

Abstract
The definitive treatment for end-stage organ failure is orthotopic transplantation. However, the demand for transplantation far exceeds the number of available donor organs. A promising tissue-engineering/regenerative-medicine approach for functional organ replacement has emerged in recent years. Decellularization of donor organs such as heart, liver, and lung can provide an acellular, naturally occurring three-dimensional biologic scaffold material that can then be seeded with selected cell populations. Preliminary studies in animal models have provided encouraging results for the proof of concept. However, significant challenges for three-dimensional organ engineering approach remain. This manuscript describes the fundamental concepts of whole-organ engineering, including characterization of the extracellular matrix as a scaffold, methods for decellularization of vascular organs, potential cells to reseed such a scaffold, techniques for the recellularization process and important aspects regarding bioreactor design to support this approach. Critical challenges and future directions are also discussed.
Keywords: organ engineering, extracellular matrix, decellularization, regenerative medicine, three-dimensional scaffold, tissue engineering
INTRODUCTION
The definitive treatment for end-stage organ failure is orthotopic transplantation. However, the critical shortage of donor organs leads to increased morbidity and mortality for tens of thousands of patients each year. Approximately 27,000 deaths occur annually in the United States alone for patients with end-stage liver disease (1), 120,000 patients as a result of chronic lung disease (2), 112,000 from end-stage kidney failure (3), and 425,000 from coronary heart disease (4). When one considers the additional patients who die waiting for heart or kidney transplants, the numbers become overwhelming. Those patients fortunate enough to receive an organ are burdened with the risk of chronic rejection and the morbidity associated with a lifelong regimen of immunosuppressant therapy. Although significant advances have been made in the development of engineered tissues such as blood vessels (4, 5), urinary bladder (6), and trachea (7, 8), none of these tissues require an intact vascular network that can be connected to the host circulation at the time of implantation. Whole-organ constructs such as heart, lung, and liver will require this type of immediate vascular supply. A successful regenerative medicine strategy for whole-organ replacement would represent a quantum leap forward in the treatment of patients with end-stage organ disease.
In recent years a promising approach for functional organ replacement has emerged. Decellularization of allogeneic or xenogeneic donor organs such as heart (9), liver (10), and lung (11–13) provides an acellular naturally occurring three-dimensional biologic scaffold material that subsequently can be seeded with either functional parenchymal cells or selected progenitor cell populations. Self-assembly of these seeded cells with the aid of a biofriendly three-dimensional matrix results in the formation of functional tissue in short-term preclinical animal models (10, 14, 15). This approach provides the opportunity for direct connection to the patient vasculature in either an orthotopic or heterotopic location. Numerous challenges for this three-dimensional organ-engineering approach remain including the determination of candidate species from which the donor organ can be harvested, optimal methods of donor organ decellularization, optimization of recellularization techniques and the most appropriate cell populations, endothelialization of the donor matrix vasculature, and determination of the appropriate use of ex-vivo bioreactors, among others.
The present manuscript provides an overview of the use of organ-derived extracellular matrix (ECM) as a biologic scaffold material, methods for decellularization and recellularization, and discussion of appropriate cell populations and bioreactor use as this approach is further investigated and developed.
EXTRACELLULAR MATRIX AS A BIOLOGIC SCAFFOLD
Biologic scaffold materials composed of ECM are commonly used to facilitate the constructive remodeling of a variety of tissues both in preclinical animal studies and in human clinical applications. The ECM used to create these scaffold materials is harvested from many different tissues including skin (16), small intestinal submucosa (17–19), urinary bladder (20–22), blood vessels (23–26), and heart valves (27–33), among others. The ECM materials are harvested and typically processed as two-dimensional scaffolds and do not require anastomosis directly to the recipient vasculature for their clinical applications. Rather, the infiltrating or seeded cell populations rely on oxygen and nutrient diffusion for survival while a supporting vascular network develops over time. The mechanisms by which naturally occurring scaffold materials promote functional-tissue reconstruction are not fully understood. There is legitimate and healthy controversy concerning the relative importance of structure versus composition of these materials, and these issues are particularly important in the context of whole-organ scaffolds prepared by techniques that involve perfusion-based decellularization of donor organs.
The composition of ECM is represented by a complex mixture of functional and structural molecules that affect a variety of cell activities. These molecules are arranged in a unique, tissue-specific, three-dimensional ultrastructure and are ideally suited to the tissue or organ from which the ECM is harvested. To make matters even more complex, the structure and composition of the ECM are constantly changing in response to the current metabolic activity of the resident cell population, the mechanical demands of the tissue, and the prevailing microenvironmental niche conditions. This concept of “dynamic reciprocity” between the ECM and the resident cell population (34) is a major advantage for the use of ECM scaffold materials over synthetic materials and emphasizes the importance of maintaining as much of the native composition and ultrastructure as possible during the preparation of these three-dimensional scaffolds.
Because the ECM is the product of the resident cell populations, it is logical that the specific composition and ultrastructural organization of the component molecules will vary depending on the source tissue/organ from which the ECM scaffold is prepared. Recent studies have suggested the potential importance of organ specificity with regard to the source of ECM that is used as a template for organ restoration. For example, ECM harvested from liver tissue may be the preferred ECM substrate for hepatocytes and even the nonparenchymal cells of the liver (35–37). ECM from the lung may be the preferred (38), or even the necessary, substrate for respiratory epithelial cells and so on. In general, however, it can be expected that ECM from all organs would consist primarily of type I collagen, glycosaminoglycans, fibronectin, laminin, and a diverse variety of growth factors. Whether the complex composition of the ECM or the tissue-specific three-dimensional spatial organization of these many molecules is more important with regard to supporting cell growth and differentiation has not been determined.
The native composition, ultrastructure, and the macroscopic three-dimensional architecture of organ-derived ECM scaffolds can be largely preserved by the appropriate use of processing steps required for decellularization of the tissue, especially if the use of harsh chaotropic agents can be avoided (39, 40). Numerous studies show that ECM scaffolds derived from specific organs retain many of the defining structures of those organs, such as the collagen IV and laminin-rich basement membrane of vascular structures. Recent studies have suggested that the ultrastructural topology and ligand landscape of the ECM provide defining “zip codes” for cells, which direct or support site-appropriate cell attachment and differentiation (11, 14, 35). Ultrastructural characteristics of the matrix appear to play important roles in modulating the behavior of cells that contact the scaffold either by regulating the cell’s ability to migrate into and attach to specific locations within the scaffold (39) or by influencing tissue-specific phenotypic differentiation (14, 35, 41). The ECM can definitively affect the differentiating pathway of human embryonic stem cells (ESCs) and selected progenitor cell populations (41–43).
In addition to the structural and mechanical functions of biologic scaffold materials, the biologic signaling activities provided by degradation products of ECM scaffolds have a marked effect on the host-remodeling response following in vivo implantation. Cell proliferation, migration, and differentiation as well as processes such as angiogenesis are all regulated in part by cell-signaling mechanisms that involve soluble molecules. As previously stated, ECM scaffold material are rich in growth factors (44, 45), bifunctional molecules such as fibronectin (46), and a variety of collagen types (18, 39), among other functional and structural moieties. A wide variety of potent and biologically important functional activity can be attributed to the degradation of the native ECM scaffold structure and the associated release of soluble bioactive cryptic peptides (47, 48). The biologic activities mediated by these matricryptic peptides depend on degradation of the intact three-dimensional structure, which is in direct contrast to the mechanical and structural properties of ECM scaffold that are dependent on an intact three-dimensional structure. Therefore, processing methods that inhibit degradation of ECM scaffold materials and the associated generation and release of these cryptic peptides, such as chemical cross-linking, can significantly alter the functional profile and the associated downstream host response and remodeling process.
In summary, the composition as well as the macroscopic and ultrastructural characteristics of three-dimensional, organ-derived biologic scaffold materials is highly complex. If properly prepared, such ECM scaffold materials can provide important microenvironmental cues necessary to support cell attachment, proliferation, and differentiation, while providing appropriate biomechanical support. These scaffold materials are in a constant state of flux and can be considered as temporary inductive site-appropriate templates to support the growth, differentiation, and function of the parenchymal cell population of each organ. To date, three-dimensional scaffolds composed of decellularized organ-specific ECM have been described for the heart (9, 49, 50), liver (10), and lungs (11, 14).
METHODS OF WHOLE-ORGAN DECELLULARIZATION
The preparation of a three-dimensional, whole-organ ECM scaffold material from an intact mammalian organ requires several processing steps, each of which can markedly affect the structure and composition of the resultant scaffold and the associated host response that these scaffolds will elicit when utilized as templates for organ reconstruction. The process of removing all cells from an organ (i.e., decellularization) while retaining the native composition and structure of the associated matrix typically involves exposure of the organ parenchymal cells to detergents, proteases, and chemicals by perfusion of the native vasculature.
The effective removal of antigenic epitopes associated with cell membranes and intracellular components of organs and tissues is necessary to avoid, or at least minimize, an adverse immune response by allogeneic or xenogeneic recipients of the ECM scaffold material. Allogeneic and xenogeneic antigens are usually recognized as foreign by the host and cause a destructive inflammatory response or overt immune-mediated rejection (51–53). However, molecules that constitute the ECM are largely and highly conserved across species lines and are well tolerated even by xenogeneic recipients (54–57). Certain antigens, such as the galactosyl Galα (1, 3) galactose moiety (i.e., gal-epitope) are present in many xenogeneic ECM materials (58, 59) and are associated with hyperacute rejection of xenogeneic whole-organ transplants. However, the matrix-associated gal-epitope antigens in ECM scaffold materials fail to activate complement or bind IgM (immunoglobulin M) antibody, presumably because of their widely scattered distribution and the relatively small amount of antigen. In addition, the presence of this antigen does not adversely affect the host-remodeling response (60). Recent work suggests that the effect of xenogeneic ECM upon the innate immune response, specifically the responding macrophages, may elicit a necessary M2 phenotype profile to support a constructive remodeling response for the scaffold (61).
The ultimate goal of organ decellularization is the removal of all cellular material without adversely affecting the composition, biologic activity, or mechanical integrity of the remaining three-dimensional matrix. Commonly used methods of decellularization include the perfusion of chemical or enzymatic agents and physical methods such as sonication, freezing, and thawing, with agitation to disrupt cell membranes, release cell contents, and facilitate the rinsing and removal of cell remnants from the ECM (Figure 1). It is exceedingly difficult to accomplish complete tissue or organ decellularization, and most ECM scaffold materials retain residual DNA (62, 63) and other cytoplasmic and nuclear material.
Figure 1.
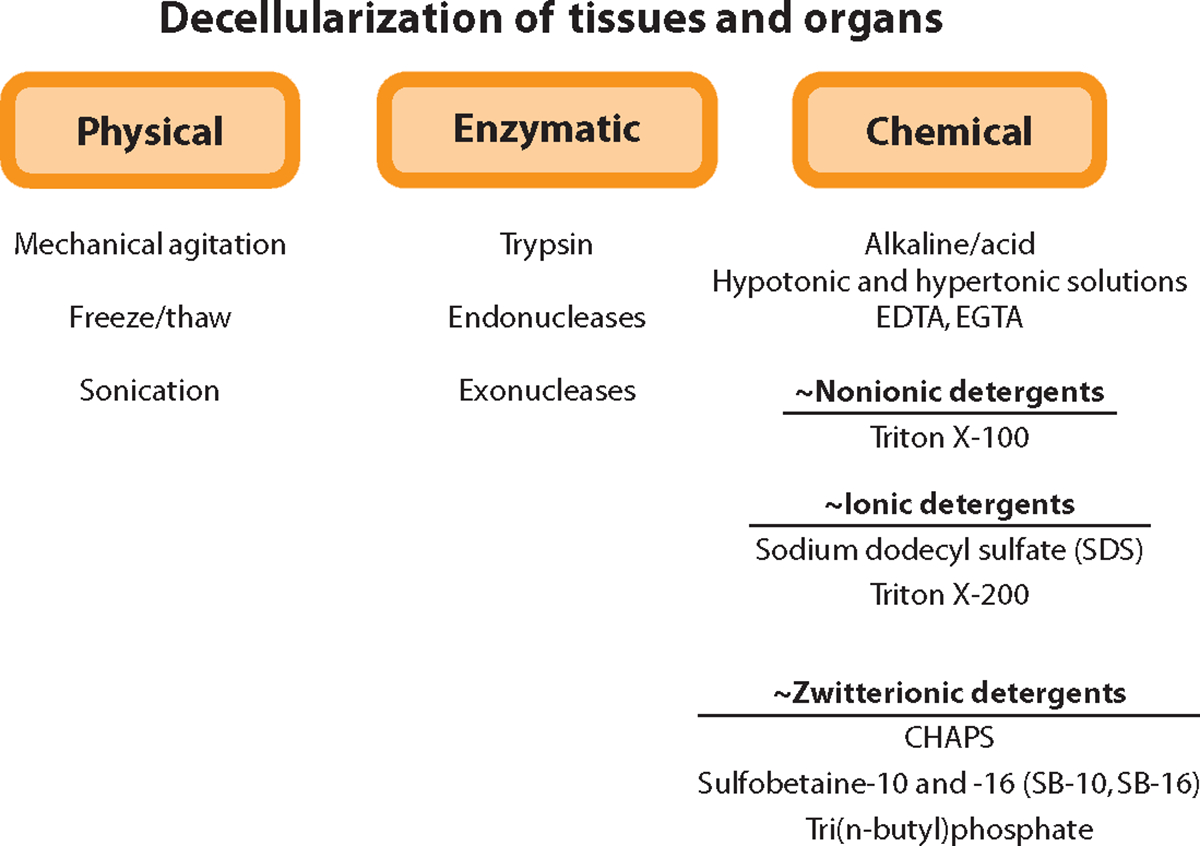
Examples of techniques used to decellularize tissues and organs. Abbreviations: CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; EDTA, ethylene diamine tetraacetic acid; EGTA, ethylene glycol tetraacetic acid.
