

Abstract
pX, the hepatitis B virus (HBV)-encoded regulator, coactivates transcription through an unknown mechanism. pX interacts with several components of the transcription machinery, including certain activators, TFIIB, TFIIH, and the RNA polymerase II (POLII) enzyme. We show that pX localizes in the nucleus and coimmunoprecipitates with TFIIB from nuclear extracts. We used TFIIB mutants inactive in binding either POLII or TATA binding protein to study the role of TFIIB-pX interaction in transcription coactivation. pX was able to bind the former type of TFIIB mutant and not the latter. Neither of these sets of TFIIB mutants supports transcription. Remarkably, the latter TFIIB mutants fully block pX activity, suggesting the role of TFIIB in pX-mediated coactivation. By contrast, in the presence of pX, TFIIB mutants with disrupted POLII binding acquire the wild-type phenotype, both in vivo and in vitro. These results suggest that pX may establish the otherwise inefficient TFIIB mutant-POLII interaction, by acting as a molecular bridge. Collectively, our results demonstrate that TFIIB is the in vivo target of pX.
Hepatitis B virus (HBV) is a small hepatotrophic DNA virus that encodes a regulatory polypeptide, pX (also called HBxAg). The open reading frame of pX (X ORF) is conserved among all mammal-specific members of the Hepadnaviridae and is essential for woodchuck hepatitis virus infectivity (7, 57). pX stimulates transcription of the HBV enhancer through an element termed E which binds a number of factors bearing the basic-leucine zipper DNA binding/dimerization domain (15, 30). However, pX also activates transcription of a vast number of promoters of other viral and cellular genes through distinct DNA cis elements, such as those binding NF-κB (51), AP1 (22, 32), and AP2 (42). Given this promiscuous behavior, a general effect on the transcription machinery has been attributed to pX (2, 26). While it was speculated that the pX effect was indirect and initiated via stimulation of signal transduction pathways (2, 13, 26, 29, 33, 34, 44), a large body of findings supports the possibility that pX directly affects transcription (9, 22, 23, 30, 36, 53, 55).
In F9 cells lacking the AP1 complex, pX is unable to activate a reporter plasmid under the regulation of an AP1 site unless c-Jun and Fos are coexpressed (22). Also, pX cannot activate a reporter bearing the UASG DNA element minimally affected by signaling without the corresponding activator. These studies revealed that the effect of pX on transcription depends on the activator activation domains. pX directly binds the VP16 activation domain, an efficient pX-responsive Gal4 chimeric activator (23). Interaction of pX with genuine cellular activators, CREB, ATF-2 (30), and p53 (16, 50, 55), has been reported. These findings link the transcriptional activity of pX to direct activator binding.
Activator-pX interactions do not guarantee transcription activation. On the contrary, they may even interfere with the interaction of the activation domain with the general transcription factors and consequently repress transcription. Therefore, additional protein-protein interactions must be mediated by pX. On the basis of deletion, insertion, and substitution analyses, two regions in the X ORF were found to be essential for transcription stimulation activity (residues 58 to 72 and 105 to 142) (1, 38, 53). Interestingly, these regions participate in distinct protein-protein interactions (46). The more amino-terminal domain was recently reported to mediate interactions with the RNA polymerase II (POLII) fifth subunit (9, 23, 28) and TFIIH (23, 28, 36, 55). The C-terminal region is required for binding to TFIIB and VP16 (23, 28). Furthermore, pX-TFIIB interaction is simultaneous with both VP16 (23) and POLII (28) interaction. The mechanism of action emerging from these observations assumes concomitant interaction of pX with both the activator and the basal transcription factors. The inclusion of pX in the transcription initiation complex may therefore modulate the formation of the transcription initiation complex. To investigate this interesting possibility, we used TFIIB mutants inactive in binding either POLII or TATA binding protein (TBP). We found that pX binds the former TFIIB mutant and rescues its defect in supporting transcription. These data strongly argue for establishment of an alternative TFIIB-POLII interaction pathway in the presence of pX, alluding to the pX target, i.e., to the transcription initiation step accelerated by pX.
MATERIALS AND METHODS
Expression vectors for the various TFIIB mutants were constructed by inserting XbaI/BamHI fragments from their corresponding pET3 bacterial expression vectors into expression vector pSG5-HA (Stratagene). For 32P labeling of pX, an oligonucleotide with the sequence CTAGCCTCCGGAGAGCTTCTCTTG was inserted into the previously described p6His-X pRSET (Invitrogen)-based vector (23) into an NheI site. The resulting recombinant bacterially expressed pX contained a substrate sequence for the protein kinase A catalytic subunit and thus was labeled with 10 μCi of [γ-32P]ATP and protein kinase (Sigma) for far-Western analysis. Far-Western analysis was performed by subjecting bacterial P-11 fractions, containing 1 μg of indicated recombinant protein, to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotting. The blot was blocked overnight and reacted with 106 cpm of 32P-labeled recombinant p6His-X (9).
We produced monoclonal antibodies against pX. Supernatants of 12 of 1,200 NS0/lymphocyte clones recognized recombinant pX in immunoblots. Four of them, designated MαX 2.2, 22.1, 26.1, and 29.6, recognized a 19.5-kDa protein in SDS-PAGE upon analysis of extracts of cells transfected with a plasmid expressing hemagglutin (HA)-tagged pX (pHA-X). MαX 2.2, 22.1, and 29.6 also immunoprecipitated native 35S-labeled pX from such extracts (data not shown). Cell extract fractionation was performed as described previously (22), with the single modification of inclusion of 0.025% Sarkosyl and 0.025% sodium deoxycholate to the high-salt nuclear extraction buffer. pX in such extracts was monitored by immunoblot analysis with the MαX monoclonal antibodies.
