

Abstract
Numerous studies have demonstrated defects in multiple metabolic pathways in Alzheimer’s disease (AD), detected in autopsy brains and in the cerebrospinal fluid in vivo. However, until the advent of techniques capable of measuring thousands of metabolites in a single sample, it has not been possible to rank the relative magnitude of these abnormalities. A recent study provides evidence that the abnormal turnover of the brain’s most abundant phospholipids: phosphatidylcholine and phosphatidylethanolamine, constitutes a major metabolic pathology in AD. We place this observation in a historical context, and discuss the implications of a central role for phospholipid metabolism in AD pathogenesis.
Keywords: Phosphatidylcholine, phosphatidylethanolamine, glycerophosphocholine, glycerophosphoethanolamine, Alzheimer’s disease, metabolome, metabolomics, lipidomics
In his pioneering paper on a patient who died with severe early dementia, Alois Alzheimer noted the presence in the brain of neurofibrillary tangles and plaques, two hallmarks of the disease that came to bear his name [1]. In a subsequent more detailed analysis [2], he described in addition the prevalence of lipid granules in glial and nerve cells, and in vascular walls in the brains of such patients. However, the role of abnormal lipid metabolism in this disease received little attention from neuropathologists and biochemists until the latter half of the century [3].
Now, a recent paper by Batra et al. [4] provides a comprehensive data set of the human cerebral cortical metabolome in autopsy samples from over 500 subjects, and highlights anew the centrality of specific lipid abnormalities in this disease. The study is a major achievement of team science and includes: 1) analyses of biorepository samples of well-phenotyped participants from 3 cohorts including the Religious Order Study and Rush Memory and Aging Project (ROS/MAP) for discovery, the Mayo Clinic for replication, and the Baltimore Longitudinal Study of Aging for comparison, 2) common unified metabolomic analytic platforms for the quantification of water-soluble compounds as well as lipids, and 3) a bioinformatic pipeline for data management and statistical analyses designed to explore the associations between tissue metabolite abundance and 8 clinical and neuropathologic traits associated with Alzheimer’s disease (AD), including final diagnosis, cognitive diagnosis, cognition, the rate of cognitive decline, global neuropathology, NIA Reagan diagnostic criteria, amyloid plaques and tangle tauopathy. The authors elected to highlight some of the novel findings including AD-linked metabolic abnormalities in bioenergetics, cholesterol metabolism, neuroinflammation, osmoregulation, and neurotransmitter pathways. However, a remarkably striking and robust finding of the study is that the abnormal turnover of the two most abundant membrane phospholipids, phosphatidylcholine (PC) and phosphatidylethanolamine (PE), constitutes the most prominent brain metabolic defect in AD. This is significant because, in addition to their role as essential structural components of cellular membranes, PC and PE act as reservoirs for second messengers, including phosphatidic acid, diacylglycerol, lysophospholipids, and eicosanoid precursors, that subserve a host of essential cellular functions. The idea that brain membrane glycerophospholipid metabolism is abnormal in AD dates to the 1980s. The initial evidence came from 31P magnetic resonance spectroscopy (MRS) studies of postmortem brains and showed elevated concentrations of glycerophosphocholine (GPC) and glycerophosphoethanolamine (GPE) in AD patients as compared to age-matched controls [5–7]. Subsequent studies employing biochemical techniques generated similar results [8, 9]. Because GPC and GPE are products of the enzymatic deacylation of PC and PE, respectively (Figure 1A), the data suggested that the metabolism of these lipids may be accelerated in AD. This notion was supported by the observation that levels of PC and PE are lower in postmortem samples from parietal and frontal cortex obtained from AD patients relative to controls [9]. Moreover, this specific pattern of changes was not seen in tissue from the brain regions of patients with Down’s syndrome, Parkinson’s disease and Huntington’s disease [8, 9], though abnormalities in brain phospholipid levels have been found in the latter disease [10, 11]. These data, and additional evidence that brain regions generally free of plaques