Combinations of these various approaches for decellularization are typically used to maximize the efficiency of the process for each tissue and organ. As stated above, organ decellularization involves delivery of these agents by vascular perfusion. Some reported perfusion protocols for organ decellularization are shown in Figure 2. As a general rule the use of detergents and chaotropic agents such as Triton X, sodium dodecyl sulfate, and deoxycholate should be minimized whenever possible to avoid damage to the ultra structure and composition of the native ECM.
Figure 2.
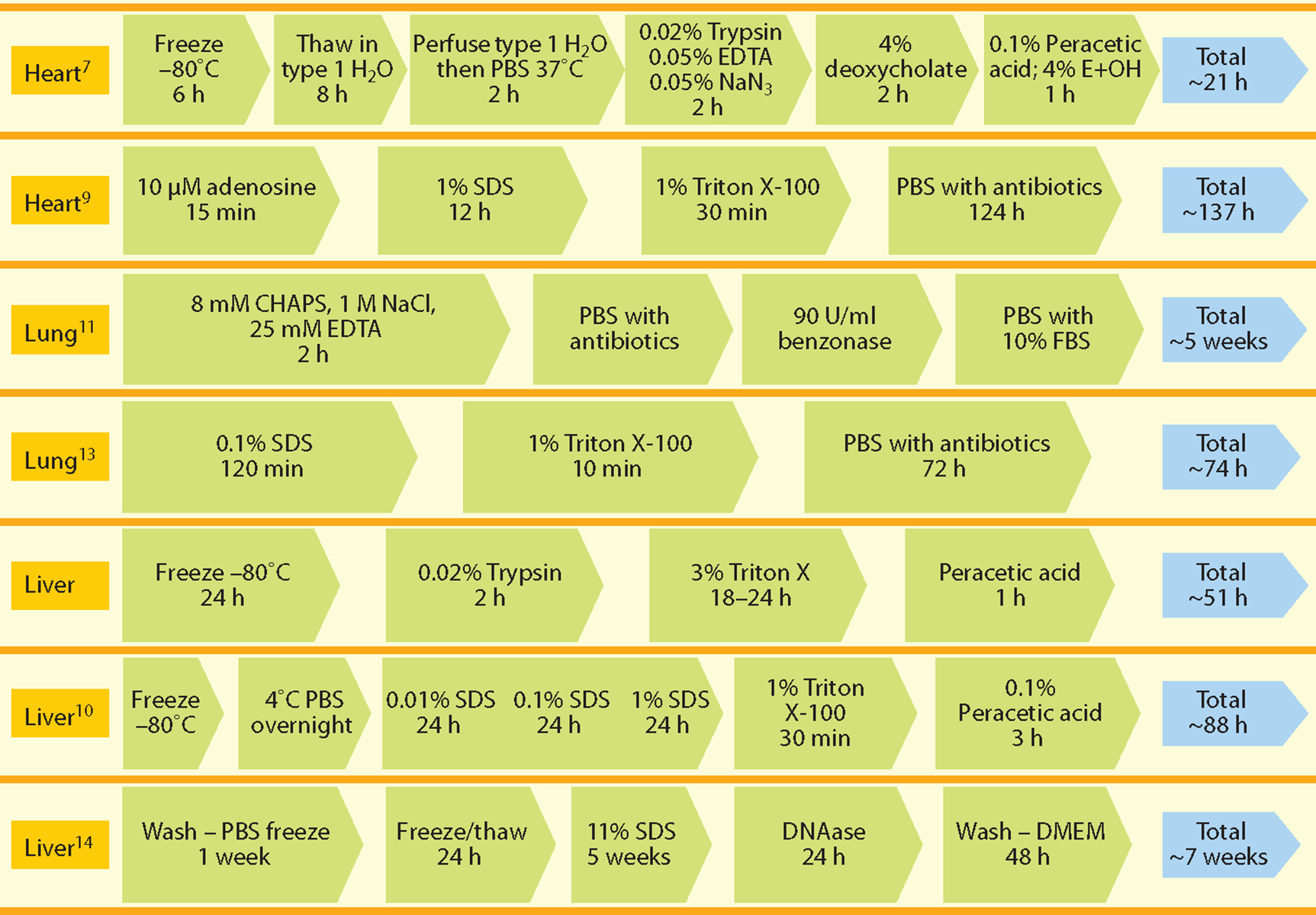
Reported perfusion protocols for decellularization of heart, liver, and lung. Abbreviations: DMEM, Dulbecco’s Modified Eagle’s Medium; EDTA, ethylene diamine tetraacetic acid; FBS, fetal bovine serum; PBS, phosphate buffered saline; SDS, sodium dodecyl sulfate.
SOURCE OF CELLS FOR WHOLE-ORGAN RECELLULARIZATION
As stated above, cells and ECM have an inherently close and dependent relationship. Therefore, the type of cells and source of cells used to repopulate an organ-specific three-dimensional ECM scaffold are critical to the eventual functionality and clinical success of the engineered construct. Engineering a complex tissue or organ requires rebuilding parenchyma, vasculature, and underlying support structures, all of which differ in cell number and cell type depending on the organ of interest. An ideal cell would be one that can proliferate or self-renew as needed and yet give rise to the heterogeneous types of cells necessary to form a functional organ or tissue. Except in isolated cases where specific organ-derived differentiated cell types can be isolated, expanded in vitro, and used, the most likely candidate to fulfill such demands is a stem or progenitor cell. The choices then become autologous versus allogeneic cells and adult versus ES or progenitor cell sources.
Autologous cells are self-derived and thus limit the potential for exposure to transmissible agents. They are less likely to be rejected or invoke an adverse immune response and thus decrease or eliminate the need for harsh immunosuppressive antirejection drug regimens. These advantages, in turn, lower the regulatory burden in first-in-human trials and appeal to patients. They also avoid the increased risk of infection and cancer and nonimmune toxic effects (64) of immunosuppression. Examples of autologous cell therapy include bone marrow cell delivery for treatment of acute illnesses such as blood-borne malignancies, acute disease states such as myocardial infarction, and chronic disorders such as osteoarthritis and multiple sclerosis (65). Unfortunately, in most organs (e.g., pancreas, lung, heart), autologous cells cannot easily be harvested or the numbers are often insufficient to be useful in a nascent tissue. Cell seeding depends on the cell type chosen and the ultimate goal. Obviously, reaching vasculature can be achieved more easily via perfusion, whereas parenchymal targets are more likely to require injection. That said, the choice of route, timing, and dose is as dependent on the organ target as it is on the cell type chosen.
Allogeneic cells, in contrast, are by definition not self-derived; however, they can often be harvested in larger quantities and from younger, healthier individuals and can be maintained or expanded in advance for an off-the-shelf use. In most regenerative-medicine applications, the decision of an autologous versus allogeneic cell source is based on criteria such as the following: (a) the number of required cells and the timeframe in which they are needed, (b) the ease of cell harvest and expansion, and (c) the ability to differentiate the needed cell types in vitro. For example, in the case of an acute injury where time is critical, an off-the-shelf cell-based product would be advantageous, whereas an autologous product that required weeks to prepare would be less useful. However, in nonemergent situations such as joint replacement where tissue-engineered constructs can be grown in vitro over weeks or months, autologous cells become a more viable option (Figures 3 and 4).
Figure 3.
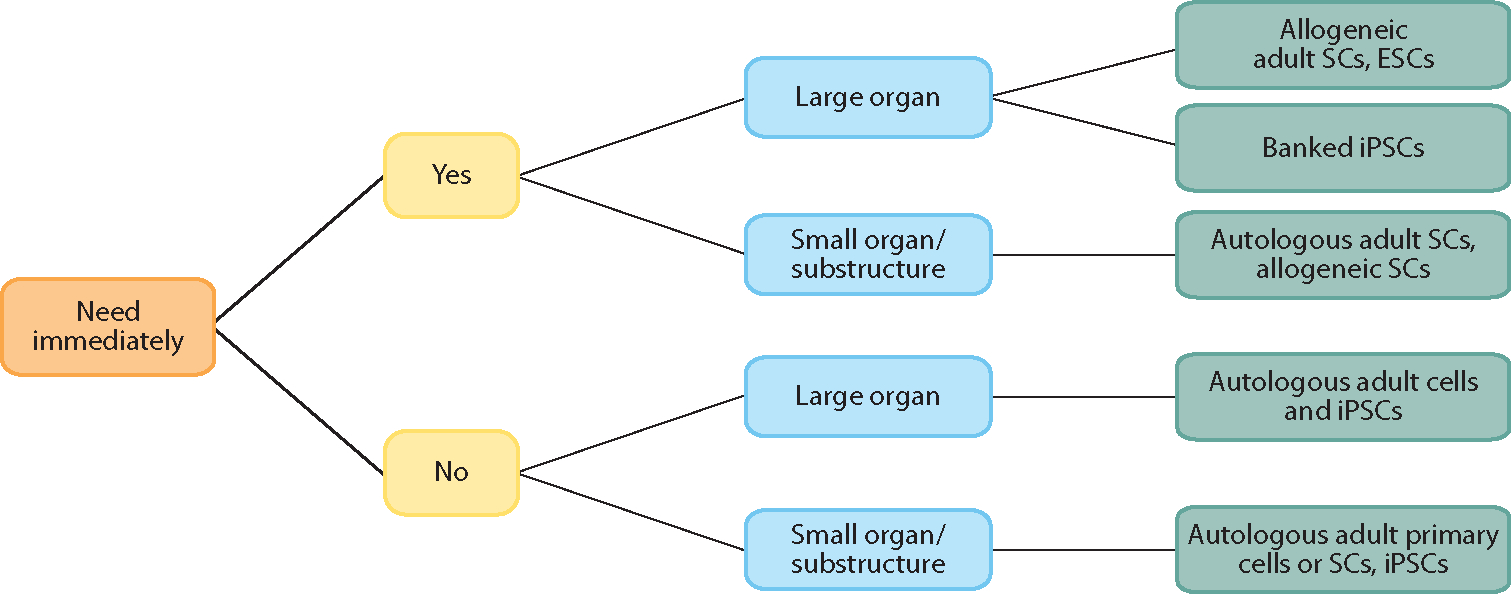
Example of a decision algorithm for selecting the type of parenchymal or stem cells to be used for the recellularization of a three-dimensional matrix scaffold. Abbreviations: ESC, embryonic stem cell; iPSC, inducible pluripotent stem cell; SC, stem cell.
Figure 4.
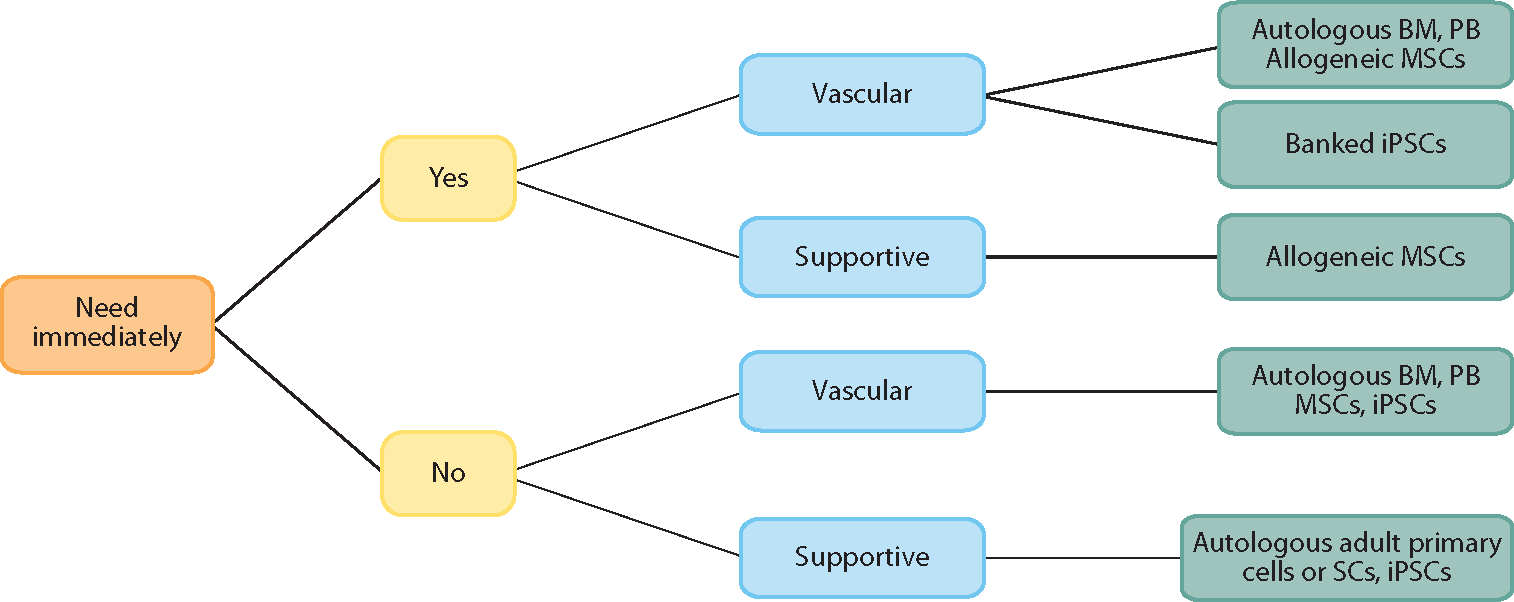
Example of a decision algorithm for selecting the type of nonparenchymal cell populations to be used in the reseeding of a three-dimensional matrix scaffold. Abbreviations: BM, bone marrow; iPSC, inducible pluripotent stem cell; MSC, marrow stromal cell; PB, peripheral blood.
