

ABSTRACT
The influenza A virus hemagglutinin protein (HA) has been studied extensively in humans but less so in other host species. Here, we compared the rates of nucleotide substitution, protein evolution, and glycosylation in the H1 and H3 head and stalk domains among five host classes (avian, canine, equine, human, and swine). For further resolution, we separately analyzed 49 lineages differentiated by host and geography. HA evolution in non-human hosts was more dynamic than expected. For example, the classical swine H1 head domain accumulated glycosylation sites in the antigenically important head domain at a rate as high as in humans. However, whereas the stalk domain was highly conserved in humans, with a low ratio of non-synonymous to synonymous changes, this ratio was approximately 2.5-fold higher in canines. Together, these findings highlight the need for further study of HA evolution in non-human hosts.
IMPORTANCE
For decades, researchers have studied the rapid evolution of influenza A viruses for vaccine design and as a useful model system for the study of host/parasite evolution. By performing an exhaustive analysis of hemagglutinin protein (HA) sequences from 49 lineages independently evolving in birds, swine, canines, equines, and humans over the last century, our work uncovers surprising features of HA evolution. In particular, the canine H3 stalk, unlike human H3 and H1 stalk domains, is not evolving slowly, suggesting that evolution in the stalk domain is not universally constrained across all host species. Therefore, a broader multi-host perspective on HA evolution may be useful during the evaluation and design of stalk-targeted vaccine candidates.
KEYWORDS: influenza A virus, phylodynamics, adaptive evolution, glycosylation, inter-species transmission, hemagglutinin
INTRODUCTION
Influenza A and B viruses are the etiological agents of seasonal influenza in humans. Unlike influenza B viruses, which are only known to infect humans and seals, influenza A viruses of H3 and H1 subtypes infect humans and a multitude of non-human animals (1). Influenza A viruses (IAVs) periodically jump between host species, presenting an ongoing pandemic threat (2, 3). Eighteen haemagglutinin (HA) subtypes in influenza A have been identified (H1–H18), of which H1 and H3 are currently circulating in human populations (4). Influenza A viruses of the H3 and H1 subtypes currently circulate in birds, humans, swine, dogs, and horses (5–11). Human seasonal IAVs infect between 10% and 20% of the human population every year, causing an estimated 290,000–650,000 deaths annually (12). IAVs also cause significant morbidity in animal populations, affecting swine and poultry production (13, 14) and the horse racing industry (15–17).
Influenza viruses of the A/H1 and A/H3 subtypes circulate in wild birds, which are regarded as the natural reservoir for IAVs (18–20). During the 20th century, influenza A/H1 and A/H3 have frequently jumped from one host to another, establishing multiple geographic- and host-specific lineages (Fig. 1).
Fig 1.
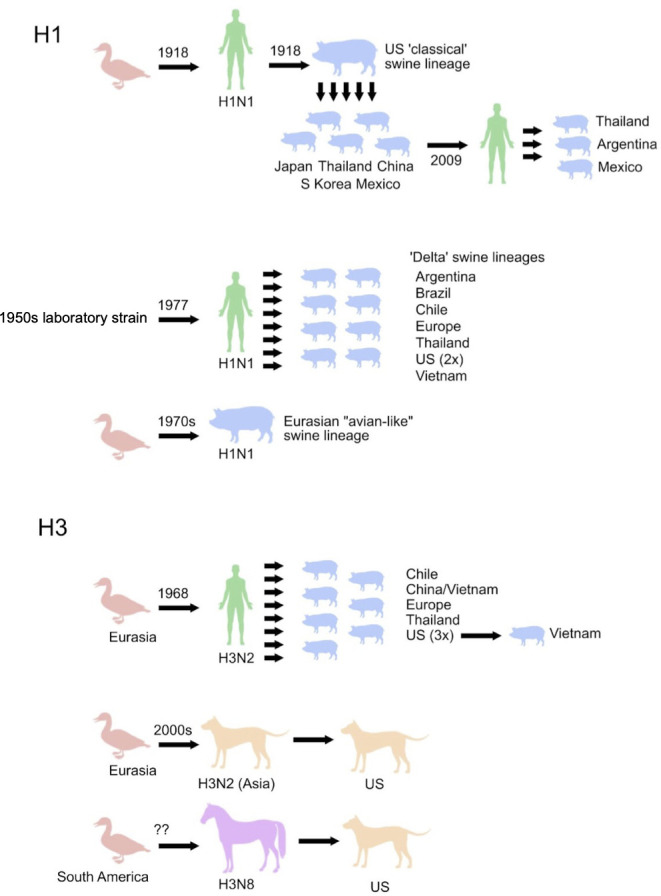
Host and location jumps of influenza H3 and H1 segments. HA jumps between host species and countries are denoted by black arrows. Question marks represent uncertainty in the timing of avian-to-equine transmission of H3N8 viruses. The evolutionary history of the other genome segments, including reassortment, is not depicted.
Spillover of avian viruses to humans caused the 1918 H1N1 (21) and 1968 H3N2 pandemics (22). Avian viruses (H3N8) also became established in equines between the 1950s and 1960s (23). Human viruses have been transmitted dozens of times to pigs, establishing several independently evolving H1N1, H1N2, and H3N2 swine lineages on multiple continents (24). In the early 2000s, two canine lineages emerged independently in the United States and in Asia through introductions from horses (H3N8) and birds (H3N2), respectively (8, 25). More recently, a H1N1 virus jumped from swine to humans and caused the 2009 H1N1 pandemic (26), with subsequent reverse zoonosis events back to pigs on multiple continents (27).
Mutations occur frequently during IAV replication by an error-prone RNA polymerase. The rate of synonymous substitution varies by host species, presumably due largely to differences in mutation rate and transmission modes (28). This is affected by host-specific factors, including the number of viral replications per unit time and biochemical differences affecting replication fidelity. The rate of non-synonymous evolution, relative to the mutation rate, is determined by the forces of natural selection. Non-synonymous mutations, which change the amino acid encoded, are frequently deleterious but may be adaptive. For example, they may diminish binding by existing antibodies (29) or improve binding to host cell receptors (30), particularly after a host switch. Research on influenza evolution and adaptation has focused on the HA, the membrane glycoprotein present on the surface of the virus which is responsible for receptor binding and membrane fusion and which is under constant selection due to the host immune response.
Each subunit of the HA homotrimer consists of two domains. The immunodominant head domain is the principal target of antibody-mediated immunity and evolves more rapidly than the stalk domain, which is relatively conserved within and among subtypes (31–33). Mutations in the HA head domain can help the virus evade antibodies, increasing its ability to infect individuals with immunity elicited by earlier strains. Some HA mutations create or eliminate glycosylation sites (34). Although glycosylations can shield HA from antibodies, they can also interfere with receptor binding and are targets of innate immunity (35, 36). Due to the different strengths of immune selection in different host species, the optimal number of glycosylations is expected to vary among them (37).
Human influenza vaccine strains are updated twice a year to reflect antigenic changes in viruses circulating globally. However, due to limitations of the global surveillance, in the influenza vaccine-manufacturing process, and in the ability to predict the dominant strains 6 months to 1 year in the future, vaccine strains are periodically mismatched to viruses in circulation, leading to reduced vaccine effectiveness (38–40). Vaccines are also available for swine (41, 42), canines (43), equines (44–46), and poultry (47), although there is no systematic surveillance to monitor their effectiveness nor standard procedure for updating strains, limiting their effectiveness and utilization (48, 49). Utilization of poultry vaccines is further reduced in the United States, European Union, and other exporters because international trade regulations prevent the export of products from vaccinated chickens. Studies of antigenic evolution rely on serological assays [hemagglutination inhibition (HI) or virus neutralization assays]. However, the assays are not always well standardized or reproducible across laboratories. Aside from US swine, there is no systematic antigenic evaluation of non-human influenza, which impedes comparison of rates of protein and antigenic evolution of influenza in non-human hosts. Although seasonal influenza vaccines rely primarily on antibodies generated against the HA head, novel vaccine approaches aim to provide broader, longer lasting protection (50) by targeting more conserved regions, including the HA stalk domain, which has not been studied as intensively to date.
Here, we compare the evolutionary dynamics of H1 and H3 head and stalk domains in humans, swine, birds, equines, and canines, including rates of synonymous and non-synonymous change, the locations of changes in three-dimensional structures, and rates and patterns of gains and losses of glycosylation sites. Our results suggest that the head domain, which evolves rapidly in humans due to selection by the immune system, also does so in other mammals, though not to the same extent. Particularly, H3 evolution within the canine clades and along the host-jump branches leading to them reveals several phenomena that appear to reflect adaptation of the protein to canine hosts.
