

Abstract
Endolysins are highly evolved bacteriophage-encoded lytic enzymes produced to damage the bacterial cell wall for phage progeny release. They offer promising potential as highly specific lytic proteins with a low chance of bacterial resistance. The diversity in lysin sequences and domain organization can be staggering. In silico analysis of bacteriophage and prophage genomes can help identify endolysins exhibiting unique features and high antibacterial activity, hence feeding the pipeline of narrow-spectrum protein antibiotics. Mycobacteriophage lysis cassettes mostly have two lytic enzymes, LysinA and LysinB. The enzyme LysinA targets peptidoglycan in the cell wall and possesses a modular architecture. LysinB typically contains a single domain and acts upon the mycolyl ester linkages in mycolyl-arabinogalactan-peptidoglycan (Payne et al., 2010). This study aimed to find novel LysinBs against Mycobacterium fortuitum. After a detailed in silico characterization of lysis cassettes from three M. fortuitum prophages, we chose to work on a LysinB (hereafter described as LysinB_MF) found in an incomplete prophage (phiE1336, 9.4 kb in strain E1336).
LysinB_MF showed low sequence similarity with any other endolysins in the database and formed a separate clade on phylogenetic analysis. LysinB_MF’s structure, extracted from the AlphaFold Protein Structure Database, demonstrated a modular architecture with two structurally distinct domains: a peptidoglycan-binding domain (PGBD) at the N-terminal and the characteristic alpha/beta hydrolase domain connected via a linker peptide. We found the alpha/beta hydrolase domain, which is the enzyme-active domain (EAD), contains the conserved Ser-Asp-His catalytic triad with a tunnel-like topology and forms intermolecular hydrogen bonds. The PGBD shows structural similarity to the cell-wall binding domain of an amidase from Clostridium acetobutylicum, hinting at its acquisition due to domain mobility. Our in silico electrostatic potential analysis suggested that PGBD might be essential to the enzyme activity. This was experimentally validated by generating a truncated version of the enzyme, which demonstrated about six-fold decreased activity compared to its native form. The antimycobacterial activity of this enzyme was also compromised in its absence. Based on our analysis, PGBD emerged as an integral constituent of enzymes with diverse functional properties and is predicted to be a conserved cross-kingdom. Overall, this study highlights the importance of mining mycobacterial prophages as a novel endolysin source. It also provides unique insights into the diverse architecture of mycobacteriophage-encoded endolysins and the importance of functional domains for their catalytic activities.
Keywords: Prophage, Endolysins, LysinB, Structure, Domains, Esterase activity
Graphical Abstract
1. Introduction
Mycobacterium infections are among the most debilitating, if not fatal (Gordon & Parish, 2018; Das et al., 2021). Mycobacterium tuberculosis, the causative agent of Tuberculosis, alone was the cause of 1.3 million deaths in the year 2022, the second-highest infectious killer after COVID-19 (Bagcchi, 2023). Infections due to nontuberculous Mycobacterium (NTM) are also becoming an emerging issue of concern, and their prevalence is increasing worldwide. While Mycobacterium fortuitum-associated pulmonary diseases are the most prevalent (Khosravi et al., 2018), the bacterium also causes extra-pulmonary infections in humans, such as cutaneous and bone lesions, which are difficult to treat, chronic, occult, resistant, and recurrent (Sethi et al., 2014; Fraga et al., 2022).
Mycobacteriophages are viruses that specifically infect Mycobacterium spp. Most mycobacteriophages code for two endolysins: LysinA and LysinB, which target the bacterial cell wall and cause lysis of their host cells at the end of their life cycle (Payne et al., 2010; Payne & Hatfull, 2012). LysinA is a peptidoglycan hydrolase, and LysinB is a mycolylarabinogalactan esterase, which hydrolyzes the mycolyl ester linkages in mycolyl-arabinogalactan-peptidoglycan layer present in mycobacteria. Since the exogenous application of endolysins has been demonstrated to show an antibacterial effect, lysins have been explored as an alternative/combination therapy with antibiotics in several studies (Fraga et al., 2019; Abouhmad et al., 2020; Hurst-Hess et al., 2023; Singh et al., 2023). While the reports on structural and functional characterization of mycobacteriophage-encoded lysins (Payne et al., 2010; Eniyan et al., 2020; Gigante et al., 2021) have provided insights into their structural diversity, these lysins are relatively less reported when compared to phage lysins against Gram-negative and Gram-positive bacteria. Besides the isolated mycobacteriophages, the genomes of nontuberculous Mycobacterium (NTM) that are enriched with prophages (Abad et al., 2023), could also serve as a promising source for diverse antibacterial proteins (Schmitz et al., 2011; Shah et al., 2023). However, these prophages are largely unannotated. In this study, we report a novel LysinB encoded by one such unique prophage in a strain of M. fortuitum, with three characteristic features: i) unlike the usual single-domain architecture of LysinB enzymes, LysinB_MF has a structurally distinct peptidoglycan binding domain (PGBD), ii) its enzyme active domain shows structural similarity to a hydrolase (amidase) of Clostridium acetobutylicum, and iii) PGBD has an important role in enzyme activity. As a first step towards understanding the molecular properties, we have characterized the lytic cassette of three M. fortuitum prophages, and carried out a detailed examination of LysinB_MF, showing the highest similarity with D29 LysinB, which we used as a bait sequence for searching LysinB sequences in M.fortuitum prophages. We have carried out an extensive sequence and structural analysis of lysis cassettes and LysinB_MF and hypothesized and biochemically characterized the importance of the PGBD in its activity.
2. Results and Discussion
2.1. Mycobacterium prophages as a source of novel lysin sequences
D29 and Ms6 LysB enzymes are the extensively studied mycolylarabinogalactan esterases with pre-clinical characterization (Payne & Hatfull, 2012; Gigante et al., 2017; Fraga et al., 2019; Hurst-Hess et al., 2023). However, to facilitate the growing interest in mycobacteriophage lysins to treat drug-resistant Mycobacterium spp., more lysins need to be discovered and investigated to understand their biological features and to develop them as novel therapeutics. Non-tubercular mycobacteria (NTM) are reported to harbour a large proportion of prophages (Abad et al., 2023). We used D29 LysB (O64205) as a bait sequence and performed BLAST to find similar but not identical LysinB sequences against all the M. fortuitum genomes in the database. The workflow for the in silico identification of LysinB enzymes is represented in Figure 1A. The top three hits for LysinB were found to be encoded by prophages of M. fortuitum strains (Table 1). In this study, we have carried out the structural and functional characterization of a unique LysinB enzyme (mentioned as LysinB_MF) encoded by a prophage, called phiE1336, present in the sequenced strain E1336 of Mycobacterium fortuitum (tax id: 1766). A detailed characterization of this prophage has been done in the later sections.
