

Abstract
The value of circulating tumor DNA (ctDNA) as a biomarker of disease activity in classic Hodgkin lymphoma (cHL) patients has not yet been well established. By profiling primary tumors and ctDNA, we identified common variants between primary tumors and longitudinal plasma samples in most of the cases, confirming high spatial and temporal heterogeneity. Although ctDNA analyses mirrored HRS cell genetics overall, the prevalence of variants shows that none of them can be used as a single biomarker. Conversely, the estimation of hGE/mL, based on measures of total ctDNA, reflects disease activity and is almost perfectly correlated with standard parameters such as PET/CT that are associated with refractoriness.
Keywords: Hodgkins lymphoma, lymphoid malignancies, minimal residual disease
1. INTRODUCTION
While most classic Hodgkin lymphoma (cHL) patients can be cured nowadays, a subgroup of patients will still relapse after first‐line treatment, some of whom will ultimately die from the disease. These relapsed/refractory (R/R) cHL cases account for 20%–30% of patients, most of whom are at an advanced stage or exhibit risk factors.
Formerly, such patients were treated with salvage chemotherapy regimens followed by autologous stem cell transplantation, but more recently, the introduction of novel agents such as brentuximab vedotin and immune checkpoint inhibitors into the treatment algorithm have modified the choice and order of salvage therapy regimens. Therefore, new tools are clearly needed to identify early on the patients who will progress after first‐line therapy, and to rationalize second‐line therapies. The clinical use and prognostic value of FDG‐PET/CT (PET/CT) during disease follow‐up and interim PET/CT, (iPET/CT) which is performed after the initiation but before the completion of treatment, varies by lymphoma subtype. Evidence supporting the prognostic value of iPET/CT is clear for cHL and, indeed, response‐adapted treatment approaches guided by PET/CT are a widely used standard of care in first‐line therapy.
Circulating tumor DNA (ctDNA) represents the cell‐free DNA released by the tumoral cell into the blood. These mainly ctDNA sequencing‐based liquid biopsies offer some advantages over classic tumor genotyping, mainly during disease monitoring and detection of minimal residual disease (MRD) in multiple cancers, including hematological malignancies [1]. Concordance between ctDNA and tumor biopsies in aggressive lymphomas is around 80%, and somewhat lower in indolent lymphomas with a low tumor burden [2]. Researchers agree that liquid biopsy could represent a major new strategy for monitoring cancer, but further studies are required to address its limitations.
The mutational landscape of the neoplastic Hodgkin and Reed–Sternberg (HRS) cells in cHL has been characterized in recent years. We and others have demonstrated recurrent somatic mutations in the components of the NF‐kB pathway (TNFAIP3, NFKBIA, NFKBIA, REL), the JAK/STAT pathway (SOCS1, PTPN1, STAT6, STAT3, CSF2RB) [3, 4, 5, 6], and regulators of immune escape, such as inactivating mutations in the gene of the MHC‐1 component B2M, the MHC‐2 transactivator (C2TA) [5, 7], and inactivating mutations in CD58 [8]. Other signaling pathways important in cHL pathogenesis include the BCR pathway (BTK, CARD11, BCL10) [3], MAPK/ERK, AP1, PI3K/AKT, and NOTCH1 [9]. Frequent mutations are also detected in epigenetic regulators such as EP300 and CREBBP, as well as TP53 mutations, and these changes have been linked to clinical outcome.
It has yet to be established whether ctDNA is a reliable biomarker of disease activity that can provide information about tumor burden in cHL patients. Furthermore, the applicability of liquid biopsies for monitoring cHL in the clinical setting needs validation. To address these matters, we profiled primary tumors and ctDNA from retrospective and prospective series of samples, with the aim of (i) reassessing the genetic landscape of the disease by checking for common markers, (ii) establishing whether ctDNA profiling can be used for qualitative and quantitative analysis of tumor‐specific somatic DNA mutations, and (iii) evaluating in a prospective cohort whether liquid biopsies can be used to monitor disease evolution.