RESULTS
Evolutionary dynamics of influenza A/H1 and A/H3 Across Host Species
A combination of Bayesian mixed effects molecular clock modeling and stochastic substitution mapping was used to estimate the rates of non-synonymous and synonymous change in several sets of positions in all lineages described above. Consistent with previous studies (33), dN/dS is higher in the head domain than the stalk domain in all hosts (Fig. 2). In both H3 and H1, the highest dN/dS in the head domain are found in the human viruses (Fig. 2; Table S10 to S13). The next highest in H3 is found in canine viruses, which unexpectedly have the highest value in the stalk. Equine viruses also have a high value for the head compared with swine and avian viruses. The dN/dS estimates tend to be lower in swine and avian viruses of either subtype. In human viruses, dN/dS is higher in H3 than H1 in both domains.
Fig 2.

dN/dS for the head and stalk domains of H1 and H3 viruses circulating in multiple hosts.
Evolution of the H3N2 and H3N8 canine lineages
We sought to investigate the high dN/dS value associated with the stalk of canine viruses. They form two clades in the H3 phylogeny. One consists of H3N8 viruses and was introduced into canines from equines in the 2000s in the United States (canine USA H3N8 lineage). The second consists of H3N2 viruses and was introduced into canines from Eurasian birds in the 2000s in Asia (canine Asia H3N2 lineage) and subsequently disseminated to the United States in around 2015, establishing the canine USA H3N2 lineage. The existence of two independently derived canine clades allows confirmation that some evolutionary patterns are related to canine hosts, rather than being idiosyncratic features of a sequence type.
A total of 14 amino acid changes were inferred to occur on the branches containing the host-switches to canines from avians or equines (Fig. 3).
Fig 3.

Amino acid changes on host-shift branches leading to canine clades
On the host-switch branch leading to the canine USA H3N8 lineage, there are five non-synonymous changes and one synonymous change. The 5:1 non-synonymous:synonymous (N:S) ratio on this branch is statistically distinguishable from the ratio in the ancestral equine lineage (366:537 = 0.68:1; P = 0.044, Fisher’s exact test) but not from the ratio in the derived canine clade (101:111 = 0.91:1; P = 0.11). Some non-synonymous changes occurred in sites with known functional importance. One of the substitutions (W222L) is located adjacent to the sialic acid binding site. I328T is a reversal of an equine-specific change: the equine sequences have mostly I and occasionally L (6%), but 97% of other H3 viruses have T, and none has I or L. This position immediately precedes Arg329, after which proteolytic cleavage splits HA into HA1 and HA2 (the peptide bond between 329 and 330). The N483T change eliminates a glycosylation site that was present in the equine lineage (confirmed by crystal structures, Protein Data Bank (PDB) entries 4uo0 and 4unw) (Fig. 3). Independent loss of this site occurred in a close equine relative of the ancestor of the canine USA H3N8 virus within about 3 years [according to the maximum clade credibility (MCC) tree, 95% highest posterior density (HPD) branch lengths] and possibly simultaneously (Fig. S11). The parallel losses are clearly distinguishable: that on the host-shift branch results from a second-position change of codon 483 from AAT (asparagine) to ACT (threonine), while the other results from a third-position change to AAG (lysine). This conclusion comes from the maximum parsimony reconstruction but is also supported by the sequences of the earliest canine USA H3N8 isolate and the affected equine isolate, which bear ACT and AAG, respectively.
The canine Asia H3N2 host-switch branch contains 9 non-synonymous and 19 synonymous changes. The 0.47:1 N:S ratio is intermediate between that in the progenitor avian Eurasia lineage (472:2,217 = 0.21:1) and that in the derived canine H3N2 clade (147:157 = 0.94). However, it is not statistically distinguishable from either due to the low absolute counts. The canine clade and the avian progenitor are clearly distinguishable from each other (P = 2e−30, Fisher’s exact test).
No changes at the same amino acid positions occur on the two host-shift branches, but in some cases (indicated by red in Fig. 3), the amino acid resulting from a change in one is identical to that inherited at the same position by the other canine clade. In two additional cases (indicated by orange), a change results in a serine at a position where the other last common ancestor (LCA) has the chemically similar threonine or vice versa. In both cases, the observed change is due to a transition mutation, whereas a change to the amino acid present in the other LCA (threonine or serine, respectively) would require a transversion of a type that is much less frequent, as determined by analysis of fourfold-degenerate synonymous sites.
A total of 21.4% of the amino acid changes on these branches result in changes of glycosylation state. This is larger than the fraction (157/3,755 or 4.1%) for the remainder of the tree (P = 0.019, Fisher’s exact test). A similar conclusion holds for the ratio of glycosylation changes to synonymous changes (P = 0.0070, Fisher’s exact test).
The non-canine ancestors of both canine clades bear two glycosylation sites near the membrane-proximal portion of HA, at positions 8 and 483 (HA2 154). One of these sites is lost in the genesis of the canine H3N2 clade (8), and the other is lost in the genesis of canine USA H3N8 (483). Canine USA H3N8 inherits a glycosylation site at position 63, which is absent from H3N2, and the canine H3N2 clade gains a glycosylation at 81. These sites lie at nearly the same position along the HA axis and may cover similar regions of the protein’s surface (Fig. S12). Additional evidence for their interchangeability comes from human H3N2, in which a gain of glycosylation at 63 is quickly followed by a loss of glycosylation at 81 in the “trunk” of the tree.
Changes within canine clades
Figure 4 shows the distribution of amino acid changes within the canine clades across the protein sequence. The variance of these counts is too large to be explained, except by extremely improbable occurrences, unless the rate at some of the sites exceeds the neutral expectation substantially. It would be explained if the counts for the seven sites with at least four changes reflected high rates, implying positive selection, rather than chance events. Unless rates at the remaining sites were mostly close to the neutral expectation or to zero, more sites would have to be subject to positive selection. Positive selection on a site might be obscured by purifying or weak selection in other parts of lineages, as appears to be the case for these changes (see below).
Fig 4.

Amino acid changes across the H3 protein of canine isolates. Total number of amino acid changes across the H3 protein of canine isolates (top). Changes in canine H3N2 (red) and canine USA H3N8 (blue) are stacked. The seven positions with at least four changes are labeled. Six of these are interface positions in all monomers of all four structures considered (PDB entries 4uo4, 6n4f, 4uo0, and 4unw). The seventh (464) is an interface position in all monomers of the two equine structures. Interface positions can be found in File S5. The seven positions evolving most rapidly in canine H3 shown on the HA structure (bottom). Three views together show all seven positions. The six protein chains (three each of HA1 and HA2) are given different light colors, and the seven positions are shown in darker versions of the same color. In panel A, three 216 and 218 residues appear (one of each on each monomer), but only one of each is labeled. In panel C, one of the HA monomers (chain C) is made invisible to reveal the buried position 107. Positions 216 and 218 in chain A are not colored in this view because they are at the interface with the invisible chain C. HA2 positions are given in parentheses. The HA structure is that of A/Equine/Richmond/07, PDB entry 4UO0.
The seven most rapidly evolving positions, those with four or more total changes in the canine clades, are 19, 107, 216, 218, 436 (HA2 107), 453 (HA2 124), and 464 (HA2 135) in the conventional H3 numbering (based on human H3 after removal of the 16-residue leader peptide). We refer to this set of positions as C7 (Fig. 4).
The locations of these seven positions in hemagglutinin crystal structures appear to lie disproportionately at interfaces between protein chains. This impression is confirmed by a quantitative analysis. We define interface residues as those for which any atom is within 5 angstroms of an atom of a different protein chain. This includes the interface between HA1 and HA2 chains derived from the same full-length HA. The set of positions meeting this criterion varies among HA crystal structures, presumably in part because of differences in the protein sequences, and also between monomers not related by crystallographic symmetry. We consider four structures: one from each canine clade (PDB entries 4uo4 for canine USA H3N8 and 6n4f for canine Asia/USA H3N2) and two of equine hemagglutinins (4uo0 and 4unw), which retain some ancestral residues that have changed in the canine USA H3N8 viruses.
Of the seven rapidly changing positions, six (all but 464) are interface positions in all monomers of all four structures. All seven are at interfaces in at least one monomer of these structures. The corresponding fractions for all positions in the protein are ~35.5% (interface in all monomers of all structures) and ~42.9% (interface in at least one). Thus, the probability that at least six of seven positions chosen at random lie at interfaces in all structures is 1.0%, and the probability that all seven do in at least some cases is less than 0.3%.