and tangles, such as the caudate and the cerebellum, are similarly affected [8], led to the hypothesis that the phospholipid defect may be specific to AD and widespread within the brain, and moreover suggested that the observed phospholipid-related changes are not simply the result of brain atrophy in areas most affected by pathologic changes. The study by Batra et al. is not only consistent with this idea but also presents compelling evidence that this abnormality is a prominent and consistent feature of the metabolic pathology in AD [4]. Indeed, Table 3 in the paper lists GPC and GPE among the top 3 compounds with levels in the dorsolateral prefrontal cortex (DLPFC) most significantly associated with each of the 8 AD-related traits listed above. Moreover, the rise in GPC and GPE levels was accompanied by reduced abundance of multiple PC and PE molecular species, particularly those carrying a polyunsaturated fatty acid (e.g. arachidonic, 20:4 and docosahexaenoic, 22:6) as shown in Figure 1B, which summarizes the most statistically significant data of the choline- and ethanolamine-containing compounds provided in the Supplementary Table 6 by Batra et al. [4]. Our examination of the transcriptomic mRNA data available on the Agora platform (https://agora.adknowledgeportal.org/genes) – derived from assays of the analogous ROS/MAP cohort participant DLPFC samples used by Batra et al. for their metabolomic studies – shows that some of the genes encoding enzymes responsible for PC/PE metabolism are differentially expressed in association with several AD-related traits (Figure 1A), suggesting that the PC/PE metabolomic defect may be the result of enzymatic abnormalities. The obvious caveat is that RNA expression need not correlate with protein levels and enzyme activities which may be modulated by the local milieu and/or post-translational modifications. Examination of the relevant proteomic data available on the Agora platform (not as complete as the transcriptomic resource) indicates a reasonable concordance between the RNA and protein levels (Figure 1A, bold text). For example mRNA, and protein expression of glycerophosphocholine phosphodiesterase 1 (GPCPD1) – which degrades GPC and GPE to choline and ethanolamine, respectively – is reduced in AD as compared to controls – a result consistent with high abundance of these compounds in AD brain as reported by earlier investigators [5–9]. Taken together the results of Batra et al. should provide strong motivation to place studies of the metabolism of choline and ethanolamine glycerophospholipids among top priorities of AD research. Some of the unanswered questions that need to be addressed include: 1) What are the enzymatic abnormalities that cause the accumulation of GPC and GPE in AD brain? 2) Which cell types are vulnerable to this pathophysiologic process? Given the large and robust size of this abnormality seen in multiple studies, these are likely to be some of the principal cortical cells, such as a specific neuronal class or oligodendrocytes. The latter possibility must be seriously considered, in view of the very high lipid content (80% of its dry matter) of the myelin produced by oligodendrocytes [12]. These cells reside in the gray and white matter and thus both of these tissue types ought to be studied; 3) In view of evidence that GPC concentrations are elevated in the CSF [13] and blood [14] of AD patients as compared to controls, would it be valuable to investigate GPC and GPE as potential biomarkers of AD and to determine whether they could be integrated with the currently used biomarkers? 4) Can the metabolomic data of Batra et al. be used to generate derivative values (ratios/classifiers) that could better discriminate the AD-related traits? 5) Is the PC/PE metabolic defect confined to the brain or is it also present in other tissues? and 6) Can this defect be investigated in vitro or using animal models of AD? Indeed, a remarkably similar pattern of metabolic changes is observed in a neuronal cell line treated with inhibitors of oxidative phosphorylation [15] – a result consistent with the data of Batra et al. reporting abnormalities of brain bioenergetics in AD [4]. Finally, it is important to note that the levels of the molecular species of PC and PE may be modulated by the supply of certain essential nutrients such as choline and fatty acids, and thus it may be possible to design nutritional strategies to prevent and/or treat the abnormal PC/PE metabolic processes in AD.
Figure 1A.
