

Abstract
Despite robust literature associating IL-31 with pruritic inflammatory skin diseases, its influence on cutaneous inflammation and on the interplay between inflammatory and neurosensory pathways remain unmapped. Here, we examined the consequences of disrupting Il31 and its receptor Il31ra in a mouse model of house dust mite (HDM)-induced allergic dermatitis. Il31-deficient mice displayed a deficit in HDM dermatitis-associated scratching, consistent with its well-established role as a pruritogen. In contrast, Il31 deficiency increased the number and proportion of cutaneous type 2 cytokine-producing CD4+ T cells and serum IgE in response to HDM prior to the onset of scratching. Furthermore, Il4ra+ monocytes and macrophages capable of fueling a feedforward type 2 inflammatory loop were selectively enriched in Il31ra-deficient HDM dermatitis skin. Thus, IL-31 is not strictly a pro-inflammatory cytokine, but rather an immunoregulatory factor that limits the magnitude of type 2 inflammatory responses in skin. Our data support a model wherein IL-31 activation of IL31RA+ pruritoceptors triggers release of calcitonin gene-related protein (CGRP), which can mediate neurogenic inflammation, inhibit CD4+ T cell proliferation, and reduce T cell production of the type 2 cytokine IL-13. Together, these results illustrate a previously-unrecognized neuroimmune pathway that constrains type 2 tissue inflammation in the setting of chronic cutaneous allergen exposure, and may explain paradoxical dermatitis flares in atopic patients treated with anti-IL31RA therapy.
One Sentence Summary:
IL31-responsive afferents that mediate allergic itch also induce neurogenic inflammation to limit type 2 inflammatory programs in skin.
Introduction:
Many cytokines are produced during type 2 skin inflammation including the IL-6/gp130-family cytokine IL-31. IL-31 stands out for its ability to directly engage its heterodimeric receptor, IL31RA/OSMRβ, on cutaneous itch-provoking sensory neurons (pruritoceptors), as well as some subsets of epithelial and hematopoietic cells (1, 2). This expression pattern links skin inflammation with neurosensory pathways that mediate itch, and suggests that IL-31 could be a promising therapeutic target in a growing number of itchy inflammatory skin diseases including atopic dermatitis (AD) and prurigo nodularis (3, 4).
Transgenic mice that constitutively overexpress Il31 in T cells (IL31Tg) develop spontaneous itch so severe that they develop scratching-induced skin lesions(1), demonstrating that T cell-derived IL-31 is sufficient to trigger pruritus. Scratching induced by hapten-mediated dermatitis is ameliorated in Il31-deficient animals, confirming that Il31 is necessary for contact hypersensitivity-associated itch(5). As intradermal or intrathecal administration of recombinant IL-31 induces acute-onset scratching(2, 6), it is likely that the pruritogenic effect of IL-31 results from direct activation of IL31RA/OSMRβ on a subset of pruritoceptive afferents marked by the calcium channel TRPV1(2). Other sensory afferent functions, such as neurogenic inflammation, have not been examined with respect to IL-31.
The skin pathology that develops spontaneously in IL31Tg animals suggests that IL-31 also has pro-inflammatory functions (1). However, contributions of IL-31 to tissue inflammation are difficult to discern in this model, because IL-31 effects on cutaneous immune cells and keratinocytes cannot be dissociated from the secondary effects of scratching-induced skin injury. In fact, enhanced rather than diminished type 2 inflammatory responses were observed in lung and gut in Il31ra-deficient mouse strains, indicating that IL-31 can function as a negative regulator of type 2 inflammation(7–9). There are multiple potential explanations for these seemingly-discordant results. First, IL31Tg skin and Il31ra-deficient lung and gut phenotypes may reflect physiological differences specific to each organ(9). Additionally, without scratching-induced skin injury as a confounder, Il31ra-deficiency may unmask inhibitory effects of IL-31 on tissue inflammation. It has also been suggested that amplified type 2 inflammatory phenotypes observed in Il31ra-deficient animals may occur as a consequence of liberating its binding partner, OSMRβ, and thus enhancing signaling via its other ligands(10). A final, untested model could invoke differential contributions of IL31RA+ neural and non-neural pathways engaged by IL-31. For example, in some patients and/or disease states, circuits activated by IL-31-dependent sensory afferents may dominate over those mediated by cutaneous epithelial or inflammatory cells (e.g. prurigo nodularis), or vice versa.
Whereas IL-31-dependent itch-sensory pathways have been well-characterized (11, 12), the contributions of IL-31 to cutaneous inflammation remain unclear. Addressing this knowledge gap is particularly important for understanding chronic AD and its variants, the clinical context in which Il31 expression is most often detected(13, 14). Here, we report the consequences of perturbing IL-31-dependent pathways by inducing chronic allergic skin inflammation in Il31-deficient mice.