This result is not due to more rapid evolution of interface residues in general. In fact, in non-canine lineages, interface positions evolve more slowly on average than non-interface positions by nearly a factor of 2. The C7 positions are also not particularly rapidly evolving in other lineages. Thus, the over-representation of interface residues among the rapidly evolving sites is even more striking than that suggested by the probabilities given above.
This phenomenon is not restricted to the seven most rapidly evolving sites. At the remaining interface positions, the average number of changes (0.44) is larger than that at non-interface positions (0.37) (P = 0.041, randomization test). In non-canine lineages, the reverse is true. The ratio of interface changes to non-interface changes, excluding the C7 sites, is higher in canines by a factor of 2.6 [P = 1e−10, 95% CI = (1.9, 3.5), Fisher’s exact test].
The structures demonstrate that changes at position 135 of HA2 (464 of HA), the only C7 position that is non-interface in some of the structures, can have large effects on inter-subunit interfaces. Changes at this position in canines are restricted to the equine-derived H3N8 clade. Both equine structures have the ancestral glycine at this position. It lies within 5 angstroms of the associated HA1 and within 5.5 angstroms of the Arg at 124 (another C7 position) of another HA2 chain. The canine USA H3N8 structure, in contrast, has an aspartate at position 135, and it is further than 5 angstroms from any other chain. The backbone configuration surrounding this position is altered, and the positioning of several nearby residues is affected. Likely as a consequence, an inter-chain bidentate salt bridge between Glu131 and Arg163, found in both equine structures, does not form in the canine structure.
Changes of Arg124 will also eliminate this salt bridge. The ancestral glycine at position 218 has backbone torsion angles, corresponding to right-handed beta-strand, that are “forbidden” for non-glycine residues, which suggests that changes there will disrupt the structure. At five of the seven positions, the ancestral residue is inferred to have changed to several different residue types, which is more consistent with disruption of favorable interactions than creation of new ones.
Synonymous rates of H1 and H3 across host species
Given the unique mechanisms that led to the emergence of the canine lineages and the high dN/dS estimate associated with their stalk domain, we sought to investigate how canine viruses evolved in the context of other human and animal lineages. Absolute rates of sequence change can be assessed because BEAST estimates branch lengths in units of time, which is possible because of the different isolation dates of the viruses. The absolute rates of synonymous change (substitutions/site/year) observed in H3 viruses in swine, human, and avian hosts were higher than those for the equine and canine lineages (Fig. 5). As expected, they were similar across codon subsets (Fig. S2). They were generally slightly lower in H1 than in H3, but patterns across host species were similar, with higher rates for swine and avian viruses (Fig. S3).
Fig 5.

Synonymous rates for all H3 and H1 sites across lineages. Viral lineages are colored by host species: green (human), blue (swine), red (avian), purple (equine), and orange (canine). Lineage estimates with wider violin plots (lower probability of observations taking a given value) are presented in Fig. S4 (H3) and Fig. S5 (H1).
Protein evolution of H3 across host species
The estimates of the ratio of non-synonymous to synonymous substitutions [dN/dS (dN/dS)] were higher in mammalian viruses than avian viruses across all subsets of the H3 sequence (Fig. 6). As expected, dN/dS was approximately 1–4 times higher in the H3 head compared with the stalk domain (Fig. S4; Table S3 to S6). Estimates for dN/dS in antigenically relevant sites in the H3 were 2.2–13.5 times higher in humans compared with canines, swine, equines, and avian species (Table S4). Second to human viruses, canine Asia H3N2 and canine USA H3N8 lineages had the highest dN/dS estimates across all sequence subsets. However, we observed a strikingly different pattern in the stalk domain. Estimates for dN/dS in the stalk domain are 1.8–7.4 times higher in canines (Table S6) compared with human, equine, swine, and avian lineages.
Fig 6.

dN/dS for all H3 sites, antigenically relevant sites, head and stalk domains across lineages. Viral lineages are colored by host species: green (human), blue (swine), red (avian), purple (equine), and orange (canine). Lineage estimates with wider violin plots (lower probability of observations taking a given value) are presented in Fig. S5.
Protein evolution of H1 across host species
Patterns of dN/dS in H1 viruses were generally similar to those observed for H3. Again, the estimates of dN/dS were higher in H1 mammalian viruses than in H1 avian viruses in all subsets of positions (Fig. 7). As expected, dN/dS was approximately 1.5–5 times higher in the H1 head compared with the stalk domain (Fig. S5; Table S7 to S9). We estimated the highest overall dN/dS for the H1 human lineage that circulated from 1918 to 1957 for all sites and head domain (Fig. 7; Table S7 and S8).
Fig 7.

dN/dS for all H1 sites, head and stalk domains across lineages. Viral lineages are colored by host species: green (human), blue (swine), and red (avian). Lineage estimates with wider violin plots (lower probability of observations taking a given value) are presented in Fig. S6.
Estimates for dN/dS for the H1 head domain were 1.5−5.8 times higher in humans than in swine and avian species (Table S8). The dN/dS of the head domain of human-like “delta” swine lineages circulating in the USA in 2005–2019 was between 1.4 and 1.9 times higher than several other swine H1 lineages, including delta, classical, and Eurasian swine viruses (Fig. 7; Table S8). The US delta swine viruses also had an elevated dN/dS in the stalk domain compared with other lineages (Fig. 7; Table S9).
Comparison of non-synonymous and antigenic rates of evolution
Rates of antigenic evolution (antigenic changes per unit time) of the corresponding human and swine HA lineages, derived from HI assays (51, 52), are compared with non-synonymous rates per unit time in Fig. S6. In all cases where confidence intervals are not too wide to permit comparison, human H3 and H1 have higher antigenic rates than their swine counterparts. Human H3 has a higher non-synonymous rate per unit time than swine H3, but human H1 has a lower rate than many types of swine H1. The large uncertainties of antigenic rate estimates for some types of swine HA limit the value of comparisons.
Glycosylation patterns across H3 lineages
We investigated trends in glycosylation for each lineage using the motif Asn-X-Ser/Thr-X, where X is any amino acid other than proline. The average yearly number of glycosylations for H3 (Fig. 8) varies markedly across host species with an average of 6.2 glycosylations in avian lineages, 7.0 in canine, 7.8 in equine, 9.2 in swine, and 11.5 in humans. While the number of glycosylations in avian, canine, and equine hosts generally remained constant over time, the number of glycosylations in human and swine viruses increased over time. The human H3N2 lineage inherited 7 glycosylations from its avian ancestor. It subsequently gained 6 glycosylations (Table 1), but the avian lineage did not gain or lose glycosylations in the decades that followed. During 1968–2019, the human H3 lineage gained an average of 1.1 glycosylation per decade, peaking at 13 glycosylations in 2017. Subsequent host-switches of H3N2 viruses from humans to swine seeded additional human-like H3 lineages in pigs. After establishment in swine, the number of glycosylations typically decreased over time to between 8 and 9 glycosylations (Fig. 8; Table 1). Two swine lineages in China and Vietnam gained glycosylations over time, but there are only a few years of data available for these lineages and long-term trends are unclear.
Fig 8.

Average number of H3 glycosylations over time. For each lineage, the average number of glycosylation sites is plotted for each calendar year for which numbers are available. The average is taken over all isolates and internal nodes (reconstructed ancestral sequences) dated to that year.
TABLE 1.
Average number and rate of accumulation of glycosylation in H3 lineages
| Lineage | Net change in number of glycosylations per year | Annual average number of glycosylations |
|---|---|---|
| Swine Chile | −0.90 | 9.17 |
| Swine USA 2017 | −0.66 | 11.26 |
| Swine USA 2012 | −0.16 | 8.30 |
| Swine North America | −0.05 | 8.23 |
| Avian Americas | 0.00 | 6.47 |
| Avian Chile | 0.00 | 6.00 |
| Canine Asia H3N2 | 0.00 | 6.96 |
| Canine USA H3N2 | 0.00 | 6.95 |
| Canine USA H3N8 | 0.00 | 7.00 |
| Swine Europe | 0.00 | 9.89 |
| SwineThailand | 0.00 | 9.35 |
| Avian Eurasia | 0.01 | 6.08 |
| Equine | 0.02 | 7.76 |
| Human | 0.11 | 11.54 |
| Swine China, Vietnam | 0.19a | 8.01 |
| Swine Vietnam | 0.21a | 9.58 |
Only 3–4 years of data is available for these lineages.