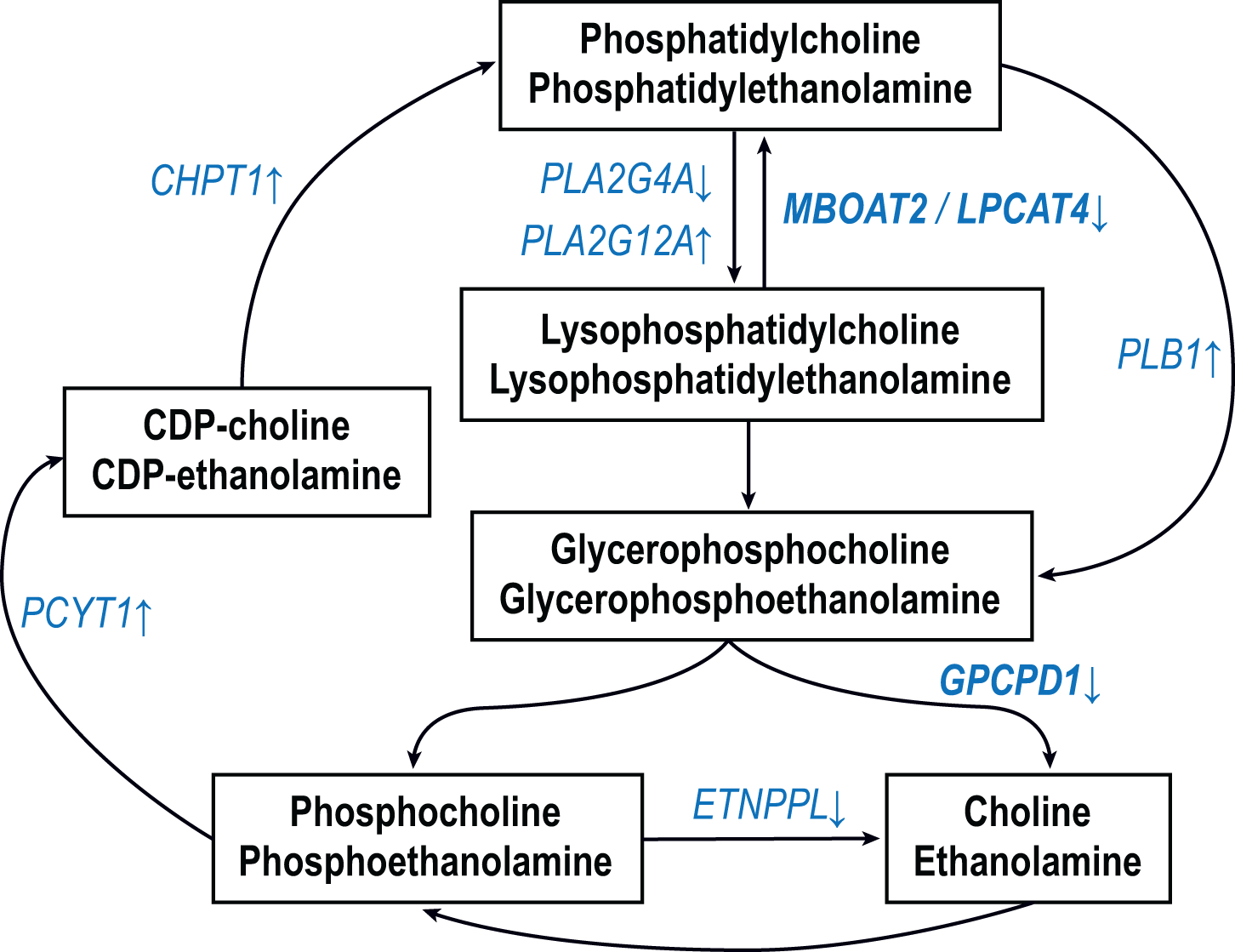
Simplified pathways of the synthesis and breakdown of phosphatidylcholine and phosphatidylethanolamine. Only the relevant enzymatic steps are shown as arrows. The ROS/MAP DLPFC transcriptomic data set was searched to determine if there are any differentially expressed genes (DEGs) in these pathways in association with AD-traits using an adjusted p value cutoff of 0.05. The DEGs thus identified (and the direction of the effect) are shown in blue font. For example, the expression of GPCPD1 was lower in AD than in controls. The traits used were: diagnosis, Braak, CERAD and cognition. In addition a ROS/MAP DLPFC proteomic data set was searched to determine if any of these DEGs were also differentially expressed proteins. This was the case for MBOAT2 and GPCPD1 (bold font).
Figure 1B.
