

Short abstract
Recent progress in predicting protein structures has revealed a surprising abundance of proteins that are significantly unfolded under physiological conditions. Unstructured, flexible polypeptides are likely to be functionally important and may cause local cytoplasmic regions to become gel-like.
Abstract
Recent progress in predicting protein structures has revealed a surprising abundance of proteins that are significantly unfolded under physiological conditions. Unstructured, flexible polypeptides are likely to be functionally important and may cause local cytoplasmic regions to become gel-like.
In his 2001 book 'Cells, Gels and the Engines of Life' [1], Gerald Pollack gives a highly entertaining and accessible account of cell biology from the standpoint of polymer chemistry. The cytoplasm is indubitably gel-like, he says, so we must expect it to have similar properties to other non-living gels: cells should swell and shrink depending on ionic conditions and undergo dramatic phase changes associated with sol-to-gel transitions. In the broad sweep of published literature on living cells there is indeed abundant support for this thesis, and Pollack is not the first to advance such views. But probably no one else has made the argument so forcefully, or taken it so far. Too far, perhaps, for when Pollack challenges the role of the phospholipid bilayer as a permeability barrier, or offers new and dramatically simplified explanations for such well-understood phenomena as action potentials, muscle contraction and mitosis, he leaves most professional biologists behind [2]. This is unfortunate, since there is much we do not know about the physical conditions existing in the cytoplasm, and here Pollack says much that is relevant - one should not throw out the baby with the bath water.
Cytoplasmic gels are associated in my mind with long flexible polymers, which is why I thought about Pollack's book recently when reading about unstructured regions of proteins. We all learned as students that proteins are made as linear chains of peptide-linked amino acids that fold up into stable, evolutionarily-determined three-dimensional structures - right? In fact, wrong! It now appears that enormous numbers of proteins are significantly unfolded under physiological conditions. Some well-characterized proteins appear to be almost completely unstructured in solution, such as the actin-binding protein thymosin and the nucleoporins involved in nuclear transport [3,4]. But many more are now known to contain significant lengths of polypeptide chain that are 'natively unfolded' under conditions of neutral pH in vitro [5]. Indeed, the true extent of this phenomenon is only just being appreciated, with the introduction of search engines such as PONDR [6,7] developed by Keith Dunker and colleagues and more recently disEMBL [8,9] developed by Linding et al. These predictors are based on neural networks trained to recognize features such as the absence of regular structures, high B factors ('temperature factors' used as a measure of how much an atom vibrates about a position specified by a crystallographic model) and missing coordinates in X-ray diffraction data sets. When applied to test sets of known proteins, the predictors with significant accuracy pick out a variety of regions referred to in other contexts as protein 'loops' or 'linkers' or 'tethers'. Set loose on entire databases, the predictors find disorder everywhere.
The abundance of unfolded sequences is astonishing. According to a recent survey, about 5% of proteins in Escherichia coli, 23% in Arabidopsis and 28% in mouse are mostly disordered [10]. Evidently many sequences of this kind are disordered only part of the time, actually folding up when they bind to their target in the cell (often another protein, but sometimes a molecule of DNA or RNA) [11]. Flexible regions are useful if you have to assemble protein molecules into a large structure, such as a ribosome or a bacterial flagellum, since they allow subunits to wriggle into place rather than being forced into rigid holes. Flexibility is also advantageous if you want to recognize another protein quickly and without binding too tightly, since entropy ensures that your dissociation will be highly favored. Another intriguing role for unstructured regions of proteins is in intracellular signaling. Extended regions of polypeptide chain allow a protein to recognize more than one target or to be modified in multiple posttranslational forms: indeed, a more specialized sequence predictor has been used to find unstructured targets of phosphorylation [12,13]. There are also many examples in which lengths of polypeptide appear to exist solely as random chains in solution, serving as tethers that constrain the diffusive encounter of different proteins (or domains of the same protein), as elastic tethers or entropic springs.
