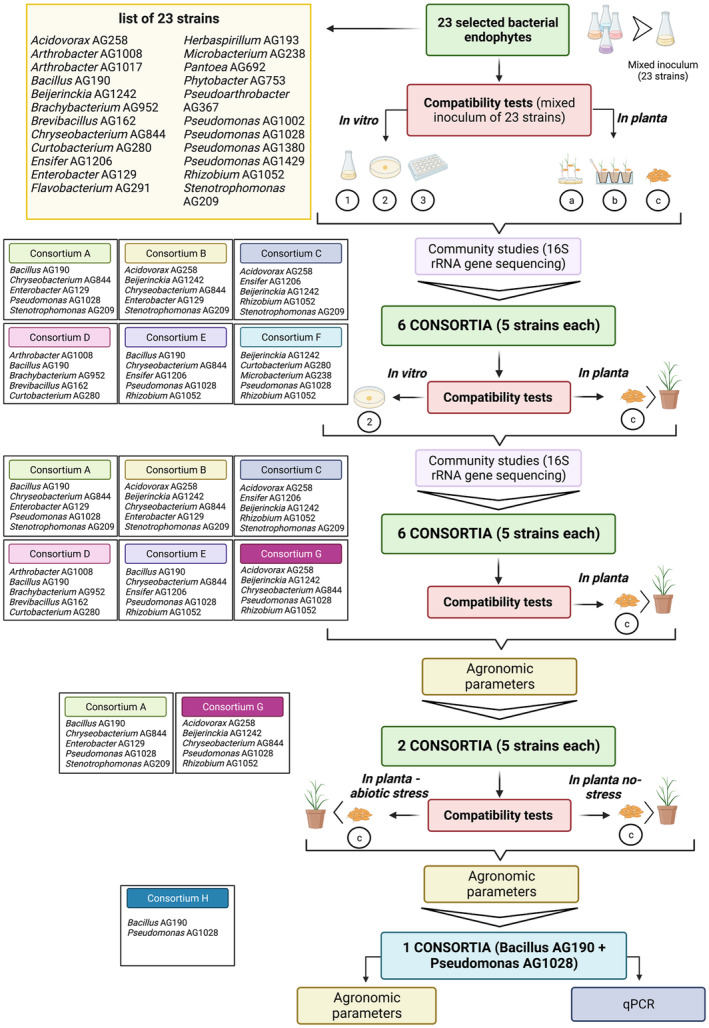
A set of 23 previously identified and characterised (as described in Section 2) rice bacterial strains have been used here in co‐growth experiments for the identification of plant‐beneficial consortia. In order to identify small beneficial consortia of a few bacterial strains, both in vitro and in planta co‐inoculation experiments have been performed (the experimental rationale is presented in Figure 1 and described in Section 2). The purpose of these co‐growth experiments was to determine which beneficial strains persisted together preventing the overgrowth of individual strains as this could be a good indication that they do not antagonise each other and can co‐colonise as well as co‐exist. We have further characterised here the 23 isolates for several in vitro plant‐beneficial phenotypes (Figure 2). These in vitro phenotypic assays have evidenced that the 23 strains display a varied and complementary set of plant‐beneficial phenotypes however it cannot be excluded that in planta strains respond differently. Most strains displayed several phenotypes in different combinations, only a few strains in vitro expressed only one or two phenotypes. The genomes of the 15/23 strains have also been sequenced here as described in Section 2 which have confirmed the classification originally established using the 16S taxonomical locus. Based on the results of the compatibility tests, the in vitro plant‐beneficial phenotypes and the genomic information, we then designed a set of 5‐strain consortia compiling together complementary strains with multiple beneficial features, potential mutualistic interactions and reduced competition.
FIGURE 2.
Diagrammatic presentation of the in vitro phenotypes of the 23 rice‐associated bacterial strains. Each bar chart represents the number of isolates that tested positive to each test. Phenotypes are arranged from the most common (Diazotroph) to the less distributed (ACC_production). The different phenotypes tested, related to PGP effects, are explained as follow: Diazotroph (ability to grow without external sources of fixed nitrogen); IAA_production (ability to synthetise indole‐3‐acetic acid); EPS_production (ability to produce exopolysaccharides); P_solubilisation (ability to solubilise phosphate); Proteolytic‐activity (production of proteolytic enzymes to breakdown proteins in smaller polypeptides); Anti_Bac (antimicrobial activity against E. coli as target); Lipolytic activity (production of extracellular lipases to hydrolyse triglycerides); ACC_production (ability to synthetise 1‐aminocyclopropane‐1‐carboxylic acid [ACC] deaminase). See text for methodologies used for the detection of these phenotypes.
Compatibility growth experiments of the 23‐bacterial‐strain‐consortia
In order to gather data on which of the 23 bacterial isolates grew and persisted in consortia (see above for rationale), a mixed inoculum of all the 23 strains was grown in several different conditions both in vitro and in planta and then detected via 16S rRNA amplicon NGS sequence determination and analysis, as explained above and in Section 2.
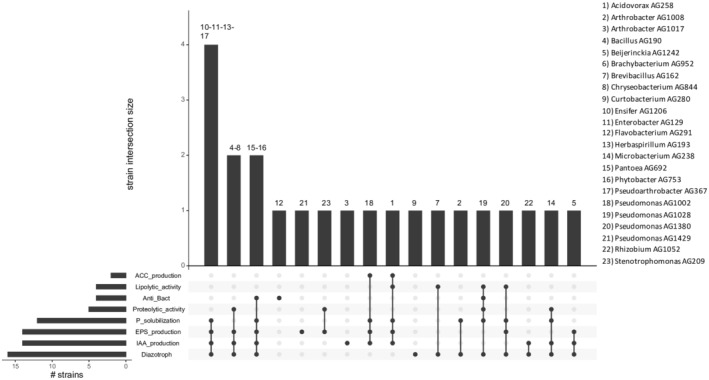
In vitro co‐growth was performed using biofilm in microtitre plates, on seeds, in co‐spotting on plates and in co‐growth in liquid media; Figure 3A depicts which bacterial strains were detected at the different growth conditions and time points. Importantly at the inoculation starting point (i.e. 0 h), all bacterial strains were detected in equal abundance suggesting that the technique used allows to unequivocally distinguish each strain. However, in vitro, some strains were detected only at 0 h such as Rhizobium AG1052 and Brachybacterium AG952, suggesting that the growth conditions or the presence of other possibly antagonistic isolates inhibit their growth or survival.