2. MATERIALS AND METHODS
2.1. Study design and clinical data
The study was planned as a prospective and retrospective screening of somatic mutations in primary lymph nodes and ctDNA, to evaluate this molecular strategy for disease monitoring and enhance early diagnosis of relapse. Formalin‐fixed paraffin‐embedded (FFPE) tumor samples (pretreatment biopsies) from 40 patients with a confirmed diagnosis of cHL were included. Patients diagnosed with cHL of any subtype were included. Most cases were treated with standard adriamycin‐based protocols, such as adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD). Clinical data are summarized in Table S1. All the samples and data were retrieved through the MD Anderson Cancer Center Madrid Biobank, in accordance with the technical and ethical procedures of the Spanish National Biobank Network and having obtained written informed consent in accordance with the Helsinki Declaration. Approval was obtained from the institutional review board.
The analysis of the results was based on evaluating the intratumoral heterogeneity in primary lymph node biopsies, as well as mapping the most frequently affected genes and signaling pathways. In addition, the temporal heterogeneity throughout the course of the disease was evaluated using ctDNA. Finally, we calculated the correlation between the standard cHL monitoring technique (PET/CT) and liquid biopsy findings.
2.2. DNA isolation
For somatic mutations identification in primary cHL tumors, duplicated aliquots of DNA were extracted from two selected HRS‐enriched regions, with a total of 80 sequenced samples. DNA was extracted with standard protocols based on proteinase K digestion and phenol/ethanol isolation.
To increase sequencing quality from FFPE samples, a DNA repair step was performed to correct cytosine‐uracil changes induced by deamination with the NEBNext FFPE DNA repair mix kit (BioLabs), following the manufacturer's instructions. After this, the samples were purified by AMPure XP Beads (Beckman Coulter) and eluted in 25 μL of nuclease‐free water.
Total cell‐free DNA was extracted from two plasma samples using Cell‐Free DNA BCT tubes (Streck). Briefly, after two consecutive centrifugations, we used the QiAamp Circulating Nucleic Acid Extraction kit (Quiagen), according to the manufacturer's instructions.
For filtering germline and polymorphic variants, gDNA was extracted from peripheral blood cells (n = 7), noninfiltrated bone marrow smears (n = 5) or nontumor tissue samples (n = 2). We used standard protocols based on proteinase K digestion and phenol/ethanol isolation (FFPE samples) or DNeasy Blood & Tissue kit (Quiagen) (for total peripheral blood).
2.3. Next generation sequencing and variant filtering
Next generation sequencing (NGS) was performed using a cHL‐specific targeted custom panel that included the 35 genes most frequently mutated in the disease, following similar protocols to those adopted in a previous study [6]. NGS was performed on an Ion Torrent S5 sequencer (Thermo Fisher, Waltham, MA). This amplicon‐based library preparation technology gives a low limit of detection of as little as 0.1% for cell‐free DNA samples. Libraries were constructed starting with 10 ng of genomic DNA. Emulsion PCR and sample enrichment was done using an initial DNA library concentration of 100 pM, quantified using the Qubit High Sensitivity DNA kit (Life Technologies).
For bioinformatic analysis and variant filtering, we used Ion Reporter and Alamut software in conjunction with ClinVar, Varsome, cBioportal, and genomeAD databases. Variants with fewer than 100 reads, with a variant allele frequency (VAF) of < 1% or > 40%, or that had previously been described as polymorphisms in the genomeAD and single nucleotide polymorphism (dbSNP) databases, were excluded. Filtered variants were reviewed and analyzed using the integrative genomics viewer (IgV), the Alamut algorithm, and reference databases such as ClinVar, Varsome, cBioportal, COSMIC (Catalogue of Somatic Mutations in Cancer), and Ensembl.