Figure 1: Analysis of prophages in M. fortuitum strains.
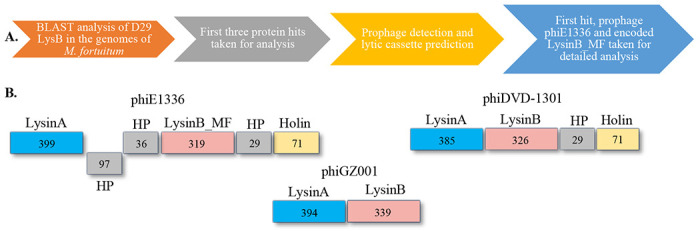
A) The workflow of prophage detection and characterization of the lytic regions. B) Lytic cassette of prophages phiE1336, phiDVD-1301 and phiGZ001.
Table 1:
Information on M. fortuitum prophages identified and characterized in the study
| S. no. | Name | Size (kb) | Completeness | GC% | Cluster similarity | M. fortuitum lysogen |
|---|---|---|---|---|---|---|
| 1 | phiE1336 | 9.4 | Incomplete | 62 | - | NZ_LZSJ01000108.1 |
| 2 | phiDVD-1301 | 39.6 | Intact | 62.8 | P | NZ_JARUNL010000131.1 |
| 3 | phiGZ001 | 38.9 | Intact | 63.4 | F1 | NZ_CP107719.1 |
We found the prophage phiE1336 genome to be incomplete (9.4 kb), however, its lysis cassette was intact. It constituted LysinA, three small hypothetical proteins, LysinB and Holin (Figure 1B). LysinB_MF, with an overall identity of 49% with D29 LysinB, is a peptidoglycan-binding domain-containing protein (WP_064866887.1). The second hit, with an overall identity of 48%, was a hypothetical protein (HP) (WP_278289780.1) from M. fortuitum strain DVD-1301 (NZ_JARUNL010000131.1). Reannotation of the genome and PHASTER prediction indicated the protein to be a LysinB, encoded by an intact prophage (hereby called phiDVD-1301). This prophage genome size was 39.6 kb with a GC content of 62.86%, similar to what is observed for other mycobacteriophages. Nucleotide BLAST analysis of the genome indicated phiDVD-1301 to have approximately 80% sequence identity with cluster P phages Tortellini (KX648391), Phayonce (KR080195) and Purky (MN096355). Annotation demonstrated the presence of a lytic cassette (Figure 1B) with LysinA (385 amino acids) as the largest protein-coding gene. LysinB (326 amino acids) from phiDVD-1301 showed 48% identity with D29 LysB, however, domain predictions for this enzyme remained unsuccessful. The third hit (WP 269975983.1) showing 36% sequence identity with D29 LysB was a hypothetical protein (HP), encoded by an M. fortuitum strain MF GZ001 (NZ CP107719.1) prophage. This prophage (hereby called phiGZ001) has a size of 38.9 kb with distinct attL (CACACTGTTCTTC) and attR (CACACTGTTCTTC) sites recognized by serine integrases (Suzuki et al., 2020). Nucleotide BLAST analysis of the genome indicated phiGZ001 to have approximately 86% sequence identity with cluster F1 phages XFactor (KT281795), Tootsieroll (MZ747519) and Blexus (MN586012). Sequence reannotation and domain analysis of the identified HP (WP_269975983.1) predicted an alpha/beta hydrolase domain. In phiGZ001, we did not find a holin gene in the lysis cassette (Figure 1B). We speculate this prophage either lyses the host independent of holin (Eniyan et al., 2020) or the lytic cassette is incomplete.
2.2. The incomplete prophage in the M. fortuitum strain E1336
PHAST analysis predicted the presence of an incomplete genome of prophage phiE1336. Its size is only 9.4 kb (Figure 2A) but the GC content (62%) is comparable to the host (Ho et al., 2012). Mycobacteriophages are grouped into clusters based on the similarities and differences in their nucleotide sequences (Hatfull, 2022). Currently, 32 clusters of mycobacteriophages have been defined, and the unclassified phages are denoted as singletons (https://phagesdb.org/). We performed nucleotide BLAST analysis to identify the possible cluster of phiE1336 and to predict phages identical to it. None of the whole genome sequences in the database showed more than 10% query coverage, a trend similar to singletons. However, considering the short genomic fragment of phiE1336, it is difficult to state conclusively whether it belongs among the singleton phages or to a cluster.
Figure 2: Characterization of phiE1366 and the encoded LysinB_MF.

A) phiE1336 has a genome size of 9.4 kb with a GC content of 62%. The genome consists of 13 ORFs, which encode for a few structural proteins and proteins of the lytic cassette B) Domain architecture of LysinB_MF from phiE1336 demonstrates distinct PGBD and EAD. C) Phylogenetic analysis of LysinB_MF to understand its evolutionary relationship. Representative sequences of LysinBs from all sub-clusters and singletons were taken for analysis. Phylogeny analysis displayed LysinB_MF forming its separate clade (in red arrow), which branched from the neighbouring Cluster V LysinB at some point. Green arrows show the clades disparate with the evolutionary relationships of mycobacteriophage clusters.
2.3. The lytic cassette in prophage phiE1336
We extracted the 9.4 kb fragment and performed annotation using RAST, which predicted it to have 13 ORFs (Figure 2A). Among the predicted ORFs, six could be assigned putative functions, which included lytic and structural proteins (Table S1). The lytic cassette begins with LysinA (44.3 kDa), a peptidoglycan hydrolase consisting of an amidase domain spanning from 171–317 and an overlapping peptidoglycan recognition protein (PGRP) region from 183–317 amino acids (Figure 2B). Co-occurrence of domains is common in phage lysins (Payne & Hatfull, 2012; Hatfull, 2018). The amidase domain targets the N-acetylmuramoyl-L-alanine linkages, which connect the sugar backbone to the peptide crosslink (Payne & Hatfull, 2012). BLAST analysis showed phiE1336 LysinA to share 71.9% identity with LysinA (gp24) from F1 sub-cluster mycobacteriophage Koella (MH316564) and 70.5% identity with LysinA (gp69) from O cluster phage Smooch (MN428052). Domain construction of lysins from these phages is similar and constitutes the active site of the amidase with an overlaying peptidoglycan recognition protein (PGRP) region.