FIGURE 3.
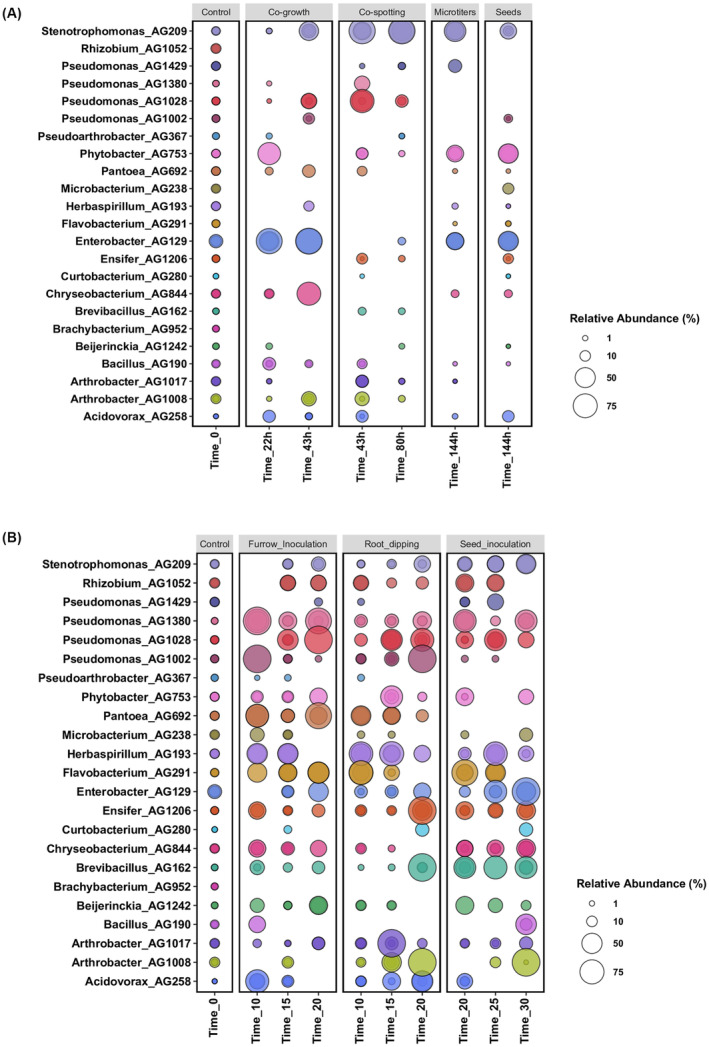
(A) Bubble plot showing the relative abundance of the 23 bacterial endophyte strains (y‐axis) used in vitro co‐growth experiments, as obtained from 16S rRNA gene sequencing analysis. The samples are arranged according to the time points (x‐axis) and subplot according to the type of compatibility growth experiments (i.e. co‐growth in liquid medium, co‐spotting in solid plates and Microtitres and Seeds biofilm growth conditions). (B) Bubble plot showing the relative abundance of each bacterial endophyte strain (y‐axis) used in planta co‐growth experiments. The samples are arranged according to the time points (x‐axis) and subplot according to the type of inoculation method used (i.e. Furrow inoculation, Root dipping and Seed inoculation). Control denotes samples collected immediately after co‐inoculum setup. The size of the bubble represents the relative abundance and the median of the percentage of at least three replicates, and colours indicate each bacterial strain used in this experiment.
In the two sessile/biofilm conditions (i.e. seed and microtitre growth) it is evident that growth on seeds resulted in a few more strains being able to persist (14 strains) compared to the microtitre condition (11 strains); interestingly, most strains (9) were shared and persisted in both conditions. In this experimental set‐up, the best biofilm performers were Stenotrophomonas AG209, Phytobacter AG753 and Enterobacter AG129.
In plate (co‐spotting) at 43 h, 13 strains were detected, while at 80 h, 11 strains were detected and among them the majority were present at low abundance levels. When grown in liquid condition (co‐growth) at 22 h, 13 strains were detected, while at 43 h of growth 10 strains were present and some of which at higher abundance compared to the previous time‐point, as for example Stenotrophomonas AG209, Pseudomonas AG1028, Enterobacter AG129 and Chryseobacterium AG844, thus being the best performing isolates. Interestingly, in vitro, there was little overlap with the strains that could persist in co‐spotting in plate with respect to liquid growth indicating that these two conditions bacteria behave very differently. Considering both the number of strains able to survive together and their abundance, the growth on plate (co‐spotting) after 43 h results proved to be the most informative.