From a business and engineering perspective, allogeneic cells have certain advantages. First, they can be isolated, expanded, characterized, and used to generate an off-the-shelf product that is available as needed. Such cells are typically proliferative, can be grown in large numbers, and can be readily modified in vitro with genes or other biologics. Allogeneic cells are often derived from a single cell source yielding a reproducible product with less lot-to-lot variability than autologous products. Finally, allogeneic cells are usually less expensive and take less time to produce than patient-specific cell products. Disadvantages of allogeneic cells include an increased risk of adventitial agent transmission, potential adverse immune reactions, and potential for widespread patient involvement should a product be contaminated. Examples of allogeneic cells used in regenerative medicine include mesenchymal bone marrow cells (for treatment of graft-versus-host disease) and allogeneic fibroblasts for cosmetic purposes, allogeneic bone marrow transplants (for almost 60 years) for blood disorders, and malignancies.
In the case of a highly regenerative organ such as liver, where organ biopsy is an option, autologous cells may be a feasible cell choice because cells can be harvested and at least minimally expanded in vitro. Unfortunately, in other organs, autologous cells cannot easily be harvested (e.g., pancreas, lung, heart) or the numbers are often insufficient to be useful in a nascent tissue. Furthermore, not all autologous cells can be expanded to large numbers in vitro either because they do not readily divide (cardiocytes, neurons) or because they are difficult to maintain in an undifferentiated proliferative state (e.g., hematopoietic bone marrow cells). For these reasons, allogeneic cells are often considered.
Regulatory issues, intellectual property constraints, and cost also impact cell choice. Autologous human cells are subject to regulation by the U.S. Food and Drug Administration if they are harvested and applied in a nonhomologous location (e.g., bone marrow cells applied in myocardium) or if they are maintained outside the body such that they are considered “manipulated.” The use of allogeneic cells is regulated in virtually all cases. In the past several years, many human stem or progenitor cell populations or the processes needed to isolate them have been patented, thus raising issues concerning the use of such cells in commercial applications. Tissue-based stem cell therapies to date have primarily been described with used autologous cell sources.
Cell Sources
To truly build an organ or tissue requires generating the specialized structures of that tissue as well as any vascular or ductal components and any supporting structures replete with cells, including populations of resident stem or progenitor cells for ongoing organ maintenance. For example, in the case of the heart, the desired cell population would include differentiated mature atrial ventricular and pacemaker myocardial cells; valvular, endocardial, and epicardial cells; arteries, veins, and capillaries; lymphatics; parasympathetic neurons; fibroblasts and myofibroblasts; and the more recently defined cardiac progenitor cell populations. Asking a single population of cells to fulfill these requirements is at present likely unreasonable. Fortunately, multiple cell choices exist.
Stem or progenitor cells utilized for most tissue-engineering approaches can be broadly categorized as ESCs, fetal cells, adult (including umbilical cord blood)-derived stem or progenitor cells, or, more recently, adult-derived inducible pluripotent stem cells (iPSCs), which have some of the properties of both adult and ESCs. Nonstem or progenitor cells used for organ engineering are usually parenchymal and supportive cells (e.g., fibroblasts) obtained from the organ of interest via biopsy or surgical harvest. Alternatively, the cell sources can include vascular cells obtained from easily accessible sources such as peripheral blood or bone marrow (e.g., endothelial cells).
Embryonic stem cells.
ESCs are pluripotent cells (i.e., can give rise to all three germ layers) derived from fertilized eggs, specifically from the inner cell mass of a blastocyst in vitro. As true stem cells, ESCs can be proliferated to large numbers without senescing. Because they are pluripotent, they can also be differentiated down virtually any cell lineage for tissue or organ engineering. Furthermore, because human ESCs are derived early after fertilization, they lack the epigenetic modifications often seen with other pluripotent stem cell populations, meaning that the genome is not yet modified by its environmental cues and thus presumably these cells are more “programmable” or responsive to their in vitro environment than more differentiated stem or progenitor cell populations. On the basis of these strengths, the first U.S. clinical study of human ESCs was initiated for therapeutic spinal cord injury in 2010 (65). To date, mouse and human ESCs have been used to engineer tissue constructs ranging from bone (66) to heart (67).
The primary disadvantages of human ESCs are the ethical debate surrounding their use, their allogeneic origin, and their propensity to give rise to teratomas if transplanted in an undifferentiated state (68, 69). Understanding the signaling cascades involved in stem-cell-fate decisions and controlling them to prevent tumorigenicity are active areas of investigation that must progress before human ESCs will be a viable target for engineering of an organ. Given these difficulties, many scientists have placed their interest in a more recently described cell type with many of the advantages of human ESCs but no ethical debate—human iPSCs (70).
Fetal cells.
Fetal stem cells are not a typical source for adult tissue engineering, but recently processes have been developed to derive human mesenchymal stem cells from fetal sources that can give rise to neuronal tissue (71) and to osteoblasts (72). The main advantage with fetal cells is that they retain their proliferative capacity while being committed to an endpoint, both rendering their differentiation easy and eliminating the formation of teratomas in vivo. Their source is controversial, however. Their extensive capability to expand in vitro may eliminate this issue, similar to ESCs; however, such in vitro–expanded fetal cells have not been tested extensively in vivo (72). As evidenced by their use in many of the papers on whole-organ tissue engineering to date, fetal cells at least provide a good starting point for preclinical studies (73).
A more accessible source of human fetal stem cells is amniotic fluid (74), which contains multiple stem or progenitor cell types and contains skin cells that can be reprogrammed to pluripotent stem cells (75). Recently methods have been developed to derive human amniotic fluid cell clones that retain the capacity to give rise to osteogenic, adipogenic, and neurogenic cell types (76). To date, amniotic fluid–derived stem cells have been used in many preclinical tissue-engineering strategies in the fields of urology (77), bone repair (78, 79), and airway repair (80) and have begun to be moved into clinical application. Given the apparent multipotency of these cells and with the advent of methods that allow for their easy derivation and expansion, the potential use of these cells in regenerative medicine (81) will likely only increase.
Inducible pluripotent stem cells.
In 2006, the stem-cell landscape changed when Yamanaka and colleagues (82) used retroviruses to express the transcription factors Oct 3/4, SOX2, c-myc, and Klf-4 in mouse fibroblasts and successfully reprogrammed the cells to yield pluripotent stem cells. Within one year, multiple groups had succeeded in reprogramming mouse fibroblasts (82–84), and within two years the production of human iPSCs was reported (70, 85).
The creation of iPSCs was heralded as changing the stem-cell landscape and eliminating the need for other cell types. But, as with all biology, the reality is less clear-cut. iPSCs overcome some of the limitations of ESCs—primarily the need for allogeneic sources (e.g., patient-specific cell lines have already been generated in several cases; for a review see Reference 86)—and do not suffer from the political constraints associated with ESCs. In addition, despite being autologous, iPSCs can be expanded to large numbers in vitro and thus would appear reasonable as a cell source for tissue-engineering applications. Indeed, iPSCs can give rise to complex tissue cell types in vitro and to chimeric animals and complex tissues in vivo.
iPSCs have at least one major disadvantage compared with their younger counterparts—epigenetic modifications of the DNA (that may have been present in the original starting cell population or arise during the transformation process), which may alter the safety profile and responsivity of the resulting cells (84). Additionally, by definition, iPSCs require transformation from a terminally differentiated state to a less differentiated and more proliferative phenotype. Thus, as with ESCs, they give rise to teratomas in vivo if transplanted in an undifferentiated state.
In summary, human iPSCs may become an ideal source for tissue engineering because they can be derived in an autologous fashion, can be grown to large numbers in vitro, and give rise to both parenchymal and supportive cells needed for complex tissue formation. Yet, similar to ESCs, they are not yet fully controllable in vitro or in vivo; thus mechanisms to control cell proliferation and phenotype will be critical to moving this cell population forward. Even so, iPSCs have recently been used in conjunction with artificial scaffolds to generate multiple tissues including blood vessels (87), airway tissues (80), and liver (88).
Adult-derived stem/progenitor cells.
Adult-derived stem or progenitor cells are those derived from any tissue after birth (including bone marrow, blood, and umbilical cord) and differ from ESCs in that they are more restricted in their proliferation and differentiation potential and cannot spontaneously give rise to complex tissues in vitro. In the field of regenerative medicine, these cells have usually been derived from biopsy material from a number of tissues including bone marrow (89–94), peripheral blood, liver (95), lung (96, 97), skeletal muscle (98), heart (99), and adipose tissue (100).
The most common source of adult stem cells is bone marrow; these cells are being used in more than 600 clinical trials at present (http://clinicaltrials.gov/ct2/results?term=bone+marrow+stem+cells&recr=Open) in diseases ranging from acute leukemia to myocardial infarction and heart failure as well as in chronic illnesses such as multiple sclerosis and osteoarthritis. In the field of tissue engineering, bone marrow has primarily been used as a source of stromal cells (89), which are mesodermal in origin and can give rise to most mesodermal phenotypes including bone (90, 91) and muscle (93) or as a source for vascular endothelial precursors, mature endothelial cells, or smooth muscle cells for rebuilding vessel structures. More recently, similar mesenchymal cells have been derived from adipose tissue (97, 101), periosteum (102), and ESCs (94, 103), thereby providing ever expanding sources for mesodermal progenitors.
Umbilical Cord Blood Cells
Recently, umbilical cord blood cells have provided a rich source of stem or progenitor cells that has been used in vivo for correction of genetic abnormalities (104) and for bone marrow transplant (105). These cells are derived from placental blood shortly after birth and are of fetal rather than maternal origin. Umbilical cord blood cells have several advantages over allogeneic bone marrow: They appear to be more primitive and thus have more potential (106) to give rise to diverse cell types needed in organ engineering. In addition, they can be transplanted under more of a genetic mismatch than can human bone-marrow-derived mononuclear cells (107).
Primary Tissue or Organ-Derived Cells
Rebuilding an organ requires both parenchymal and nonparenchymal cell sources. Ideally both would be derived from nonimmunogenic sources and would give rise to mature functioning tissue. Thus, a simple source of cells to rebuild any given organ would seem to be that actual organ. However, most adult organs either do not contain cells that are sufficiently proliferative to generate the numbers of cells needed for rebuilding the whole structure or do not contain cells that can be easily harvested.
Organ-Derived Progenitors
Organ-derived stem or progenitor cells are another choice of autologous adult cells that can be expanded and used for organogenesis in vitro. The major progenitors relevant to these discussions are cells derived from heart (99), lung (96), liver (95), pancreas (108), and adipose tissue (97, 101). Bone marrow (89–94), mononuclear, mesenchymal, as well as umbilical cord blood (106) cells will also likely continue to be potent examples of these adult-derived progenitors with a role in organogenesis. However, all these cells, whether autologous or allogeneic, must be expanded to provide the numbers of cells needed for organogenesis and thus are not considered primary cells.
An exception to this requirement for cell expansion prior to the use of autologous cells may be the use of primary bone marrow or peripheral blood to derive vascular or other supportive cell components for complex organs. Bone marrow and peripheral blood are easily accessible and contain large numbers of stem or progenitor cells that can be proliferated in vitro. For this reason they are utilized in a number of clinical studies for endogenous cell therapy but also could provide a source of cells for rebuilding the vascular network in nascent organs. Re-lining of organ vessels with an autologous endothelial cell population to minimize exposure of the recipient’s blood to foreign proteins after transplant is particularly desirable. Furthermore, as bone marrow and blood continue to be good sources of stem or progenitor cells, their use for deriving parenchymal cell components in liver (92, 94), lung (96), and heart has been demonstrated and will likely continue.