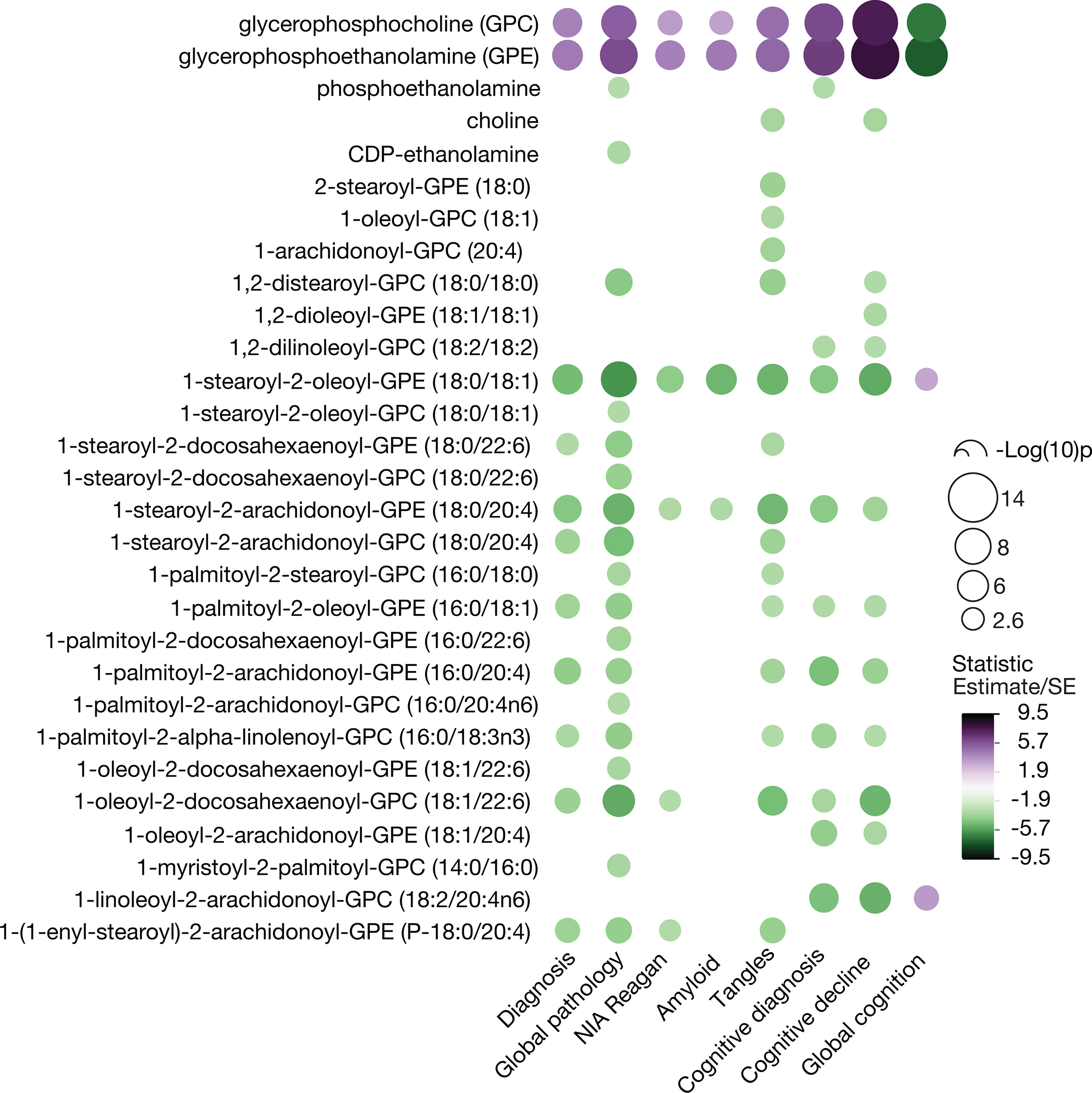
Association of the levels of choline and ethanolamine compounds with AD traits. Data from the Supplementary Table 6 in Batra et al. derived from the analyses of the DLPFC of the ROS/MAP cohort. The data were filtered to include only the most statistically-significant compounds (statistic >3 or <−3). The unadjusted p values for this set are <0.0026 [−Log(10)p=2.6].