In planta co‐growth: was performed using different ways of inoculation consisting in furrow inoculation, root dipping inoculation and seed inoculation. All these co‐growth conditions of the 23 strains provided a considerable amount of data which is presented in Figure 3B. In the furrow inoculation, at the last time point (20 days) many strains were abundantly present such as Pseudomonas, Phytobacter, Pantoea, Flavobacterium and Enterobacter. Similarly, in root dipping inoculation, at 20 days several strains (15) could be detected with various abundancies with Pseudomonas, Enterobacter, Ensifer, Chryseobacterium, Arthrobacter and Brevibacillus strains being the most abundant. Seed inoculation also resulted mainly in the enrichment of Pseudomonas, Enterobacter, Bacillus and Arthrobacter strains. However, some strains were not detected in any of the in planta co‐growth conditions, such as Brachybacterium AG952 and in a very low‐abundance Pseudoarthrobacter AG367, suggesting that these two strains are not competitive in the in planta conditions tested in this study. Interestingly, Pantoea AG692 was undetected in all the three time points when inoculated on seeds, while it was detected in high abundance in the other two conditions of in planta inoculation. This result suggests that also the type of inoculation is important for the survival for some of the strains.
In planta, a more rich and diverse community can persist in comparison to the in vitro growth conditions; these could be due to the more varied growth conditions; it must be noted that regardless sterile soil was used, most likely there is also a background of bacterial strains originating from the seeds. It is also noted that in all the in planta inoculation conditions, several strains displayed similar appearances in the three time points, only in a few cases they were detectable in the earlier time points and not at 20 or 30 days.
The results obtained from this first 23‐strains compatibility test suggested that the growth on plate (co‐spotting) proved to be the most informative in vitro condition, while in planta the seed inoculation method resulted to be the best performing one. Regarding the bacterial strains and their ability to co‐exist, the majority resulted to be able to co‐live, nevertheless in different abundance and time, while only Pseudoarthrobacter AG367 and Brachybacterium AG952 were significantly lost both in vitro and in planta experiments and Pantoea AG692 was undetected in all the three time points when inoculated on seeds.
Design of a subset of 5‐bacterial‐strain‐consortia and compatibility growth experiments
As a next step, it was of interest to generate consortia having fewer bacterial strains since it is more manageable as well as practical as a PPB consortia. Following the results of the co‐growth compatibility experiments above using the 23 rice‐associated bacterial strains, a set of six 5‐strain (designated as A, B, C, D, E and F) consortia were designed and further tested for their co‐colonisation and persistence. Figure 1 depicts the composition of the six 5‐strains consortia. For the choice of strains, the co‐existence in vitro and in planta data in the different growth conditions presented above was taken into consideration as well as the in vitro phenotypic assays as depicted in Figure 2 (as mentioned above, it was of interest to design consortia that also have a complementary set of in vitro phenotypes). Consortium A for example contained the four strains (AG209, AG1028, AG129 and AG844) which were the most abundant and stable in liquid co‐growth experiments (see above). It is noted that AG209 and AG129 of this consortium A co‐grew very well in the two biofilm modes of growth; these latter two strains were also included in consortium B together with AG258 and AG1242 which have complementary in vitro phenotypes and with strain AG844 which persisted very well in liquid co‐growth. Consortia C and D was a combination of strains which persisted well either in vitro or in planta with one strain with a good set of in vitro phenotypes. Consortium E contained strains AG190, AG844, AG1206 and AG1028 which persisted well in planta using different inoculation methods. Consortium F on the other hand, apart from strain AG1028, consisted of strains which did not persist well in most conditions tested however was a good combination of strains exhibiting different in vitro phenotypes. In order to determine their co‐colonisation behaviour, the consortia were then grown in two different conditions, one in vitro (co‐spotting on plates) and one in planta (seed inoculation), as explained in the Section 2. Figure 4 illustrates the detection of the strains in the consortia in the two growth conditions; in plate co‐spotting (Figure 4A), in all six consortia the 5‐strains could be detected after 24 h, in some cases the abundance increases or decreases when compared to time 0 but not significantly. In particular, we noticed that Stenotrophomonas, Pseudomonas, Enterobacter and Chryseobacterium co‐grew in a comparable abundance. On the other hand, Microbacterium, Curtobacterium, Brachybacterium and Beijerinckia were all detected in a very low abundance, suggesting that either the laboratory conditions used or the presence of antagonists limited their growth. In summary, the consortium A and E displayed the best level of compatibility and co‐growth when inoculated as a co‐spot in plate, as all the isolates were detected in a similar abundance and comparable rate of growth, while the consortium F presented an over‐growth of Pseudomonas AG1028 and a reduction in abundance and presence of all the other co‐inoculated strains.
FIGURE 4.
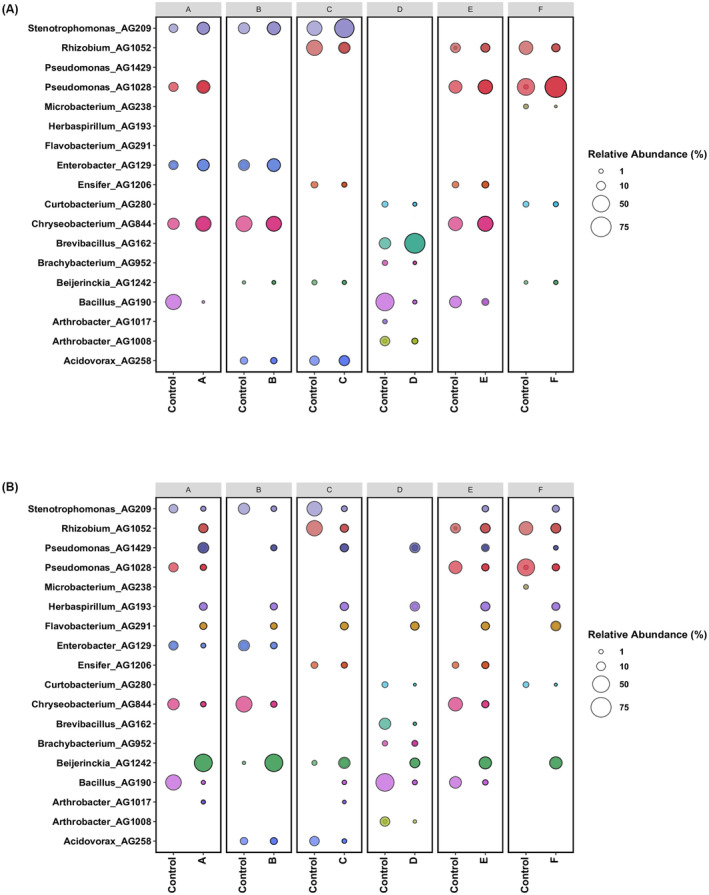
(A) Bubble plot showing the relative abundance of each bacterial endophyte strain (y‐axis) used in vitro co‐spotting growth experiments after 24 h, as obtained from 16S rRNA gene sequencing analysis. The samples are arranged and subplot according to each specific consortium (i.e. Consortium A, B, C, D, E, F) (x‐axis). (B) Bubble plot showing the relative abundance of each bacterial endophyte strain (y‐axis) used in planta compatibility growth experiments after 15 dpi. The seed inoculation method has been used in this compatibility growth experiments of 5‐bacterial‐strain‐consortia. The samples are arranged and subplot according to each specific consortium (i.e. Consortium A, B, C, D, E, F) (x‐axis). Control denotes samples collected immediately after co‐inoculum setup. The size of the bubble represents the relative abundance and the median of the percentage of at least three replicates, and colours indicate each bacterial strain used in this experiment.