Cell-Specific Hurdles in Organ Engineering
The hurdles associated with obtaining large numbers of well-defined, controllable cells are not specific to organ or tissue engineering with biologic scaffolds. The existence of whole-organ biologic scaffolds makes the possibility of whole-organ engineering feasible but in turn raises questions: Can billions of cells can be grown with fidelity? Can they be differentiated appropriately? Can they be delivered to appropriate locales? Finally, can they be induced to survive, mature, and function properly? ESCs and iPSCs offer the potential for large-scale cell proliferation, but they are limited by an inability to control cell fate and benign tumor formation if undifferentiated cells are delivered. Scientific efforts are under way worldwide to define the appropriate differentiation and purification profiles to overcome these limitations. Adult-derived cells have a more limited proliferation and differentiation profile but can often be derived in an autologous fashion. For smaller engineering problems such as organ substructures, adult-derived stem cells may be ideal; however, the need to produce large numbers of cells in a defined time period will likely limit their use. For any autologous cell product, the costs of patient-specific cell processing will have to be considered, but patient acceptance of an autologous product is likely to be greater than for an identical allogeneic product. Both allogeneic and autologous cell-based tissue-engineered constructs are being evaluated in human clinical studies for repair of cartilage or bone defects, as skin substitutes, for vascular repair, or for regeneration of periodontal tissue. In a recent case report, autologous bone marrow cells were seeded onto a decellularized tracheal matrix and transplanted as a tracheal substitute in a young female patient (109). The successful use of cells to construct these simpler tissues suggests that tissue-engineered complex tissues and organs are now within reach.
METHODS OF RECELLULARIZATION
The methods employed in recellularization of whole-organ scaffolds are typically adaptations of techniques from a wide range of procedures including traditional cell culture, tissue-engineering methods, cell-transplantation therapies, and isolated-organ perfusion. The recellularization process can be considered in two major steps: cell seeding where the goal is redistribution of cells similar to their in vivo spatial configuration, followed by perfusion culture, which is typically utilized to prepare the cells for in vivo function.
Cell Seeding
The first challenge in recellularization of decellularized whole-organ scaffolds is its repopulation with an appropriate mixture and number of cells, and placement of these cells to necessary niches within the scaffold to match the native distribution as much as possible.
Cell types.
The types and numbers of cells necessary for recellularization vary significantly with the organ. The parenchymal cells, the cell types responsible for the specific functions of the organ, are of obvious necessity, e.g., hepatocytes in liver, cardiomyocytes in heart, epithelium in lung, etc. In addition, nonparenchymal cells such as fibroblasts and endothelial cells enhance the functional phenotype of the parenchymal cells and contribute to the organization of the cellular architecture of the tissue (110–113). Of primary interest and commonly utilized in tissue engineering are endothelial cells and fibroblasts. Endothelial cells are necessary to provide a nonthrombotic barrier for the decellularized organ matrix and assure that blood flow in vivo is confined to the vascular spaces and the parenchymal cells are protected from the shear stress created by the flow (114, 115). Thrombosis can be a major challenge with decellularized three-dimensional ECM scaffolds. Thrombosis has been a relatively manageable issue with decellularized heart valves after transplantation in vivo where only the outer surface of the structure is a blood interface (116, 117). However, extensive antithrombotic medication has been required for the three-dimensional liver scaffold (10). It should be noted that a major advantage of whole-organ scaffolds is the presence of intact blood vessel networks, but full utilization of this vascular system to direct flow within the tissue in vivo requires proper endothelialization. Functional long-term endothelialization is yet to be demonstrated, although initial attempts have been promising in showing endothelial cell attachment (9, 10). Strategies to improve the endothelialization of three-dimensional organ constructs are under investigation (118). Moreover, re-endothelialization can occur naturally in vivo after transplantation of decellularized tissues such as the skin (119, 120), so it is plausible than initial endothelialization need be present only long enough for in vivo re-endothelialization to occur.
Another important nonparenchymal cell type in tissue engineering is the fibroblast, which secretes and remodels the ECM and improves parenchymal cell functions in cocultures. For instance, coculture of hepatocytes with fibroblasts or endothelial cells significantly increases liver-specific functions of the individual cell types (121, 122). Recently, it was shown that fibroblasts are also involved in the electrical properties of the myocardium by coupling with the cardiomyocytes and aiding in the propagation of the electrical stimuli over longer distances (123, 124). Although fibroblasts are recruited as necessary in vivo, they may also prove important during in vitro organ culture to allow or facilitate remodeling and cellular organization.
Depending on the organ, other cell types are also important, if not necessary, for functional recellularization. To date, the increased complexity of dealing with multiple cell types has limited coculture studies mostly to the use of endothelial cells and fibroblasts. Dealing with a mixture of cells is a combinatorial problem, and as in the case of tissue-engineered bladders, serendipity may be necessary to find the proper seeding and culture protocol (125).
Cell numbers.
The number of cells initially required for seeding the decellularized matrix is a design parameter that is dependent on the organ type. For instance, the biomechanical duties of the heart and lungs require a functional whole organ at the time of implantation. These duties also likely require a high percentage of original cell types and numbers to be immediately present. By contrast, organs with primarily metabolic functions, such as the liver and pancreas, can be deployable with only a small percentage of their native cell mass. The liver of an adult (70 kg) is estimated to contain approximately 2.8 × 1011 hepatocytes, or 4 × 109 cells per kilogram of body weight (126). Although the cell mass required to support an animal model of hepatic failure has not been systematically determined, clinical improvement has been seen in hepatocyte transplantation studies using less than 10% of the host’s liver mass. Intrasplenic transplantation of 2.5 × 107 allogeneic rat hepatocytes (~5% of the liver mass in this model) prolonged the survival and improved blood chemistry of anhepatic rats (127), and similar small successes have been possible with transplantation of 1–10% of the native liver mass in rats (128) and humans (129). Assuming that the minimum cell mass necessary to support a patient with acute liver failure is approximately 5–10% of the total liver weight sets the requirement to approximately 1010 cells for humans and approximately 50–100 × 106 cells for the rat model. Well over 20% repopulation of liver mass with ~50% of actual packing density has been reported (10).
In contrast, the heart is a dense organ with approximately 108 cardiomyocytes/cm3 (114). To achieve a fully functional whole heart, the three-dimensional heart matrix needs to be nearly fully recellularized at the time of implantation. Given that these numbers are quite large for stem/progenitor-cell-based techniques and may raise practicality issues as discussed earlier, an important question is whether to use such a large number of fully mature cells or instead start with seeding a small number of proliferative cells and allow them to expand inside the matrix during in vitro culture. A combination of the above approaches may prove to be the answer. The use of larger numbers of stem and progenitor cells may also require long-term (i.e., weeks) organ culture for proliferation and maturation processes to complete.
Seeding strategies.
Seeding techniques currently employed in recellularization of whole-organ grafts are essentially adaptations of the approaches employed in cell-transplantation therapies, where cells are either injected directly into the organ or injected into the circulation with the expectation that the cells will home to the injury site. These techniques include intramural injection of cells (9) or infusion of cells into the vasculature followed by continuous perfusion (10, 11). In the report for whole-rat heart recellularization (9), recellularization occurred with 50–75 × 106 neonatal cardiac cells delivered in five injections of 200 μl into the anterior left ventricle with a seeding efficiency of approximately 50%. This approach resulted in approximately 34% of recellularization proximal to the injection sites with decreasing percentages distally. Furthermore, 2 × 107 endothelial cells were placed by direct infusion into the aorta, which yielded 550.7 ± 99 cells per mm2 on the endocardial surface and 264.8 ± 49.2 cells per mm2 within the vascular tree after 7 days of perfusion culture. Through the use of a continuous perfusion seeding approach, liver recellularization with 50–200 × 106 hepatocytes was demonstrated (10). Hepatocytes were infused into the portal vasculature in four separate injections, which was followed by continuous perfusion. The number of cells used in this recellularization process corresponds to 5–20% of the rat-liver cell mass with approximately 50% recellularization of the organ matrix as only some of the liver lobes were used. The seeding efficiency was more than 95%. Comparison of multiple injections versus bolus seeding in the liver by histology demonstrated that low-dose multiple seeding followed by continuous perfusion resulted in a more uniform distribution of cells throughout the matrix with fewer blockages of the vessels (10). The perfusion seeding strategy was also used for rat-lung recellularization (11). One hundred million neonatal epithelial cells were seeded into the airway compartment followed by an overnight static culture to promote adherence. Lungs were then perfused through the vasculature or ventilated through the airways. Thirty million endothelial cells were seeded via injection into the vasculature. Ventilation during administration of cells enabled diffuse endothelial cell seeding. The seeding regimen resulted in satisfactory coverage of the decellularized matrix. A similar approach by Ott et al. (13) resulted in a parenchymal fractional volume as well as number, size, and fractional volume of alveoli equal or similar to values in native lungs. Table 1 provides some pros and cons of direct injection versus perfusion recellularization strategies.
Table 1.
Alternatives in parenchymal cell-seeding strategies
| Site | Injection | Perfusion |
|---|---|---|
| Local | Global | |
| Delivery into parenchyma | Easy to control seeding site; easy to avoid parenchymal cells in vascular spaces | Requires that the cells are pushed into the parenchyma by flow; potentially damaging |
| Ease of distribution | Multiple injections necessary | Simple; reported to enable cell repopulation similar to native tissue (13) |
| Potential cell injury | High cell density at injection site, potentially leading to aggregate formation and necrotic cores | Continuous exposure to shear during seeding |
As whole-organ engineering of vascularized tissues represents a new approach, the optimal seeding method has yet to be determined. Limited initial experience indicates perfusion seeding is preferable owing to enhanced distribution as well as practicality if the objective is full-organ matrix repopulation. However, the prolonged exposure to shear within the perfusion system may prove harmful. However, if progenitor cells rather than mature cells are infused and they are subsequently induced to expand and mature within the grafts, the need for perfect and complete seeding may not be necessary.
In addition to injection versus perfusion and number of doses, another variable to be determined in optimization of seeding strategies is cell concentration. For the liver, high concentrations of cells could lead to extensive cell death or occlusion of the vascular spaces (10). Evaluation of alternate delivery routes for the cells (venous versus arterial, direct versus retrograde, etc.) remains to be done. Similar optimizations are also likely to be necessary for the nonparenchymal cells seeded in decellularized organ scaffolds. The approach has been sequential for the liver (10), whereas simultaneous dual circuit perfusion has been employed for lungs with vascular perfusion for endothelium and trachea for epithelium (11, 13). To date, there are no studies comparing alternative approaches in terms of seeding efficiency to provide optional coverage. Such studies are a necessary step in development of the techniques in whole-organ engineering.
Culture System Characteristics
Depending on whether the strategy is to achieve full repopulation in vitro or in vivo, the culture conditions for an engineered whole organ may need to support cell survival and function for as long as several weeks to promote engraftment and rearrangement. In vitro, endothelialization of decellularized blood vessels has been shown to take two weeks (130); hence, two weeks in vitro culture is a reasonable initial estimate. If stem/progenitor cells are used as the starting population, with expansion and maturation to be performed within the graft, more than one month may be necessary.
Although the cellular compartment of an engineered whole organ is expected not to be as complex and dense as the native organ, capturing only the minimum components necessary, the required size or mass of the construct to provide clinically relevant function is still quite large. As such, delivery of nutrients to the cells inside the scaffold and removal of harmful products from within remain challenging tasks. Indeed, the delivery/removal problem has been a major bottleneck in development of vascularized tissues in vitro as well as their survival in vivo (131). The need for a constant supply of nutrients renders perfusion the in vitro culture method of choice for all highly vascular organs. Perfusion is achieved via the use of bioreactors (132–134) as well as suitable perfusion medium, potentially an oxygen carrier, and, for some organs, a biophysical stimulation.
Bioreactor.
Whole-organ culture systems are based on extracorporeal organ perfusion devices. Normothermic machine perfusion, also called organ culture, has been used as an isolated organ model for nearly a century (135), and it has been used as a preservation modality prior to the invention of cold storage solutions (136). This method is receiving renewed interest as a strategy to expand the donor pool by reanimating the marginal donors (137).
Although the presence of existing systems creates an easily accessible blueprint for system design, in addition to providing availability of all the necessary components from mouse to human scale, by no means does it offer a readily available perfect solution. As a preservation modality, machine perfusion of highly vascular organs is limited in duration, with reports of successful preservation (established by transplantation) ranging from a few hours for the heart (138), to 12 h for the lung (139), to 20 h for the liver (140). Early reports in recellularized whole-organ scaffolds report successful preservation on the order of weeks [four weeks for the heart (9)], although these recellularized organs do not contain the entire cellular mass and have not been required to replace native organ function. It is unknown whether existing bioreactor systems are capable of maintaining a fully recellularized three-dimensional organ scaffold. Moreover, in culture conditions some adult cells do dedifferentiate; for instance, dedifferentiation occurs within a few weeks for hepatocytes (141), creating another potential time limitation. It is likely, though not proven, that coculture in the native three-dimensional ECM scaffold will reduce or at least delay such issues.