Results:
Decreased spontaneous scratching in HDM-treated IL31KO reveals a selective sensory deficit
We generated Il31-deficient mice from Il31tm1e(EUCOMM)Wtsi C57Bl6/N ES cells provided by the European Conditional Mouse Mutagenesis (EUCOMM) Program(15) (Fig. 1A and S1A–C). Consistent with previous reports(1, 16), we detected Il31 mRNA in in vitro-differentiated Th1- and Th2-polarized CD4+ T cell subsets from C57Bl/6 wildtype (WT) mice (Fig. S1E, filled bars) and used this in vitro system to confirm that Il31 mRNA was undetectable in CD4+ T cells from Il31null/null mice (hereafter ‘IL31KO’), validating the mutant allele (Fig. S1E, unfilled bars). Similarly, intracellular staining with an IL-31-specific antibody detected robust IL-31 production in restimulated WT Th1 and Th2 cultured cells, but none in IL31KO cultures (Fig. 1B). Expression of lineage-defining cytokines in Th1 (IFN-γ) and Th2 (IL-4, IL-13) cells was unaffected by Il31 deficiency (Fig. S1E), indicating that lineage specification of Th cells does not depend on IL-31. In Th2 cultures, IL-31+ cells were enriched among IL-4/IL-13 producing cells (Fig. 1C), demonstrating that IL-31 is a component of the Th2 effector cytokine program.
Fig. 1. HDM-sensitized IL31KO mice display a deficit in spontaneous scratching.
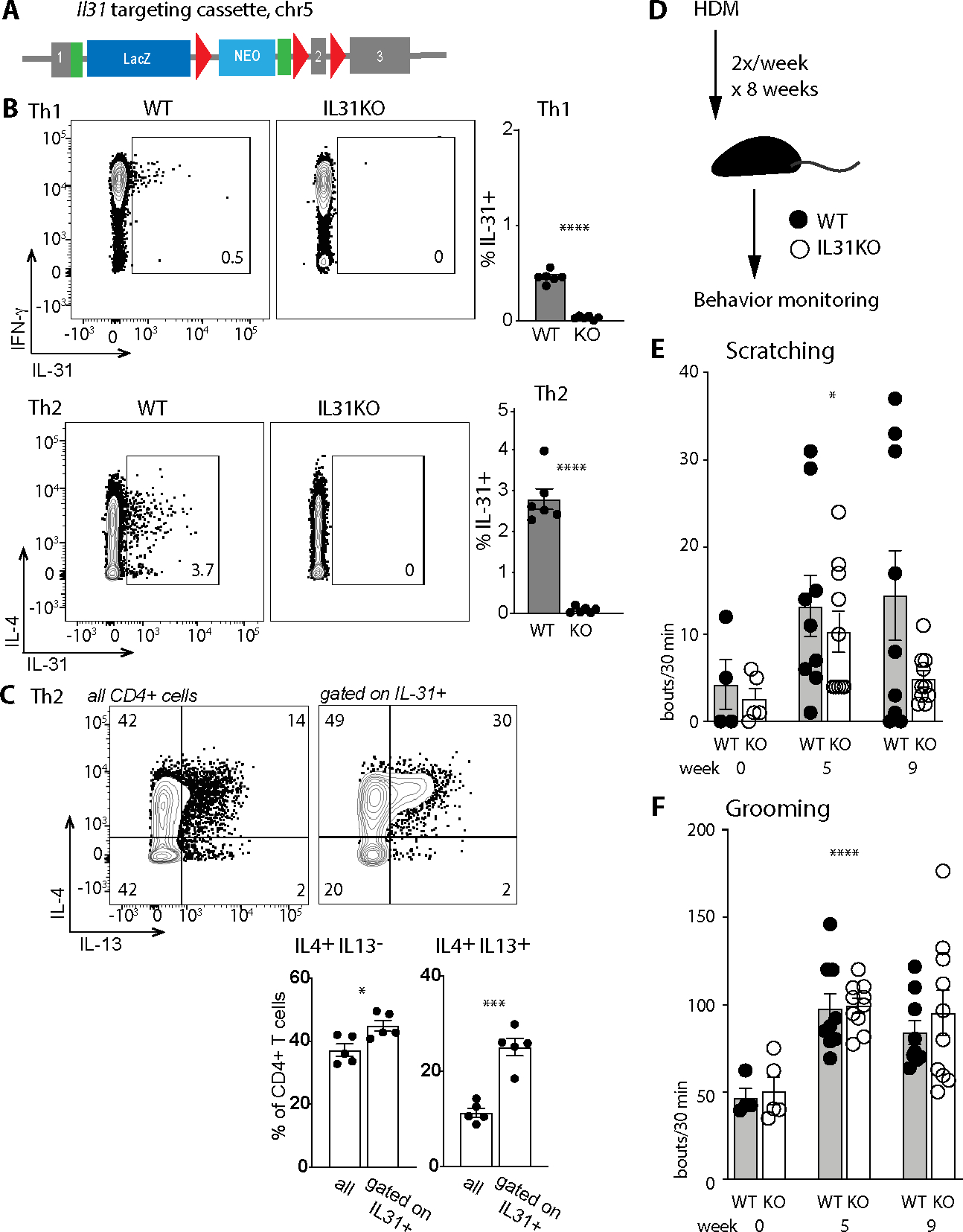
(A) Schematic of the Il31 targeting construct (Il31tm1e(EUCOMM)Wtsi, MGI 1923649). The transgene interrupts the endogenous 3-exon Il31 locus (mm10 ch5:123480419–123482049, minus strand) at position 123481232, introducing loxp sites flanking exon 2. Genetic cross of this transgenic mouse to a β-actin Cre recombinase-expressing strain results in a frameshift mutation and generation of a nonsense transcript (see also Fig. S1).