When co‐inoculated in the seed, in all the six consortia the 5‐strains could be detected after 15 days, however with varying abundance and predominance when compared to time 0 (Figure 4B). There was a reduction in 16S amplicon reads for many of the strains and sequences were also detected which did not belong to the inoculated consortia; this is likely to be due to other bacteria being present in the seed microbiome. However, most strains which belonged to the genera of the consortia were detected in the roots after 15 days, albeit at lower levels. Interestingly, strain Beijerinckia AG1242 significantly increased in abundance in consortia; strains from this genus were also detected when un‐inoculated indicating that they were present as part of the seed microbiome. Similarly, strains Pseudomonas AG1429, Herbaspirillum AG193 and Flavobacterium AG291 were always detected in the roots of rice plants at 15 days, probably due to the seed microbiome. In many inoculation experiments, sequence reads belonging to certain strains were only detected if they were part of the consortia as for example with consortia A, E and F with Pseudomonas AG1028, with consortia A and B for Enterobacter AG129, with consortia C and E for Ensifer AG1206, with consortia A and B for Chryseobacterium AG844, with consortium D for Brevibacillus AG952, with consortia A, D and E for Bacillus AG190, with consortia D for Arthrobacter AG1008 and with consortia B and C for Acidovorax AG258. This is evidence that many of the strains could colonise well only in the presence of other strains which most likely indicating inter‐species interactions. In summary, just like the in vitro growth, also in planta the chosen consortia resulted in the 5 strains in most cases being able to colonise and persist up to 15 days indicating that the choice of strains following the 23 strains inoculum studies performed above, proved effective.
In planta assays inoculated with 5‐bacterial‐strain‐consortia
Following the compatibility experiments of the six 5‐strain consortia above, it was now of interest to perform in planta assays in order to determine their potential plant growth‐promoting properties. For these experiments, we kept five of the six 5‐strain consortia used in the compatibility assays above, namely A, B, C, D, and E since the 5‐strains all colonised well in the conditions tested and saw no reason to exclude them. On the other hand, the consortium F consisted of strains which did not persist well in most conditions tested and for this reason it was not taken into consideration for further studies. Consequently, we designed one new 5‐strain consortium (consortia G, Figure 1) making use of the data of the 5‐strain consortia compatibility studies; for this consortia G we mixed some of the strains which best colonised and persisted as outliers. Seed inoculated rice plants were grown independently with the six different consortia and dry biomass was scored after 20 and 40 days (Figure 5A,B). Interestingly, consortium A displayed a significant shoot biomass increase after 20 days (Figure 5A) when compared to the un‐inoculated control while consortium A and G displayed a significant increase of shoot biomass after 40 days (Figure 5B). Consortia A had also an increase in root biomass at 40 days (Figure 5B). It was concluded that the two 5‐strain consortia (A and G) promoted plant growth under the conditions tested.
FIGURE 5.
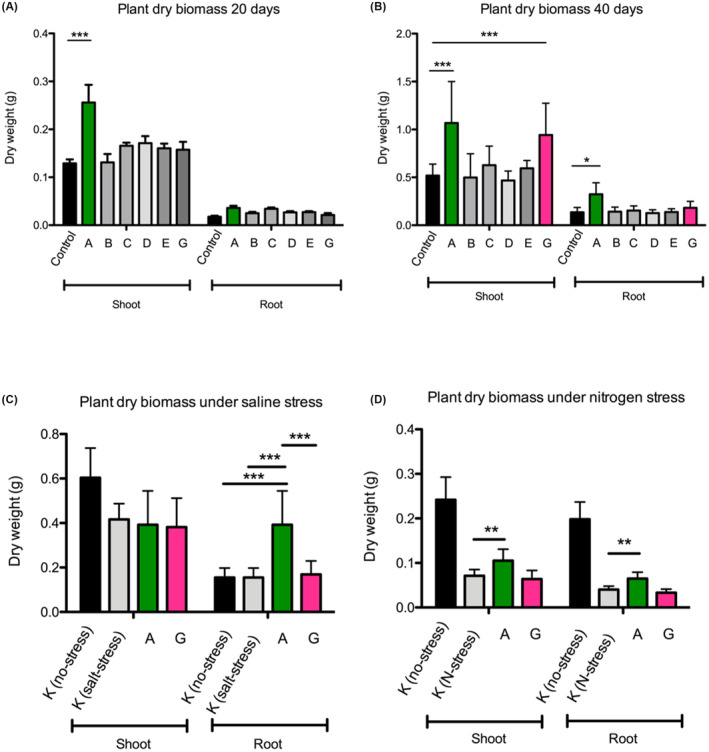
Plant growth promotion assays performed on rice of the 5‐strain bacterial consortia under normal growth conditions (A at 20 days post inoculation and B at 40 days post inoculation) and under abiotic stress conditions (C at 20 days post inoculation under salt stress and D at 20 days post inoculation under nitrogen stress). All data are presented as means ± SEM. GraphPad Prism 8.2 (GraphPad Software, Inc.) was used to perform all statistical analyses. *p < 0.05, **p < 0.01, ***p < 0.001, as indicated. The error bars indicate SDs. See text for all details.