While various temperature ranges are being investigated for organ preservation, the ideal temperature for recellularized three-dimensional ECM organ scaffolds is likely to be in the physiological (also referred to as normothermic) or near physiological temperatures [32°C has been tested for kidney perfusion (142)] to ensure that the cells are able to attach and self-assemble. Physiological temperatures are obviously the first place to start, unless proven otherwise. Culture within an incubator (10, 12) or a water-jacketed medium reservoir (9, 13) have both been used to achieve temperature regulation.
The core of any perfusion system is a mechanical power source to drive the media through the graft. For organ-perfusion systems, common methods are centrifugal and positive-displacement pumps, although a ventilator has been used for the lung recellularization system (11). An important note here is that, whereas continuous perfusion has been used without any issues, endothelial cells prefer pulsatile flow (15). Full endothelialization of a three-dimensional scaffold may require pulsatile flow and careful consideration of the flow rate. Near physiological flow rates/pressures are the common and logical first guess (10–12). However, parenchymal cells are typically protected from shear stresses by the endothelium. Some cells, especially hepatocytes, are very fragile and susceptible to shear stress; hence, in the absence of this protective layer, flow well below in vivo values may be necessary. For hepatocytes, a shear stress of 0.23 dynes/cm2 has been noted to correspond to high viability (143), and this value is probably a good conservative estimate for all other cell types. However, flow within a decellularized organ, which is a sponge-like structure, may easily become turbulent; hence, even with low flow rates, some cells may be subjected to high local shear stresses. Flow and shear analysis within a decellularized graft has not been elucidated beyond confirming that significant flow occurs through the vascular spaces (10). Hence, if significant cell death is observed early during culture, shear is a very likely cause.
The other component of a bioreactor system that is likely to be necessary is an oxygenator to ensure sufficient oxygen presence, as discussed below in detail. Another component that has been used in isolated organ perfusion settings is a dialyzer. The dialyzer is necessary to maintain transplantability of perfused livers (144); its duties are likely the elimination of toxic by-products such as bilirubin from the perfusion circuit. Whether such additional units are necessary in three-dimensional cellularized scaffold systems remains to be determined. Table 2 compares alternative bioreactor approaches.
Table 2.
Bioreactor operational alternatives
| Alternative 1 | Alternative 2 | |
|---|---|---|
| Temperature | Physiological (9–13) | Subnormothermic (32°C) (142) |
| Flow regime | Pulsatile (9, 15) | Continuous (10) |
| Flow rate | Physiological (9–13) | Minimum needed for sufficient nutrient delivery (exact values not determined) |
| Shear | Unregulated; shear observed at physiological flow rates within grafts does not appear to induce major cell death (143) | 0.23 dynes/cm2 suggested for hepatocyte viability (9–13) |
| Perfusate | Cell culture based | Growth factor supplementation may enhance rearrangement |
| Oxygenation | Oxygenated media (9–11, 13) | Ventilator for lungs (12) |
| Oxygen carrier | Erythrocytes (144) | Hemoglobin based (149); note that none of the whole-organ recellularization studies so far has required an oxygen carrier, although in normothermic organ perfusion it is absolutely necessary (144) |
| Organ culture duration | Approximately two weeks is necessary for endothelium formation (130) | Up to four weeks has been reported for the heart (9) |
| Biophysical stimuli | Ventilation for lungs (11–13), electrical for heart (9), shear for endothelial realignment (118) |
Perfusate.
The perfusate used in the culture of engineered whole organs has been chosen on the basis of the culture media used in cell culture of the constituent cell types (9–12). Although this approach has generally succeeded in achieving the primary goal of viable organ culture in the order of weeks, it is unknown if the selections are optimal. For example, hepatocyte culture media (e.g., C+H and Willams E) have been in use for ~30 years with few changes and are considered the de facto gold standards. However, with these media the cells typically start to dedifferentiate after a few weeks and lose their hepatocyte-specific functions such as cytochrome P450 (CYP450) activities (141). Furthermore, the media have nonphysiological levels of some supplements (e.g., insulin is ~1000-fold higher than in vivo in C+H), and the impact of these artificial levels when the cells are transplanted is little known. Because preconditioning in hyperphysiological levels of insulin can cause steatosis in cultured hepatocytes when exposed to plasma, their use in bioartificial reactors is problematic (145), and similar problems may arise when the recellularized grafts are transplanted if they are improperly conditioned.
A unique factor in whole-organ scaffolds is the presence of ECM, which, as mentioned above, is likely to provide additional factors to support cell function and viability (146). Investigating the impact of these ECM components on the cells, e.g., cell-cell interactions between different cell types and their rearrangement, remains a little investigated area that could offer significant scientific insights for advancing tissue engineering.
Oxygen delivery.
Poor oxygen delivery is probably the single most important reason that tissue engineering of the highly vascularized, highly metabolically active organs has not been successful so far (131). Traditional tissue-engineering techniques have failed to provide tools to support the growth of thick, dense layers of tissue in vitro and/or in vivo because the oxygen delivery is usually limited by diffusion. The metabolic demand for oxygen is 27.6 nmol/[mg protein.min] for cardiomyocytes (147) and 18 nmol/[mg protein.min] for hepatocytes (148), both of which cannot be met by diffusional supply of oxygen across thick tissues. The whole-organ scaffolds with their nature-built vascular networks are ideal for engineering such tissues because they avoid the diffusion limits by perfusion, enabling preparation of thick and dense tissue constructs. However, in the absence of capillaries, it may still be necessary to supply oxygen at levels higher than the atmospheric saturation levels in the perfusate to support the survival of the cells. To achieve this, the basic approach involves gassing the perfusion medium with an oxygen/carbon dioxide mixture in a membrane-type oxygenator that is part of the bioreactor system. The gassing of the perfusion fluid with 95% oxygen and 5% carbon dioxide (carbon dioxide is necessary to maintain the pH of the perfusate) mixture results in oxygen partial pressure of approximately 360 mm Hg (10). Should higher oxygen-carrying capacity be required owing to high numbers of cells, oxygen carriers, e.g., perfluorocarbons (149), red blood cells with a hematocrit around 20% (144), or whole blood (150) can be utilized; the most ideal solution likely is red blood cells because artificial oxygen carriers tend to get oxidized quickly (149) and whole blood introduces immune components that, if activated, may initiate a systemic inflammatory response (151).
Biophysical stimuli.
In vitro conditioning of cells, especially maturation of progenitor-type cells, requires exposure to biophysical stimuli in a bioreactor setting mimicking the in vivo environment during development. For example, it is known that exposure of endothelial cells to shear stresses created by fluid flow stimulates the alignment of the cells in the direction of the flow similar to its in vivo arrangement (118). In addition, functional myocardium is known for its ability to propagate electrical impulses and to respond to these impulses by synchronized contractions that generate forces for pumping blood. Cardiac constructs engineered under electrical stimuli or mechanical stretch in vitro exhibited propagation velocities similar to those observed in the native heart and responded to external electrical stimuli by synchronized contractions (9, 152, 153). During development of lung tissue, mechanical stretch induced by breathing is an important factor in the differentiation of the epithelium, and intermittent ventilation of the engineered lungs with air has beneficial effects such as survival of distal alveoli, clearance of the epithelial secretions from the airway tree, and proliferation and differentiation of the epithelium (11). Therefore, the bioreactor design for engineering whole organs necessitates the inclusion of elements that mimic the biophysical environment in vivo to induce maturation and function of the tissue.
Endpoints
Several examples of engineered simple organ constructs have emerged in the past several years. Recently, more complex organs, notably heart (9), liver (10), and lung (12), have been engineered from decellularized organ matrices and animal cells. These studies provide early insights not only into the power of the technology, but also into the functional endpoints that may be achievable. If organs are to be truly capable of rescue or repair, they must function as a physiologic enhancement or replacement of the damaged tissue. With this in mind, the types of endpoints that may be required of a given organ depend on its function. Simple endpoints achieved in proof-of-principle studies and more advanced endpoints that will be expected of organs generated in vitro are summarized in Table 3. Essentially, it will be necessary to show that organs created in the laboratory can survive for the extended periods needed (years in the case of a chronic disease), meet the functional demands of that individual either via transplant or extracorporeal use, and demonstrate that the nascent organ does not contribute to disease progression. These demands are significant, but based on early data, success appears feasible.
Table 3.
Endpoints for tissue-engineered whole organs
| Organ | Simplest cells | Minimal function | Advanced endpoints |
|---|---|---|---|
| Heart | Cardiocytes | Contractility | Pumping against afterload, drug responsivity, conduction systems, pacemaker activity, working valves |
| Liver | Hepatocytes | Albumin, urea production | Drug metabolism, metabolic regulation, coagulation factor synthesis |
| Kidney | Renal medulla cells | Urine production | Blood filtration, renin angiotensin system, blood pressure maintenance |
| Lung | Alveolar type 2 cells | O2 exchange | Respiration |
| Pancreas | Islet a,p-cells | Insulin/glucagon secretion | Pancreatic digestive enzymes, endocrine hormone production |
WHOLE-ORGAN TISSUE ENGINEERING: NEXT STEPS
There is much cause for excitement about having a scaffold that has intact vascular spaces and perfect material composition and biocompatibility, in particular for tissue engineering of highly vascular organs. However, none of the whole-organ grafts produced to date have been used to replace or support function in vivo for more than a few hours, nor have they been expected to cure a diseased animal. In vitro function was demonstrated extensively (9–13), including a whole blood reperfusion as a surrogate model of short-term transplantation (10). The next milestone to be reached is in vivo graft viability and function of mid-term (days to weeks) and long-term (months) in healthy animal models, assessed via maintained structural integrity of the matrix, healthy remodeling by the body, viability of the cells, as well as other appropriate organ-specific functions. Assuming success, the next step will be success in various animal models of organ failure.
To create in vivo functional grafts, a recellularized graft must be viable in vivo. More than two decades of experience in tissue engineering indicate this goal is not so easily achieved (154, 155). Perhaps the most appropriate insights about the challenges ahead come from skin grafts constructed of decellularized human skin, such as Alloderm, which has been in use for two decades and has been immensely successful (156). Skin is a relatively simple tissue that is essentially a two-dimensional layer with no complex microarchitecture as, for instance, the liver sinusoids. Furthermore, the ease of access makes it easy to assess engraftment and viability and to replace the graft if necessary. However, a skin graft typically involves extensive scarring that may significantly decrease quality of life for survivors (157). The blood-vessel ingrowth from the host may take up to several days to weeks to vascularize sufficiently and perfuse an acellular graft (158), during which scar-tissue formation creates large patches of scars between areas that are properly perfused and heal (157). Approaches to slow scar formation while enhancing progenitor recruitment, for instance via growth factors, are ongoing, although no major breakthroughs have happened at the clinical level (157). Stated differently, vascularization has been a rate-limiting step for tissue engineering. A nutrient delivery system has to be in place such that the healthy regenerative response can outpace scar formation.
For organs such as heart, liver, lungs, or pancreas, extensive fibrosis would mean failure, especially if the scar tissue further inhibits vascularization. Moreover, because abdominal organ grafts are not as easy to observe, remove, or replace as are skin grafts, probabilities for loss of graft, and possibly the patient, are non-negligible. Indeed, all the initial studies have limited the in vivo observation of the transplanted organs to a few hours, well short of any fibrotic activity. Therefore, a major question for whole-organ tissue engineering is the in vivo outcome. This question involves multiple components that include the following concerns: (a) What happens to the decellularized organ matrix in the absence of recellularization; is there extensive fibrosis or is it readily remodeled by the body? (b) What percentage of the cells placed in vitro survive? (c) Is there significant recruitment of progenitor cells from the body as well as repopulation of the organ in vivo?
The last question brings forth an interesting, if controversial, train of thought: If remodeling and repopulation of the organ are possible in vivo, then it is possible to avoid trying to regrow the cells and original architecture in bioreactors and instead simply transplant the acellular organ scaffold (or in certain cases just a part of it, such as one lobe of the liver) in the patient and let it grow with the patient’s own cells, thereby also avoiding any immunological problems. Although heterotopic transplantation is not a commonly considered approach for heart or lung, and indeed this scenario may not be feasible for these organs, liver and pancreas cells are transplanted in alternate sites clinically (159, 160), which provides a feasible approach. Of course, each tissue is different, and actual experimental evaluations of such alternatives remain as targets for exploration.
The source of the organs for decellularization is also an issue. Use of porcine organs would vastly simplify logistics, enable precise quality control for obtaining decellularized grafts, avoid spread of human viruses, and indeed appears to be the chosen approach for some tissues. Even though any xenogeneic material creates the potential for immune complications, few have been observed thus far (161). Perhaps surprisingly, human organs for decellularization are widely available, because only the organs in near-pristine condition are used for transplantation. The number of donor organs that are unrecovered, for instance, those available as donors after cardiac death, is estimated to vastly exceed the demand for transplantation (162, 163). However, adult human organs are often somewhat damaged, and their recovery comes along with a plethora of ethical questions. Still, it is fortunate that alternate options exist, and determination of the best approach can be done when investigating clinical applications.