(B) Representative flow cytometry plots for intracellular cytokine staining of in vitro-differentiated Th1 and Th2 WT and IL31KO lymph node CD4+ T cells. Gates indicate percent IL-31+ of live CD4+ cells.
(C) Representative flow cytometry plots of IL-4 and IL-13 expression in in vitro-differentiated WT Th2 CD4+ T cells (left panel all cells; right panel gated on IL-31+ cells as in B).
(D) Schematic of the 8-week chronic HDM epicutaneous sensitization strategy; behavior monitoring was performed in week 0 (before treatment), week 5, and week 9.
(E) Spontaneous scratching bouts (hindpaw) per 30 minute window, measured serially in a cohort of WT and IL31KO animals at week 0 (before treatment), week 5, and week 9 (after completion of treatment), ANOVA p= 0.0196 (*).
(F) Spontaneous grooming bouts (forepaw) per 30 minutes, scored in the same videos as B. ANOVA p<0.0001 (****).
Black circles, WT; open circles, IL31KO. For B-C, data are representative of 3 independent experiments and datapoints reflect biological replicates, n=3 mice per genotype per condition. For D-F, data are pooled from 2 independent experiments, each with n ≥4 mice per genotype. Each dot represents an individual mouse. Statistical significance was determined by unpaired 2-tailed student’s t-test (B-C) or one-sided Welch’s ANOVA (E-F). Only significant p-values are noted. For all experiments, ns = p>0.05, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. Error bars show mean +/−SD.
To evaluate the contributions of endogenous IL-31 in vivo, we employed mouse models of dermatitis. Skin-innervating IL31RA+ pruritoceptors constitute the major non-hematopoietic population of IL-31-responsive cells. Therefore, to determine whether IL-31 is a necessary contributor to allergen-induced pruritoception, we evaluated behaviors attributable to itching (hindpaw scratching) versus grooming (forepaw wiping)(17) in a chronic Dermatophagoides farinae house dust mite (HDM) skin allergy model (Fig. 1D)(18, 19). Baseline scratching and grooming frequency did not differ between IL31KO and WT mice (Fig. 1E–F, week 0). In WT animals, HDM treatment induced an increase in scratching at week 5 and the majority of mice continued to scratch through week 9, consistent with previous reports(20). IL31KO animals also displayed increased spontaneous scratching at week 5, but by week 9 the frequency of scratching in these mice fell to near-baseline (Fig. 1E). In contrast, grooming frequency of IL31KO and WT mice tracked together throughout the HDM-treatment period (Fig. 1F), increasing similarly in HDM-treated animals of both genotypes. From these results, we conclude that IL-31 is not required for HDM dermatitis-induced scratching in the acute phase, but contributes later, when dermatitis and scratching have become chronic.
Il31 limits CD4+ T cell cytokine production and systemic markers of type 2 inflammation in HDM dermatitis
We assessed HDM-induced skin inflammation earlier, at week 5, a time point at which expression of Il31 and its receptor subunits (Il31ra and Osmr) were detectable in treated skin (Fig. S2A–B) and scratching-induced skin injury was not yet appreciable. HDM treatment induced IL-31 protein expression in a subset of WT skin CD4+ T cells (Fig. S2C, 2nd row middle column). Within the myeloid compartment, IL-31 was detected in a few eosinophils (SiglecF+CD11c- cells; Fig. S2D, left column) and CD11c+MHCII+ dendritic cells (Fig. S2D, middle column), but no cKit+ mast cells (Fig. S2D, right column). Histopathologic features of HDM-induced dermatitis included acanthosis, epithelial hypergranulosis, and mixed dermal infiltrates (Fig. 2A–B). All of these metrics increased in HDM-treated IL31KO and IL31RAKO compared to WT, with no significant differences between IL31KO and IL31RAKO skin (Fig. 2B). HDM induced comparable expansion of cutaneous CD45+ hematopoietic cells including eosinophils and T cell subsets (CD4, CD8 and dermal γδ T cells) in IL31KO and WT mice, as measured by flow cytometry (Fig. 2C and Fig. S2E; see Fig. S3 for gating strategy). HDM treatment also triggered selective expansion and/or influx of cutaneous Foxp3−CD4+ T-effector cells as compared to Foxp3+CD4+ regulatory T cells (Tregs); again this was comparable in WT and KO mice (Fig. S2F).