Indirect immunofluorescent staining was performed by published methods (20), with a preference for paraformaldehyde fixation. To improve the sensitivity and signal of the staining, a mixture of three monoclonal antibodies was used for indirect immunofluorescent staining. The cells were transfected; 24 h after a wash, they were plated on glass coverslips and allowed to grow for an additional 24 h and then extracted, fixed, and blocked (49). The slips were consequently incubated with the indicated immunoglobulin G (IgG) antibodies in 1:300 dilutions in phosphate-buffered saline–Tween 20 with 10% normal goat serum for 1 h in a room temperature humid chamber, washed twice briefly and twice with 30-min incubations, and then incubated for 30 min with the secondary antibody or with phalloidin where indicated. Following washing as described above, the slips were reacted briefly with Hoechst 33258 and analyzed by confocal microscopy. In triple staining, Hoechst 33258 labeled the DNA in the nucleus (blue), fluorescein isothiocyanate-conjugated phalloidin labeled the actin filaments in the cytoplasm (green), and rhodamine-conjugated goat anti-mouse serum was used to detect the pX-bound monoclonal antibodies (red). The advantage of transient transfection is that in every field, up to 1 in 10 cells is positive, helping us to distinguish specific staining from nonspecific staining.
Transient cotransfection of SK-Hep1 cells with the G5 luciferase reporter construct (0.2 μg in a total of 1.5 μg of DNA in 1.5-cm-diameter plates) plus expressors of TFIIB mutants or pX (50 ng of expressor) was performed in duplicate (22). Cells were grown for 48 h and harvested for luciferase assays. For analysis of HBV gene expression, HepG2 cells were transfected with 15 μg of plasmid 2XHBV per 10-cm-diameter dish. The plasmid contained a dimer of HBV DNA ligated head to tail at the unique EcoRI site. RNA and protein extraction was done by using TRI reagent (Molecular Research Center Inc.). For the coimmunoprecipitation experiments, 10-cm-diameter plates (three for each sample) of either COS or 293T cells were transfected with 4 μg of indicated expression vectors, in a total of 20 μg of DNA, as described previously (22). Thirty-six hours after the wash, nuclear extract proteins (1.5 mg in 0.3 ml) were diluted twofold in salt-free nuclear extraction buffer, to reduce the salts, and incubated with 10 μl of protein A-G resin (Santa Cruz) cross-linked to the indicated IgG (dimethylpimelimidate protocol [20]), with 8 h of rotation at 4°C. After collection of unbound proteins, the resin was washed five times with 1 ml of buffer, each after a 5-min incubation period. The bound proteins were eluted with the corresponding epitope peptide and then subjected to SDS-PAGE and immunoblotting.
In vitro transcription and preparation of recombinant proteins were performed as described previously (23), with the modification that the template 5xUASG-MLP-U-less 112 (56) was used.
RESULTS
pX is preferentially enriched in the nuclear extract.
To determine the cellular distribution of pX, cytosolic and nuclear extracts of cells transiently transfected with a pX expression plasmid were monitored with pX-specific monoclonal antibody MαX 26.1 IgG (Fig. 1). Solubilization of the cytoplasmic membrane in hypotonic buffer recovered low amounts of pX (lane 1). However, a significantly higher amount of pX was detected in a high-salt nuclear extract fraction (lane 3). pX nuclear extraction was further improved by inclusion of the anionic detergents sodium deoxycholate and Sarkosyl in the high-salt buffer (lane 2). This signal was highly pX specific, as it was absent from a nuclear extract of mock-transfected cells (lane 4). These results suggest that pX is preferentially enriched in the nuclear extract.
FIG. 1.
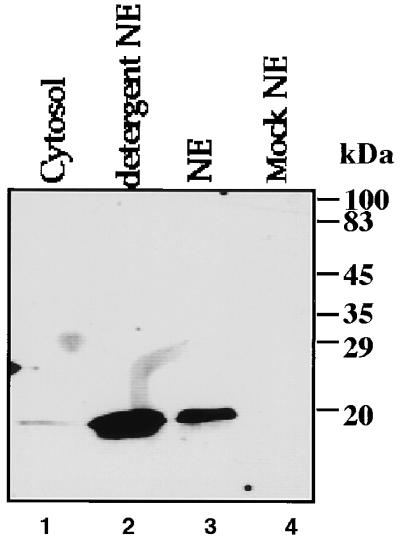
Localization of pX in nuclear extracts (NE). (A) Nuclear and cytoplasmic fractions of either pX (lanes 1 to 3) or mock (lane 4)-transfected cells were analyzed by SDS-PAGE and immunoblotting with anti-pX monoclonal IgG.
Nuclear localization of pX by indirect immunofluorescent staining.
To further determine the cellular distribution of pX, we stained cells with pX-specific monoclonal antibodies MαX 22.1 and 29.6 IgG. Nuclear or cytoplasmic localization was determined by staining the nuclear chromatin blue and the cytoplasmic actin filaments green. By labeling the MαX in red, staining of nuclei of a subset of cells transiently transfected with pX expressor plasmid was seen (Fig. 2A and B); untransfected cells, seen in the same field, served as negative controls. The staining was pX specific, based on its absence in mock-transfected cells (not shown). A similar nuclear staining pattern was achieved with anti-HA or anti-flag immunostaining of cells transfected with expressor plasmids of either HA-tagged or flag-tagged pX (data not shown). However, we cannot exclude the possibility that a cytoplasmic pX either is unrecognized by the anti-pX antibody or escaped the cells during treatment. Treating the cells with hypotonic buffer including Triton X-100 (as in Fig. 1) before fixation did not affect the nuclear staining of pX, arguing against lost cytoplasmic pX. These results imply that a significant fraction of pX is in the nucleus.