The avian H3 lineages have stably maintained the lowest number of glycosylations (6–7) in both the Americas and Eurasia. The equine lineage maintained a low glycosylation average of 7 for approximately 30 years after emergence, but in the late 1980s, it gained a single glycosylation that has been maintained as of 2019 (Fig. 8). One site was lost in the genesis of canine H3N8, which mostly maintained the resulting glycosylation state. The avian-origin canine H3N2 clade has also maintained 7 glycosylations, like the Eurasian avian lineage from which it derived (Fig. 8), though one of the glycosylations is at a different position.
In H3, glycosylations are accumulated in the HA structural domains at different rates across lineages (Figs. S7 and S8; Table S14 and S15). Glycosylations were mainly gained and lost in the HA stalk domain in Avian viruses circulating in the Americas (Fig. S8), with no net change in the number (Table S15). There were gains of 1–2 glycosylations in the stalk domain of swine viruses circulating in China and Vietnam. The number of glycosylations in the HA head domain of human viruses increased by 3 during the period between late-1990s and early-2000s and was overall stable during approximately 15 years, followed by the loss of at least 1 glycosylation. The stalk domain of the human lineage kept a stable number of 5 glycosylations from its emergence to 2010 but since then has gained 2 glycosylations.
Glycosylation patterns across H1 lineages
The average number of glycosylations (Fig. 9; Table S16; File S4) varies across H1 lineages in a manner similar to H3. The avian lineages have maintained a stable, lower number of 7 glycosylations over time (Fig. 9). Like human H3, the human 1918–1957 H1 lineage, which is thought to have originated in birds, initially had 7 glycosylations. By 1957, the lineage had 11 glycosylations, a net gain of 4 (Fig. 9; Table S16). As expected, the human 1977–2009 H1 lineage, which began with a virus genetically similar to H1N1 from the 1950s, inherited 11 glycosylations. This number dropped to 10 by the time the virus went extinct in 2009 during the swine-origin H1N1 pandemic. The currently circulating swine-origin H1N1 pandemic (PDM) lineage in humans inherited a relatively low number of glycosylations from swine (n = 8), but this number increased to 9 within a decade (Fig. 9; Table S16). Surprisingly, the initial classical swine virus circulating in North America experienced a dramatic gain in glycosylations since 1970. The virus emerged in US swine during the 1918 pandemic with 7 glycosylations, like early human H1N1, but this number increased to 11 by 2019, reaching the same maximum as humans. In contrast, the Eurasian avian-like H1 swine lineage only gained 1 glycosylation in the decades since the virus switched hosts from birds to pigs in the late 1970s. The swine delta lineages (Fig. 9) resulted from host jumps from Human1977-2009 viruses. The average number of glycosylations in swine delta lineages and PDM lineages has remained relatively stable over time (Table S16). There have been multiple spillovers of human pandemic H1N1 into swine since 2009. Between 1992 and 2005, six of the swine lineages (DTswEUR1994, DTswVNM, DTswUSA2005OK, DTswCHLRancagua, DTswCHLMelipilla, and EAswEUR) have mostly lost glycosylation at position 94 (File S4).
Fig 9.

Average number of H1 glycosylations over time. For each lineage, the average number of glycosylation sites is plotted for each calendar year for which numbers are available. The average is taken over all isolates and internal nodes (reconstructed ancestral sequences) dated to that year.
As in H3, the accumulation of glycosylations in H1 occurred more frequently in the head domain in the human lineages (1.5–5.8 times more for H1 human lineages than in other swine and avian lineages) (Fig. S9 and S10; Table S17 and S18), whereas swine tended to gain and lose glycosylation at similar proportions in either HA domain, irrespective of subtype.
No gains or losses in glycosylation were observed in the stalk domain of human H1 lineages, avian, and pandemic swine viruses (Fig. S11).
DISCUSSION
Studying the evolution of influenza sequences in non-human hosts is important for several reasons: the burden of non-human infections, the ever-present possibility of a jump to human hosts, the knowledge gained about viral evolution in general, and the light it sheds on evolution of human influenza. A molecular evolutionary approach may be particularly informative for non-human influenza for which we are lacking antigenic data.
To study the evolution of H1 and H3 in all major hosts, we first built time-scaled phylogenetic trees for representative sets of coding sequences. Previous studies have paved the way for this type of analysis and have shown that independent rates of molecular evolution are necessary for reliable phylodynamic inference of influenza A viruses across hosts (53). However, a host-specific molecular model does not account for other sources of evolutionary rate variation. In this work, we apply an extension of this model by combining lineage-specific rates of evolution with random effects that accommodate within-lineage variation of the evolutionary rate. We determined rates of non-synonymous and synonymous change in the sequences and in portions corresponding to the two domains of HA. We also reconstructed the specific changes that occurred on all branches of the tree.
The ratio of non-synonymous to synonymous changes, relative to the ratio expected in the absence of selection, reflects the selective forces acting on the protein sequence. Figure 2 shows this quantity for all hosts for the head and stalk of H1 and H3, and Tables S10 to S13 compare them among hosts. In all cases, the avian dN/dS is lower than all of the mammalian values. Where the mammalian values are statistically distinguishable from each other, the human value is highest, except for the special case of canine stalk. Where differences exist, they might reflect differences in the strength of purifying selection, differences in positive selection, or some combination of the two.
High dN/dS on the head of human HA has long been attributed to selection for antigenic change, which allows escape from antibodies elicited by previous infections. Values for the head in other mammalian hosts are indistinguishable from human dN/dS or intermediate between human and avian values. The simplest explanation of this pattern is that there is selection for antigenic change in these other mammals as well, albeit weaker, but little or none in avians. Influenza infection in birds differs from that in mammals, mainly affecting the gastrointestinal tract, and birds may be less likely to be reinfected with the same subtype in their lifetimes. These differences, and other possible explanations of weak antigenic selection, may be due to the long association of these subtypes with avian hosts (5).
The N:S ratio on the H1 and H3 stalks is also higher in mammalian than avian viruses. The very high canine value can be explained by adaptations to canines, discussed further below. The higher value in other mammals might reflect weaker purifying selection or the operation of positive selection. There is no obvious reason for purifying selection on the stalk to be generally weaker in mammals, but the possibility cannot be excluded. Adaptive evolution of the stalk might involve escape from rare anti-stalk antibodies. The same force responsible for higher mammalian rates in the head would then also explain higher rates in the stalk. This possibility would be a concern for universal influenza vaccines designed to elicit antibodies that target the HA stalk (54).
Some significant differences exist in N:S ratios among lineages of the same subtype that infect the same host (Table S5, S6, S8, and S9). The dN/dS of the head is lower in the 2009 human H1N1 pandemic lineage than in the other human H1N1 lineages. This likely reflects the small amount of time since the introduction of the 2009 lineage into humans and the consequent weakness of selection for antigenic change over much of its history (55). The vaccine strain for 2009pdm was not updated until 2016 because of a paucity of antigenic change (56). There is significant variation among both H3 and H1 swine lineages on both the head and the stalk. Differences in agricultural practices and swine vaccination rates over time and among countries may explain much of this variation.
We analyzed changes at 131 H3 positions commonly described as “antigenically relevant” to allow comparison to other results (57–60). These positions are based on dubious premises: that all changes in the membrane-distal portion of the HA head that became widespread among human H3N2 were caused by selection for antigenic change and that regions of the protein not observed to have changed are not bound by antibodies. Analysis based on these sites can lead to circular reasoning, since positions that have changed in one part of the tree are more likely to change in other parts, even if the changes are not due to antigenic selection. This potential for this effect is highest in the human lineage, since these sites were selected based on human virus data.
Estimates of the rate of antigenic change, based on laboratory measurements, are available for human and swine H1 and H3 (51, 52). Large uncertainties for several of the swine rates limit the conclusions that can be drawn from comparisons ( Fig. S7). In all cases where a conclusion is possible, the rate of antigenic change in human viruses is larger than that in swine. This is consistent with the generally higher non-synonymous:synonymous ratio on the head of human viruses and its interpretation as an indication of stronger selection for antigenic change. Perhaps more appropriate is consideration of the rate of non-synonymous change per unit time. By this measure, human H3 again has a higher rate than swine H3, but human H1 is slower than most types of swine H1. The implication is that there is more antigenic change, on average, for a non-synonymous change in humans than swine H1. This is expected if there is stronger selection for antigenic change. Even in the absence of such selection, HA sequences, like those of most proteins, are expected to change over time. This will result in some antigenic change but less than that due to amino acid changes selected for the antigenic difference that they cause.