Fig. 2. Increased cytokine-producing CD4+ T cells distinguish dermal infiltrates in HDM-treated IL31KO animals.
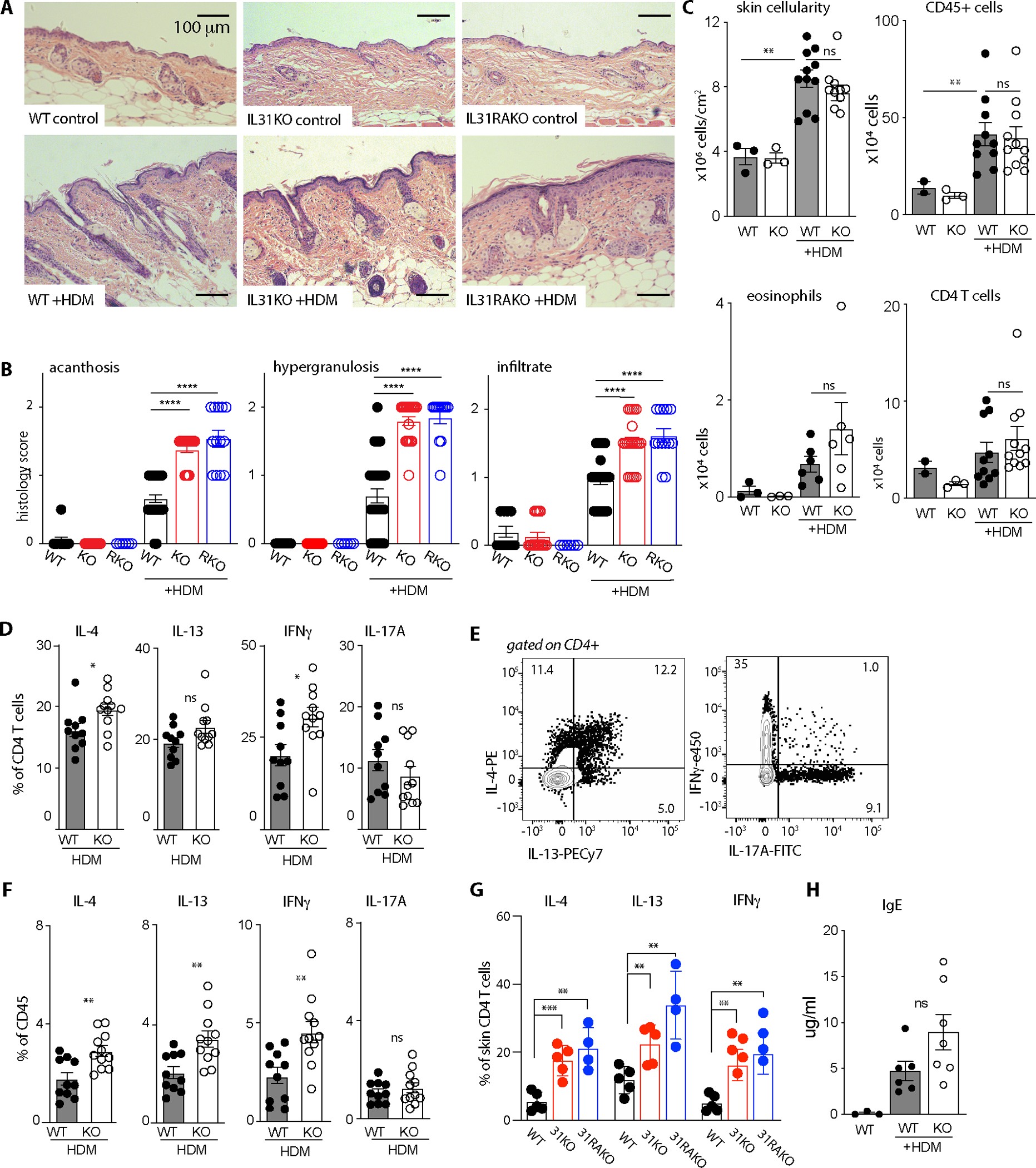
(A) Representative H&E-stained tissue sections from control and HDM-treated dorsal neck skin at 10x magnification. Scale bar = 100 μm
(B) Acanthosis (epithelial proliferation), epithelial hypergranulosis, and dermal inflammatory infiltrate scores for WT, IL31KO (KO), and IL31RAKO (RKO) mouse skin sections, control- or HDM-treated as indicated. Scoring metric: 0= none, 0.5=mild, 1=mild-moderate, 1.5= moderate, 2=severe. One-way Brown-Forsythe ANOVA p<0.0001 for all metrics; post-test pairwise p-values as indicated.
(C) Flow cytometry-based quantification of total cellularity (p<0.0001), CD45+ (hematopoietic) cells (p<0.05), CD11b+SiglecF+ eosinophils, and CD4+ T cells (p<0.01). Cells shown per cm2 section of dorsal neck skin. Representative of >3 experiments, with n=3 controls and n≥6 for HDM-treated animals. See Fig. S3 for gating strategy.