2.4. Correlation between 18‐FDG‐PET and haploid genomic equivalent per milliliter (hGE/mL)
Deauville Scores (DSs) were obtained for each patient after reviewing the reports from the Nuclear Medicine Department and patients’ medical records. Total ctDNA was quantified and treated as a continuous variable, calculating the haploid genomic equivalent per milliliter (hGE/mL) of plasma, determined as the product of total ctDNA in pg/mL, and the mean VAF was divided by 3.3 pg per DNA molecule, as previously described [10].
3. RESULTS
This approach enabled us to detect mutations in primary cHL tumors in 81% of the samples. All analyzed DNA libraries had a mean length of 116 bp with a range from 90 to 169 bp. The mean average base coverage depth was 2115x, with a 92% uniformity of base coverage.
Overall, our results confirmed the previously described mutational distribution [5, 6]. We identified common mutations affecting TP53, CARD11, EP300, STAT6, and PIK3CD (Figure 1A and Table S2), among other genes. The TP53 and CARD11 variants were the most frequent alterations. As has often been described, we did not find any single gene or specific variant common to enough patients to warrant it being proposed as a specific cHL biomarker. Instead, we found a heterogeneous spectrum of alterations that potentially affect the function of the most relevant pathways in the pathogenesis of cHL (Figure 1A). By performing two independent analyses of each tumor sample, we also corroborated the great spatial intratumoral heterogeneity that characterizes this disease, whereby the tumors presented shared variants in 13 patients (32.5%), and individual mutations were noted in 27 patients (67.5%) (Figure 1B). All the mutations detected are listed in Tables S2 and S3.
FIGURE 1.
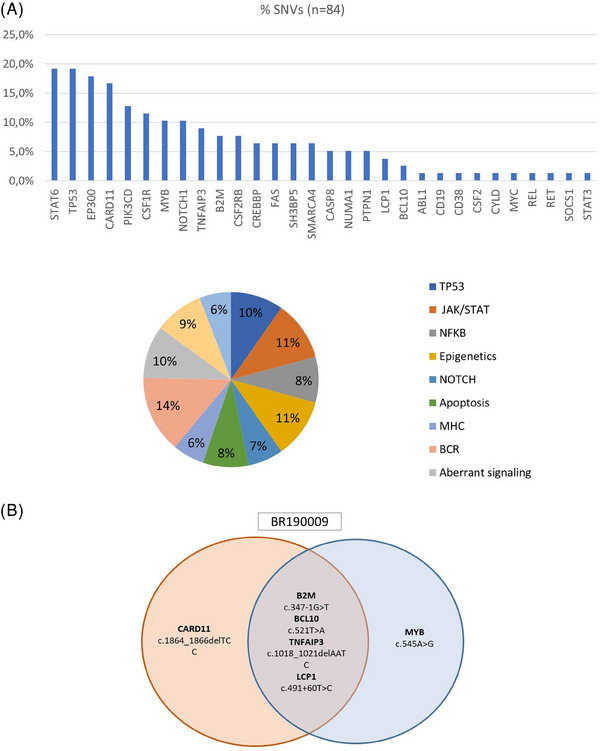
(A) Classic Hodgkin lymphoma (cHL) is a disease characterized by the concurrent dysregulation of several oncogenic pathways. (B) Mutational analyses in primary tumors. Representative example showing great intratumor heterogeneity between two tumor regions. Detailed information on all variants detected in all tumor tissues and plasma samples can be found in Table S1.
We also carried out a prospective longitudinal analysis by using liquid biopsy for disease monitoring in those patients with sufficient available plasma in samples extracted from peripheral blood (n = 17) and normal germline genomic gDNA for variant filtering (n = 14). With these series, we completed the study of 55 samples, detecting tumor mutations in 89% of ctDNA samples. The mean average base coverage depth was 1236x, with 78% uniformity of base coverage. There were no major variations in the sample collection times and protocols between patients, collecting samples at diagnosis or before treatment (basal or pretreatment samples), at mid‐treatment (between the 3rd and 4th ABVD cycles), samples at the end of treatment, and samples in follow‐up visits.