The two small HPs between LysinA and LysinB of phiE1336 (Figure 1B) are of size 10.6 kDa and 3.9 kDa and do not possess a recognizable domain. Neither of the HPs shows any significant similarity to other sequences in the database. This arrangement of the lytic cassette, with HPs between Lysin A and LysinB, appears unique for phiE1336 among the other prophages characterized in this study (Figure 1B). The other reported mycobacterium prophages, such as phiMAV1 and MabR, constitute lytic regions with LysinA and LysinB present together (Payne et al., 2010; Cote et al., 2022).
Upstream to the two HPs in the cassette are the genes encoding for mycolylarabinogalactan esterase (LysinB_MF), another small HP (3.1 kDa), and holin (Figure 2B). LysinB_MF is a multidomain enzyme with a molecular weight of 35.3 kDa. While we could not precisely predict the function of the 3.1 kDa protein, BLAST analysis showed similar proteins to have an ATP binding activity. The holin gene encodes for a protein of size 7.4 kDa, which shares 61.1% identity with the holin from cluster I2 mycobacteriophage Che9c. Comparable to other phage-encoded holin, this, too, consists of two transmembrane helices, which could destabilize the membrane proton motive force of the host in a time-dependent manner (Pollenz et al., 2022).
2.4. LysinB_MF
Although the information on mycobacteriophage-encoded mycolylarabinogalactan esterase (LysinB) has been discussed earlier in several studies (Payne et al., 2010; Korany et al., 2019; Abouhmad et al., 2020), reports on the characterization of a multidomain LysinB is limited (Gigante et al., 2021). We found LysinB_MF to be a modular enzyme composed of two structurally distinct domains connected by a linker peptide (Figure 2B). The enzyme (319 amino acids) is comprised of two functionally defined regions: PGBD and the alpha/beta hydrolase domain (Figure S1). The PGBD is present at the N-terminal region of LysinB_MF and spans from amino acids 39–70, and a linker of 14 amino acids connects it to the catalytic enzyme-active domain (EAD) (Figure 2B). The EAD, like most mycolylarabinogalactan esterases, is comprised of an alpha/beta hydrolase domain that spans right after the linker until the C-terminal end, covering a substantial part of the enzyme. Through BLAST analysis, we found LysinB from a V cluster mycobacteriophage Cosmo (KP027195) to be the closest predicted homologue of LysinB_MF. The sequences showed a high query coverage of 98% and 56.43% identity (e-value 3e-116). Besides this, we could not find duplicate or identical proteins to LysinB_MF in the database. Therefore, to examine the evolutionary relationship of LysinB_MF, we performed the phylogenetic analysis using LysinB sequences from singletons and mycobacteriophages belonging to all the sub-clusters (Figure 2C). There were 187 sequences in the repository file prepared with at least one sequence and a maximum of three. The multi-FASTA file was subjected to sequence alignment and then processed using the iTOL to generate a rooted phylogenetic tree based on the maximum likelihood method.
Interestingly, we found a separate clade for LysinB_MF, as observed for LysinBs from singleton phages (Figure 2C). The LysinB_MF sequence appeared to have branched from the cluster V endolysins and had a branch length of 0.79. A PGBD in these LysinBs remains unannotated or lacking, although conserved residues in the N-terminal region were observed in the multiple sequence file. It appears Cluster V LysinBs and LysinB_MF may have had a common ancestor at some point; however, they diverged into forming their clade, and LysinB_MF retained the PGBD function.
Though the phylogenetic analysis gave us some clue about the evolutionary relationship of LysinB_MF, what was interesting to note was the unconventional grouping of endolysins from different sub-clusters in the same clade (shown in green arrows, Figure 2C). Phylogenetic analysis of tape-measure proteins in mycobacteriophage genomes distinctly shows separate clades, called clusters or even specific sub-clusters (Jacobs-Sera et al., 2012). Phages belonging to the same cluster are grouped into the same clade. However, while studying LysinB, we observed sequences belonging to entirely different clusters, if not sub-clusters, forming a common sub-clade and demonstrating the possibility of a common ancestor at some point in the evolutionary history. LysinB from clusters A7 and K6; F1, F4 and F5; B6 and B12; P1, P3 and I1, respectively, were present in the same clade (Figure 2C). In some instances, the evolution of LysinB appeared to be independent of the evolution of mycobacteriophages. Mycobacteriophage-encoded LysinBs, therefore, emerge as enzymes with a complex evolutionary pattern, and their lineage requires a greater analysis.
2.5. LysinB_MF has a complex enzyme active domain (EAD)
To analyze the structural information of LysinB_MF, we attempted to characterize the 3D model of the enzyme available in the AlphaFold Protein Structure Database (AFDB: A0A1A0NU50). The structure of this enzyme was available as a peptidoglycan-binding protein derived from the M. fortuitum strain described earlier. The protein structure showed a high model confidence (pLDDT > 90) and was taken forward for further analysis (Figure 3).
Figure 3: Structural analysis of LysinB_MF.
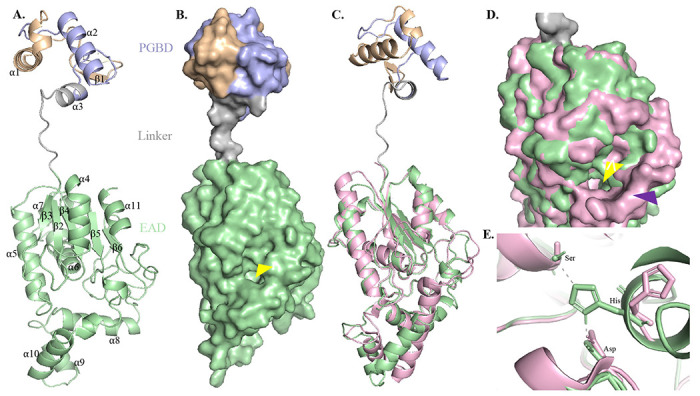
A) Three-dimensional (3D) model of LysinB_MF demonstrates a multi-domain structure comprising two structurally distinct domains: the PGBD (39-70), a short linker region followed by the EAD, an α/β hydrolase (85-317). B) The surface model of LysinB_MF demonstrates a bulky C-terminal end with a distinct active-site groove formed with residues Ser-Asp-His. The groove of the EAD, indicated with a yellow arrow, displays a tunnel-like topology. C) LysinB_MF can be superimposed on D29 LysB (in pink) with an overall RMSD of 0.56. Structural alignment of the two endolysins shows similar active-site arrangement, although PGBD in D29 Lysin B is absent. D) Active-site conformation in LysinB_MF and D29 LysinB. LysinB_MF shows a narrow but deeper topology (shown with a yellow arrow). D29 LysB displays a larger edge, having a funnel-like conformation (shown with a purple arrow). E) Active site residues in LysinB_MF (Ser156-Asp236-His305) and D29 LysB (S82-Asp166-His240).