(D) Percent of HDM-treated skin CD4+ T cells that express IL-4, IL-13, IFN-γ, or IL-17A, as determined by flow cytometry after intracellular antibody staining. Pooled from 2 independent experiments including n=3 for controls and n≥6 mice for HDM. Significance was determined by unpaired 2-tailed student’s t-test.
(E) Concatenated flow cytometry plots demonstrate gating for IL-4, IL-13 (left), IFN-γ, and IL-17A intracellular cytokine stains (right).
(F) Cytokine expression data from D normalized by percent of CD45+ cells per sample.
(G) Percent of HDM-treated WT, IL31KO, and IL31RAKO skin CD4+ T cells that express IL-4 (p<0.01), IL-13 (p<0.01), or IFN-γ ( p<0.01) as determined by flow cytometry after intracellular antibody staining.
(H) ELISA-based quantification of serum IgE in WT or after 8 weeks of control- or HDM-treatment in WT and IL31KO. p<0.01.
All data points reflect unique biological replicates. Error bars displayed as mean +/−SD. For all experiments, ns = p>0.05, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. One-sided Welch’s ANOVA with multiple comparisons testing was performed except when indicated. WT, black circles; IL31KO, open circles.
In analogous models of allergic airway inflammation induced by intranasal HDM administration, CD4+ T cell-mediated inflammation is characterized by mixed Th1/Th2/Th17 infiltrates(21). Similarly, topical HDM treatment of dorsal neck skin resulted in expansion and/or recruitment of Th2, Th1 and Th17 cells in both WT and IL31KO mice (Fig. S2G). Importantly, however, HDM-treated IL31KO mice generated significantly greater proportions of IL-4 and IFN-γ- producing CD4+ T cells compared to WT controls (Fig. 2D–E, see Fig. S4 for full gating strategy). When calculated as a percentage of CD45+ cells to correct for variation in skin CD4+ T cell numbers in individual animals, IL-4-, IL-13-, and IFN-γ-producing cells all increased in HDM-treated IL31KO compared to WT (Fig. 2F). An increase in CD4+ T cell cytokine production was also observed in HDM-treated IL31RAKO (Fig. 2G). In control vehicle-treated animals of all genotypes, there were fewer CD4+ T cells and therefore very few cytokine-producing T cells (Fig. S2G).
To assess signs of systemic type 2 inflammation, we also measured serum IL-4 and IgE in HDM-treated WT and IL31KO animals. Serum IL-4 was above the limit of detection in 5 of 12 treated IL31KO mice but in none of 13 treated WT mice. This trend was reflected in the abundance of serum IgE, the production of which is dependent on IL-4. Serum IgE increased in HDM-treated WT animals compared to controls, and to an even greater degree in HDM-treated IL31KO animals (Fig. 2H). Overall, these results illustrate a role for IL-31 in restraining allergen-induced skin inflammation and IgE hyperproduction.
Taken together, the IL31KO phenotype of increased IL-4, IL-13 and IFN-γ cytokine production and increased serum IgE indicates that endogenous IL-31 attenuates skin inflammation. Given that we did not observe effects on the frequency of multiple IL31RA+ immune cell populations, these data suggest that IL-31 may act via other IL31RA+ populations such as neurons, to subsequently influence CD4+ effector T cell cytokine production.
Disproportionate expansion of M2 macrophages in HDM-treated Il31ra-deficient skin
IL-4- and IL-13-producing CD4+ T cells communicate bidirectionally with IL4RA+ type 2 myeloid cells to fuel type 2 tissue inflammation. Some of these myeloid cells also express IL31RA(1). Therefore, to characterize IL-31-dependent effects on type 2 signaling circuits involving cutaneous myeloid populations, including those directly responsive to IL-4/13, we performed single cell RNA-sequencing on sorted CD45+ cells from the skin of HDM-treated WT and Il31ra-deficient (IL31RAKO) mice. After concatenating WT and IL31RAKO sequencing files, we resolved a combined total of 16284 cells, 47% lymphoid (7509) and 53% (8775) myeloid (Fig. 3A). Within the myeloid compartment, we observed a 1.72-fold enrichment of IL31RAKO cells (5549 KO, 3226 WT), consistent with the exacerbated cutaneous inflammatory response to topical HDM observed in IL31KO animals by flow cytometry.
Fig. 3. Selective expansion of cutaneous M2 macrophage subsets in HDM-treated IL31RAKO skin.