In planta assays under abiotic stress inoculated with the two best‐performing 5‐strain PGP consortia
It was then of interest to assay the two best performing 5‐strain consortia (A and G as described above and in Figure 5A,B) in planta under two abiotic stress conditions; i.e. nitrogen and saline stress as described in Section 2. Figure 5C depicts that consortium A displayed a significant root dry weight biomass increase in salt stress conditions when compared to the un‐inoculated control. Interestingly, under nitrogen stress at 20‐day post‐inoculation, consortium A resulted in a significant increase in root dry weight as well as shoot dry weight biomass (Figure 5D). It was concluded that consortium A resulted in improving plant growth under two abiotic stress conditions under the conditions tested.
Design of a 2‐strain Pseudomonas fulva AG1028 and Bacillus megaterium AG1090 consortium and root colonisation assay
As a last experiment, it was decided to perform trials with a two‐strain consortium consisting of Pseudomonas AG1028 (this is a P. fulva strain) and Bacillus AG190 (this is a B. megaterium strain) since they colonised well and were part of the best performing PGP consortium A, as described in the experiment above. An additional motive for performing experiments with this two‐strain consortium were (i) members of the genera Pseudomonas and Bacillus are known to be beneficial rhizosphere colonisers, (ii) several microbial products have been developed using strains of these two genera, and (iii) a two strain consortium is more manageable and applicable as a microbial bioinoculant product in agriculture. Rice plants were inoculated with the two strains and it was observed that after 20 days there was a significant increase in root as well as in shoot biomass indicating a beneficial effect on the plant (Figure 6B). In order to monitor the presence and colonisation levels of the two strains, plants were inoculated with either the single strains or both strains and their presence was determined via qPCR method performed on DNA samples isolated from the roots as described in the Experimental Procedures section (Figure 6A). This novel qPCR method for the detection of these two specific strains was devised here and proved to be very effective. The results at 24 h showed that the DNA copies of P. fulva AG1028 when inoculated alone were significantly (p = 0.037) more abundant compared to the co‐inoculum. Similarly, the DNA copies of the strain B. megaterium AG190 had the same tendency, and a significant difference (p = 0.0035) was reached between the single inoculum roots and co‐inoculum roots. The results of the 10th day post‐inoculation however indicated that the DNA copies of the strain P. fulva AG1028 when co‐inoculated, increased significantly (p < 0.0001), while both in the single inoculum and in the co‐inoculum the strain B. megaterium AG190 decreased in the abundance level. The results at the 15th day post‐inoculation showed that the DNA copies of strain P. fulva AG1028 had the same tendency as at the 10th day, notably, displaying a high abundance and reaching a significant level (p < 0.0001); however, at the 20th day, results indicated that the DNA copies of both bacteria decreased and nearly failed to be detected. In summary, these results showed that the two strains can promote plant growth and most notably, in the first 15 days, the ability of P. fulva AG1028 to colonise the root is significantly affected by the presence of B. megaterium AG190.
FIGURE 6.
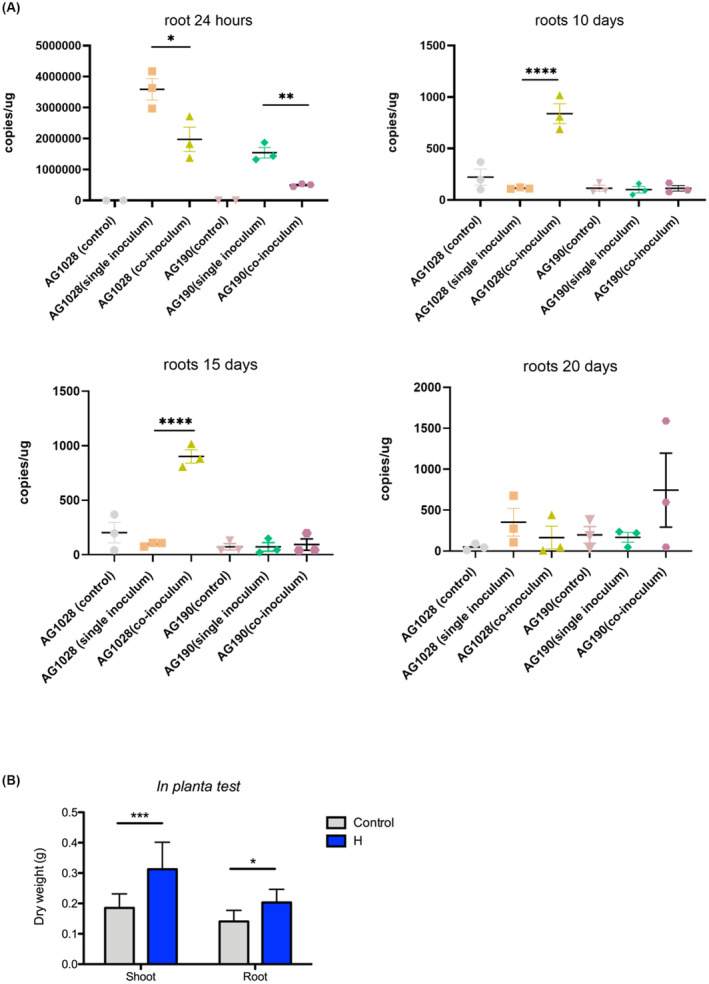
(A) qPCR analysis for the presence of Pseudomonas fulva AG1028 and Bacillus megaterium AG190 in rice roots. Bacterial DNA copies/μg of root grown in Hoagland's solution with three different treatments with AG190 (B. megaterium) and AG1028 (P. fulva). Inoculated 7‐days young rice plant roots samples collected 24 h, 10, 15, and 20 days. Control means non‐inoculated; single inoculum means just AG190 (B. megaterium) or AG1028 (P. fulva) inoculated, and co‐inoculum means AG190 (B. megaterium) and AG1028 (P. fulva) co‐inoculated. Three biological replicates were used in each group. Two‐way analysis of variance (ANOVA) with Tukey's post hoc multiple comparison test was used for multigroup comparison analysis. All data are presented as means ± SEM. GraphPad Prism 8.2 (GraphPad Software, Inc.) was used to perform all statistical analyses. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as indicated. The error bars indicate SDs. (B) Plant growth promotion assays performed on rice of the 2‐strain bacterial consortia (P. fulva AG1028 and B. megaterium AG190) under normal growth conditions at 20 days post inoculation. See text for all details.