Given their abundance, you might suppose that virtually every corner of a cell is full of writhing polypeptide chains. Well, it could be so... at least if we are allowed to generalize from one particularly well-understood volume of cytoplasm. The polar cluster of receptors that in Escherichia coli and related bacteria detects attractants and repellents contains five kinds of chemotaxis receptors associated on their cytoplasmic domains with six other proteins involved in downstream signaling. All of the proteins are known (we have a complete inventory), all have been sequenced, and atomic structures are available for all of the major domains [14]. The protein molecules - perhaps 20,000 in number -form a distinct 'compartment' of the cell, about 300 nm in diameter and perhaps 30 nm deep; and many of them have unstructured regions. The histidine kinase CheA, a large protein by bacterial standards, has two tethers per monomer (four per dimer) which link different domains and undoubtedly play a crucial part in the catalytic cycle. The recently discovered structure of CheZ, the protein that dephosphorylates CheYp and is associated with the receptor cluster, includes two unstructured regions, one long and the other short. Most impressive of all, the carboxy-terminal regions of the major receptor types, Tar and Tsr, consist of a length of perhaps 30 amino acids that so far as anyone can tell are without structure. These provide flexible tethers for enzymes involved in receptor methylation, namely CheR and CheB (Figure 1) [15].
Figure 1.
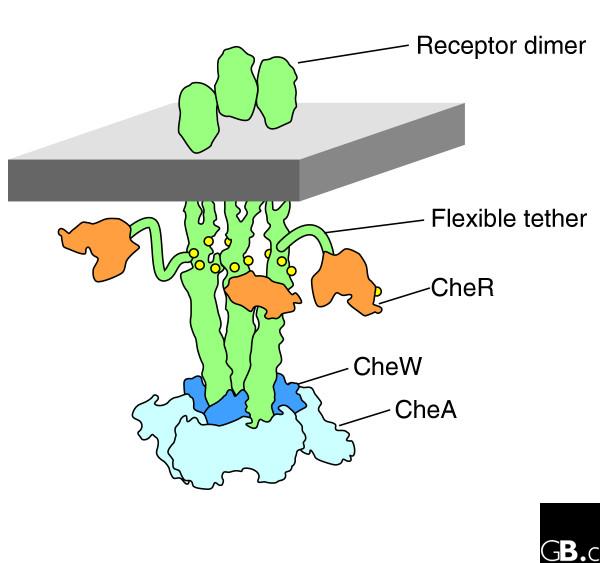
Flexible tails in chemotaxis receptors. Bacterial chemotaxis receptors such as Tsr and Tar are transmembrane homodimers with long α-helical coiled-coil domains. Sets of three dimers are associated at their cytoplasmic ends with downstream signaling proteins such as CheA and CheW. Carboxy-terminal regions of the receptors consist of about 30 amino acids with no obvious secondary structure and are thought to serve as flexible tethers for the methylating enzyme CheR and the demethylating enzyme CheB (not shown). Methylation sites are indicated by small circles. A typical cluster of receptors on the bacterial surface might contain several thousand such receptors in close proximity.
Quite apart from the functions of these various lengths of polypeptide there is the issue of how they are all crammed together. The concentration of proteins in the cytoplasm (20-30% by weight) is close to that at which some globular proteins crystallize. If these molecules are in addition flexing and writhing through the effects of thermal energy then a rich set of possible behaviors emerges. For example, if flexible polymer chains come close enough to make contact and restrict each other's motion then the 'solution' will inevitably become gel-like. Given the density of receptors and associated proteins in the polar cluster, it seems unavoidable that this criterion will be fulfilled, in at least some regions. It also follows that if, in the course of signal transmission, these same flexible chains move from a randomly diffusing state to one in which they fold up against a neighboring globular protein, physical conditions might change very quickly. Local regions of gel could rapidly transform into a sol-like state, and vice versa. The consequences of such transformations are difficult to predict, but they could be quite dramatic. We should at least consider some of the phenomena rehearsed in Pollack's book [1], such as changes in ionic balance, or the generation of localized currents of fluid.