Whole-organ tissue engineering is a potential breakthrough in vascularized tissue engineering, with an ability to leapfrog the current approaches with synthetic biomaterials and their associated obstacles. The number of landmark papers demonstrating feasibility in various organs since the first paper on heart was published underlines the enthusiasm. However, it is important not to claim victory prematurely and create overoptimistic expectations until indisputable success in animal models of organ failure is demonstrated.
ACKNOWLEDGMENTS
K.U. thanks Basak Uygun for discussions and refinements regarding this review; his work on this topic is supported by grants from the National Institutes of Health (R00-DK080942), the National Science Foundation (CBET-0853569), and the Shriners Hospitals for Children. This work has been supported in part by The Medtronic Foundation, the National Heart Lung and Blood Institute’s Progenitor Cell Biology Consortium #1U01HL100407-01, and the American Heart Association’s Jon Holden DeHaan Cardiac Myogenesis Research Center #AHA0970499N.
Glossary
- ECM
extracellular matrix
- ESC
embryonic stem cell
- iPSC
inducible pluripotent stem cell
Footnotes
DISCLOSURE STATEMENT
Dr. Taylor holds a financial interest in Miromatrix, Inc., and is entitled to sales royalties through the University of Minnesota for products related to the research described in this paper. This relationship has been reviewed and managed by the University in accordance with its conflict of interest policies.
LITERATURE CITED
- 1.United States Renal Data System (USRDS). 2009. http://www.usrds.org/ [PubMed]
- 2.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, et al. 2010. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121:e46–e215 [DOI] [PubMed] [Google Scholar]
- 3.Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B. 2009. Deaths: final data for 2006. Natl. Vital Stat. Rep. 57:1–134 [PubMed] [Google Scholar]
- 4.Shin’oka T, Imai Y, Ikada Y. 2001. Transplantation of a tissue-engineered pulmonary artery. N. Engl. J. Med. 344:532–33 [DOI] [PubMed] [Google Scholar]
- 5.L’Heureux N, McAllister TN, de la Fuente LM. 2007. Tissue-engineered blood vessel for adult arterial revascularization. N. Engl. J. Med. 357:1451–53 [DOI] [PubMed] [Google Scholar]
- 6.Atala A, Bauer SB, Soker S, Yoo JJ, Retik AB. 2006. Tissue-engineered autologous bladders for patients needing cystoplasty. Lancet 367:1241–46 [DOI] [PubMed] [Google Scholar]
- 7.Macchiarini P, Jungebluth P, Go T, Asnaghi MA, Rees LE, et al. 2008. Clinical transplantation of a tissue-engineered airway. Lancet 372:2023–30 [DOI] [PubMed] [Google Scholar]
- 8.Macchiarini P, Walles T, Biancosino C, Mertsching H. 2004. First human transplantation of a bioengineered airway tissue. J. Thorac. Cardiovasc. Surg. 128:638–41 [DOI] [PubMed] [Google Scholar]
- 9.Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, et al. 2008. Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart. Nat. Med. 14:213–21 [DOI] [PubMed] [Google Scholar]
- 10.Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, et al. 2010. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat. Med. 16:814–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen TH, Calle EA, Zhao L, Lee EJ, Gui L, et al. 2010. Tissue-engineered lungs for in vivo implantation. Science 329:538–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price AP, England KA, Matson AM, Blazar BR, Panoskaltsis-Mortari A. 2010. Development of a decellularized lung bioreactor system for bioengineering the lung: the matrix reloaded. Tissue Eng. Part A 16:2581–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ott HC, Clippinger B, Conrad C, Schuetz C, Pomerantseva I, et al. 2010. Regeneration and orthotopic transplantation of a bioartificial lung. Nat. Med. 16:927–33 [DOI] [PubMed] [Google Scholar]
- 14.Cortiella J, Niles J, Cantu A, Brettler A, Pham A, et al. 2010. Influence of acellular natural lung matrix on murine embryonic stem cell differentiation and tissue formation. Tissue Eng. Part A 16:2565–80 [DOI] [PubMed] [Google Scholar]
- 15.Niklason LE, Gao J, Abbott WM, Hirschi KK, Houser S, et al. 1999. Functional arteries grown in vitro. Science 284:489–93 [DOI] [PubMed] [Google Scholar]
- 16.Chen RN, Ho HO, Tsai YT, Sheu MT. 2004. Process development of an acellular dermal matrix (ADM) for biomedical applications. Biomaterials 25:2679–86 [DOI] [PubMed] [Google Scholar]
- 17.Badylak SF, Lantz GC, Coffey A, Geddes LA. 1989. Small intestinal submucosa as a large diameter vascular graft in the dog. J. Surg. Res. 47:74–80 [DOI] [PubMed] [Google Scholar]
- 18.Badylak SF, Tullius R, Kokini K, Shelbourne KD, Klootwyk T, et al. 1995. The use of xenogeneic small intestinal submucosa as a biomaterial for Achilles tendon repair in a dog model. J. Biomed. Mater. Res. 29:977–85 [DOI] [PubMed] [Google Scholar]
- 19.Kropp BP, Eppley BL, Prevel CD, Rippy MK, Harruff RC, et al. 1995. Experimental assessment of small intestinal submucosa as a bladder wall substitute. Urology 46:396–400 [DOI] [PubMed] [Google Scholar]
- 20.Chen F, Yoo JJ, Atala A. 1999. Acellular collagen matrix as a possible “off the shelf” biomaterial for urethral repair. Urology 54:407–10 [DOI] [PubMed] [Google Scholar]
- 21.Freytes DO, Badylak SF, Webster TJ, Geddes LA, Rundell AE. 2004. Biaxial strength of multilaminated extracellular matrix scaffolds. Biomaterials 25:2353–61 [DOI] [PubMed] [Google Scholar]
- 22.Gilbert TW, Stolz DB, Biancaniello F, Simmons-Byrd A, Badylak SF. 2005. Production and characterization of ECM powder: implications for tissue engineering applications. Biomaterials 26:1431–35 [DOI] [PubMed] [Google Scholar]
- 23.Conklin BS, Richter ER, Kreutziger KL, Zhong DS, Chen C. 2002. Development and evaluation of a novel decellularized vascular xenograft. Med. Eng. Phys. 24:173–83 [DOI] [PubMed] [Google Scholar]
- 24.Dahl SL, Koh J, Prabhakar V, Niklason LE. 2003. Decellularized native and engineered arterial scaffolds for transplantation. Cell Transplant. 12:659–66 [PubMed] [Google Scholar]
- 25.Schmidt CE, Baier JM. 2000. Acellular vascular tissues: natural biomaterials for tissue repair and tissue engineering. Biomaterials 21:2215–31 [DOI] [PubMed] [Google Scholar]
- 26.Uchimura E, Sawa Y, Taketani S, Yamanaka Y, Hara M, et al. 2003. Novel method of preparing acellular cardiovascular grafts by decellularization with poly(ethylene glycol). J. Biomed. Mater. Res. A 67:834–37 [DOI] [PubMed] [Google Scholar]
- 27.Bader A, Schilling T, Teebken OE, Brandes G, Herden T, et al. 1998. Tissue engineering of heart valves—human endothelial cell seeding of detergent acellularized porcine valves. Eur. J. Cardiothorac. Surg. 14:279–84 [DOI] [PubMed] [Google Scholar]
- 28.Booth C, Korossis SA, Wilcox HE, Watterson KG, Kearney JN, et al. 2002. Tissue engineering of cardiac valve prostheses I: development and histological characterization of an acellular porcine scaffold. J. Heart Valve Dis. 11:457–62 [PubMed] [Google Scholar]
- 29.Grauss RW, Hazekamp MG, Oppenhuizen F, van Munsteren CJ, Gittenberger-de Groot AC, DeRuiter MC. 2005. Histological evaluation of decellularised porcine aortic valves: matrix changes due to different decellularisation methods. Eur. J. Cardiothorac. Surg. 27:566–71 [DOI] [PubMed] [Google Scholar]
- 30.Kasimir MT, Rieder E, Seebacher G, Silberhumer G, Wolner E, et al. 2003. Comparison of different decellularization procedures of porcine heart valves. Int. J. Artif. Organs 26:421–27 [DOI] [PubMed] [Google Scholar]
- 31.Korossis SA, Booth C, Wilcox HE, Watterson KG, Kearney JN, et al. 2002. Tissue engineering of cardiac valve prostheses II: biomechanical characterization of decellularized porcine aortic heart valves. J. Heart Valve Dis. 11:463–71 [PubMed] [Google Scholar]
- 32.Rieder E, Kasimir MT, Silberhumer G, Seebacher G, Wolner E, et al. 2004. Decellularization protocols of porcine heart valves differ importantly in efficiency of cell removal and susceptibility of the matrix to recellularization with human vascular cells. J. Thorac. Cardiovasc. Surg. 127:399–405 [DOI] [PubMed] [Google Scholar]
- 33.Schenke-Layland K, Vasilevski O, Opitz F, Konig K, Riemann I, et al. 2003. Impact of decellularization of xenogeneic tissue on extracellular matrix integrity for tissue engineering of heart valves. J. Struct. Biol. 143:201–8 [DOI] [PubMed] [Google Scholar]
- 34.Bissell MJ, Aggeler J. 1987. Dynamic reciprocity: How do extracellular matrix and hormones direct gene expression? Prog. Clin. Biol. Res. 249:251–62 [PubMed] [Google Scholar]
- 35.Sellaro TL, Ranade A, Faulk DM, McCabe GP, Dorko K, et al. 2010. Maintenance of human hepatocyte function in vitro by liver-derived extracellular matrix gels. Tissue Eng. Part A 16:1075–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin P, Chan WC, Badylak SF, Bhatia SN. 2004. Assessing porcine liver-derived biomatrix for hepatic tissue engineering. Tissue Eng. 10:1046–53 [DOI] [PubMed] [Google Scholar]
- 37.Sellaro TL, Ravindra AK, Stolz DB, Badylak SF. 2007. Maintenance of hepatic sinusoidal endothelial cell phenotype in vitro using organ-specific extracellular matrix scaffolds. Tissue Eng. 13:2301–10 [DOI] [PubMed] [Google Scholar]
- 38.Lin YM, Zhang A, Rippon HJ, Bismarck A, Bishop AE. 2010. Tissue engineering of lung: the effect of extracellular matrix on the differentiation of embryonic stem cells to pneumocytes. Tissue Eng. Part A 16:1515–26 [DOI] [PubMed] [Google Scholar]
- 39.Brown B, Lindberg K, Reing J, Stolz DB, Badylak SF. 2006. The basement membrane component of biologic scaffolds derived from extracellular matrix. Tissue Eng. 12:519–26 [DOI] [PubMed] [Google Scholar]
- 40.Sacks MS, Gloeckner DC. 1999. Quantification of the fiber architecture and biaxial mechanical behaviour of porcine intestinal submucosa. J. Biomed. Mater. Res. 46:1–10 [DOI] [PubMed] [Google Scholar]
- 41.Gong J, Sagiv O, Cai H, Tsang SH, Del Priore LV. 2008. Effects of extracellular matrix and neighboring cells on induction of human embryonic stem cells into retinal or retinal pigment epithelial progenitors. Exp. Eye Res. 86:957–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosokawa T, Betsuyaku T, Nishimura M, Furuyama A, Katagiri K, Mochitate K. 2007. Differentiation of tracheal basal cells to ciliated cells and tissue reconstruction on the synthesized basement membrane substratum in vitro. Connect. Tissue Res. 48:9–18 [DOI] [PubMed] [Google Scholar]
- 43.Hosokawa T, Betsuyaku T, Odajima N, Suzuki M, Mochitate K, et al. 2008. Role of basement membrane in EMMPRIN/CD147 induction in rat tracheal epithelial cells. Biochem. Biophys. Res. Commun. 368:426–32 [DOI] [PubMed] [Google Scholar]
- 44.Voytik-Harbin SL, Brightman AO, Kraine MR, Waisner B, Badylak SF. 1997. Identification of extractable growth factors from small intestinal submucosa. J. Cell Biochem. 67:478–91 [PubMed] [Google Scholar]
- 45.Hodde JP, Record RD, Liang HA, Badylak SF. 2001. Vascular endothelial growth factor in porcine-derived extracellular matrix. Endothelium 8:11–24 [DOI] [PubMed] [Google Scholar]
- 46.Hodde JP, Record R, Tullius R, Badylak SF. 2002. Fibronectin peptides mediate HMEC adhesion to porcine-derived extracellular matrix. Biomaterials 23:1841–48 [DOI] [PubMed] [Google Scholar]