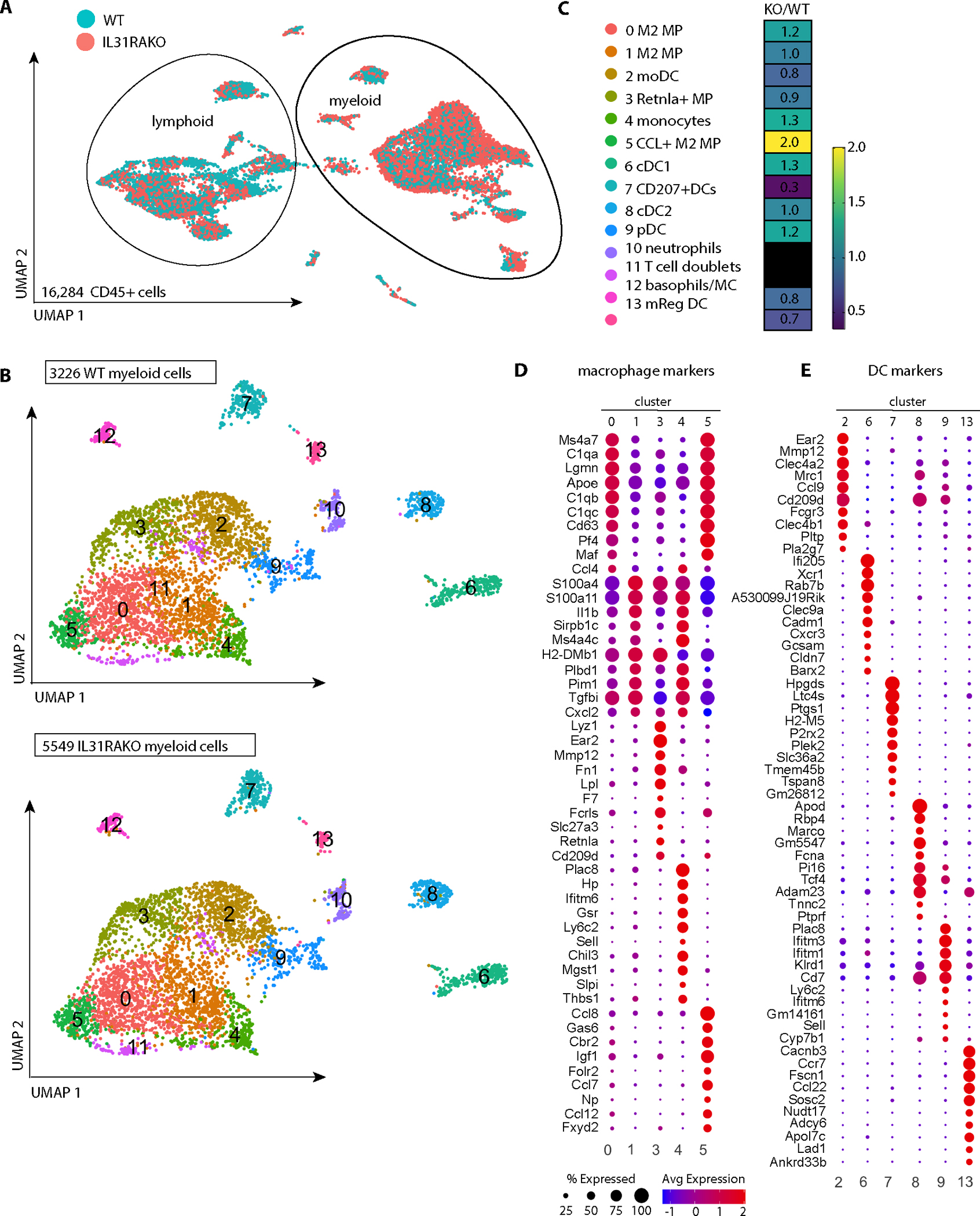
(A) UMAP plot of merged WT and IL31RAKO scRNA-seq files (combined total 16,284 cells) demonstrates overall clustering by lineage (lymphoid vs myeloid). Individual cells are displayed by genotype using the Seurat shuffle command. Turquoise = WT cells; salmon = IL31RAKO cells. IL31RAKO and WT scRNA-seq files were each generated from sorted CD45+ cells pooled from n=4 animals per group.
(B) UMAP plot after subclustering the myeloid cells indicated in A. Top: WT cells; Bottom: IL31RAKO cells. Cell identities of the 13 myeloid cell clusters (inset box) were determined by inspection of cell marker genes identified by differential gene expression (DEG) analysis (see Fig. S5).
(C) Heat map depicting proportional representation of IL31KO and WT cells in each myeloid cluster, where numbers reflect the ratio of IL31RAKO to WT cells per cluster after normalization to correct for the 1.72-fold greater number of total cells resolved in the IL31RAKO sequencing file.
(D) Dot plots depict expression of the top 10 DEGs for each macrophage cluster mapped across all macrophage clusters (clusters 0,1,3,4,5).
(E) Dot plots depict expression of the top 10 DEGs for each dendritic cell cluster (clusters 2,6,7,8,9,13) mapped across all DC clusters. As indicated by the legend, the diameter of each circle indicates the proportion of cells in that cluster that express the gene, and the heat map indicates average expression per cell in that cluster by read count.