The EAD of LysinB_MF comprises the α/β sandwich belonging to the α/β hydrolase family and contains the catalytic triad Ser-Asp-His. This domain spanned from amino acid 85–317, covering a major portion of the enzyme and demonstrated a highly twisted structure, consisting of a C-terminal linker region from 246-310. The active site of LysinB_MF displayed four β-strands (β2–β5), surrounded from all sides by α-helices (α4-α6). A surface model of the enzyme demonstrated the active-site groove with a tunnel-like topology (Figure 3B). A similar topology is observed in D29 LysB, which serves as a binding pocket for substrates such as several p-NP derivatives, trehalose monomycolate and mAGP from mycobacterial cell wall (Payne et al., 2010). A DALI search with high confidence (DALI Z-score 29.7) identified the structural relationship of LysinB_MF with the tertiary structure of D29 LysB (PDB accession 3HC7). The two esterases could be superimposed with an overall RMSD of 0.56 (153 Cα atoms) (Figure 3C). Both the structures displayed similar β-sheet conformation around the active site surrounded by the helices. Given the high structural similarity observed between LysinB_MF and D29 LysinB, the topology of D29 LysB’s binding pocket, unlike LysinB_MF, showed an edge with a wider circumference and a funnel-like appearance (Figure 3D). Furthermore, a bird’s eye view and analysis of the conserved active-site residues (Ser82, Asp166 & His240 in D29 LysB and Ser156, Asp236 & His305 in LysinB_MF) (Figure 4A and 4D) of these enzymes displayed notable differences. Unlike their counterparts, the residues Asp166 in D29 LysB and His305 in LysinB_MF were part of alpha-helices in these enzymes, which added to the differences in their groove topology. Also, unlike D29 LysB, the active-site residue His305 in LysinB_MF was found to form polar contacts with residues Ser156 and Asp236 (Figure 3E). These bonds are known to provide stability by acting as electrostatic attractive forces which influence the charge redistribution in the enzyme and are responsible for the uptake of electrons (pull) from the active site towards the amino acid residues (Qiu et al., 2018). Also, in the case of serine proteases with similar Ser-Asp-His catalytic triads, the removal of hydrogen bonds resulted in the partial release of the active-site residues in molecular dynamic simulations and a decrease in the overall stability of the transition states, which affects the enzyme’s turnover number (kcat) (Zheng et al., 2006).
Figure 4: Conserved residues in LysinB_MF.
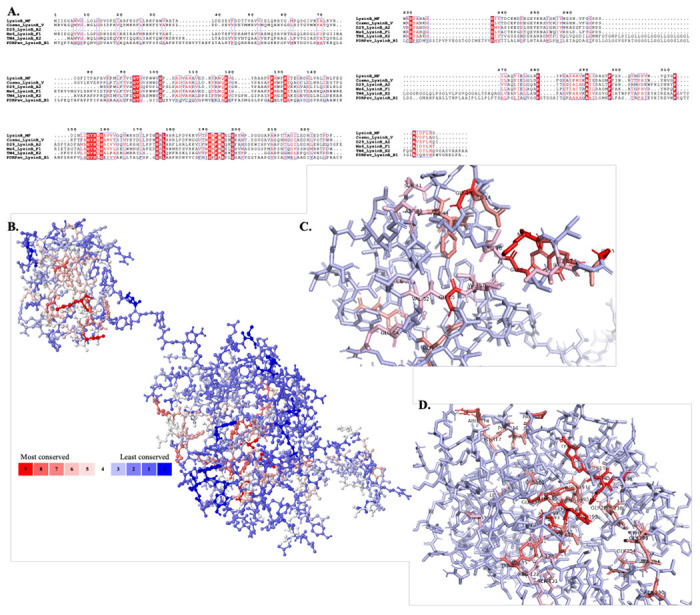
A) Multiple sequence alignment of LysinB enzymes from various clusters demonstrate key residues in PGBD and EAD. The EAD displays the conserved active-site residues Ser-Asp-His and the GXP motif. B) Analysis using POLYVIEW-MM identified and mapped conserved residues in the tertiary structure of LysinB_MF. A scale of 0-9 is used to quantify the conservation index, where 0 indicates the least conserved and nine is the most. Exposed residues in the structure of LysinB_MF are found to be least conserved among all others, while most of the highly conserved residues are found buried. C) Bird’s eye view of PGBD displays conserved amino acids. Ser41, Tyr43 Phe44, Asp45, Val52, Glu54, Gln56, Gly66 and Ile67 are a few highly conserved residues in this domain found both in sequence alignment and in structural analysis. D) Bird’s eye view of EAD displays conserved amino acids. The active site Ser156, Asp236 & His305 and Gly191 & Pro193 from the GXP motif were highly conserved in LysinBs.
2.6. The PGBD of LysinB_MF is structurally similar to an amidase
In most of the mycolylarabinogalactan esterases reported earlier (Fraga et al., 2019; Abouhmad et al., 2020; Eniyan et al., 2020; Singh et al., 2023), an extended N-terminal region consisting of a PGBD hasn’t been reported. It is a rare feature that has been observed in Ms6 LysinB (Gigante et al., 2021) and our analysis has predicted only seven out of sixty LysinB sequences to contain PGBD (unpublished data). Region 7–71 in LysinB_MF was interpreted as a PGBD-like superfamily, with region 39–70 predicted to be the functional PGBD (highlighted in light blue in Figure 2A). Only a single β-sheet in this region is present in the enzyme’s N-terminal end, and the rest of the structure appeared to be highly twisted, dominated by three α-helices. The annotated PGBD is comprised of two α helices (α2 and α3), and it tapers to form the linker region connecting to the EAD. The multiple sequence alignment and structural analysis of LysinB_MF demonstrated several conserved residues in this region (Figure 4), which are also found in LysinBs without the N-terminal PGBD. The amino acids with the highest score in the conservation index are mostly buried in the enzyme and could be involved in the functional properties of the PGBD (Figures 4A–4C).