FIG. 2.

Nuclear localization of pX and TFIIB. (A and B) Indirect immunofluorescent staining of cells transiently transfected with pX expressor. Blue labels DNA in the nucleus; green labels actin filaments in the cytoplasm; red labels pX. (C) Double indirect immunofluorescent staining of transiently transfected cells. Red indicates TFIIB staining, and green indicates pX staining.
Next we performed double indirect immunofluorescent staining, visualizing mouse anti-pX with fluorescein isothiocyanate-conjugated goat anti-mouse and rabbit anti-TFIIB with rhodamine-conjugated goat anti-rabbit (Fig. 2C). Green staining localizes pX, red staining localizes TFIIB, and yellow or orange staining represents colocalization.
Endogenous TFIIB coimmunoprecipitates with pX.
To further show that pX is associated with TFIIB in vivo, we used coimmunoprecipitation assays. Cells were transfected with a plasmid expressing either HA-tagged or native pX. Nuclear extracts were prepared and immunoprecipitated with the monoclonal HA-specific antibody. Immunoprecipitates were analyzed by anti-TFIIB immunoblotting (Fig. 3A). A protein with the predicted size of TFIIB was detected in cell extracts transfected with pHA-X (lane 1) but not in the untagged pX-transfected cells (lane 2). These results indicate that TFIIB precipitation is mediated by epitope-tagged pX and suggest in vivo physical pX-TFIIB interaction.
FIG. 3.

pX binds endogenous as well as C34,37S TFIIB mutants but not R290E mutant. (A) Distinct pX-transfected cell extracts (HA tagged or not, as indicated) were immunoprecipitated) (IP) with anti-HA (α-HA), and the resulting proteins were analyzed by SDS-PAGE and anti-TFIIB immunoblotting. (B) Nuclear extracts (5%) of pHA-TFIIB and pflag-X-transfected cells, as well as fractions that were immunoprecipitated with α-flag, were analyzed by anti-HA immunoblotting. (C) The level of coimmunoprecipitation of the wild-type TFIIB was compared to that of C34,37S mutant. (D) Recombinant GST-X, wt TFIIB, and TFIIB mutants were resolved by SDS-PAGE, blotted, and reacted with 32P-labeled recombinant pX. Shown is the resulting autoradiogram.
The C34,37S, but not R290E, TFIIB mutant coimmunoprecipitates with pX.
TFIIB contains structural motifs with known functions (Fig. 4). To map the TFIIB regions involved in pX binding, we used TFIIB mutants in the pX coimmunoprecipitation assays. TFIIB substitution mutants each defective in a specific transcription factor interaction, were generated. (5, 19, 37). Mutations in an amino-terminal zinc finger of TFIIB (C34S and C37S) abrogate POLII binding (5), substitution of R290E in the carboxy terminus abrogated acidic activator binding, and substitution of a basic stretch in the hinge region between a tandem repeat (cyclin fold; K189, 200E) abrogated both TBP and acidic activator binding (19). We cotransfected vectors expressing TFIIB mutants as HA-tagged proteins with a flag-tagged pX expression vector. The resulting nuclear extracts were first immunoprecipitated with anti-flag antibody and then analyzed by immunoblotting with anti-HA (Fig. 3B and C). No HA-TFIIB was detected in the extract of either untransfected cells (Fig. 3B lane 1) or cells transfected only with the pflag-X expression vector (lane 2), demonstrating the specificity of our assays. Interestingly, the C34,37S HA-TFIIB mutant was efficiently immunoprecipitated with pflag-X (Fig. 3B and C, lane 3), whereas the K189,200E (lane 4) and R290E (lane 5) HA-TFIIB mutants were poorly detected. The level of pX interaction with the TFIIB C34,37S mutant was comparable to that of wild-type (wt) TFIIB (Fig. 3C). The same extracts (5% of each sample) were analyzed by anti-HA immunoblotting, directly revealing comparable levels of protein expression (Fig. 3B and C, bottom panels). These results indicate the presence of stable pX binding to the C34,37S TFIIB mutant. Thus, the TFIIB zinc finger motif is not important for pX binding.
FIG. 4.

Schematic structures of the TFIIB and pX ORFs. The pX regions required for specific binding to RPB5 of POLII (9) and to TFIIB (23) are designated with the residues at the boundaries. The TFIIB regions of the zinc finger and the direct repeat are shown as dark boxes. Also designated are the regions of TFIIB required for interaction with POLII and TBP (5, 19, 37).
pX directly binds the C34S TFIIB mutant.
To test if pX binding to the C34S TFIIB mutant is direct and not mediated by a third, unknown partner, we used far-Western analysis (28). We resolved glutathione S-transferase (GST)–X and TFIIB wt and mutant recombinant proteins by SDS-PAGE, blotted them, and reacted the membrane with 32P-labeled recombinant pX (Fig. 3C). As reported previously, the recombinant pX probe has pX binding activity and binds GST-X (31). pX binds wt TFIIB (Fig. 3D, lane 2) and the C34S TFIIB mutant (lane 4) efficiently but the R290E TFIIB mutant very poorly (lane 3). Similar amounts of proteins were loaded, as confirmed by Ponceau S staining of the blot (data not shown). Also, pX binds the C34,37S but not the K189,200E TFIIB mutant (not shown). Thus, substitution abrogating the TFIIB amino-terminal zinc finger does not abolish direct pX binding, while substitutions in the C-terminal direct repeat, R290E, and K189,200E do.