Unsupervised subclustering analysis of the 8775 myeloid cells identified 13 distinct populations, including 5 monocyte-macrophage clusters, 6 dendritic cell (DC) clusters, and a co-cluster of mast cells and basophils (Fig. 3B–C; see Fig. S5 for cluster marker genes). IL31RAKO and WT cells were represented in all 13 clusters, but IL31RAKO cells were overrepresented in all but one: cluster 7 (Cd207+ DCs). To compare proportional representation of IL31RAKO and WT cells per cluster, we normalized to the overall ratio of IL31RAKO to WT myeloid cells (a factor of 1.72). After normalization, IL31RAKO cells were still overrepresented in multiple myeloid clusters, including two large macrophage populations (cluster 5, 2.0-fold) and cluster 0, 1.2-fold) that together account for 2,101 cells, or 23.9% of total myeloid cells sequenced (Fig. 3C).
Among DC subsets, IL-4-responsive plasmacytoid DCs (pDCs) (cluster 9, 1.2-fold) and cDC1s (cluster 6, 1.3-fold) were also modestly enriched, whereas mRegDCs (22) (cluster 13, 0.7-fold) were relatively depleted in IL31RAKO skin (Fig. 3C). HDM response factor Clec10a (23) was expressed by multiple macrophage and DC subsets, but unaffected by IL31RA deficiency (Fig. S6A). We only resolved Il31ra and Osmr transcripts on rare cells (Fig. S6B). Oncostatin M (Osm) was expressed in cells of multiple clusters, with no appreciable differences between IL31RAKO and WT (Fig. S6C).
We used differentially expressed gene (DEG) analysis to define markers for monocyte-macrophage and dendritic cell clusters (Fig. 3D–E). The macrophage subsets specifically enriched in IL31RAKO skin (clusters 0 and 5) were highly similar to one another, and distinct from other macrophage clusters (Fig. 3D). Genes whose selective expression drove this clustering included Ms4a7, Lgmn, Apoe, Cd63, Pf4 (Cxcl4), and Maf (Fig. 3D). Cluster 0 and 5 cells also expressed high levels of all 3 subunits of the complement component C1q (C1qa, C1qb, and C1qc), reflective of IL-4-driven type 2 macrophage (M2) differentiation (24). Additional cluster 0 and 5 markers were enriched in other macrophages (clusters 1 and 3) and monocytes (cluster 4), but at lower read counts and in smaller percentages of cells (Fig. 3D).
We next examined macrophage subsets for expression of M2 differentiation markers. Transcripts encoding the canonical M2 marker Arg1 were rare (Fig. S6D), and the M1 marker Nos2 was undetectable. However, multiple macrophage clusters contained a large proportion of cells that expressed Mrc1 (mannose receptor, CD206) and Sepp1, genes also associated with M2 macrophage differentiation (Fig. 4A)(25), suggesting that most macrophages in HDM-treated skin adopt an M2 fate. Nearly all cluster 5 macrophages expressed abundant Mrc1 and Sepp1, and additional M2 markers (hemoglobin scavenger receptor Cd163 and Folr2) were also enriched in this population (25) (Fig. 4A). Retnla (Fizz1) was enriched in cluster 3 macrophages (Fig. 4B), which were equally represented in IL31RAKO and WT samples (Fig. 3C). Altogether, these scRNA-seq results highlight heterogeneous subsets of M2 macrophages in HDM-induced dermatitis skin, most limited by IL-31.
Fig. 4. Macrophage subsets enriched in HDM-treated IL31RAKO skin are distinguished by transcripts encoding M2 markers, CCR2 ligands and IL4RA.