Considering the unavailability of structural information about the PGBD in a mycobacteriophage-encoded LysinB and failure to acquire the same through structural similarity analysis based on the native structure of LysinB_MF, we separately built the PGBD and analyzed it further. The tertiary structure of PGBD was prepared using LysinB_MF as a template, and it had a QMEANDisCo Global score of 0.64 ± 0.11, which ensured high quality (Figure S2A). We then performed structural similarity prediction using DALI, which predicted 20 possible neighbours, and these were visually sorted based on their Z-score. Interestingly, the top scoring structure (Z-score from DALI 8.9) found similar to the PGBD of LysinB_MF was an enzyme with amidase activity (PDB accession code 4XXT). This zinc-dependent amidase is a PGBD-containing protein from Clostridium acetobutylicum (ATCC 824). To analyze the structural similarity to PGBD of LysinB_MF, we downloaded the protein 3D file from the PDB database and carried out structural alignment using PyMOL. The PGBDs from the amidase and LysinB_MF could be superpositioned with an overall RMSD as low as 0.46, even though the sequences do not show identical residues (Figure S2B). Also, amidases (or lysozyme-like domains) possessing a peptidoglycan-binding and hydrolyzing function are usually the features of LysinA enzymes. Due to the unavailability of the 3D structure of a LysinA in the PDB database, we could not examine the structural similarity of PGBD specifically to a mycobacteriophage-encoded N-acetylmuramoyl-L-alanine amidase. However, it is noteworthy that structural similarity of PGBD of LysinB_MF with high confidence was achieved with an amidase from a bacterial species. We speculate that domain mobility could be a reason behind the generation of modular proteins, and they play a crucial role in their functioning (Basu et al., 2009).
2.7. PGBD is essential for the esterase activity of LysinB_MF
To understand the role of PGBD in the activity of LysinB_MF, we first computationally generated its truncated version (LysinB_MFΔPGBD), lacking the N-terminal domain (Figures 5A and 5B). Using eF-surf, a comparative analysis of the native and the truncated structures indicated a change in the electrostatic potential of the active site. The catalytic tunnel appeared more electronegative in LysinB_MFΔPGBD than its native structure (Figures 5C and 5D). A change in electrostatic potential could impact the interactions between an enzyme and its substrate and, hence, the enzyme’s activity. The importance of electrostatics in enzyme-mediated catalysis has been demonstrated for amylases where a change in the charge near the active site residues resulted in an enzyme with drastically reduced activity (Nielsen et al., 1999). To find out if the deletion of PGBD also hindered the in vitro activity of LysinB_MF, we purified the complete (LysinB_MF) and the truncated enzyme (LysinB_MFΔPGBD) as recombinant proteins (Figures 5E–5F). The lipolytic assay of the truncated protein yielded low Ca2+ precipitation (Figure 5H). The in vitro esterase assay also revealed the truncated LysinB_MFΔPGBD (Figures 5A, 5B and 5E) to show about a six-fold decrease in activity when compared with the activity of the whole enzyme (Figure 5G), indicating PGBD’s role in enzyme activity. A similar effect has also been observed for an N-acetylmuramoyl-L-alanine amidase from bacteriophage PSA, where truncation of the cell wall binding domain led to a several-fold decrease in enzyme activity (Korndorfer et al., 2006). The stabilizing effect of PGBD in both mycolylarabinogalactan esterase (LysinB) and N-acetylmuramoyl-L-alanine amidase (LysinA) is significant and requires more investigation.
Figure 5: PGBD is essential for the optimum activity of LysinB_MF.

A) Domain architecture and comparison of native LysinB_MF and truncated LysinB_MFΔPGBD (EAD). B) The structure of LysinB_MF without the PGBD (LysinB_MFΔPGBD) was built using the native structure as a template. LysinB_MFΔPGBD structure comprises the linker region (in grey) and the EAD (in green). C-D) Comparison of the intact LysinB_MF and LysinB_MFΔPGBD demonstrates a change in electrostatic potential in the active site (indicated with a yellow arrow). E) SDS-PAGE (12%) analysis of purified LysinB_MF and LysinB_MFΔPGBD. Lane 1: protein molecular mass standards. Lane 2: purified recombinant protein LysinB_MF (36.3kDa). Lane 3: LysinB_MFΔPGBD (26 kDa). F) Immunoblot of N-terminal His-tagged LysinB_MF. Lane 1: protein molecular mass standards. Lane 2: N-terminal His-tagged LysinB_MF. G) A white zone indicates Ca2+-precipitation by LysinB_MF and LysinB_MFΔPGBD (5 μg each). H) Esterase activity of whole LysinB_MF and LysinB, determined by the PNP release assay (n=6). I) Spot test for intrinsic activity of LysinB_MF and LysinB_MFΔPGBD against M. smegmatis and M. fortuitum. All the assays were performed using 5 μg of each enzyme preparation.
2.8. Antimycobacterial activity of LysinB_MF and LysinB_MFΔPGBD
Given the complex, hydrophobic cell envelope of mycobacterial spp., the ability of an endolysin to cause cell lysis on external application is highly desired since it obviates the need for an outer membrane permeabilizer (OMP) or a membrane-destabilizer. This property also makes lysins more efficacious candidates as antibiotics (enzybiotics) or as an adjunct to antibiotics. To check whether LysinB_MF possessed intrinsic cell lysis activity, we performed a lysin susceptibility test on M. fortuitum and M. smegmatis. A zone of clearance on both the mycobacterium species was observed, indicating its ability to lyse mycobacterial cells unaided. Considering the significant decrease in LysinB_MFΔPGBD activity observed in the esterase assay (Figure 5H), we tested its cell lysis activity against M. fortuitum (and M. smegmatis) and, unlike whole LysinB_MF, did not find clearance zone on the bacterial lawn (Figures 5I). It appears that the PGBD not only supports the enzymatic activity of LysinB_MF but might also have a role in destabilizing the cell wall of M.fortuitum and M.smegmatis. The cross-species activity of the PGBD has not been reported for a mycobacteriophage lysin. It will be exciting to identify the PGBD-interacting region in the mycobacterial cell wall.