TFIIB regulates HBV gene expression.
To examine the role of TFIIB in HBV gene expression, we cotransfected HepG2 cells with HBV DNA and either wt TFIIB or the C34,37S mutant and performed Northern analysis. We used a wt HBV DNA and a viral genome with a mutation at the X ORF (Xm HBV). Western analysis had confirmed that the latter does not produce pX (data not shown). Overproduction of wt TFIIB has only mild effect on the level of the RNA expression by the Xm HBV (Fig. 5, lanes 1 to 4). In contrast, overproduction of the C34,37S TFIIB mutant significantly reduced production of the HBV transcripts (lanes 5 to 7). The latter finding is in accordance with the dominant negative function of the TFIIB mutants (11, 12, 19, 37). Remarkably, when a wt HBV DNA was used, the C34,37S TFIIB mutant, capable of pX binding, displayed no negative effect (lanes 8 to 10). Northern analysis confirmed that in all cases the expected amount of TFIIB RNA was produced, and Western analysis confirmed the production of the corresponding proteins. These data suggest that physiological amounts of pX might abrogate the negative effect of the C34,37S TFIIB mutant.
FIG. 5.

Effect of TFIIB on HBV transcription. HepG2 cells were transfected with plasmids that contain two tandem HBV full-length DNA sequences, either wt (lanes 8 and 9) or mutant with a stop codon at position 27 of the X ORF (Xm HBV; lanes 1 to 7). Cells were cotransfected with increasing amounts of plasmids (simian virus 2) that express the production of wt and mutant HA-tagged TFIIB as indicated. A 32P-labeled HBV DNA probe was used to detect the known viral transcripts, a TFIIB-specific DNA probe was used to detect the level of expression of the cotransfected TFIIB RNA, and a GAPDH probe was used to quantitate the amount of RNA per lane. For Western analysis, an HA-specific antibody (αHA) was used.
The R290E, but not C34,37S, TFIIB mutant blocks transcription coactivation by pX.
Next we examined the effect of an increasing amount of TFIIB mutants on the ability of Gal4-p53 to activate transcription of the G5 luciferase reporter (Fig. 6A). The C34,37S and R290E TFIIB mutants equally inhibited transcription activation in a dose-dependent manner. Under these conditions, overproduction of wt TFIIB has a mild repression activity (data not shown). A more inhibitory effect was observed with the K189,200E mutant, possibly reflecting a difference in the interaction defect of this mutant. We tested the functional ramifications of the pX-TFIIB interactions by comparing the activities of these TFIIB mutants in response to coexpression of pX (22, 23). We assayed the effect of increasing amounts of pX on transcription activation by Gal4-p53 and, as expected, found a dose-dependent coactivation (Fig. 6B). The R290E mutant abrogated the ability of pX to coactivate transcription, suggesting the role of TFIIB in pX-mediated coactivation. Interestingly, the C34,37S TFIIB mutant did not inhibit transcription coactivation by pX. Comparable levels of these TFIIB mutant proteins were expressed upon transfection (Fig. 3B), and both were localized to the nucleus, as confirmed by immunostaining (data not shown).
FIG. 6.

The R290E TFIIB mutant blocks transcription coactivation by pX. (A) The indicated mutant TFIIB expressors were cotransfected (increasing amounts) with constant amounts of the Gal4-p53 and the Gal4-luciferase reporter. Luciferase activity is presented relative to activator and reporter only. (B) Increasing amounts of the pX expressor were examined as for panel A, with or without the TFIIB mutants as indicated. Luciferase activity, relative to activator and reporter only, in the absence of ectopic pX is presented as fold activation.
The C34,37S, but not R290E, TFIIB mutant becomes transcriptionally active with pX.
The C34,37S TFIIB mutant has pX binding activity and thus should competitively inhibit the interaction of pX with endogenous TFIIB, yet it did not abolish pX-mediated coactivation (Fig. 6). Also, in the context of the intact HBV, this mutant has no dominant negative effect (Fig. 5). Thus, we asked whether the TFIIB C34,37S mutant acquires wt activity in the presence of pX. We assayed the effect of pX on increasing amounts of ectopic TFIIB mutants. Interestingly, the effect of pX increased in response to cotransfected C37S TFIIB mutant (Fig. 7A). Furthermore, this effect is more dramatic with a double mutant (C34,37S). This is in contrast to the transcriptional repressive effect of these TFIIB mutants in the absence of pX (Fig. 6A). These results suggest that TFIIB mutants lacking the zinc finger motif are functionally active in a pX-dependent manner. This in vivo pX-binding-dependent gain of function of a TFIIB mutant lacking POLII binding raises the possibility that transcription coactivation by pX requires TFIIB binding.
FIG. 7.