DISCUSSION
A new agricultural transformation is underway which aims at improving crop yields while using reduced levels of agrochemicals; finding alternatives for plant disease control and plant‐growth promotion (PGP) are very important targets. Microorganisms have long been applied as inoculants for biocontrol or biostimulation as possible alternatives to agrochemicals with narrow success (Romano et al., 2020). The functionality and persistence of inoculated microbes in the microbiome is likely to be dependent on the environment as well as microbe‐microbe interactions that take place within the plant microbiota (Venturi & Bez, 2021). It is now of interest to design more complex microbial communities consisting of several microbial strains; this approach is in its infancy and future studies need to take into consideration ecological processes which drive and maintain microbiota.
In this study, we used a set of 23 rice‐beneficial bacteria with the aim to identify which strains within this subset were able to co‐exist in vitro as well as in planta growth conditions. Following the co‐growth in different conditions, visualisation of the presence of the strains was made possible via community 16S amplicon library sequencing indicating which bacterial strains co‐colonised and co‐grew and thus likely being compatible to share the same niche. Smaller 5‐strain consortia which are more manageable and applicable as a microbial bioinoculant product, were then designed and further tested for growth conditions resulting in the identification of two 5‐strain consortia with PGP potential. In addition, a two‐strain consortium based on a B. megaterium and a P. fulva strain was also devised as both of these strains constantly emerged during the co‐inoculation studies and strains belonging to these species are known to be PGP.
The approach used here relies on having a good starting set of PGP isolates; results have identified two 5‐strain consortia, one (consortium A) consisting of a Bacillus, Chryseobacterium, Eneterobacter, Pseudomonas and Stenotrophomonas strain which colonises well and displayed PGP activities also in two conditions of abiotic stress (saline and nitrogen limiting conditions) and another (consortium G) consisting of Acidovorax, Beijerinckia, Chryseobacterium, Pseudomonas and Rhizobium strains which co‐exist well and displayed PGP activities in no‐stress conditions. We then took the Pseudomonas and Bacillus strains (P. fulva and B. megaterium) from the consortium A, showing the best PGP activities in both stress and no‐stress conditions, and performed colonisation and PGP and qPCR experiments; the reason being that practically a 2‐strain consortium is more manageable and applicable as a bioinoculant in the field. In addition, microbial products consisting of Pseudomonas or Bacillus strains exist as they are known to be good and beneficial rhizosphere colonisers. This binary consortium performed well under the conditions we tested and colonisation studies have evidenced that most likely they positively interact. Importantly, the use of the qPCR technique for the detection and quantification of the two strains forming the consortium resulted to be a valuable method when applied to moderately complex systems. In fact, unlike the 16S rRNA amplicon sequencing technique which shows considerable bias for certain bacterial species due to PCR primer mismatches, variations in the number of copies for the 16S rRNA gene, and bias introduced in the bioinformatics analysis, the quantitative PCR resulted to be more accurate in the quantification of specific bacterial targets (Dreier et al., 2022). This approach might open up new possibilities for studying simple microbial communities as the case of SynComs or small and well defined consortia. Moreover, it provided valuable insights into the temporal dynamics emphasising the importance of considering the interplay between different microbial species and their likely implications for plant‐microbe interactions.
It is now of importance to determine the behaviour of these three PGP consortia identified here in pertinent wild conditions. A diverse microbial consortium is likely to contain a larger amount of plant‐beneficial functions increasing functional diversity and redundancy (Hu et al., 2016); the three consortia which displayed PGP features (two 5‐strain and the Pseudomonas‐B. megaterium binary consortia) consist of bacteria which do display an array of different in vitro plant‐beneficial phenotypes (Figure 2). Importantly, the specific combination of the strains forming these three best consortia was important for the positive plant effect of the inoculum. In fact, when some of the strains where mixed in different combinations, did not all result to have PGP activities for the plants. Multi‐strain consortia can therefore possibly perform better due to ecological complementarity of the plant‐beneficial functions that are provided by the consortium and by other effects arising via microbe‐microbe interactions (Hassani et al., 2018; Venturi & Bez, 2021).