The number and locations of glycosylation sites vary across the HA trees, especially on the head domain. Glycosylation of the head can shield the protein from antibodies (61–63). It also comes with various costs (35, 63, 64). The optimal glycosylation state reflects a trade-off between the costs and the benefits, both of which may vary considerably among hosts and due to other factors.
As previously described (34, 63, 65), there has been net acquisition of glycosylations on the head since the introductions of H1 and H3 to humans in 1918 and 1968. This phenomenon is attributed to the selection for the shielding effects of glycosylation. The 2009 pandemic H1N1 may be in the early stages of a similar trajectory, having gained a head glycosylation after a few years. In contrast, avian H1 and H3 have generally maintained just one glycosylation on the head over long periods of time.
Equine and canine H3 typically have two or three head glycosylations. Swine H3 lineages converge to around four, despite being derived from human progenitors with a variable number. Loss of glycosylation is common in the swine lineages derived from highly glycosylated human H3 and on the host-switch branches leading to them. Presumably, this reflects weaker antigenic pressure in swine compared with humans, such that the costs of glycosylation outweigh its benefits, leading to selection for loss.
The pattern in swine H1 is similar in that the average number of head glycosylations is intermediate between the low number in avians and the higher numbers attained over time in humans. There is, however, considerable variation among swine lineages, mostly ranging from a single head glycosylation (like avian viruses) to three. The head has no glycosylations for a few years in the avian-derived EAswEUR. As discussed above, the N:S ratio also varies significantly among swine lineages.
The overall pattern of head glycosylation is similar to that of the non-synonymous to synonymous rate on the head:highest in human viruses, lowest in avian, and intermediate in non-human mammals. Differences in antigenic pressure among hosts would explain both patterns.
Considering only the total number of glycosylations hides the complexity of their evolution. This complexity is exemplified by position 94 of H1. Loss of this glycosylation site occurs in the ancestors of most isolates of six swine lineages. The site is present throughout most of the tree, including the avian lineages, so it would seem to have a cost that is specific to swine. Nevertheless, it is regained six times in EAswEUR, suggesting that it has a net advantage in swine under some circumstances. It is never regained, however, in EAswCHN, which descends from EAswEUR. The explanation may be that glycosylation at position 94 provides protection against antibodies in those swine that have received certain vaccines (49, 66). Vaccination of swine is not common in China (67).
The sequence data sets compiled in this study were subjected to substantial downsampling to mitigate the computational burden of Bayesian phylogenetic inference. The sequences used were chosen so as to represent the diversity through space, time, and host as well as possible. Some of the branches connecting viruses with different hosts were long, perhaps reflecting a lack of sampling of the earliest viruses infecting the new host. This made the characterization of early adaptation events difficult, particularly for swine and canine lineages.
This work sheds light on the evolution of influenza viruses circulating in a variety of non-human hosts, which has not been studied as well as human influenza evolution. Higher dN/dS on the HA head in all mammalian hosts compared with the avian host and the retention or accumulation of large numbers of head glycosylations in some swine lineages (as occurs in human HA) suggest that mammals in general impose greater selection for antibody escape. Mammals also had higher dN/dS ratios in the stalk domain compared with birds. The reasons for this require further investigation, but it may reflect antigenic evolution in the stalk in mammals. In addition, evolutionary analysis of the sequences identified host adaptation in canines involving a particular phenotype. These results, based entirely on sequence data, provide information about influenza evolution in non-human hosts and place human influenza evolution in a larger context.
MATERIALS AND METHODS
Data set assembly
We downloaded all available hemagglutinin sequences for influenza A H3 and H1 from GenBank and downloaded specific geographical isolates, not available in GenBank, from GISAID on 8 December 2018. The sequences were aligned using the MAFFT (68) multiple sequence alignment tool. The aligned sequences were manually edited and cleaned in AliView version 1.26 software (69). The resulting data set was trimmed at the 5′ and 3′ ends to include solely the coding sequence. This data set was subjected to multiple iterations of phylogeny reconstruction using FastTree version 1.0 (70) with a general time-reversible (GTR) model and exclusion of outlier sequences whose genetic divergence and sampling date were incongruent using TempEst (71).
We defined lineages from the same host and geographical region and only considered those circulating for at least 3 years in the region (Table S1). Each lineage includes only viruses that infect a single host species or closely related host species. Each inferred introduction into a new species creates a new lineage. In some cases, what could be a lineage is subdivided on the basis of geography. Besides the H1 pandemic lineage circulating in humans since 2009, we defined two human H1 lineages circulating during the periods of 1918–1957 and 1977–2009. The H1 lineage reintroduced in 1977 derives directly from the viruses circulating in 1957, so studying the two lineages together would disrupt the temporal signal and consequently the molecular clock model, due to the lack of accumulation of mutations during the 20-year period where H1 did not circulate (72–75). For the non-avian lineages, we excluded sequences with mixed subtypes, as marked by “Hx” or “mixed.” For some analysis, we combine lineages for the same host.
The number of sequences available makes a full Bayesian inference using all of them impractical due to the computations required. For this reason, we analyzed a representative subset, which excluded those that cover less than 75% of the coding sequence with unambiguous bases and retained only one of identical sequences collected in the same country on the same day or in the same month or year in the case of incomplete date information.
Next, we applied a subsampling strategy that selected a specific number of sequences per country per time, in order to obtain similarly sized lineages that represented the overall circulating diversity. When sequences had the same country and collection date, we chose, in a decreasing order of priority, those that were longer, those that had at least the month of collection rather than just the year, those with a smaller number of internal gaps with lengths not divisible by 3 (codons), and those with full dates of collection (https://github.com/jlcherry/SAMPI). We combined all subsampled lineages for each subtype and performed a final iteration of phylogeny reconstruction and exclusion of outlier sequences as described above. The final H3 data set consisted of 873 sequences from viruses isolated between 1963 and 2019 from the following hosts: avian (n = 178), human (n = 101), swine (n = 370), equine (n = 87), and canine (n = 137). For H1, there were 964 sequences, with collection dates between 1918 and 2009, from avian (n = 136), human (n = 186), and swine (n = 642) hosts. The H3 sequences were divided into 16 lineages and the H1 sequences into 33 (Fig. S1; Table S1; Files S1 and S2).
Phylogenetic analysis
Phylogenetic relationships were inferred separately for the H3 and H1 data sets, with a Bayesian phylogenetic approach using Markov chain Monte Carlo (MCMC) available via the BEAST v1.10.5 package (76) and the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD, USA (http://biowulf.nih.gov).
We divided the sequences into lineages based on host, geography, and a preliminary phylogeny inferred using FastTree with a GTR substitution model (77), retaining clusters with a minimum of 10 sequences and a minimum of three different collection years . Some lineages are paraphyletic with respect to other lineages. We partitioned the coding sequences into first, second, and third codon positions and applied a separate Hasegawa-Kishino-Yano 85 [HKY85 (78)] substitution model with gamma-distributed rate variation among sites to all partitions (79). To infer time-scaled trees, we apply a mixed-effects molecular clock model that combines a host-specific local clock (53) with random effects that allow for branch-specific rate variation similar to an uncorrelated relaxed clock model. We specified a Bayesian Skygrid coalescent tree prior (80). For sequences with an incomplete date of viral collection available, the lack of tip date precision was accommodated by sampling uniformly across a 1-year window from 1 January to 31 December. If the month of collection was available, the lack of tip date precision was accommodated by sampling uniformly across a 1-month window from day 1 to day 28, 30, or 31.
We applied an approach that relies on counting methods, set in a Bayesian framework, to estimate lineage-specific synonymous (synonymous substitutions/all sites/year) and non-synonymous (non-synonymous substitutions/all sites/year) substitution rates as well as their ratio relative to the neutral expectation (dN/dS) in large data sets, while integrating over the posterior distribution of phylogenies and ancestral reconstructions to quantify the uncertainty on the lineage-specific estimates (81).
The ratios that we refer to as such are not, strictly speaking, dN/dS, and the sequence changes are not properly referred to as substitutions, because many of the changes have not in any sense gone to fixation and never will. We nevertheless continue a looser use of these terms that has become common in similar contexts. Terminology aside, these changes may have characteristics that differ from those that reflect the full effects of selection because they have come to predominate in their lineages.