2.9. Conservation of PGBD across different kingdoms
Through protein truncation and enzymatic assays, we demonstrated the integral role of PGBD in LysinB_MF activity (Figures 5H- 5I). However, this feature is not constant in all other endolysins. While in the case of bacteriophage PSA, removing the cell-wall binding domain of endolysin demonstrated a similar decrease in enzymatic activity (Korndorfer et al., 2006), the lytic activity of a Bacillus anthracis prophage endolysin was enhanced when the C-terminal cell-wall binding domain was removed (Low et al., 2005). PGBD appears to have diverse functionality; however, a proper understanding of the conservation and importance of PGBD is limited. To comprehend the importance of this functional domain in other organisms, we analyzed all the sequences present in the UniProt database against the Pfam entry for the PGBD (PF01471). There are approximately 122,000 protein sequences consisting of this domain. From our curated analysis of these sequences based on TaxId from different organisms, we found this domain is conserved across all kingdoms (Figure 6). Among the prokaryotes, PF01471 was predicted to be the highest in E. coli (Figure 6), where it is present in LD-transpeptidases, which are involved in cell wall adaptation to stress and for providing outer membrane stability (Aliashkevich & Cava, 2022). In M. fortuitum, PF01471 was found chiefly in enzymes with a PG hydrolase activity. In the evolutionary Tree of Life in higher kingdoms, with Arabidopsis thaliana as an example, our curated search predicted 27 UniProt protein matches against PF01471 (Figure 6). Most of the matched proteins in this species were found to be zinc-dependent endo-peptidases or matrix metalloproteinases, which are involved in root development and water uptake (Mishra et al., 2021). In higher animals, including humans, we received 54 UniProt protein matches against PF01471, which, notably, was higher than most prokaryotes analyzed in this study (Figure 6). In humans, a diverse group of essential enzymes possess a PGBD, including matrix metalloproteinases (also observed in A. thaliana), enzymes involved in the degradation of extracellular matrix and development of human bones such as Stromelysin-2 (Bord et al., 1998) and collagenases, which are implicated in the turnover of connective tissue matrix constituents (Knauper et al., 1996). The cross-kingdom conservation of the short PGBD observed here would require a deeper analysis and is beyond the scope of our study.
Figure 6: Cross-kingdom conservation of a PGBD.

The UniProt database was screened against the Pfam ID of PGBD (PF01471), which resulted in approximately 122,000 protein hits with this domain. The curated database of hits was then individually screened against TaxIDs from various organisms across all kingdoms.
3. Conclusion
We have carried out an in silico analysis of three prophages in M. fortuitum, a medically important NTM. The analysis includes extraction, annotation and sequence characterization of prophages, followed by the structural analysis of the lysis cassettes. In this study, a detailed analysis of one of the three prophages, ‘phiE1336’ and its encoded LysinB (LysinB_MF) was done. Besides the catalytic alpha/beta domain and a C-terminal linker region, LysinB_MF has an extended N-terminal, containing a peptidoglycan-binding domain. A conservation analysis predicted this domain in diverse enzymes encoded by several organisms. Though LysinB_MF is encoded by an incomplete prophage, which should be incapable of forming phage progeny on induction, its in vitro esterase activity is greater than the reported LysinB enzymes. It will be worthwhile to investigate whether this highly active, prophage-derived LysinB enzyme has a role in the host M.fortuitum life cycle. Based on our in silico analysis, we predicted PGBD’s role in the enzyme’s activity and validated it experimentally. In addition, we also observed a loss of the intrinsic antibacterial activity in the truncated enzyme.
In summary, the diversity in domain organization of lysins in the prophage-encoded lysis cassettes and the efficient in vitro and antimycobacterial activity of LysinB_MF highlights the importance of prophages as a source of novel efficacious endolysins, which could be developed as promising anti-mycobacterial protein antibiotics.
4. Materials and Methods
4.1. In silico identification of LysinB_MF and characterization
Lysin B from D29 (GenBank: AAC18452.1) was blasted (Altschul et al., 1990) against all the genomes available in the NCBI database for M. fortuitum (tax id: 1766), possibly harbouring prophages. The M. fortuitum genome with the top lysin hits (LysinB_MF) was subjected to PHAST (Zhou et al., 2011) and to annotate and fix frameshifts in these genomes, the RAST server was used. The CGview tool (Grant & Stothard, 2008) was used to build the genome map of the first hit, phiE1336. Cluster-wise sequences of LysinB were downloaded from the Actinobacteriophage Database (https://phagesdb.org/), and domains of those lysins were predicted using InterProScan 5 (Quevillon et al., 2005). ClustalW (Thompson et al., 1994) and ESPript (https://espript.ibcp.fr) (Robert & Gouet, 2014) were used to generate alignment files which were then used to create phylogenetic trees using Interactive Tree Of Life (iTOL) v5 (Letunic & Bork, 2021). The tertiary structure of LysinB_MF was downloaded from the AlphaFold Protein Structure Database (Varadi et al., 2023) and visualized using PyMOL (https://pymol.org/2/). The POLYVIEW-MM tool was used to predict conserved residues in the tertiary structure of LysinB_MF (Porollo & Meller, 2010). The DALI server was used to find structures similar to the LysinB_MF (Holm, 2022) and eF-surf was used to predict the electrostatic potential of the enzyme (Nielsen et al., 1999). Conservation analysis was done by analyzing the Pfam entry of PGBD (PF01471) against all the proteins available in the UniProt database (https://www.uniprot.org/). TaxIDs were used to detect the presence of this domain across several species using the InterPro classification of protein family website (https://www.ebi.ac.uk/interpro/).
4.2. Cloning, overexpression and purification of recombinant LysinB_MF and LysinB_MFΔPGBD
The LysinB_MF gene was synthesized and cloned in the pET28a vector by Biomatik Corporation, Kitchener, Ontario. To clone the truncated version of LysinB_MF lacking the PGBD (LysinB_MFΔPGBD), internal primers were designed to PCR amplify the EAD and the amplicon was ligated to the pET28a vector. The recombinant constructs pET28a-LysinB_MF and pET28a-LysinB_MFΔPGBD were overexpressed in BL21(DE3) and C41(DE3) strains of E. coli, respectively. For soluble expression, induction with 0.1 mM IPTG and overnight incubation at 16°C was done. The induced culture pellet was collected by centrifugation at 8,000 rpm for ten minutes at 4°C and sonicated at 90% amplitude with 30 seconds each on or off pulse for ten cycles (Eniyan et al., 2020; Das & Bajpai, 2023). The cell lysate was centrifuged at 13,000 rpm for 45 minutes at 4°C, and the supernatant was considered the soluble fraction.