Funding:
Supported by the National Institutes of Health grants: RF1AG057768, P30AG072978, U19AG068753.
Footnotes
Competing interests: The authors declare that they have no competing interests.
References
- [1].Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin 64, 146–148. [Google Scholar]
- [2].Alzheimer A (1911) Über eigenartige Krankheitsfälle des späteren Alters. Zeitschrift für die gesamte Neurologie und Psychiatrie 4, 356–385. [Google Scholar]
- [3].Foley P (2010) Lipids in Alzheimer’s disease: A century-old story. Biochim Biophys Acta 1801, 750–753. [DOI] [PubMed] [Google Scholar]
- [4].Batra R, Arnold M, Worheide MA, Allen M, Wang X, Blach C, Levey AI, Seyfried NT, Ertekin-Taner N, Bennett DA, Kastenmuller G, Kaddurah-Daouk RF, Krumsiek J, Alzheimer’s Disease Metabolomics C (2022) The landscape of metabolic brain alterations in Alzheimer’s disease. Alzheimers Dement. (available on 2024–01-13) DOI: 10.1002/alz.12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barany M, Chang YC, Arus C, Rustan T, Frey WH (1985) Increased glycerol-3-phosphorylcholine in post-mortem Alzheimer’s brain. Lancet 1, 517. [DOI] [PubMed] [Google Scholar]
- [6].Pettegrew JW, Panchalingam K, Moossy J, Martinez J, Rao G, Boller F (1988) Correlation of phosphorus-31 magnetic resonance spectroscopy and morphologic findings in Alzheimer’s disease. Arch Neurol 45, 1093–1096. [DOI] [PubMed] [Google Scholar]
- [7].Miatto O, Gonzalez RG, Buonanno F, Growdon JH (1986) In vitro 31P NMR spectroscopy detects altered phospholipid metabolism in Alzheimer’s disease. Can J Neurol Sci 13, 535–539. [DOI] [PubMed] [Google Scholar]
- [8].Blusztajn JK, Lopez Gonzalez-Coviella I, Logue M, Growdon JH, Wurtman RJ (1990) Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer’s disease but not of Down’s syndrome patients. Brain Res 536, 240–244. [DOI] [PubMed] [Google Scholar]
- [9].Nitsch RM, Blusztajn JK, Pittas AG, Slack BE, Growdon JH, Wurtman RJ (1992) Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci USA 89, 1671–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mastrokolias A, Pool R, Mina E, Hettne KM, van Duijn E, van der Mast RC, van Ommen G, t Hoen PA, Prehn C, Adamski J, van Roon-Mom W (2016) Integration of targeted metabolomics and transcriptomics identifies deregulation of phosphatidylcholine metabolism in Huntington’s disease peripheral blood samples. Metabolomics 12, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Phillips GR, Hancock SE, Jenner AM, McLean C, Newell KA, Mitchell TW (2022) Phospholipid Profiles Are Selectively Altered in the Putamen and White Frontal Cortex of Huntington’s Disease. Nutrients 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].O’Brien JS, Sampson EL (1965) Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res 6, 537–544. [PubMed] [Google Scholar]
- [13].Walter A, Korth U, Hilgert M, Hartmann J, Weichel O, Hilgert M, Fassbender K, Schmitt A, Klein J (2004) Glycerophosphocholine is elevated in cerebrospinal fluid of Alzheimer patients. Neurobiology of aging 25, 1299–1303. [DOI] [PubMed] [Google Scholar]
- [14].Jia L, Yang J, Zhu M, Pang Y, Wang Q, Wei Q, Li Y, Li T, Li F, Wang Q, Li Y, Wei Y (2022) A metabolite panel that differentiates Alzheimer’s disease from other dementia types. Alzheimers Dement 18, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Farber SA, Slack BE, Blusztajn JK (2000) Acceleration of phosphatidylcholine synthesis and breakdown by inhibitors of mitochondrial function in neuronal cells: a model of the membrane defect of Alzheimer’s disease. FASEB J 14, 2198–2206. [DOI] [PubMed] [Google Scholar]