A microbial consortium can also induce changes in the composition and functioning of the resident microbiome (Hu et al., 2021; Mallon et al., 2018; Xiong et al., 2017). These effects are beginning to be reported and at present are very poorly understood. For example, Hu et al. (2021) showed via using a consortium of Pseudomonas spp. an alteration in the microbiome, increasing the abundance of rare taxa and overall microbiome biodiversity. This colonisation success of the Pseudomonas spp. consortia and microbiome shift was then associated with improved plant growth, nutrient assimilation and biocontrol of pathogens. Multispecies PPB consortia can therefore improve both the inoculant establishment in the plant microbiome and the variety of beneficial effects microbes can provide to the plant (Hu et al., 2017, 2021).
A few single‐strain inoculants have proved very successful in agriculture (e.g. based on Trichoderma, mycorrhiza or rhizobia) however we need to now consider multistrain consortia as many microbe‐mediated plant‐beneficial effects are determined by community‐level interactions; this presents an opportunity for microbial engineering of plant microbiomes (Kaul et al., 2021). In the last few years, a strategy has been introduced based on synthetic communities (SynComs) aimed at understanding microbe‐microbe and plant‐microbe interactions (de Souza et al., 2020; Liu et al., 2019). Alternatively, computational tools allow the prediction of keystone microbial species through topological networks and these can have a direct and/or indirect effects on microbiome assembly and can optimise their persistence upon inoculation in an agricultural setting. These co‐occurrence network approaches can be generalised to identify keystone microbes providing a way to identify which strains to isolate in order to then experimentally test them (Trivedi et al., 2020). Identifying few strain bacterial beneficial consortia as presented in this study can also be used in studying plant‐bacteria interactions/responses.
In summary, this study had the aim to set‐out a series of experiments in order to identify possible bacterial consortia from a larger subset of PGP rice bacterial isolates. Three possible PGP consortia have been identified which now merit further greenhouse and in‐field studies in order to assess PGP and microbiome effects. This is a bottom‐up approach different from the top‐down direction of designing microbial consortia by using next‐generation sequencing microbiome bioinformatics approaches.
AUTHOR CONTRIBUTIONS
Yixu Wang: Conceptualization (equal); data curation (equal); writing – original draft (equal); writing – review and editing (equal). Rebeca Fuzinatto Dall’Agnol: Conceptualization (equal); data curation (equal); methodology (equal); writing – original draft (equal); writing – review and editing (equal). Iris Bertani: Conceptualization (equal); methodology (equal); visualization (equal); writing – review and editing (equal). Cristina Bez: Conceptualization (equal); data curation (equal); writing – original draft (equal); writing – review and editing (equal). Vittorio Venturi: Conceptualization (equal); funding acquisition (equal); investigation (equal); project administration (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
Supporting information
Data S1.
ACKNOWLEDGMENTS
We thank Sarah Seaton and Shib Basu from Indigo Ag for technical suggestions and careful reading of the manuscript. The bacterial genome sequences reported here were sequenced at the DOE Joint Genome Institute (Lawrence Berkeley National Laboratory, Berkeley, CA, USA) under a CSP project having a JGI Proposal Id: 503198. No funding information provided.
Wang, Y. , Dall’Agnol, R.F. , Bertani, I. , Bez, C. & Venturi, V. (2024) Identification of synthetic consortia from a set of plant‐beneficial bacteria. Microbial Biotechnology, 17, e14330. Available from: 10.1111/1751-7915.14330
Yixu Wang and Rebeca Fuzinatto Dall'Agnol contributed equally.
Contributor Information
Cristina Bez, Email: bez@icgeb.org.
Vittorio Venturi, Email: venturi@icgeb.org.
DATA AVAILABILITY STATEMENT
The raw sequencing data discussed in this publication have been deposited in NCBI's Sequence Read Archive (SRA) and are accessible through Bioproject. ID PRJNA956463.
REFERENCES
- Bertani, I. , Abbruscato, P. , Piffanelli, P. , Subramoni, S. & Venturi, V. (2016) Rice bacterial endophytes: isolation of a collection, identification of beneficial strains and microbiome analysis. Environmental Microbiology Reports, 8, 388–398. [DOI] [PubMed] [Google Scholar]
- Better, M. , Lewis, B. , Corbin, D. , Ditta, G. & Helinski, D.R. (1983) Structural relationships among Rhizobium meliloti symbiotic promoters. Cell, 35, 479–485. [DOI] [PubMed] [Google Scholar]
- Bolyen, E. , Rideout, J.R. , Dillon, M.R. , Bokulich, N.A. , Abnet, C.C. , Al‐Ghalith, G.A. et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B.J. , McMurdie, P.J. , Rosen, M.J. , Han, A.W. , Johnson, A.J. & Holmes, S.P. (2016) DADA2: high‐resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compant, S. , Cambon, M.C. , Vacher, C. , Mitter, B. , Samad, A. & Sessitsch, A. (2021) The plant endosphere world—bacterial life within plants. Environmental Microbiology, 23, 1812–1829. [DOI] [PubMed] [Google Scholar]
- Compant, S. , Samad, A. , Faist, H. & Sessitsch, A. (2019) A review on the plant microbiome: ecology, functions, and emerging trends in microbial application. Journal of Advanced Research, 19, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza, R.S.C. , Armanhi, J.S.L. & Arruda, P. (2020) From microbiome to traits: designing synthetic microbial communities for improved crop resiliency. Frontiers in Plant Science, 11, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado‐Baquerizo, M. , Maestre, F.T. , Reich, P.B. , Jeffries, T.C. , Gaitan, J.J. , Encinar, D. et al. (2016) Microbial diversity drives multifunctionality in terrestrial ecosystems. Nature Communications, 7, 10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier, M. , Meola, M. , Berthoud, H. , Shani, N. , Wechsler, D. & Junier, P. (2022) High‐throughput qPCR and 16S rRNA gene amplicon sequencing as complementary methods for the investigation of the cheese microbiota. BMC Microbiology, 22, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouda, S.K. & Saranga, H. (2018) Sustainable supply chains for supply chain sustainability: impact of sustainability efforts on supply chain risk. International Journal of Production Research, 56, 5820–5835. [Google Scholar]