Recombinant LysinB_MF and LysinB_MFΔPGBD were purified by Ni-NTA affinity chromatography. Briefly, the soluble fraction containing the His6-tagged protein was kept for binding with the Ni-NTA matrix for 2 hours at 4 °C, and the bound resin was packed in a column. After packing and discarding the flow-through, the column was gradient-washed with buffer containing 20-40 mM imidazole and the bound protein was eluted at 200-250 mM imidazole concentrations. The eluted fractions were analyzed using 12% SDS-PAGE (Laemmli, 1970) and the purified proteins were dialyzed with a buffer containing 50 mM Tris (pH 7.9) and 150 mM NaCl. The concentration of each protein was estimated by Bradford assay (Bradford, 1976). For immunoblotting, the protein(s) in the gel were transferred to a PVDF membrane using a Mini-PROTEAN Tetra Cell (Bio-Rad, United States). After blocking the membrane with 3% BSA at 4°C overnight, the membrane was incubated with mouse anti-His6 antiserum (Santacruz Biotech, USA) for 4 hours at a dilution of 1:2000. The membrane was washed once with PBS and twice with PBS-Tween, followed by incubation with IgG HRP-conjugated antibody at a dilution of 1:10000 for 45 minutes at 4 °C. The blot was developed by using 3, 3’-Diaminobenzidine (DAB-H2O2) as the substrate.
4.3. Assay for the esterase activity of LysinB
The micro-titre plate-based p-Nitrophenol butyrate (pNPB) esterase assay (Fraga et al., 2019) was performed to estimate the activity of LysinB_MF and LysinB_MFΔPGBD. The reaction conditions for both the recombinant proteins were kept constant: 5 μg of enzyme 10 mM pNPB in 25 mM Tris buffer (pH 7.2) in a reaction volume of 200 μl at 37°C for 30 minutes. Absorbance at 410 nm was measured to determine the release of p-nitrophenol (PNP). In the control reaction, enzyme buffer was added in place of the enzymes. The experiment was done twice in three sets, and the absorbance value (A410) was plotted as mean ± SD.
4.4. Lipolytic activity of LysinB
The enzyme activity was also observed by CaCl2 precipitation. Five μg each of LysinB_MF and LysinB_MFΔPGBD was spot-tested on Middlebrook 7H10 plates containing 1mM CaCl2 and 1% (v/v) Tween-20. Precipitation, confirming lipolytic activity, was observed with 10 min incubation at room temperature.
4.5. Spot Test of LysinB
For the antimycobacterial activity, the spot test on bacterial lawns was performed. Briefly, Mycobacterium smegmatis strain mc2155 and M. fortuitum (NIHJ 1615) were grown for 48 hours at 37°C using Middlebrook 7H9 broth (HiMedia, India), supplemented with 0.4% glycerol. Bacterial lawn in 7H10 Middlebrook agar plates containing 0.05% Tween 80 plates were prepared by spreading 0.5 mL of M. smegmatis and M. fortuitum using 7H10 soft agar. Five μg each of LysinB_MF and LysinB_MFΔPGBD were added to the prepared lawn as spots and the plates were incubated at 37°C.
Supplementary Material
Acknowledgement
We thank the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India, for supporting the project (SERB PROJECT EMR/2017/004051). Acharya Narendra Dev College (ANDC), University of Delhi, is acknowledged for providing the infrastructural facilities and ELITE fellowship to RD. KN received the Innovation in Science Pursuit for Inspired Research (INSPIRE) fellowship from the Department of Science and Technology (DST), India; RA received the Senior Research Fellowship (SRF) from the University Grants Commission (UGC), New Delhi, India. The graphical abstract was created using BioRender.com (https://biorender.com).
Funding
This study was supported by the funding provided to the project investigator UB by the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India (SERB PROJECT EMR/2017/004051).
Footnotes
Ethics approval and consent to participate.
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
This published article and its supplementary information files include all data generated or analyzed during this study.
References
- Abad L, Gauthier CH, Florian I, Jacobs-Sera D, Hatfull GF. 2023. The heterogeneous and diverse population of prophages in Mycobacterium genomes. mSystems 8(5): e0044623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouhmad A, Korany AH, Grey C, Dishisha T, Hatti-Kaul R. 2020. Exploring the Enzymatic and Antibacterial Activities of Novel Mycobacteriophage Lysin B Enzymes. Int J Mol Sci 21(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliashkevich A, Cava F. 2022. LD-transpeptidases: the great unknown among the peptidoglycan cross-linkers. Febs Journal 289(16): 4718–4730. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215(3): 403–410. [DOI] [PubMed] [Google Scholar]
- Bagcchi S. 2023. WHO’s Global Tuberculosis Report 2022. Lancet Microbe 4(1): e20. [DOI] [PubMed] [Google Scholar]
- Basu MK, Poliakov E, Rogozin IB. 2009. Domain mobility in proteins: functional and evolutionary implications. Brief Bioinform 10(3): 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bord S, Horner A, Hembry RM, Compston JE. 1998. Stromelysin-1 (MMP-3) and stromelysin-2 (MMP-10) expression in developing human bone: potential roles in skeletal development. Bone 23(1): 7–12. [DOI] [PubMed] [Google Scholar]
- Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Cote J, Welch C, Kimble M, Archambault D, Ross JC, Orellana H, Amero K, Bourett C, Daigle A, Hutchison KW, et al. 2022. Characterization of the cluster MabR prophages of Mycobacterium abscessus and Mycobacterium chelonae. G3 (Bethesda) 12(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Bajpai U. 2023. Functional characterization of a DNA-dependent AAA ATPase in a F-cluster mycobacteriophage. Virus Res 323: 198957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Eniyan K, Bajpai U. 2021. Computational identification and characterization of antigenic properties of Rv3899c of Mycobacterium tuberculosis and its interaction with human leukocyte antigen (HLA). Immunogenetics 73(5): 357–368. [DOI] [PubMed] [Google Scholar]
- Eniyan K, Sinha A, Ahmad S, Bajpai U. 2020. Functional characterization of the endolysins derived from mycobacteriophage PDRPxv. World J Microbiol Biotechnol 36(6): 83. [DOI] [PubMed] [Google Scholar]
- Fraga AG, Trigo G, Murthy RK, Akhtar S, Hebbur M, Pacheco AR, Dominguez J, Silva-Gomes R, Goncalves CM, Oliveira H, et al. 2019. Antimicrobial activity of Mycobacteriophage D29 Lysin B during Mycobacterium ulcerans infection. PLoS Negl Trop Dis 13(8): e0007113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga K, Maireles M, Jordan M, Soldevila L, Murillo O. 2022. Mycobacterium fortuitum osteomyelitis of the cuboid bone treated with CERAMENT G and V: a case report. J Bone Jt Infect 7(4): 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante AM, Hampton CM, Dillard RS, Gil F, Catalao MJ, Moniz-Pereira J, Wright ER, Pimentel M. 2017. The Ms6 Mycolyl-Arabinogalactan Esterase LysB is Essential for an Efficient Mycobacteriophage-Induced Lysis. Viruses 9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante AM, Olivenca F, Catalao MJ, Leandro P, Moniz-Pereira J, Filipe SR, Pimentel M. 2021. The Mycobacteriophage Ms6 LysB N-Terminus Displays Peptidoglycan Binding Affinity. Viruses 13(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SV, Parish T. 2018. Microbe Profile: Mycobacterium tuberculosis: Humanity’s deadly microbial foe. Microbiology (Reading) 164(4): 437–439. [DOI] [PubMed] [Google Scholar]