pX recovers TFIIB activity of the C34,37S mutant that it binds. (A) Increasing amounts of the TFIIB mutants were cotransfected with the Gal4-p53 and the Gal4-luciferase reporter, with or without pX expressor as indicated. Luciferase activity in the presence of pX relative to activity in the absence of pX is presented as fold pX effect. Cytoplasmic fractions of cells transfected as indicated were analyzed by luciferase assay. Two different pX mutants, Δ67-86 and Δ105-154, were used. (B) DNA templates bearing the U-less cassette of 112 bases, under the regulation of adenovirus type 5 major late promoter TATA and UASG elements, were used for in vitro-reconstituted transcription reactions. All reactions contained the DEA and DEB fractions, as described in reference 23. Two human TFIIB proteins (30 ng of either wt [hTFIIB] or C34,37S [hTFIIBm]), 10 ng of Gal4-VP16 protein, and 10 ng of recombinant pX were added to the reactions where indicated. Transcription products were purified and analyzed by denaturing gel electrophoresis.
Recovery of the C34,37S TFIIB protein by pX is obtained in in vitro transcription.
Although unlikely, it is possible that an indirect effect of pX in the cells activates the C34,37S TFIIB mutant. To rule out this possibility, we performed in vitro transcription assays using recombinant pX and TFIIB proteins and partially purified HeLa fractions which are absolutely dependent on exogenous TFIIB for transcription in vitro (23). We compared the abilities of wt and C34,37S TFIIB proteins to enable transcription. Indeed, transcription was not obtained in the absence of TFIIB (Fig. 7B, lane 1), even in the presence of pX and activator (lane 4). However, inclusion of wt recombinant TFIIB induced efficient transcription (lane 2), which increased with addition of recombinant pX (lane 3). In contrast, the C34,37S TFIIB mutant was significantly defective in supporting transcription under these conditions (lane 5). Remarkably, the addition of pX together with the C34,37S TFIIB mutant partially recovered transcription (lane 6).
pX-TFIIB interaction is not sufficient to rescue TFIIB zinc finger mutants.
Two-codon insertion mutations along the X ORF have defined two separate regions necessary for its transactivation function (38). We used these mutants to define their coactivation functions and obtained similar patterns (Fig. 8A). Interestingly, however, in this assay, pX mutants at one of these two important regions behave as dominant negative and display repression activity. Region 105–142 was reported to play a role in binding of pX to TFIIB and VP16 in vitro (23, 28). Mutations at this region generated pX mutants (M11 to M14) with repression activity (Fig. 8A). Here we examined the in vivo TFIIB binding activity of pX deletion mutants by coimmunoprecipitation with the two HA-TFIIB mutants. pX mutant Δ105-154 failed to bind both TFIIB mutants (Fig. 8B, lanes 2 and 5), correlating TFIIB binding with pX coactivation function. In contrast, pX mutant Δ67-86 significantly bound the C34,37S TFIIB mutant (lane 1). This mutant, however, was unable to recover the activity of the C34,37S TFIIB mutant (Fig. 7A). Region 58–72 was previously shown to be involved in POLII binding (9). Thus, it is likely that rescue of zinc finger TFIIB mutants by pX requires simultaneous pX interaction with both TFIIB and POLII. These results imply that pX may stabilize POLII TFIIB interaction, an effect which in the case of the C34,37S TFIIB mutant becomes the essential bridge between these cellular proteins.
FIG. 8.

Mutational analysis of pX. (A) A set of two-codon insertion mutants of pX (M1 to M14) was used in a coactivation assay; fold coactivation and repression by pX mutants was calculated and is shown along the X ORF at the corresponding regions. For this assay, the Gal4-luciferase reporter was cotransfected with Gal4-p53 activator with or without pX expressor plasmids. Also shown are the regions involved in POLII and TFIIB binding. (B) pHA-TFIIB- and pflag-X (and corresponding mutant)-transfected cell extracts were immunoprecipitated with anti-flag and analyzed by anti-TFIIB immunoblotting.
DISCUSSION
We previously showed TFIIB-, TFIIH-, and POLII-selective retention to a recombinant pX column (23). Homogenous general transcription factors, and RNA POLII, do not support transcription activation due to the lack of TBP-associated factors (TAFs) and other coactivators (8, 35, 52), but they do so in the presence of pX (23). Among this minimal set of cellular transcription factors required for pX response in vitro, only the POLII enzyme and TFIIB were found to bind pX (23). In this work, we tested the involvement of TFIIB in mediating coactivation by pX.
The TFIIB ORF (Fig. 4) is composed of an amino-terminal zinc finger, responsible for POLII interactions (5) through either POLII subunits (28, 45) or TFIIF (19), and a carboxy-terminal repeat, responsible for interaction with DNA and TBP. TFIIB-pX interaction was further analyzed in a set of coimmunoprecipitation experiments. Endogenous TFIIB interacts with pX in transiently pX-transfected cells. To delineate the regions of TFIIB required for pX binding, we tested the effect of various TFIIB point mutations, C34,37S in the zinc finger, K189,200E in the hinge region within the direct repeat, and R290E in the carboxy-terminal VP16 binding site, on pX binding. Whereas the C34,37S TFIIB mutant bound pX, the R290E and K189,200E TFIIB mutants did not. This selective binding argues for the specificity of TFIIB-pX interaction. TFIIB is known to interact with a number of activators and at least three components of the basal transcription machinery, TBP, TFIIH, and POLII (5, 19, 37).