- Hassani, M.A. , Duran, P. & Hacquard, S. (2018) Microbial interactions within the plant holobiont. Microbiome, 6, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Wei, Z. , Friman, V.P. , Gu, S.H. , Wang, X.F. , Eisenhauer, N. et al. (2016) Probiotic diversity enhances rhizosphere microbiome function and plant disease suppression. MBio, 7. doi: 10.1128/mbio.01790-01716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Weidner, S. , Friman, V.P. , Xu, Y.C. , Shen, Q.R. & Jousset, A. (2017) Probiotic Pseudomonas communities enhance plant growth and nutrient assimilation via diversity‐mediated ecosystem functioning. Soil Biology and Biochemistry, 113, 122–129. [Google Scholar]
- Hu, J. , Yang, T. , Friman, V.P. , Kowalchuk, G.A. , Hautier, Y. , Li, M. et al. (2021) Introduction of probiotic bacterial consortia promotes plant growth via impacts on the resident rhizosphere microbiome. Proceedings of the Biological Sciences, 288, 20211396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul, S. , Choudhary, M. , Gupta, S. & Dhar, M.K. (2021) Engineering host microbiome for crop improvement and sustainable agriculture. Frontiers in Microbiology, 12, 635917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. et al. (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Research, 41, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Watts, D.B. , Kloepper, J.W. , Adesemoye, A. & Feng, Y. (2019) Effect of plant growth‐promoting Rhizobacteria at various nitrogen rates on corn growth. Agricultural Sciences, 10, 1542–1565. [Google Scholar]
- Liu, Y.X. , Qin, Y. & Bai, Y. (2019) Reductionist synthetic community approaches in root microbiome research. Current Opinion in Microbiology, 49, 97–102. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugtenberg, B. & Kamilova, F. (2009) Plant‐growth‐promoting rhizobacteria. Annual Review of Microbiology, 63, 541–556. [DOI] [PubMed] [Google Scholar]
- Lundberg, D.S. , Lebeis, S.L. , Paredes, S.H. , Yourstone, S. , Gehring, J. , Malfatti, S. et al. (2012) Defining the core Arabidopsis thaliana root microbiome. Nature, 488, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick, H. , Rahnavard, A. , McIver, L.J. , Ma, S. , Zhang, Y. , Nguyen, L.H. et al. (2021) Multivariable association discovery in population‐scale meta‐omics studies. PLoS Computational Biology, 17, e1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon, C.A. , Le Roux, X. , van Doorn, G.S. , Dini‐Andreote, F. , Poly, F. & Salles, J.F. (2018) The impact of failure: unsuccessful bacterial invasions steer the soil microbial community away from the invader's niche. The ISME Journal, 12, 728–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P.J. & Holmes, S. (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One, 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes, R. , Garbeva, P. & Raaijmakers, J.M. (2013) The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiology Reviews, 37, 634–663. [DOI] [PubMed] [Google Scholar]
- Menendez, E. & Garcia‐Fraile, P. (2017) Plant probiotic bacteria: solutions to feed the world. AIMS Microbiology, 3, 502–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.H. (1972) Experiments in molecular genetics. Cold Spring Harbor, New York, USA: Cold Spring Harbor Laboratory. [Google Scholar]
- Minchev, Z. , Kostenko, O. , Soler, R. & Pozo, M.J. (2021) Microbial consortia for effective biocontrol of root and foliar diseases in tomato. Frontiers in Plant Science, 12, 756368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, U.G. & Sachs, J.L. (2015) Engineering microbiomes to improve plant and animal health. Trends in Microbiology, 23, 606–617. [DOI] [PubMed] [Google Scholar]
- Romano, I. , Ventorino, V. & Pepe, O. (2020) Effectiveness of plant beneficial microbes: overview of the methodological approaches for the assessment of root colonization and persistence. Frontiers in Plant Science, 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, B.K. & Kapoor, K.K. (1999) Inoculation with phosphate‐solubilizing microorganisms and a vesicular‐arbuscular mycorrhizal fungus improves dry matter yield and nutrient uptake by wheat grown in a sandy soil. Biology and Fertility of Soils, 28, 139–144. [Google Scholar]
- Trivedi, P. , Leach, J.E. , Tringe, S.G. , Sa, T. & Singh, B.K. (2020) Plant‐microbiome interactions: from community assembly to plant health. Nature Reviews. Microbiology, 18, 607–621. [DOI] [PubMed] [Google Scholar]
- Venturi, V. & Bez, C. (2021) A call to arms for cell‐cell interactions between bacteria in the plant microbiome. Trends in Plant Science, 26, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Vurukonda, S.S. , Vardharajula, S. , Shrivastava, M. & Sk, Z.A. (2016) Enhancement of drought stress tolerance in crops by plant growth promoting rhizobacteria. Microbiological Research, 184, 13–24. [DOI] [PubMed] [Google Scholar]
- Wang, C.J. , Yang, W. , Wang, C. , Gu, C. , Niu, D.D. , Liu, H.X. et al. (2012) Induction of drought tolerance in cucumber plants by a consortium of three plant growth‐promoting rhizobacterium strains. PLoS One, 7, e52565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, W. , Guo, S. , Jousset, A. , Zhao, Q. , Wu, H. , Li, R. et al. (2017) Bio‐fertilizer application induces soil suppressiveness against Fusarium wilt disease by reshaping the soil microbiome. Soil Biology and Biochemistry, 114, 238–247. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The raw sequencing data discussed in this publication have been deposited in NCBI's Sequence Read Archive (SRA) and are accessible through Bioproject. ID PRJNA956463.