The MCMC chain was run separately at least 20 times for each of the data sets and for at least 50 million iterations with variables recorded every 50,000 iterations, using the BEAGLE (82) library to improve computational performance (Files S1 and S2). All parameters reached convergence (effective sample sizes greater than 200), as assessed visually using Tracer v.1.7.1, with statistical uncertainty reflected in values of the 95% HPD. At least 10% of the chain was removed from the beginning as burn-in. MCC trees were summarized using TreeAnnotator v1.10.5 and visualized in FigTree v1.4.4.
We summarized the synonymous and non-synonymous rates and dN/dS estimates for all protein sites (AS), head and stalk domains (83), and a set of 131 positions claimed to constitute the antigenically relevant sites for H3 (ARS, analyzed only for H3) (84, 85). All sites investigated are presented in Table S2. We evaluated the statistical significance of differences in dN/dS by applying Fisher’s exact test to the counts of synonymous and non-synonymous changes in each pair of lineages inferred from a most parsimonious reconstruction on the MCC tree. We then applied the Benjamini-Hochberg Procedure to find the set of pairwise comparisons giving a false discovery rate of 5%.
Ancestral sequence reconstruction
Ancestral state reconstruction of nucleotides was performed on the MCC tree using maximum parsimony, implemented as a Python program (File S3 at https://github.com/jlcherry/HA_Evolution). In cases where the reconstruction was ambiguous, it was disambiguated arbitrarily for simplicity. Changes to the protein sequence were inferred from translations of the reconstructed nucleotide states. Raw ratios of non-synonymous to synonymous changes (N:S) were calculated from the reconstructions.
Glycosylation patterns
We scanned all sequences (ancestral and contemporaneous) for each data set to identify potential N-linked glycosylated sites, based on the motif Asn-X-Ser/Thr-X (N-X-S/T-X), where X is any amino acid other than proline (86) using a program written in Python. We then plotted the number of glycosylated sites over time for each lineage and estimated their rate of accumulation or loss.
ACKNOWLEDGMENTS
We thank Caitlin Bowers at Dartmouth College for the help with the compilation of the genetic data sets and Emma Wagner at Dartmouth College and James R. Otieno at NIH/FIC for the assistance with making figures in R. The opinions expressed in this article are those of the authors and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
National Institutes of Health, Intramural Research Program of the National Library of Medicine (J.L.C.).
European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme grant agreement no. ~725422 - ReservoirDOCS (P.L.).
EU grant 874850 MOOD (P.L.).
Wellcome Trust through project 206298/Z/17/Z - ARTIC Network (P.L.).
National Institutes of Health grant R01 AI153044 (P.L.).
National Institutes of Health grant U19 AI135995 (P.L.).
Center for Research on Influenza Pathogenesis (CRIP), an NIAID Centers of Excellence for Influenza Research and Surveillance (CEIRS) contract # HHSN272201400008C (N.S.T., S.M.K., and M.I.N.).
Center for Research on Influenza Pathogenesis and Transmission (CRIPT), a NIAID Centers of Excellence for Influenza Research and Response (CEIRR) contract # 75N93019R00028 (N.S.T., S.M.K., and M.I.N.)
Conceptualization: N.S.T., M.I.N., and J.L.C.; methodology: N.S.T., P.L., M.I.N., and J.L.C.; investigation: S.M.K., N.S.T., M.I.N., and J.L.C.; visualization: N.S.T., M.I.N., and J.L.C.; supervision: P.L., M.I.N., and J.L.C.; writing—original draft: N.S.T., M.I.N., and J.L.C.; writing—review & editing: N.S.T., P.L., M.I.N., and J.L.C.
The authors declare that they have no competing interests.
Contributor Information
Nidia S. Trovão, Email: nidiastrovao@gmail.com.
Joshua L. Cherry, Email: jcherry@ncbi.nlm.nih.gov.
Paul Keim, Northern Arizona University, Flagstaff, Arizona, USA.
DATA AVAILABILITY
No new data were generated or analyzed in support of this research. The XML-format files containing the data and model parametrization are provided in Files S2 and S3.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/mbio.02649-23.
Supplemental file legends and Fig. S1.
H1 BEAST xml.
H3 BEAST xml.
Fig. S2 to S12 and Tables S1 to S18.
Glycosylation gains and losses according to the H3 MCC tree hierarchy.
Glycosylation gains and losses according to the H1 MCC tree hierarchy.
Interface positions in four H3 structures.
Maximum clade credibility tree for H3 colored by lineages.
Maximum clade credibility tree for H1 colored by lineages.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Johnson KEE, Song T, Greenbaum B, Ghedin E. 2017. Getting the flu: 5 key facts about influenza virus evolution. PLoS Pathog 13:e1006450. doi: 10.1371/journal.ppat.1006450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Widdowson M-A, Bresee JS, Jernigan DB. 2017. The global threat of animal influenza viruses of zoonotic concern: then and now. J Infect Dis 216:S493–S498. doi: 10.1093/infdis/jix331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mostafa A, Abdelwhab EM, Mettenleiter TC, Pleschka S. 2018. Zoonotic potential of influenza A viruses: a comprehensive overview. Viruses 10:497. doi: 10.3390/v10090497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gamblin SJ, Skehel JJ. 2010. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J Biol Chem 285:28403–28409. doi: 10.1074/jbc.R110.129809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179. doi: 10.1128/mr.56.1.152-179.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song D, Kang B, Lee C, Jung K, Ha G, Kang D, Park S, Park B, Oh J. 2008. Transmission of avian influenza virus (H3N2) to dogs. Emerg Infect Dis 14:741–746. doi: 10.3201/eid1405.071471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parrish CR, Murcia PR, Holmes EC. 2015. Influenza virus reservoirs and intermediate hosts: dogs, horses, and new possibilities for influenza virus exposure of humans. J Virol 89:2990–2994. doi: 10.1128/JVI.03146-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Voorhees IEH, Dalziel BD, Glaser A, Dubovi EJ, Murcia PR, Newbury S, Toohey-Kurth K, Su S, Kriti D, Van Bakel H, Goodman LB, Leutenegger C, Holmes EC, Parrish CR. 2018. Multiple incursions and recurrent epidemic fade-out of H3N2 canine influenza A virus in the United States. J Virol 92:e00323-18. doi: 10.1128/JVI.00323-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guan L, Shi J, Kong X, Ma S, Zhang Y, Yin X, He X, Liu L, Suzuki Y, Li C, Deng G, Chen H. 2019. H3N2 avian influenza viruses detected in live poultry markets in China bind to human-type receptors and transmit in guinea pigs and ferrets. Emerg Microbes Infect 8:1280–1290. doi: 10.1080/22221751.2019.1660590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trovão NS, Nelson MI. 2020. When pigs fly: pandemic influenza enters the 21st century. PLoS Pathog 16:e1008259. doi: 10.1371/journal.ppat.1008259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olguin-Perglione C, Barrandeguy ME. 2021. An overview of equine influenza in South America. Viruses 13:888. doi: 10.3390/v13050888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. World Health Organization . 2018. Influenza (seasonal) fact sheet. Available from: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal)
- 13. Boni MF, Galvani AP, Wickelgren AL, Malani A. 2013. Economic epidemiology of avian influenza on smallholder poultry farms. Theor Popul Biol 90:135–144. doi: 10.1016/j.tpb.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hill EM, House T, Dhingra MS, Kalpravidh W, Morzaria S, Osmani MG, Brum E, Yamage M, Kalam MA, Prosser DJ, Takekawa JY, Xiao X, Gilbert M, Tildesley MJ. 2018. The impact of surveillance and control on highly pathogenic avian influenza outbreaks in poultry in Dhaka division, Bangladesh. PLoS Comput Biol 14:e1006439. doi: 10.1371/journal.pcbi.1006439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kemen MJ, Frank RA, Babish JB. 1985. An outbreak of equine influenza at a harness horse racetrack. Cornell Vet 75:277–288. [PubMed] [Google Scholar]
- 16. Sack A, Cullinane A, Daramragchaa U, Chuluunbaatar M, Gonchigoo B, Gray GC. 2019. Equine influenza virus—A neglected, reemergent disease threat. Emerg Infect Dis 25:1185–1191. doi: 10.3201/eid2506.161846 [DOI] [Google Scholar]