By taking advantage of the distinctive behavior of the TFIIB mutants, we investigated pX’s mechanism of action. Reporter transient transfection assays of these TFIIB mutants with pX show that coexpression of the former affects transcription coactivation by the latter. The R290E and K189,200E TFIIB mutants are equally inhibitory in both the presence and absence of pX, thus completely abrogating transcription coactivation by pX. This negative dominant effect of TFIIB mutants implies that TFIIB binding is a crucial step in transcription coactivation by pX; i.e., TFIIB is a molecular target of pX. Therefore, it was unexpected that the C34,37S TFIIB mutant did not inhibit transcription when coexpressed with pX. In addition, increasing its amounts enhances the effect of pX. This effect of pX on the C34,37S TFIIB mutant is direct, as it was reconstituted in in vitro transcription reactions. A possible explanation is that pX complements a TFIIB mutant-containing transcription complex by recruiting the otherwise missing POLII, by acting as a molecular bridge (Fig. 9). In agreement with this model, pX mutants at the POLII binding region failed to rescue the TFIIB mutants that lack an intact zinc finger. Also, a ternary pX-TFIIB-RPB5 (a POLII subunit) complex was described recently (28), providing strong support for the proposed model.
FIG. 9.

Schematic model for pX function, describing the mode of interaction of POLII (the RPB5 subunit), TFIIB, and pX, in the absence of pX (A), in the presence of pX (B), and in the presence of pX and a TFIIB mutant that has no functional zinc finger (ZnF) (C). The model is based on a report by Lin et al. (28) that TFIIB interacts with RPB5 through the zinc finger motif and that TFIIB and pX interact with different RPB5 regions.
Homogenous general transcription factors, and RNA POLII, do not support transcription activation due to the lack of TAFs and other coactivators (8, 35, 52), but they do so in the presence of pX (23). TBP mutants that poorly respond to activators and poorly bind TAFII250 (4, 47, 48) exhibit wt activity in the presence of pX (21). Remarkably, ts13, a cell line temperature sensitive for TAFII250 function (54), exhibiting growth arrest at the restrictive temperature (41), is rescued by pX expression (21). The ability of pX to suppress the phenotype of mutations at TBP and TAFII250 implies that pX circumvents the requirement for a holo-TFIID complex for transcription activation. We propose that these different roles of pX, i.e., TAF substitution and bridging POLII to TFIIB, are related. Indeed, bridging POLII and TFIIB was also attributed to TAFs. TAFII40 binds both TFIIB and acidic activators (18, 25). In yeast, TAFII30 is associated with TFIIF, POLII, and the SWI-SNF coactivator complex (6, 24). Also, antibodies directed against the RAP30 (TFIIF small subunit) binding domain of TAFII100 inhibit the in vitro transcriptional activity of TFIID (14). Finally TAFII250 requires its RAP74 (TFIIF large subunit) binding domain in order to complement the ts13 cell line with TAFII250 function (39).
Transcription of the pregenomic RNA is an essential intermediate in HBV reverse transcription (17). The small (3.2-kb) circular DNA bears multiple (about four) promoters (43). This means that elongating POLII must traverse active promoters, thus dissociating transcription initiation complexes from the DNA (3). The overall efficiency of HBV gene expression and replication may thus be limited to the rate of transcription complex reassociation to the DNA. This may explain why HBV has acquired an ORF (X) that encodes a protein that facilitates recruitment of the transcription machinery. Alternatively, pX may enable the assembly of transcription complexes on HBV promoters containing free TBP instead of holo-TFIID, complexes with lower affinity for DNA (10, 27, 40).
ACKNOWLEDGMENTS
We thank D. Reinberg for the TFIIB mutant expression vectors, L. Runkel and H. Schaller for the X two-codon insertion mutants, and E. Schejter and D. Vaizel-Ohayon for help and advice in indirect immunofluorescent staining. We thank S. Budilovski for technical assistance.
This work was supported by the Leo and Julia Forcheimer Center for Molecular Genetics.
REFERENCES
- 1.Arii M, Takada S, Koike K. Identification of three essential regions of hepatitis B virus X protein for trans-activation function. Oncogene. 1992;7:397–403. [PubMed] [Google Scholar]
- 2.Benn J, Schneider R J. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci USA. 1994;91:10350–10354. doi: 10.1073/pnas.91.22.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhargava P. Dynamics of interaction of RNA polymerase II with nucleosomes. II. During read-through and elongation. Protein Sci. 1993;2:2246–2258. doi: 10.1002/pro.5560021224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryant G O, Martel L S, Burley S K, Berk A J. Radical mutations reveal TATA-box binding protein surfaces required for activated transcription in vivo. Genes Dev. 1996;10:2491–2504. doi: 10.1101/gad.10.19.2491. [DOI] [PubMed] [Google Scholar]