As previously reported [11, 12], the results of the ctDNA analyses mirrored HRS cell genetics overall. We identified common variants between primary tumors and longitudinal plasma samples in eight of 14 cases. However, and consistent with the tumor heterogeneity, there were also high levels of temporal heterogeneity, so that several variants detected in the primary tumors were not detected in some of the blood samples, some variants reappeared when ctDNA levels rose during follow‐up, and new variants could be detected that were probably related to newly emerging clones (Figure 2A). Again, no single or common genetic marker can be proposed to cover all patients (Table S3).
FIGURE 2.
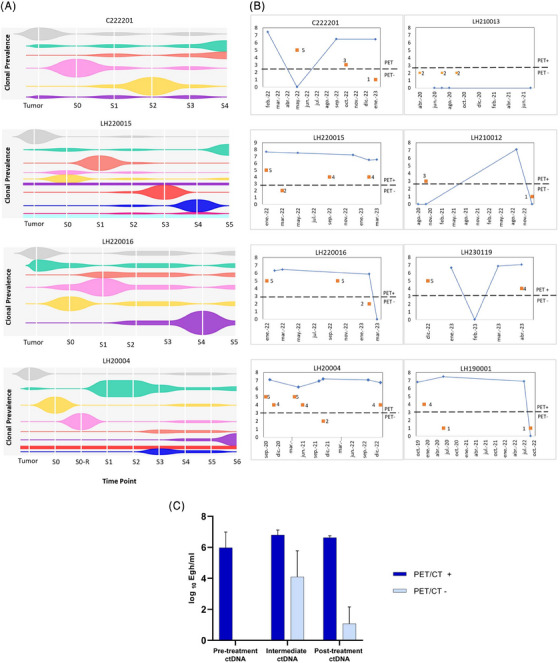
(A) Clonal evolution data from four representative cases; plots show clonal prevalence vertically (variant allele frequency), time points horizontally (sampling times), and the height of each clone reflects its proportionate prevalence at each sampling time point. Figures were created using the TimeScape R package (https://bioconductor.org/packages/release/bioc/html/timescape.html), clustering the variants observed in each clone and their allele prevalences over time (see Supplemental Table). (B) Comparison between iPET/CT (DS of 1–5, considering a DS of >3 to be a positive iPET/CT result) and ctDNA quantification showing similar trends over time. (C) Correlation between ctDNA and iPET/CT (two‐way ANOVA, p = 0.022); error bars indicate the standard error of the mean (SEM). iPET, interim FDG‐PET/CT; CT, computed tomography; DS, Deauville score.
Finally, we thirdly carried out the quantitative longitudinal analyses in those patients with at least three available blood samples that were informative of the mutational changes during and after treatment, as well as the germline DNA (n = 9).
Given the short follow‐up period of this series, we used iPET/CT and after treatment PET/CT as surrogates for early treatment failure. According to clinical guidelines, responses were assessed by PET/CT after 3 cycles of chemotherapy (iPET/CT) and 4 ± 1 weeks after the end of the treatment. DSs were obtained for each patient after reviewing the reports from the Nuclear Medicine Department and patients’ medical records, and ctDNA values from liquid biopsies were compared with DS values (Figure 2B). In most cases, the two parameters had similar temporal variation, with a significant statistical correlation between total ctDNA assessed in plasma and PET/CT variations over time (Figure 2C and Table S4). It is of note that one patient (C222201) showed persistently high ctDNA values despite obtaining a negative PET/CT at the end of the treatment.
4. DISCUSSION
ctDNA forms the basis of liquid biopsy, and offers some advantages over classical tumor genotyping, mainly during disease monitoring. The concordance between ctDNA and tumor biopsies in aggressive lymphomas is about 80%, and somewhat lower in indolent lymphomas with a low tumor burden 2. For these reasons, researchers agree that liquid biopsy could represent a major new strategy for monitoring cancer, but further studies are required to address its limitations.