A reversible association of flexible regions might also contribute to receptor function, allowing influences to spread across the cluster. Recent experiments show that the chemotaxis receptors in E. coli are not independent agents but that they cooperate, one consequence being that the sensitivity, or gain, of the system to small increments of attractants is much higher than it would otherwise be [16]. Various theories have been proposed to account for this effect, including models based on conventional allostery - the idea that the conformation of one receptor can influence that of its neighbors, rather like the influence of one domain of a hemoglobin molecule on another [17]. Another idea that has been suggested is that the signaling capabilities of receptors might depend on a change in the flexibility, or degree of order, of the receptors. An analysis of X-ray diffraction patterns of the bacterial serine receptor suggests that this is more highly ordered when completely methylated than in the half-methylated state [18]. Given that methylation normally accompanies an increase in activity in the system, this led to the suggestion that structural order and signaling activity are correlated. Binding of a ligand could therefore cause a change in receptor flexibility that then spreads within the cluster like discrete conformational changes.
Changes in protein flexibility associated with ligand binding have now been reported in a wide variety of other systems. The binding of the small molecule biotin to the bacterial protein streptavidin, for example, occurs with enormous affinity (estimated at 1013-1014 M). Analysis of this interaction using deuterium exchange with backbone hydrogens shows that as binding occurs, the streptavidin structure becomes better packed: there is a benefit in enthalpy but a cost in entropy [19]. In fact, from a thermodynamic standpoint, biotin binds very tightly precisely because it promotes better packing of the streptavidin structure. Moreover, a review of ligand-protein associations shows that a trade-off between enthalpy and entropy is very widespread. Some receptors work like the biotin-streptavidin system in that binding promotes the protein structure; the catalytic efficiency of many enzymes can also be increased in this way. But there are also instances in which a ligand binds to a protein with unfavorable enthalpy but favorable entropy change. Examples include delta opioid and thyrotropin receptors, adenosine G-protein-coupled receptors, and ionic channels receptors gated by 5-HT, serotonin, and GABA [20]. In all these cases, protein structure is in fact loosened by ligand binding, and a relatively weak association is driven mainly by entropic changes. Williams and colleagues [20] relate these changes to a previously described 'thermodynamic discrimination' shown by many different kinds of receptors: if agonist binding is predominantly enthalpy-driven then antagonist binding will be entropy-driven, and vice versa. Put simply: if you want two ligands to bind to the same receptor but produce radically different effects, you can achieve this through their effects on the structure of the receptor. Allow one to tighten up the structure, leading to better packing within the protein itself and conceivably increasing the degree of oligomerization; let the other ligand loosen the structure and perhaps reduce oligomerization.
I was sternly rebuked by a referee of an earlier version of this article for terminological inexactitude in using the terms 'flexibility', disorder', 'unstructured chains' and so on. Yes, I admit it: crystallographic temperature factors are not the same thing as structural disorder; nor are atomic vibrations about a fixed point the same as the writhing motions of flexible chains. But I would have thought them different parts of the same fish. Surely an amino acid with a less well-defined position in a protein in a crystal is more likely to flail around if that same molecule is diffusing freely in solution? And surely all kinds of motion will contribute to that long balance sheet of movements that constitutes changes in entropy? In the case of bacterial chemotaxis receptors, for example, we not only have an increase in apparent order with methylation, but cross-linking studies also indicate that periplasmic domains form a tighter structure in the absence of attractant [21]. Taken together with the extensive evidence from other kinds of receptors, this is at least consistent with the idea that attractant binding is entropy-driven whereas repellent binding is enthalpy-driven. Following this thread, we see that attractants might loosen up the structure not only of the receptor itself but also of any complexes made by the receptors and neighboring proteins. In particular, we can imagine interactions between receptors and the crucial kinase CheA being lessened, thereby accounting for the dramatic fall in downstream signaling produced by attractants. (There is indeed experimental evidence that the binding of CheA to receptors is reduced by attractant [22].) Repellents, in this picture, would do the opposite: lateral interactions between receptors should be strengthened and the activity of CheA associated with the receptors should rise. It is all very different from the conventional view of conformational changes between rigidly defined structures - it is more coarse-grained and mesoscopic. But perhaps propagating regions of disorder are a useful way to think about these large multiprotein structures? It would certainly encourage investigators to find out exactly what regions of the proteins are involved in entropic changes and whether, in the case of the bacterial polar cluster, the various unstructured loops linkers and tethers play a part. It is hard to say conclusively what happens at this point - but it all does seem reminiscent of the sol-to-gel transitions championed by Pollack [1].