- 47.Brennan EP, Reing J, Chew D, Myers-Irvin JM, Young EJ, Badylak SF. 2006. Antibacterial activity within degradation products of biological scaffolds composed of extracellular matrix. Tissue Eng. 12:2949–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarikaya A, Record R, Wu CC, Tullius B, Badylak S, Ladisch M. 2002. Antimicrobial activity associated with extracellular matrices. Tissue Eng. 8:63–71 [DOI] [PubMed] [Google Scholar]
- 49.Taylor DA. 2009. From stem cells and cadaveric matrix to engineered organs. Curr. Opin. Biotechnol. 20:598–605 [DOI] [PubMed] [Google Scholar]
- 50.Wainwright JM, Czajka CA, Patel UB, Freytes DO, Tobita K, et al. 2010. Preparation of cardiac extracellular matrix from an intact porcine heart. Tissue Eng. Part C Methods 16:525–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erdag G, Morgan JR. 2004. Allogeneic versus xenogeneic immune reaction to bioengineered skin grafts. Cell Transplant. 13:701–12 [DOI] [PubMed] [Google Scholar]
- 52.Gock H, Murray-Segal L, Salvaris E, Cowan P, D’Apice AJ. 2004. Allogeneic sensitization is more effective than xenogeneic sensitization in eliciting Gal-mediated skin graft rejection. Transplantation 77:751–53 [DOI] [PubMed] [Google Scholar]
- 53.Ross JR, Kirk AD, Ibrahim SE, Howell DN, Baldwin WM 3rd, Sanfilippo FP. 1993. Characterization of human anti-porcine “natural antibodies” recovered from ex vivo perfused hearts—predominance of IgM and IgG2. Transplantation 55:1144–50 [DOI] [PubMed] [Google Scholar]
- 54.Bernard MP, Chu ML, Myers JC, Ramirez F, Eikenberry EF, Prockop DJ. 1983. Nucleotide sequences of complementary deoxyribonucleic acids for the pro alpha 1 chain of human type I procollagen. Statistical evaluation of structures that are conserved during evolution. Biochemistry 22:5213–23 [DOI] [PubMed] [Google Scholar]
- 55.Bernard MP, Myers JC, Chu ML, Ramirez F, Eikenberry EF, Prockop DJ. 1983. Structure of a cDNA for the pro alpha 2 chain of human type I procollagen. Comparison with chick cDNA for pro alpha 2(I) identifies structurally conserved features of the protein and the gene. Biochemistry 22:1139–45 [DOI] [PubMed] [Google Scholar]
- 56.Constantinou CD, Jimenez SA. 1991. Structure of cDNAs encoding the triple-helical domain of murine alpha 2 (VI) collagen chain and comparison to human and chick homologues. Use of polymerase chain reaction and partially degenerate oligonucleotide for generation of novel cDNA clones. Matrix 11:1–9 [DOI] [PubMed] [Google Scholar]
- 57.Exposito JY, D’Alessio M, Solursh M, Ramirez F. 1992. Sea urchin collagen evolutionarily homologous to vertebrate pro-alpha 2(I) collagen. J. Biol. Chem. 267:15559–62 [PubMed] [Google Scholar]
- 58.McPherson TB, Liang H, Record RD, Badylak SF. 2000. Galalpha(1,3)Gal epitope in porcine small intestinal submucosa. Tissue Eng. 6:233–39 [DOI] [PubMed] [Google Scholar]
- 59.Raeder RH, Badylak SF, Sheehan C, Kallakury B, Metzger DW. 2002. Natural anti-galactose alpha1,3 galactose antibodies delay, but do not prevent the acceptance of extracellular matrix xenografts. Transplant. Immunol. 10:15–24 [DOI] [PubMed] [Google Scholar]
- 60.Daly K, Stewart-Akers A, Hara H, Ezzelarab M, Long C, et al. 2009. Effect of the alphaGal epitope on the response to small intestinal submucosa extracellular matrix in a nonhuman primate model. Tissue Eng. Part A 15:3877–88 [DOI] [PubMed] [Google Scholar]
- 61.Badylak SF, Valentin JE, Ravindra AK, McCabe GP, Stewart-Akers AM. 2008. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng. Part A 14:1835–42 [DOI] [PubMed] [Google Scholar]
- 62.Derwin KA, Baker AR, Spragg RK, Leigh DR, Iannotti JP. 2006. Commercial extracellular matrix scaffolds for rotator cuff tendon repair. Biomechanical, biochemical, and cellular properties. J. Bone Joint Surg. Am. 88:2665–72 [DOI] [PubMed] [Google Scholar]
- 63.Gilbert TW, Freund JM, Badylak SF. 2009. Quantification of DNA in biologic scaffold materials. J. Surg. Res. 152:135–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halloran PF. 2004. Immunosuppressive drugs for kidney transplantation. N. Engl. J. Med. 351:2715–29 [DOI] [PubMed] [Google Scholar]
- 65.Geron Corp. 2010. Safety study of GRNOPC1 in spinal cord injury. http://clinicaltrials.gov/show/NCT01217008
- 66.Marot D, Knezevic M, Novakovic GV. 2010. Bone tissue engineering with human stem cells. Stem. Cell Res. Ther. 1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kofidis T, de Bruin JL, Hoyt G, Ho Y, Tanaka M, et al. 2005. Myocardial restoration with embryonic stem cell bioartificial tissue transplantation. J. Heart Lung Transplant. 24:737–44 [DOI] [PubMed] [Google Scholar]
- 68.Nussbaum J, Minami E, Laflamme MA, Virag JA, Ware CB, et al. 2007. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J. 21:1345–57 [DOI] [PubMed] [Google Scholar]
- 69.Lees JG, Lim SA, Croll T, Williams G, Lui S, et al. 2007. Transplantation of 3D scaffolds seeded with human embryonic stem cells: biological features of surrogate tissue and teratoma-forming potential. Regen. Med. 2:289–300 [DOI] [PubMed] [Google Scholar]
- 70.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, et al. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–72 [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Shen W, Sun B, Lv C, Dou Z. 2011. Plasticity of marrow mesenchymal stem cells from human first-trimester fetus: from single-cell clone to neuronal differentiation. Cell. Reprogram. 13(1):57–64 [DOI] [PubMed] [Google Scholar]
- 72.Zhang ZY, Teoh SH, Teo EY, Khoon Chong MS, Shin CW, et al. 2010. A comparison of bioreactors for culture of fetal mesenchymal stem cells for bone tissue engineering. Biomaterials 31:8684–95 [DOI] [PubMed] [Google Scholar]
- 73.Oertel M, Shafritz DA. 2008. Stem cells, cell transplantation and liver repopulation. Biochim. Biophys. Acta 1782:61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Decembrini S, Cananzi M, Gualdoni S, Battersby A, Allen N, et al. 2011. Comparative analysis of the retinal potential of embryonic stem cells and amniotic fluid-derived stem cells. Stem Cells Dev. 20(5):851–63 [DOI] [PubMed] [Google Scholar]
- 75.Galende E, Karakikes I, Edelmann L, Desnick RJ, Kerenyi T, et al. 2010. Amniotic fluid cells are more efficiently reprogrammed to pluripotency than adult cells. Cell Reprogram. 12:117–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Phermthai T, Odglun Y, Julavijitphong S, Titapant V, Chuenwattana P, et al. 2010. A novel method to derive amniotic fluid stem cells for therapeutic purposes. BMC Cell Biol. 11:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aboushwareb T, Atala A. 2008. Stem cells in urology. Nat. Clin. Pract. Urol. 5:621–31 [DOI] [PubMed] [Google Scholar]
- 78.Antonucci I, Pantalone A, De Amicis D, D’Onofrio S, Stuppia L, et al. 2009. Human amniotic fluid stem cells culture onto titanium screws: a new perspective for bone engineering. J. Biol. Regul. Homeost. Agents 23:277–9 [PubMed] [Google Scholar]
- 79.Peister A, Deutsch ER, Kolambkar Y, Hutmacher DW, Guldberg RE. 2009. Amniotic fluid stem cells produce robust mineral deposits on biodegradable scaffolds. Tissue Eng. Part A 15:3129–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roomans GM. 2010. Tissue engineering and the use of stem/progenitor cells for airway epithelium repair. Eur. Cell Mater. 19:284–99 [DOI] [PubMed] [Google Scholar]
- 81.Cananzi M, Atala A, De Coppi P. 2009. Stem cells derived from amniotic fluid: new potentials in regenerative medicine. Reprod. Biomed. Online 18(Suppl. 1):17–27 [DOI] [PubMed] [Google Scholar]
- 82.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–76 [DOI] [PubMed] [Google Scholar]
- 83.Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, et al. 2007. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448:318–24 [DOI] [PubMed] [Google Scholar]
- 84.Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, et al. 2007. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem. Cell 1:55–70 [DOI] [PubMed] [Google Scholar]
- 85.Vogel G, Holden C. 2007. Developmental biology. Field leaps forward with new stem cell advances. Science 318:1224–25 [DOI] [PubMed] [Google Scholar]
- 86.Lengner CJ. 2010. iPS cell technology in regenerative medicine. Ann. NY Acad. Sci. 1192:38–44 [DOI] [PubMed] [Google Scholar]
- 87.Kusuma S, Gerecht S. 2010. Engineering blood vessels using stem cells: innovative approaches to treat vascular disorders. Expert Rev. Cardiovasc. Ther. 8:1433–45 [DOI] [PubMed] [Google Scholar]
- 88.Xu D, Wang F, Gu H, Wang J, Guo Q, et al. 2010. Hybrid cells differentiate to hepatic lineage cells and repair oxidative damage. Cell Mol. Biol. Lett. 15:451–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sawada R, Ito T, Tsuchiya T. 2007. Safety evaluation of tissue engineered medical devices using normal human mesenchymal stem cells. Yakugaku Zasshi 127:851–56 (In Japanese) [DOI] [PubMed] [Google Scholar]
- 90.de Peppo GM, Sjovall P, Lenneras M, Strehl R, Hyllner J, et al. 2010. Osteogenic potential of human mesenchymal stem cells and human embryonic stem cell-derived mesodermal progenitors: a tissue engineering perspective. Tissue Eng. Part A 16:3413–26 [DOI] [PubMed] [Google Scholar]
- 91.Chanda D, Kumar S, Ponnazhagan S. 2010. Therapeutic potential of adult bone marrow-derived mesenchymal stem cells in diseases of the skeleton. J. Cell Biochem. 111:249–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, et al. 1999. Bone marrow as a potential source of hepatic oval cells. Science 284:1168–70 [DOI] [PubMed] [Google Scholar]
- 93.Caimi PF, Reese J, Lee Z, Lazarus HM. 2010. Emerging therapeutic approaches for multipotent mesenchymal stromal cells. Curr. Opin. Hematol. 17:505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grompe M 2005. Bone marrow-derived hepatocytes. Novartis Found. Symp. 265:20–27; discussion 28–34, 92–97 [PubMed] [Google Scholar]
- 95.Alison MR. 2005. Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev. 1:253–60 [DOI] [PubMed] [Google Scholar]
- 96.Krause DS. 2008. Bone marrow-derived cells and stem cells in lung repair. Proc. Am. Thorac. Soc. 5:323–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jezierska-Wozniak K, Nosarzewska D, Tutas A, Mikolajczyk A, Oklinski M, Jurkowski MK. 2010. Use of adipose tissue as a source of mesenchymal stem cells. Postepy Hig. Med. Dosw. 64:326–32 [PubMed] [Google Scholar]
- 98.Li Y, Pan H, Huard J. 2010. Isolating stem cells from soft musculoskeletal tissues. J. Vis. Exp. doi: 10.3791/2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Parmacek MS. 2006. Cardiac stem cells and progenitors: developmental biology and therapeutic challenges. Trans. Am. Clin. Climatol. Assoc. 117:239–55; discussion 55–56 [PMC free article] [PubMed] [Google Scholar]
- 100.Obokata H, Kojima K, Westerman K, Yamato M, Okano T, et al. 2011. The potential of stem cells in adult tissues representative of the three germ layers. Tissue Eng. Part A. 17:607–15 [DOI] [PubMed] [Google Scholar]
- 101.Quirici N, Scavullo C, de Girolamo L, Lopa S, Arrigoni E, et al. 2010. Anti-L-NGFR and -CD34 monoclonal antibodies identify multipotent mesenchymal stem cells in human adipose tissue. Stem. Cells Dev. 19:915–25 [DOI] [PubMed] [Google Scholar]
- 102.Ringe J, Leinhase I, Stich S, Loch A, Neumann K, et al. 2008. Human mastoid periosteum-derived stem cells: promising candidates for skeletal tissue engineering. J. Tissue Eng. Regener. Med. 2:136–46 [DOI] [PubMed] [Google Scholar]