- Grant JR, Stothard P. 2008. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res 36(Web Server issue): W181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF. 2018. Mycobacteriophages. Microbiol Spectr 6(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF. 2022. Mycobacteriophages: From Petri dish to patient. PLoS Pathog 18(7): e1010602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YS, Adroub SA, Aleisa F, Mahmood H, Othoum G, Rashid F, Zaher M, Ali S, Bitter W, Pain A, et al. 2012. Complete genome sequence of Mycobacterium fortuitum subsp. fortuitum type strain DSM46621. J Bacteriol 194(22): 6337–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. 2022. Dali server: structural unification of protein families. Nucleic Acids Res 50(W1): W210–W215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst-Hess K, Walz A, Yang Y, McGuirk H, Gonzalez-Juarrero M, Hatfull GF, Ghosh P, Ojha AK. 2023. Intrapulmonary Treatment with Mycobacteriophage LysB Rapidly Reduces Mycobacterium abscessus Burden. Antimicrob Agents Chemother 67(6): e0016223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing G, et al. 2012. On the nature of mycobacteriophage diversity and host preference. Virology 434(2): 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi AD, Mirsaeidi M, Farahani A, Tabandeh MR, Mohajeri P, Shoja S, Hoseini Lar KhosroShahi SR. 2018. Prevalence of nontuberculous mycobacteria and high efficacy of d-cycloserine and its synergistic effect with clarithromycin against Mycobacterium fortuitum and Mycobacterium abscessus. Infect Drug Resist 11: 2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. 1996. Biochemical characterization of human collagenase-3. J Biol Chem 271(3): 1544–1550. [DOI] [PubMed] [Google Scholar]
- Korany AH, Abouhmad A, Bakeer W, Essam T, Amin MA, Hatti-Kaul R, Dishisha T. 2019. Comparative Structural Analysis of Different Mycobacteriophage-Derived Mycolylarabinogalactan Esterases (Lysin B). Biomolecules 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korndorfer IP, Danzer J, Schmelcher M, Zimmer M, Skerra A, Loessner MJ. 2006. The crystal structure of the bacteriophage PSA endolysin reveals a unique fold responsible for specific recognition of Listeria cell walls. J Mol Biol 364(4): 678–689. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259): 680–685. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2021. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49(W1): W293–W296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low LY, Yang C, Perego M, Osterman A, Liddington RC. 2005. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J Biol Chem 280(42): 35433–35439. [DOI] [PubMed] [Google Scholar]
- Mishra LS, Kim SY, Caddell DF, Coleman-Derr D, Funk C. 2021. Loss of Arabidopsis matrix metalloproteinase-5 affects root development and root bacterial communities during drought stress. Physiol Plant 172(2): 1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JE, Beier L, Otzen D, Borchert TV, Frantzen HB, Andersen KV, Svendsen A. 1999. Electrostatics in the active site of an alpha-amylase. Eur J Biochem 264(3): 816–824. [DOI] [PubMed] [Google Scholar]
- Payne K, Sun Q, Sacchettini J, Hatfull GF. 2010. Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Molecular Microbiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne KM, Hatfull GF. 2012. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One 7(3): e34052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollenz RS, Bland J, Pope WH. 2022. Bioinformatic characterization of endolysins and holin-like membrane proteins in the lysis cassette of phages that infect Gordonia rubripertincta. PLoS One 17(11): e0276603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porollo A, Meller J. 2010. POLYVIEW-MM: web-based platform for animation and analysis of molecular simulations. Nucleic Acids Res 38(Web Server issue): W662–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Azofra LM, MacFarlane DR, Sun C. 2018. Hydrogen bonding effect between active site and protein environment on catalysis performance in H2-producing [NiFe] hydrogenases. Physical Chemistry Chemical Physics. [DOI] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. 2005. InterProScan: protein domains identifier. Nucleic Acids Res 33(Web Server issue): W116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42(Web Server issue): W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JE, Ossiprandi MC, Rumah KR, Fischetti VA. 2011. Lytic enzyme discovery through multigenomic sequence analysis in Clostridium perfringens. Appl Microbiol Biotechnol 89(6): 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Arora S, Gupta V, Kumar S. 2014. Cutaneous Mycobacterium fortuitum Infection: Successfully Treated with Amikacin and Ofloxacin Combination. Indian J Dermatol 59(4): 383–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Das R, Chavan B, Bajpai U, Hanif S, Ahmed S. 2023. Beyond antibiotics: phage-encoded lysins against Gram-negative pathogens. Front Microbiol 14: 1170418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Gangakhedkar R, Thakur HS, Raman SK, Patil SA, Jain V. 2023. Mycobacteriophage D29 Lysin B exhibits promising anti-mycobacterial activity against drug-resistant Mycobacterium tuberculosis. Microbiol Spectr 11(6): e0459722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Yoshikawa M, Imamura D, Abe K, Eichenberger P, Sato T. 2020. Compatibility of Site-Specific Recombination Units between Mobile Genetic Elements. iScience 23(1): 100805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22): 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M, Bertoni D, Magana P, Paramval U, Pidruchna I, Radhakrishnan M, Tsenkov M, Nair S, Mirdita M, Yeo J, et al. 2023. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZL, Ye MQ, Zuo ZY, Liu ZG, Tai KC, Zou GL. 2006. Probing the importance of hydrogen bonds in the active site of the subtilisin nattokinase by site-directed mutagenesis and molecular dynamics simulation. Biochem J 395(3): 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39(Web Server issue): W347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This published article and its supplementary information files include all data generated or analyzed during this study.