- 17. Cullinane A, Gahan J, Walsh C, Nemoto M, Entenfellner J, Olguin-Perglione C, Garvey M, Huang Fu TQ, Venner M, Yamanaka T, Barrandeguy M, Fernandez CJ. 2020. Evaluation of current equine influenza vaccination protocols prior to shipment, guided by OIE standards. Vaccines (Basel) 8:107. doi: 10.3390/vaccines8010107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clark L, Hall J. 2006. Avian influenza in wild birds: status as reservoirs, and risks to humans and agriculture. Ornithol Monogr 60:3. doi: 10.1642/0078-6594(2006)60[3:AIIWBS]2.0.CO;2 [DOI] [Google Scholar]
- 19. Blagodatski A, Trutneva K, Glazova O, Mityaeva O, Shevkova L, Kegeles E, Onyanov N, Fede K, Maznina A, Khavina E, Yeo S-J, Park H, Volchkov P. 2021. Avian influenza in wild birds and poultry: dissemination pathways, monitoring methods, and virus ecology. Pathogens 10:630. doi: 10.3390/pathogens10050630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uyeki TM, Peiris M. 2019. Novel avian influenza A virus infections of humans. Infect Dis Clin North Am 33:907–932. doi: 10.1016/j.idc.2019.07.003 [DOI] [PubMed] [Google Scholar]
- 21. Taubenberger JK, Morens DM. 2006. Influenza: the mother of all pandemics. Emerg Infect Dis 12:15–22. doi: 10.3201/eid1201.050979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jester BJ, Uyeki TM, Jernigan DB. 2020. Fifty years of influenza A(H3N2) following the pandemic of 1968. Am J Public Health 110:669–676. doi: 10.2105/AJPH.2019.305557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murcia PR, Wood JLN, Holmes EC. 2011. Genome-scale evolution and phylodynamics of equine H3N8 influenza A virus. J Virol 85:5312–5322. doi: 10.1128/JVI.02619-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anderson TK, Macken CA, Lewis NS, Scheuermann RH, Van Reeth K, Brown IH, Swenson SL, Simon G, Saito T, Berhane Y, Ciacci-Zanella J, Pereda A, Davis CT, Donis RO, Webby RJ, Vincent AL, Lowen AC. 2016. A phylogeny-based global nomenclature system and automated annotation tool for H1 hemagglutinin genes from swine influenza A viruses. mSphere 1:e00275-16. doi: 10.1128/mSphere.00275-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collins PJ, Vachieri SG, Haire LF, Ogrodowicz RW, Martin SR, Walker PA, Xiong X, Gamblin SJ, Skehel JJ. 2014. Recent evolution of equine influenza and the origin of canine influenza. Proc Natl Acad Sci U S A 111:11175–11180. doi: 10.1073/pnas.1406606111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mena I, Nelson MI, Quezada-Monroy F, Dutta J, Cortes-Fernández R, Lara-Puente JH, Castro-Peralta F, Cunha LF, Trovão NS, Lozano-Dubernard B, Rambaut A, van Bakel H, García-Sastre A. 2016. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife 5:e16777. doi: 10.7554/eLife.16777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nelson MI, Vincent AL. 2015. Reverse zoonosis of influenza to swine: new perspectives on the human-animal interface. Trends Microbiol 23:142–153. doi: 10.1016/j.tim.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanada K, Suzuki Y, Gojobori T. 2004. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol Biol Evol 21:1074–1080. doi: 10.1093/molbev/msh109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van de Sandt CE, Kreijtz JHCM, Rimmelzwaan GF. 2012. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses 4:1438–1476. doi: 10.3390/v4091438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi Y, Wu Y, Zhang W, Qi J, Gao GF. 2014. “Enabling the “host jump”: structural determinants of receptor-binding specificity in influenza A viruses”. Nat Rev Microbiol 12:822–831. doi: 10.1038/nrmicro3362 [DOI] [PubMed] [Google Scholar]
- 31. Bouvier NM, Palese P. 2008. The biology of influenza viruses. Vaccine 26 Suppl 4:D49–53. doi: 10.1016/j.vaccine.2008.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shaw M, Palese P. 2013. Fields virology. 6th ed, p 1151–1185. Lippincott Williams Wilkins, Phila PA. [Google Scholar]
- 33. Kirkpatrick E, Qiu X, Wilson PC, Bahl J, Krammer F. 2018. The influenza virus hemagglutinin head evolves faster than the stalk domain. Sci Rep 8:10432. doi: 10.1038/s41598-018-28706-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altman MO, Angel M, Košík I, Trovão NS, Zost SJ, Gibbs JS, Casalino L, Amaro RE, Hensley SE, Nelson MI, Yewdell JW. 2019. Human influenza A virus hemagglutinin glycan evolution follows a temporal pattern to a glycan limit. mBio 10:e00204-19. doi: 10.1128/mBio.00204-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reading PC, Tate MD, Pickett DL, Brooks AG. 2007. Glycosylation as a target for recognition of influenza viruses by the innate immune system. Adv Exp Med Biol 598:279–292. doi: 10.1007/978-0-387-71767-8_20 [DOI] [PubMed] [Google Scholar]
- 36. Kosik I, Ince WL, Gentles LE, Oler AJ, Kosikova M, Angel M, Magadán JG, Xie H, Brooke CB, Yewdell JW. 2018. Influenza A virus hemagglutinin glycosylation compensates for antibody escape fitness costs. PLoS Pathog 14:e1007141. doi: 10.1371/journal.ppat.1007141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watanabe Y, Bowden TA, Wilson IA, Crispin M. 2019. Exploitation of glycosylation in enveloped virus pathobiology. Biochim Biophys Acta Gen Subj 1863:1480–1497. doi: 10.1016/j.bbagen.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taubenberger JK, Kash JC. 2010. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 7:440–451. doi: 10.1016/j.chom.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osterholm MT, Kelley NS, Manske JM, Ballering KS, Leighton TR, Moore KA. 2022. The compelling need for game- changing influenza vaccines. Available from: https://www.cidrap.umn.edu/compelling-need-game-changing-influenza-vaccines
- 40. Yamayoshi S, Kawaoka Y. 2019. Current and future influenza vaccines. Nat Med 25:212–220. doi: 10.1038/s41591-018-0340-z [DOI] [PubMed] [Google Scholar]
- 41. van Reeth K, Ma W. 2013. Swine influenza virus vaccines: to change or not to change—that’s the question, p 173–200. In Richt JA, Webby RJ (ed), Swine influenza. Springer, Berlin, Heidelberg. [DOI] [PubMed] [Google Scholar]
- 42. Vincent AL, Perez DR, Rajao D, Anderson TK, Abente EJ, Walia RR, Lewis NS. 2017. Influenza A virus vaccines for swine. Vet Microbiol 206:35–44. doi: 10.1016/j.vetmic.2016.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodriguez L, Nogales A, Reilly EC, Topham DJ, Murcia PR, Parrish CR, Martinez Sobrido L. 2017. A live-attenuated influenza vaccine for H3N2 canine influenza virus. Virology 504:96–106. doi: 10.1016/j.virol.2017.01.020 [DOI] [PubMed] [Google Scholar]
- 44. Chambers TM. 2014. A brief introduction to equine influenza and equine influenza viruses. Methods Mol Biol Clifton NJ 1161:365–370. doi: 10.1007/978-1-4939-0758-8 [DOI] [PubMed] [Google Scholar]
- 45. Tabynov K, Kydyrbayev Z, Ryskeldinova S, Assanzhanova N, Kozhamkulov Y, Inkarbekov D, Sansyzbay A. 2014. Safety and immunogenicity of a novel cold-adapted modified-live equine influenza virus vaccine. Aust Veterinary J 92:450–457. doi: 10.1111/avj.12248 [DOI] [PubMed] [Google Scholar]
- 46. Na W, Yeom M, Yuk H, Moon H, Kang B, Song D. 2016. Influenza virus vaccine for neglected hosts: horses and dogs. Clin Exp Vaccine Res 5:117–124. doi: 10.7774/cevr.2016.5.2.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swayne DE. 2009. Avian influenza vaccines and therapies for poultry. Comp Immunol Microbiol Infect Dis 32:351–363. doi: 10.1016/j.cimid.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 48. Nelson MI, Holmes EC. 2007. The evolution of epidemic influenza. Nat Rev Genet 8:196–205. doi: 10.1038/nrg2053 [DOI] [PubMed] [Google Scholar]