References
- Pollack GH. Cells, Gels and the Engines of Life. Seattle WA: Ebner & Sons; 2001. [Google Scholar]
- Stossel TP. Manifesto for a cytoplasmic revolution. Science. 2001;293:611. [Google Scholar]
- Domanski M, Hertzog M, Coutant J, Gutsche-Perelroizen I, Bontems F, Carlier MF, Guittet E, van Heijenoort C. Coupling of folding and binding of thymosin beta4 upon interaction with monomeric actin monitored by nuclear magnetic resonance. J Biol Chem. 2004;279:23637–23645. doi: 10.1074/jbc.M311413200. [DOI] [PubMed] [Google Scholar]
- Denning DP, Patel SS, Uversky V, Fink AL, Rexach M. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc Natl Acad Sci USA. 2003;100:2450–2455. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Gillespie JR, Fink AL. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- PONDR (Predictions of Natural Disordered Regions) http://www.pondr.com
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- disEMBL (Intrinsic Protein Disorder Prediction) http://dis.embl.de
- Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically unstructured proteins: reassessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- Iakoucheva LM, Radivojac P, Brown CJ, O'Conner TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DISPHOS (Disorder Enhanced Phosphorylation) http://www.ist.temple.edu/DISPHOS
- Bourret RB, Stock AM. Molecular information processing: lessons from bacterial chemotaxis. J Biol Chem. 2002;277:9625–9628. doi: 10.1074/jbc.R100066200. [DOI] [PubMed] [Google Scholar]
- Li M, Hazelbauer GL. Adaptational assistance in clusters of bacterial chemoreceptors. Mol Microbiol. [DOI] [PubMed]
- Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- Bray D, Duke T. Conformatonal spread: the propagation of allosteric states in large multipotein complexes. Annu Rev Biophys Biomol Struct. 2004;33:53–73. doi: 10.1146/annurev.biophys.33.110502.132703. [DOI] [PubMed] [Google Scholar]
- Kim S-H, Wang W, Kim KK. Dynamic and clustering model of bacterial chemotaxis receptors: structural basis for signaling and high sensitivity. Proc Natl Acad Sci USA. 2002;99:11611–11615. doi: 10.1073/pnas.132376499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DH, Stephens E, Zhou M. Ligand binding energy and catalytic efficiency from improved packing within receptors and enzymes. J Mol Biol. 2003;329:389–399. doi: 10.1016/S0022-2836(03)00428-5. [DOI] [PubMed] [Google Scholar]
- Williams DH, O'Brien DP, Sandercock AM, Stephens E. Order changes within receptor systems upon ligand binding: receptor tightening/oligomerisation and the interpretation of binding parameters. J Mol Biol. 2004;340:373–383. doi: 10.1016/j.jmb.2004.04.056. [DOI] [PubMed] [Google Scholar]
- Homma M, Shiomi D, Homma M, Kawagishi I. Attractant binding alters arrangement of chemoreceptor dimers within its cluster at a cell pole. Proc Natl Acad Sci USA. 2004;101:3462–3467. doi: 10.1073/pnas.0306660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Weis RM. Covalent modification regulates ligand binding to receptor complexes in the chemosensory system of Escherichia coli. Cell. 2000;100:357–365. doi: 10.1016/S0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]