The usefulness of liquid biopsies is evident for monitoring non‐Hodgkin lymphomas, where circulating ctDNA levels are associated with tumor volume and MRD [2]. However, the higher levels of intratumoral and temporal heterogeneity in cHL make it extremely difficult to use specific mutations as single biomarkers. There are no earlier reports proposing the quantitative measurement of ctDNA using the hGE/mL, independent of specific variants as an alternative surrogate biomarker in cHL, which is clinically relevant and could identify patients at higher risk of early progression.
Since previous reports have not identified a common single recurrent mutation that characterizes cHL tumors, monitoring cHL in ctDNA may be controversial. One frequent alteration in the SOCS1 gene, which is detected in approximately 50% of cHL cases [4], has been proposed as a potential biomarker, but common variants consist of truncating mutations and indels, which are technically challenging to detect in plasma. The XPO1 E571K mutation has been repeatedly detected in both tissue and plasma samples and has also been proposed as a biomarker for use in the diagnosis and detection of MRD [13, 14, 15]. However, the detection frequency of the XPO1 in primary tumors was significantly lower [15] and was described only in 10%–15% of ctDNA patients samples [15, 16]. Here, w confirm that no specific gene mutation contributes uniquely to the pathogenesis of the disease. Rather, a set of alterations in different essential pathways is associated with the characteristic phenotype of HRS cells and, consequently, the pathogenesis of the disease.
In conclusion, we confirm the feasibility of monitoring cHL using ctDNA from liquid biopsies coupled with simple NGS procedures based on cHL‐specific targeted panels. ctDNA mirrored HRS cell genetics, showing clonal evolution and capturing the known spatial and longitudinal heterogeneity, but it is impossible to propose unique genetic markers of the disease. Total ctDNA quantification based on estimates of hGE/mL is a good surrogate for disease activity, since it probably reflects the tumor burden and is almost perfectly correlated with parameters such as PET/CT that are associated with refractoriness. Whether this trend is correlated with clinical outcomes and survival remains unclear and needs to be confirmed in longitudinal series with longer follow‐up, in the context of well‐designed clinical trials.
AUTHOR CONTRIBUTIONS
SF designed the study, performed the research, and wrote the manuscript. LC, ED, and SF performed the research. LC, LR, ME, JLS and CM collected the clinical data. SF and VM statistically analyzed and interpreted the data. JFG designed the study, analyzed and interpreted the data, obtained funding for the research, and wrote the manuscript. All the authors critically reviewed and approved the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
ETHICS STATEMENT
All the samples and data were collected through the MD Anderson Cancer Center Madrid Biobank, in accordance with the technical and ethical procedures of the Spanish National Biobank Network and having obtained written informed consent according to the Helsinki Declaration. Approval was obtained from the institutional review board (CEIm H. Ramón y Cajal, reference 445/22).
PATIENT CONSENT STATEMENT
Written informed consent was obtained from all patients
CLINICAL TRIAL REGISTRATION
The authors have confirmed clinical trial registration is not needed for this submission.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors are very grateful to all the technicians and nursing staff of the MDACC Blood Collection Clinics for their invaluable help with the logistics. They also acknowledge MD Anderson Cancer Center Madrid Biobank (B.0000745) for sample and clinical data management. This work was supported by the Instituto de Salud Carlos III (ISCIII), through the project PI19/00083 (co‐funded by European Regional Development Fund/European Social Fund “A way to make Europe”/“Investing in your future”), the project PI22/00556 (co‐funded by the European Union), and a Roche Foundation Research Grant. V.M. is a recipient of a PFIS predoctoral contract from ISCIII‐AES‐2020 (FI20/00184).
Fernández S, Cereceda L, Díaz E, Figueroa S, Reguera L, Menéndez V, et al. Circulating tumor DNA for monitoring classic Hodgkin lymphoma patients: Correlation with FDG‐PET/CT. eJHaem. 2024;5:70–75. 10.1002/jha2.826
DATA AVAILABILITY STATEMENT
NGS data are available at GEO under accession number GSE244989.