- 103.Evseenko D, Zhu Y, Schenke-Layland K, Kuo J, Latour B, et al. 2010. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 107:13742–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prasad VK, Kurtzberg J. 2010. Transplant outcomes in mucopolysaccharidoses. Semin. Hematol. 47:59–69 [DOI] [PubMed] [Google Scholar]
- 105.Doan PL, Chao NJ. 2010. Advances in cord blood transplants in adults. F1000 Med. Rep. 2:12; doi: 10.3410/M2-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zaibak F, Bello P, Kozlovski J, Crombie D, Ang H, et al. 2009. Unrestricted somatic stem cells from human umbilical cord blood grow in serum-free medium as spheres. BMC Biotechnol. 9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gyurkocza B, Rezvani A, Storb RF. 2010. Allogeneic hematopoietic cell transplantation: the state of the art. Expert Rev. Hematol. 3:285–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bonner-Weir S, Sharma A. 2002. Pancreatic stem cells. J. Pathol. 197:519–26 [DOI] [PubMed] [Google Scholar]
- 109.Bader A, Macchiarini P. 2010. Moving towards in situ tracheal regeneration: the bionic tissue engineered transplantation approach. J. Cell Mol. Med. 14:1877–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Morin O, Normand C. 1986. Long-term maintenance of hepatocyte functional activity in coculture: requirements for sinusoidal endothelial cells and dexamethasone. J. Cell Physiol. 129:103–10 [DOI] [PubMed] [Google Scholar]
- 111.Brutsaert DL. 2003. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 83:59–115 [DOI] [PubMed] [Google Scholar]
- 112.Hermanns MI, Unger RE, Kehe K, Peters K, Kirkpatrick CJ. 2004. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: development of an alveolo-capillary barrier in vitro. Lab. Investig. 84:736–52 [DOI] [PubMed] [Google Scholar]
- 113.Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, et al. 2004. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 304:1338–40 [DOI] [PubMed] [Google Scholar]
- 114.Vunjak-Novakovic G, Tandon N, Godier A, Maidhof R, Marsano A, et al. 2010. Challenges in cardiac tissue engineering. Tissue Eng. Part B Rev. 16:169–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nahmias Y, Berthiaume F, Yarmush ML. 2007. Integration of technologies for hepatic tissue engineering. Adv. Biochem. Eng. Biotechnol. 103:309–29 [DOI] [PubMed] [Google Scholar]
- 116.Lichtenberg A, Tudorache I, Cebotari S, Suprunov M, Tudorache G, et al. 2006. Preclinical testing of tissue-engineered heart valves re-endothelialized under simulated physiological conditions. Circulation 114:I559–65 [DOI] [PubMed] [Google Scholar]
- 117.Kasimir MT, Weigel G, Sharma J, Rieder E, Seebacher G, et al. 2005. The decellularized porcine heart valve matrix in tissue engineering: platelet adhesion and activation. Thromb. Haemost. 94:562–67 [DOI] [PubMed] [Google Scholar]
- 118.Lovett M, Lee K, Edwards A, Kaplan DL. 2009. Vascularization strategies for tissue engineering. Tissue Eng. Part B Rev. 15:353–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Menon NG, Rodriguez ED, Byrnes CK, Girotto JA, Goldberg NH, Silverman RP. 2003. Revascularization of human acellular dermis in full-thickness abdominal wall reconstruction in the rabbit model. Ann. Plastic Surg. 50:523–27 [DOI] [PubMed] [Google Scholar]
- 120.Schechner JS, Crane SK, Wang F, Szeglin AM, Tellides G, et al. 2003. Engraftment of a vascularized human skin equivalent. FASEB J. 17:2250–56 [DOI] [PubMed] [Google Scholar]
- 121.Bhatia SN, Balis UJ, Yarmush ML, Toner M. 1998. Microfabrication of hepatocyte/fibroblast cocultures: role of homotypic cell interactions. Biotechnol. Prog. 14:378–87 [DOI] [PubMed] [Google Scholar]
- 122.Jindal R, Nahmias Y, Tilles AW, Berthiaume F, Yarmush ML. 2009. Amino acid-mediated heterotypic interaction governs performance of a hepatic tissue model. FASEB J. 23:2288–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kamkin A, Kiseleva I, Isenberg G. 2003. Activation and inactivation of a nonselective cation conductance by local mechanical deformation of acutely isolated cardiac fibroblasts. Cardiovasc. Res. 57:793–803 [DOI] [PubMed] [Google Scholar]
- 124.Kamkin A, Kiseleva I, Lozinsky I, Scholz H. 2005. Electrical interaction of mechanosensitive fibroblasts and myocytes in the heart. Basic Res. Cardiol. 100:337–45 [DOI] [PubMed] [Google Scholar]
- 125.Aschheim K 2006. Anthony Atala. Nat. Biotechnol. 24:1311–11 [DOI] [PubMed] [Google Scholar]
- 126.Sussman NL, Kelly JH. 1993. Artificial liver: a forthcoming attraction. Hepatology 17:1163–64 [DOI] [PubMed] [Google Scholar]
- 127.Arkadopoulos N, Lilja H, Suh KS, Demetriou AA, Rozga J. 1998. Intrasplenic transplantation of allogeneic hepatocytes prolongs survival in anhepatic rats. Hepatology 28:1365–70 [DOI] [PubMed] [Google Scholar]
- 128.Kobayashi N, Miyazaki M, Fukaya K, Inoue Y, Sakaguchi M, et al. 2000. Transplantation of highly differentiated immortalized human hepatocytes to treat acute liver failure. Transplantation 69:202–7 [DOI] [PubMed] [Google Scholar]
- 129.Bilir BM, Guinette D, Karrer F, Kumpe DA, Krysl J, et al. 2000. Hepatocyte transplantation in acute liver failure. Liver Transplant. 6:32–40 [DOI] [PubMed] [Google Scholar]
- 130.Deutsch M, Meinhart J, Vesely M, Fischlein T, Groscurth P, et al. 1997. In vitro endothelialization of expanded polytetrafluoroethylene grafts: a clinical case report after 41 months of implantation. J. Vasc. Surg. 25:757–63 [DOI] [PubMed] [Google Scholar]
- 131.Kulig KM, Vacanti JR. 2004. Hepatic tissue engineering. Transplant. Immunol. 12:303–10 [DOI] [PubMed] [Google Scholar]
- 132.Ratcliffe A, Niklason LE. 2002. Bioreactors and bioprocessing for tissue engineering. In Reparative Medicine: Growing Tissues and Organs, ed. Sipe JD, Kelley CA, McNicol LA, pp. 210–15. New York: NY Acad. Sci. [DOI] [PubMed] [Google Scholar]
- 133.Martin I, Smith T, Wendt D. 2009. Bioreactor-based roadmap for the translation of tissue engineering strategies into clinical products. Trends Biotechnol. 27:495–502 [DOI] [PubMed] [Google Scholar]
- 134.Martin I, Wendt D, Heberer M. 2004. The role of bioreactors in tissue engineering. Trends Biotechnol. 22:80–86 [DOI] [PubMed] [Google Scholar]
- 135.Carrel A, Lindbergh CA. 1935. The culture of whole organs. Science 81:621–23 [DOI] [PubMed] [Google Scholar]
- 136.Lee CY, Mangino MJ. 2009. Preservation methods for kidney and liver. Organogenesis 5:105–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Feng S 2010. Donor intervention and organ preservation: Where is the science and what are the obstacles? Am. J. Transplant. 10:1155–62 [DOI] [PubMed] [Google Scholar]
- 138.Cobert ML, West LM, Jessen ME. 2008. Machine perfusion for cardiac allograft preservation. Current Opin. Organ Transplant. 13:526–30 [DOI] [PubMed] [Google Scholar]
- 139.Cypel M, Yeung JC, Hirayama S, Rubacha M, Fischer S, et al. 2008. Technique for prolonged normothermic ex vivo lung perfusion. J. Heart Lung Transplant. 27:1319–25 [DOI] [PubMed] [Google Scholar]
- 140.Brockmann J, Reddy S, Coussios C, Pigott D, Guirriero D, et al. 2009. Normothermic perfusion: a new paradigm for organ preservation. Ann. Surg. 250:1–6 [DOI] [PubMed] [Google Scholar]
- 141.Elaut G, Henkens T, Papeleu P, Snykers S, Vinken M, et al. 2006. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr. Drug Metab. 7:629–60 [DOI] [PubMed] [Google Scholar]
- 142.Brasile L, Stubenitsky BM, Booster MH, Lindell S, Araneda D, et al. 2002. Overcoming severe renal ischemia: the role of ex vivo warm perfusion. Transplantation 73:897–901 [DOI] [PubMed] [Google Scholar]
- 143.Park J, Li Y, Berthiaume F, Toner M, Yarmush ML, Tilles AW. 2008. Radial flow hepatocyte bioreactor using stacked microfabricated grooved substrates. Biotechnol. Bioeng. 99:455–67 [DOI] [PubMed] [Google Scholar]
- 144.Tolboom H, Pouw R, Uygun K, Tanimura Y, Izamis ML, et al. 2007. A model for normothermic preservation of the rat liver. Tissue Eng. 13:2143–51 [DOI] [PubMed] [Google Scholar]
- 145.Chan C, Berthiaume F, Lee K, Yarmush ML. 2003. Metabolic flux analysis of hepatocyte function in hormone- and amino acid-supplemented plasma. Metab. Eng. 5:1–15 [DOI] [PubMed] [Google Scholar]
- 146.Badylak SE. 2002. The extracellular matrix as a scaffold for tissue reconstruction. Semin. Cell Dev. Biol. 13:377–83 [DOI] [PubMed] [Google Scholar]
- 147.Yamada T, Yang JJ, Ricchiuti NV, Seraydarian MW. 1985. Oxygen consumption of mammalian myocardial cells in culture: measurements in beating cells attached to the substrate of the culture dish. Anal. Biochem. 145:302–7 [DOI] [PubMed] [Google Scholar]
- 148.Rotem A, Toner M, Tompkins RG, Yarmush ML. 1992. Oxygen uptake rates in cultured rat hepatocytes. Biotechnol. Bioeng. 40:1286–91 [DOI] [PubMed] [Google Scholar]
- 149.Nahmias Y, Kramvis Y, Barbe L, Casali M, Berthiaume F, Yarmush ML. 2006. A novel formulation of oxygen-carrying matrix enhances liver-specific function of cultured hepatocytes. FASEB J. 20:2531–33 [DOI] [PubMed] [Google Scholar]
- 150.Qiu Y, Hearse DJ. 1992. Comparison of ischemic vulnerability and responsiveness to cardioplegic protection in crystalloid-perfused versus blood-perfused hearts. J. Thorac. Cardiovasc. Surg. 103:960–68 [PubMed] [Google Scholar]
- 151.Vohra HA, Whistance R, Modi A, Ohri SK. 2009. The inflammatory response to miniaturised extracorporeal circulation: a review of the literature. Mediat. Inflamm. 2009:707042; doi: 10.1155/2009/707042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bursac N, Loo Y, Leong K, Tung L. 2007. Novel anisotropic engineered cardiac tissues: studies of electrical propagation. Biochem. Biophys. Res. Commun. 361:847–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Radisic M, Fast VG, Sharifov OF, Iyer RK, Park H, Vunjak-Novakovic G. 2009. Optical mapping of impulse propagation in engineered cardiac tissue. Tissue Eng. Part A 15:851–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lysaght MJ, Hazlehurst AL. 2004. Tissue engineering: the end of the beginning. Tissue Eng. 10:309–20 [DOI] [PubMed] [Google Scholar]
- 155.McAllister TN, Dusserre N, Maruszewski M, L’Heureux N. 2008. Cell-based therapeutics from an economic perspective: primed for a commercial success or a research sinkhole? Regenerat. Med. 3:925–37 [DOI] [PubMed] [Google Scholar]
- 156.van der Veen VC, van der Wal MB, van Leeuwen MC, Ulrich MM, Middelkoop E. 2009. Biological background of dermal substitutes. Burns 36:305–21 [DOI] [PubMed] [Google Scholar]
- 157.Bottcher-Haberzeth S, Biedermann T, Reichmann E. 2010. Tissue engineering of skin. Burns 36:450–60 [DOI] [PubMed] [Google Scholar]
- 158.Laschke MW, Vollmar B, Menger MD. 2009. Inosculation: connecting the life-sustaining pipelines. Tissue Eng. Part B Rev. 15:455–65 [DOI] [PubMed] [Google Scholar]
- 159.Strom SC, Fisher RA, Thompson MT, Sanyal AJ, Cole PE, et al. 1997. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 63:559–69 [DOI] [PubMed] [Google Scholar]
- 160.Shapiro AMJ, Lakey JRT, Ryan EA, Korbutt GS, Toth E, et al. 2000. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 343:230–38 [DOI] [PubMed] [Google Scholar]
- 161.Badylak SF. 2004. Xenogeneic extracellular matrix as a scaffold for tissue reconstruction. Transplant. Immunol. 12:367–77 [DOI] [PubMed] [Google Scholar]
- 162.Safar P 1988. Clinical death symposium. Crit. Care Med. 16:919–20 [DOI] [PubMed] [Google Scholar]
- 163.Abt PL, Desai NM, Crawford MD, Forman LM, Markmann JW, et al. 2004. Survival following liver transplantation from non-heart-beating donors. Ann. Surg. 239:87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]