- 49. Yoo SJ, Kwon T, Lyoo YS. 2018. Challenges of influenza A viruses in humans and animals and current animal vaccines as an effective control measure. Clin Exp Vaccine Res 7:1–15. doi: 10.7774/cevr.2018.7.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sautto GA, Kirchenbaum GA, Ross TM. 2018. Towards a universal influenza vaccine: different approaches for one goal. Virol J 15:17. doi: 10.1186/s12985-017-0918-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bedford T, Suchard MA, Lemey P, Dudas G, Gregory V, Hay AJ, McCauley JW, Russell CA, Smith DJ, Rambaut A. 2014. Integrating influenza antigenic dynamics with molecular evolution. Elife 3:e01914. doi: 10.7554/eLife.01914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lewis NS, Russell CA, Langat P, Anderson TK, Berger K, Bielejec F, Burke DF, Dudas G, Fonville JM, Fouchier RA, et al. 2016. The global antigenic diversity of swine influenza A viruses. Elife 5:e12217. doi: 10.7554/eLife.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Worobey M, Han G-Z, Rambaut A. 2014. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 508:254–257. doi: 10.1038/nature13016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park J-K, Xiao Y, Ramuta MD, Rosas LA, Fong S, Matthews AM, Freeman AD, Gouzoulis MA, Batchenkova NA, Yang X, Scherler K, Qi L, Reed S, Athota R, Czajkowski L, Han A, Morens DM, Walters K-A, Memoli MJ, Kash JC, Taubenberger JK. 2020. Pre-existing immunity to influenza virus hemagglutinin stalk might drive selection for antibody-escape mutant viruses in a human challenge model. Nat Med 26:1240–1246. doi: 10.1038/s41591-020-0937-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma Y, Liu K, Yin Y, Qin J, Zhou Y-H, Yang J, Li S, Poon LLM, Zhang C. 2020. The phylodynamics of seasonal influenza A/H1N1Pdm virus in China between 2009 and 2019. Front. Microbiol 11. doi: 10.3389/fmicb.2020.00735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tewawong N, Prachayangprecha S, Vichiwattana P, Korkong S, Klinfueng S, Vongpunsawad S, Thongmee T, Theamboonlers A, Poovorawan Y. 2015. Assessing antigenic drift of seasonal influenza A(H3N2) and A(H1N1)pdm09 viruses. PLoS One 10:e0139958. doi: 10.1371/journal.pone.0139958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee M-S, Chen J-E. 2004. Predicting antigenic variants of influenza A/H3N2 viruses. Emerg Infect Dis 10:1385–1390. doi: 10.3201/eid1008.040107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tharakaraman K, Raman R, Stebbins NW, Viswanathan K, Sasisekharan V, Sasisekharan R. 2013. Antigenically intact hemagglutinin in circulating avian and swine influenza viruses and potential for H3N2 pandemic. Sci Rep 3:1822. doi: 10.1038/srep01822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Broecker F, Liu STH, Sun W, Krammer F, Simon V, Palese P. 2018. Immunodominance of antigenic site B in the hemagglutinin of the current H3N2 influenza virus in humans and mice. J Virol 92:e01100-18. doi: 10.1128/JVI.01100-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bullard BL, DeBeauchamp J, Pekarek MJ, Petro-Turnquist E, Vogel P, Webby RJ, Weaver EA. 2022. An epitope-optimized human H3N2 influenza vaccine induces broadly protective immunity in mice and ferrets. NPJ Vaccines 7:65. doi: 10.1038/s41541-022-00492-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wrigley NG, Brown EB, Daniels RS, Douglas AR, Skehel JJ, Wiley DC. 1983. Electron microscopy of influenza haemagglutinin-monoclonal antibody complexes. Virology 131:308–314. doi: 10.1016/0042-6822(83)90499-3 [DOI] [PubMed] [Google Scholar]
- 62. Skehel JJ, Stevens DJ, Daniels RS, Douglas AR, Knossow M, Wilson IA, Wiley DC. 1984. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc Natl Acad Sci U S A 81:1779–1783. doi: 10.1073/pnas.81.6.1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abe Y, Takashita E, Sugawara K, Matsuzaki Y, Muraki Y, Hongo S. 2004. Effect of the addition of oligosaccharides on the biological activities and antigenicity of influenza A/H3N2 virus hemagglutinin. J Virol 78:9605–9611. doi: 10.1128/JVI.78.18.9605-9611.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gallagher P, Henneberry J, Wilson I, Sambrook J, Gething MJ. 1988. Addition of carbohydrate side chains at novel sites on influenza virus hemagglutinin can modulate the folding, transport, and activity of the molecule. J Cell Biol 107:2059–2073. doi: 10.1083/jcb.107.6.2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun S, Wang Q, Zhao F, Chen W, Li Z. 2011. Glycosylation site alteration in the evolution of influenza A (H1N1) viruses. PLoS One 6:e22844. doi: 10.1371/journal.pone.0022844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mancera Gracia JC, Pearce DS, Masic A, Balasch M. 2020. Influenza A virus in swine: epidemiology, challenges and vaccination strategies. Front Vet Sci 7:647. doi: 10.3389/fvets.2020.00647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kong W, Ye J, Guan S, Liu J, Pu J. 2014. Epidemic status of swine influenza virus in China. Indian J Microbiol 54:3–11. doi: 10.1007/s12088-013-0419-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298. doi: 10.1093/bib/bbn013 [DOI] [PubMed] [Google Scholar]
- 69. Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. doi: 10.1093/bioinformatics/btu531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rambaut A, Lam TT, Max Carvalho L, Pybus OG. 2016. Exploring the temporal structure of heterochronous sequences using tempest (formerly path-O-Gen). Virus Evol 2:vew007. doi: 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakajima K, Desselberger U, Palese P. 1978. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature 274:334–339. doi: 10.1038/274334a0 [DOI] [PubMed] [Google Scholar]
- 73. Kendal AP, Noble GR, Skehel JJ, Dowdle WR. 1978. “Antigenic similarity of influenza A(H1N1) viruses from epidemics in 1977–1978 to “scandinavian” strains isolated in epidemics of 1950–1951”. Virology 89:632–636. doi: 10.1016/0042-6822(78)90207-6 [DOI] [PubMed] [Google Scholar]
- 74. Wertheim JO. 2010. The re-emergence of H1N1 influenza virus in 1977: a cautionary tale for estimating divergence times using biologically unrealistic sampling dates. PLoS One 5:e11184. doi: 10.1371/journal.pone.0011184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Carter RW, Sanford JC. 2012. A new look at an old virus: patterns of mutation accumulation in the human H1N1 influenza virus since 1918. Theor Biol Med Model 9:42. doi: 10.1186/1742-4682-9-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. 2018. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4:vey016. doi: 10.1093/ve/vey016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Price MN, Dehal PS, Arkin AP. 2010. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hasegawa M, Kishino H, Yano T. 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174. doi: 10.1007/BF02101694 [DOI] [PubMed] [Google Scholar]
- 79. Shapiro B, Rambaut A, Drummond AJ. 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol 23:7–9. doi: 10.1093/molbev/msj021 [DOI] [PubMed] [Google Scholar]
- 80. Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA. 2013. Improving bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol Biol Evol 30:713–724. doi: 10.1093/molbev/mss265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lemey P, Minin VN, Bielejec F, Kosakovsky Pond SL, Suchard MA. 2012. A counting renaissance: combining stochastic mapping and empirical bayes to quickly detect amino acid sites under positive selection. Bioinformatics 28:3248–3256. doi: 10.1093/bioinformatics/bts580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ayres DL, Cummings MP, Baele G, Darling AE, Lewis PO, Swofford DL, Huelsenbeck JP, Lemey P, Rambaut A, Suchard MA. 2019. BEAGLE 3: improved performance, scaling, and usability for a high-performance computing library for statistical phylogenetics. Syst Biol 68:1052–1061. doi: 10.1093/sysbio/syz020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Krammer F, Palese P. 2013. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr Opin Virol 3:521–530. doi: 10.1016/j.coviro.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wiley DC, Wilson IA, Skehel JJ. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378. doi: 10.1038/289373a0 [DOI] [PubMed] [Google Scholar]
- 85. Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. 1999. Predicting the evolution of human influenza A. Science 286:1921–1925. doi: 10.1126/science.286.5446.1921 [DOI] [PubMed] [Google Scholar]
- 86. Mellquist JL, Kasturi L, Spitalnik SL, Shakin-Eshleman SH. 1998. The amino acid following an ASN-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37:6833–6837. doi: 10.1021/bi972217k [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental file legends and Fig. S1.
H1 BEAST xml.
H3 BEAST xml.
Fig. S2 to S12 and Tables S1 to S18.
Glycosylation gains and losses according to the H3 MCC tree hierarchy.
Glycosylation gains and losses according to the H1 MCC tree hierarchy.
Interface positions in four H3 structures.
Maximum clade credibility tree for H3 colored by lineages.
Maximum clade credibility tree for H1 colored by lineages.
Data Availability Statement
No new data were generated or analyzed in support of this research. The XML-format files containing the data and model parametrization are provided in Files S2 and S3.