- 5.Buratowski S, Zhou H. Functional domains of transcription factor TFIIB. Proc Natl Acad Sci USA. 1993;90:5633–5637. doi: 10.1073/pnas.90.12.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cairns B R, Henry N L, Kornberg R D. TFG/TAF30/ANC1, a component of the yeast SWI/SNF complex that is similar to the leukemogenic proteins ENL and AF-9. Mol Cell Biol. 1996;16:3308–3316. doi: 10.1128/mcb.16.7.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H S, Kaneko S, Girones R, Anderson R W, Hornbuckle W E, Tennant B C, Cote P J, Gerin J L, Purcell R H, Miller R H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol. 1993;67:1218–1226. doi: 10.1128/jvi.67.3.1218-1226.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J L, Attardi L D, Verrijzer C P, Yokomori K, Tjian R. Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- 9.Cheong J, Yi M, Lin Y, Murakami S. Human RPB5, a subunit shared by eukaryotic nuclear RNA polymerases, binds human hepatitis B virus X protein and may play a role in X transactivation. EMBO J. 1995;14:143–150. doi: 10.1002/j.1460-2075.1995.tb06984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang C M, Ge H, Wang Z, Hoffmann A, Roeder R G. Unique TATA-binding protein-containing complexes and cofactors involved in transcription by RNA polymerases II and III. EMBO J. 1993;12:2749–2762. doi: 10.1002/j.1460-2075.1993.tb05936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colgan J, Ashali H, Manley J L. A direct interaction between a glutamine-rich activator and the N terminus of TFIIB can mediate transcriptional activation in vivo. Mol Cell Biol. 1995;15:2311–2320. doi: 10.1128/mcb.15.4.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colgan J, Wampler S, Manley J L. Interaction between a transcriptional activator and transcription factor IIB in vivo. Nature. 1993;362:549–553. doi: 10.1038/362549a0. [DOI] [PubMed] [Google Scholar]
- 13.Doria M, Klein N, Lucito R, Schneider R J. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995;14:4747–4757. doi: 10.1002/j.1460-2075.1995.tb00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubrovskaya V, Lavigne A C, Davidson I, Acker J, Staub A, Tora L. Distinct domains of hTAFII100 are required for functional interaction with transcription factor TFIIF beta (RAP30) and incorporation into the TFIID complex. EMBO J. 1996;15:3702–3712. [PMC free article] [PubMed] [Google Scholar]
- 15.Faktor O, Shaul Y. The identification of hepatitis B virus X gene responsive elements reveals functional similarity of X and HTLV-I tax. Oncogene. 1990;5:867–872. [PubMed] [Google Scholar]
- 16.Feitelson M A, Zhu M, Duan L X, London W T. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene. 1993;8:1109–1117. [PubMed] [Google Scholar]
- 17.Ganem D, Varmus H E. The molecular biology of the hepatitis B viruses. Annu Rev Biochem. 1987;56:651–693. doi: 10.1146/annurev.bi.56.070187.003251. [DOI] [PubMed] [Google Scholar]
- 18.Goodrich J A, Hoey T, Thut C J, Admon A, Tjian R. Drosophila TAFII40 interacts with both a VP16 activation domain and the basal transcription factor TFIIB. Cell. 1993;75:519–530. doi: 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- 19.Ha I, Roberts S, Maldonado E, Sun X, Kim L U, Green M, Reinberg D. Multiple functional domains of human transcription factor IIB: distinct interactions with two general transcription factors and RNA polymerase II. Genes Dev. 1993;7:1021–1032. doi: 10.1101/gad.7.6.1021. [DOI] [PubMed] [Google Scholar]
- 20.Harlow E, Lane D. Antibodies, a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 21.Haviv, I., and Y. Shaul. pX, the HBV encoded coactivator, suppresses the phenotypes of TBP and TAFII250 mutants. Submitted for publication. [DOI] [PMC free article] [PubMed]
- 22.Haviv I, Vaizel D, Shaul Y. The X protein of hepatitis B virus coactivates potent activation domains. Mol Cell Biol. 1995;15:1079–1085. doi: 10.1128/mcb.15.2.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haviv I, Vaizel D, Shaul Y. pX, the HBV-encoded coactivator, interacts with components of the transcription machinery and stimulates transcription in a TAF-independent manner. EMBO J. 1996;15:3413–3420. [PMC free article] [PubMed] [Google Scholar]
- 24.Henry N L, Campbell A M, Feaver W J, Poon D, Weil P A, Kornberg R D. TFIIF-TAF-RNA polymerase II connection. Genes Dev. 1994;8:2868–2878. doi: 10.1101/gad.8.23.2868. [DOI] [PubMed] [Google Scholar]
- 25.Hori R, Pyo S, Carey M. Protease footprinting reveals a surface on transcription factor TFIIB that serves as an interface for activators and coactivators. Proc Natl Acad Sci USA. 1995;92:6047–6051. doi: 10.1073/pnas.92.13.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kekule A S, Lauer U, Weiss L, Luber B, Hofschneider P H. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature. 1993;361:742–745. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- 27.Lieberman P M, Berk A J. A mechanism for TAFs in transcriptional activation: activation domain enhancement of TFIID-TFIIA-promoter DNA complex formation. Genes Dev. 1994;8:995–1006. doi: 10.1101/gad.8.9.995. [DOI] [PubMed] [Google Scholar]
- 28.Lin Y, Nomura T, Cheong J, Dorjsuren D, Iida K, Murakami S. Hepatitis B virus x protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J Biol Chem. 1997;272:7132–7139. doi: 10.1074/jbc.272.11.7132. [DOI] [PubMed] [Google Scholar]
- 29.Luber B, Lauer U, Weiss L, Hohne M, Hofschneider P H, Kekule A S. The hepatitis B virus transactivator HBx causes elevation of diacylglycerol and activation of protein kinase C. Res Virol. 1993;144:311–321. doi: 10.1016/s0923-2516(06)80047-6. [DOI] [PubMed] [Google Scholar]
- 30.Maguire H F, Hoeffler J P, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 31.Murakami S, Cheong J, Ohno S, Matsushima K, Kaneko S. Transactivation of human hepatitis B virus X protein, HBx, operates through a mechanism distinct from protein kinase C and okadaic acid activation pathways. Virology. 1994;199:243–246. doi: 10.1006/viro.1994.1119. [DOI] [PubMed] [Google Scholar]