REFERENCES
- 1. Buedts L, Vandenberghe P. Circulating cell‐free DNA in hematological malignancies. Haematologica. 2016;101(9):997–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fernández‐Miranda I, Pedrosa L, Llanos M, Franco FF, Gómez S, Martín‐Acosta P, et al. Monitoring of circulating tumor DNA predicts response to treatment and early progression in follicular lymphoma: results of a prospective pilot study. Clin Cancer Res. 2023;29(1):209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mata E, Díaz‐López A, Martín‐Moreno AM, Sánchez‐Beato M, Varela I, Mestre MJ, et al. Analysis of the mutational landscape of classic Hodgkin lymphoma identifies disease heterogeneity and potential therapeutic targets. Oncotarget. 2017;8(67):111386–111395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K, et al. Mutations of the tumor suppressor gene SOCS‐1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho‐STAT5 accumulation. Oncogene. 2006;25(18):2679–2684. [DOI] [PubMed] [Google Scholar]
- 5. Reichel J, Chadburn A, Rubinstein PG, Giulino‐Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed‐Sternberg cells. Blood. 2015;125(7):1061–1072. [DOI] [PubMed] [Google Scholar]
- 6. Mata E, Fernández S, Astudillo A, Fernández R, García‐Cosío M, Sánchez‐Beato M, et al. Genomic analyses of microdissected Hodgkin and Reed‐Sternberg cells: mutations in epigenetic regulators and p53 are frequent in refractory classic Hodgkin lymphoma. Blood Cancer J. 2019;9(3):7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471(7338):377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schneider M, Schneider S, Zühlke‐Jenisch R, Klapper W, Sundström C, Hartmann S, et al. Alterations of the CD58 gene in classical Hodgkin lymphoma. Genes Chromosomes Cancer. 2015;54(10):638–645. [DOI] [PubMed] [Google Scholar]
- 9. Weniger MA, Küppers R. Molecular biology of Hodgkin lymphoma. Leukemia. 2021;35(4):968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bourbon E, Alcazer V, Cheli E, Huet S, Sujobert P. How to obtain a high quality ctDNA in Lymphoma Patients: preanalytical tips and tricks. Pharmaceuticals. 2021;14(7):617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131(22):2413–2425. [DOI] [PubMed] [Google Scholar]
- 12. Vandenberghe P, Wlodarska I, Tousseyn T, Dehaspe L, Dierickx D, Verheecke M, et al. Non‐invasive detection of genomic imbalances in Hodgkin/Reed‐Sternberg cells in early and advanced stage Hodgkin's lymphoma by sequencing of circulating cell‐free DNA: a technical proof‐of‐principle study. Lancet Haematol. 2015;2(2):e55–65. [DOI] [PubMed] [Google Scholar]
- 13. Camus V, Viennot M, Lévêque E, Viailly PJ, Tonnelet D, Veresezan EL, et al. Circulating tumor DNA in primary mediastinal large B‐cell lymphoma versus classical Hodgkin lymphoma: a retrospective study. Leuk Lymphoma. 2022;63(4):834‐844. [DOI] [PubMed] [Google Scholar]
- 14. Camus V, Stamatoullas A, Mareschal S, Viailly PJ, Sarafan‐Vasseur N, Bohers E, et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell‐free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica. 2016;101(9):1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Camus V, Viennot M, Lequesne J, Viailly PJ, Bohers E, Bessi L, et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: a prospective study. Haematologica. 2021;106(1):154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bessi L, Viailly PJ, Bohers E, Ruminy P, Maingonnat C, Bertrand P, et al. Somatic mutations of cell‐free circulating DNA detected by targeted next‐generation sequencing and digital droplet PCR in classical Hodgkin lymphoma. Leuk Lymphoma. 2019;60(2):498–502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
NGS data are available at GEO under accession number GSE244989.