- 32.Natoli G, Avantaggiati M L, Chirillo P, Costanzo A, Artini M, Balsano C, Levrero M. Induction of the DNA-binding activity of c-jun/c-fos heterodimers by the hepatitis B virus transactivator pX. Mol Cell Biol. 1994;14:989–998. doi: 10.1128/mcb.14.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natoli G, Avantaggiati M L, Chirillo P, De M E, Collepardo D, Falco M, Balsano C, Levrero M. Modulation of intracellular signal transduction pathways by the hepatitis B virus transactivator pX. J Hepatol. 1995;22:14–20. [PubMed] [Google Scholar]
- 34.Natoli G, Avantaggiati M L, Chirillo P, Puri P L, Ianni A, Balsano C, Levrero M. Ras- and Raf-dependent activation of c-jun transcriptional activity by the hepatitis B virus transactivator pX. Oncogene. 1994;9:2837–2843. [PubMed] [Google Scholar]
- 35.Parvin J D, Shykind B M, Meyers R E, Kim J, Sharp P A. Multiple sets of basal factors initiate transcription by RNA polymerase II. J Biol Chem. 1994;269:18414–18421. [PubMed] [Google Scholar]
- 36.Qadri I, Maguire H F, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc Natl Acad Sci USA. 1995;92:1003–1007. doi: 10.1073/pnas.92.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts S G, Ha I, Maldonado E, Reinberg D, Green M R. Interaction between an acidic activator and transcription factor TFIIB is required for transcriptional activation. Nature. 1993;363:741–744. doi: 10.1038/363741a0. [DOI] [PubMed] [Google Scholar]
- 38.Runkel L, Fischer M, Schaller H. Two-codon insertion mutations of the HBx define two separate regions necessary for its trans-activation function. Virology. 1993;197:529–536. doi: 10.1006/viro.1993.1626. [DOI] [PubMed] [Google Scholar]
- 39.Ruppert S, Tjian R. Human TAFII250 interacts with RAP74: implications for RNA polymerase II initiation. Genes Dev. 1995;9:2747–2755. doi: 10.1101/gad.9.22.2747. [DOI] [PubMed] [Google Scholar]
- 40.Sauer F, Hansen S K, Tjian R. DNA template and activator-coactivator requirements for transcriptional synergism by Drosophila bicoid. Science. 1995;270:1825–1828. doi: 10.1126/science.270.5243.1825. [DOI] [PubMed] [Google Scholar]
- 41.Sekiguchi T, Nohiro Y, Nakamura Y, Hisamoto N, Nishimoto T. The human CCG1 gene, essential for progression of the G1 phase, encodes a 210-kilodalton nuclear DNA-binding protein. Mol Cell Biol. 1991;11:3317–3325. doi: 10.1128/mcb.11.6.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seto E, Mitchell P J, Yen T S. Transactivation by the hepatitis B virus X protein depends on AP-2 and other transcription factors. Nature. 1990;344:72–74. doi: 10.1038/344072a0. [DOI] [PubMed] [Google Scholar]
- 43.Shaul Y. Regulation of HBV transcription. Boca Raton, Fla: CRC Press; 1991. [Google Scholar]
- 44.Su F, Schneider R J. Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of Rel-related proteins. J Virol. 1996;70:4558–4566. doi: 10.1128/jvi.70.7.4558-4566.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Z W, Tessmer A, Hampsey M. Functional interaction between TFIIB and the Rpb9 (Ssu73) subunit of RNA polymerase II in Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:2560–2566. doi: 10.1093/nar/24.13.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takada S, Koike K. Three sites of the hepatitis B virus X protein cooperatively interact with cellular proteins. Virology. 1994;205:503–510. doi: 10.1006/viro.1994.1671. [DOI] [PubMed] [Google Scholar]
- 47.Tansey W P, Herr W. The ability to associate with activation domains in vitro is not required for the TATA box-binding protein to support activated transcription in vivo. Proc Natl Acad Sci USA. 1995;92:10550–10554. doi: 10.1073/pnas.92.23.10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tansey W P, Ruppert S, Tjian R, Herr W. Multiple regions of TBP participate in the response to transcriptional activators in vivo. Genes Dev. 1994;8:2756–2769. doi: 10.1101/gad.8.22.2756. [DOI] [PubMed] [Google Scholar]
- 49.Todorov I T, Attaran A, Kearsey S E. BM28, a human member of the MCM2-3-5 family, is displaced from chromatin during DNA replication. J Cell Biol. 1995;129:1433–1445. doi: 10.1083/jcb.129.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish J A. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol. 1995;69:1851–1859. doi: 10.1128/jvi.69.3.1851-1859.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Twu J S, Chu K, Robinson W S. Hepatitis B virus X gene activates kappa B-like enhancer sequences in the long terminal repeat of human immunodeficiency virus 1. Proc Natl Acad Sci USA. 1989;86:5168–5172. doi: 10.1073/pnas.86.13.5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyree C M, George C P, Lira D L, Wampler S L, Dahmus M E, Zawel L, Kadonaga J T. Identification of a minimal set of proteins that is sufficient for accurate initiation of transcription by RNA polymerase II. Genes Dev. 1993;7:1254–1265. doi: 10.1101/gad.7.7a.1254. [DOI] [PubMed] [Google Scholar]
- 53.Unger T, Shaul Y. The X protein of the hepatitis B virus acts as a transcription factor when targeted to its responsive element. EMBO J. 1990;9:1889–1895. doi: 10.1002/j.1460-2075.1990.tb08315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang E H, Tjian R. Promoter-selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250. Science. 1994;263:811–814. doi: 10.1126/science.8303298. [DOI] [PubMed] [Google Scholar]
- 55.Wang X W, Forrester K, Yeh H, Feitelson M A, Gu J R, Harris C C. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci USA. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zawel L, Kumar K P, Reinberg D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- 57.Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68:2026–2030. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]