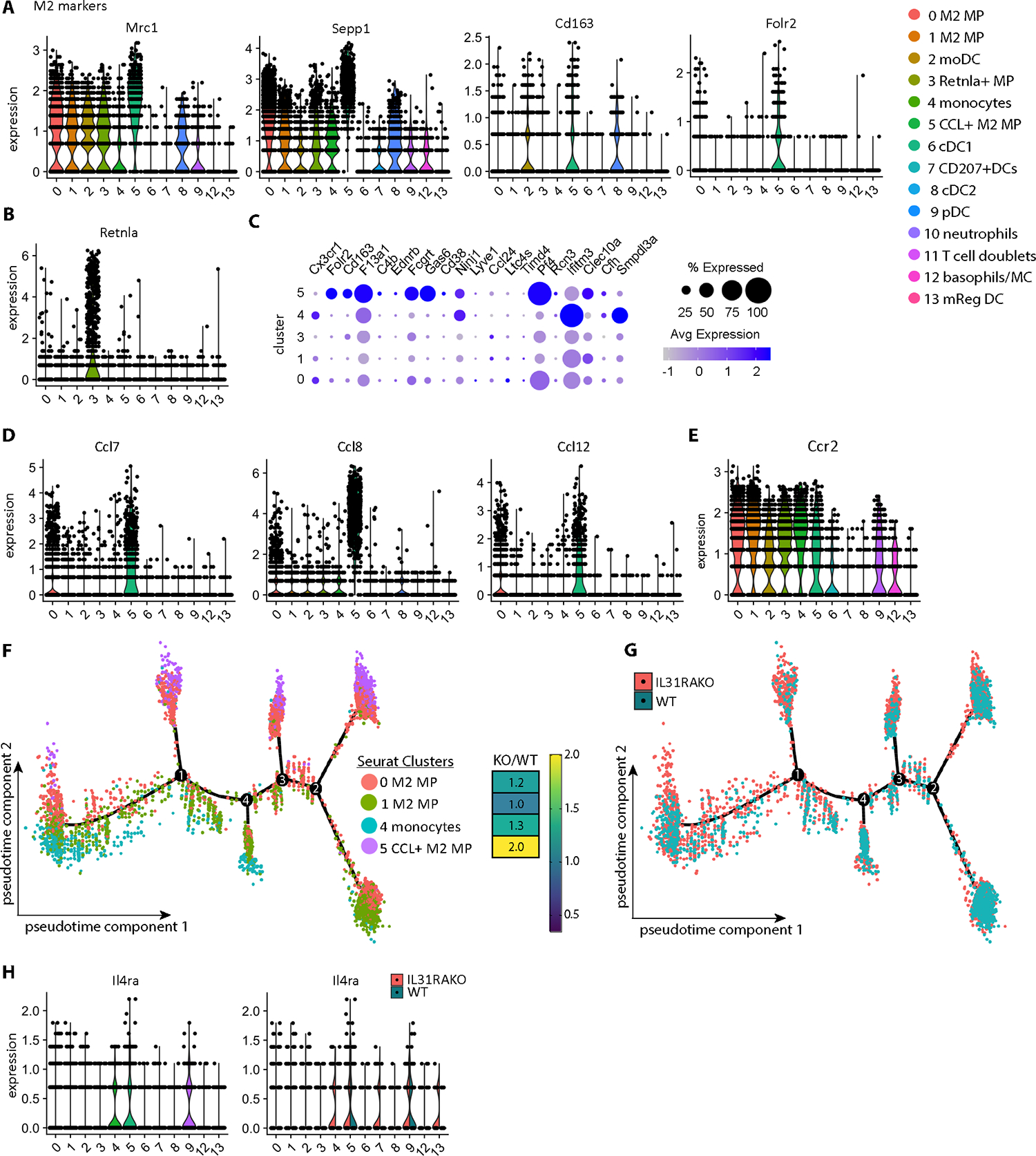
(A) Violin plots depicting expression of M2 markers Mrc1, Sepp1, Cd163, and Folr2 mapped across myeloid clusters demonstrate enriched expression in multiple macrophage clusters, including 0 and 5.
(B) Violin plot depicting expression of M2 marker gene Rentla (Fizz1).
(C) Dot plot depicting expression of markers for Cx3cr1low tissue macrophages* in our scRNAseq macrophage clusters. *Genelist extracted from Chakarov et al, Table S3(26)).
(D) Additional cluster 5 markers include Ccl7, Ccl8, and Ccl12, CCR2 ligands encoded in a chemokine cluster on mouse chromosome 11.
(E) Ccr2 is broadly expressed by macrophages (clusters 0,1,3,4,5), monocyte-derived DCs (cluster 2), pDCs (cluster 9), and basophils/mast cells (cluster 12).
(F) Monocle pseudotime plot depicts the developmental trajectory between monocyte and macrophage subpopulations (Seurat clusters 0, 1, 4, 5). DEG markers and branchpoints (1, 2, 3, 4) computationally defined by Monocle are available in Data file S1. Dots indicate individual cells colored to reflect their Seurat cluster identity.
(G) Monocle pseudotime plot as in F. Dots indicate individual cells colored to reflect genetic identify (WT or IL31RAKO).
(H) Violin plot (left) highlights expression of Il4ra in cluster 5 (CCL+ macrophages) and cluster 9 (pDCs). Split violin plots (right) reveal additional Il4ra expression in IL31RAKO cluster 4 (monocytes), cluster 7 (Cd207+DCs) and cluster 13 (mReg DCs) cells.
IL-31 dampens a feed-forward loop that amplifies M2 macrophages
The expanded macrophage populations in Il31-deficient skin likely derive from both tissue-resident macrophages and from recruited monocytes that subsequently differentiate in skin. Based upon common expression of Maf, Mrc1, Sepp1, and Cd163 in previously-described Cx3cr1low tissue-resident macrophages, we hypothesize that clusters 0 and/or 5 most closely approximate this subset(26–28). Cluster 5 macrophages are also enriched in expression of additional tissue-resident macrophage signature genes (Fig. 4C) (26).
However, cluster 5-specific DEG markers also included Mertk ligand Gas6 (Fig. S6E), and Ccl7, Ccl8, and Ccl12 (Fig. 4D; see also Fig. 3D). The latter encode monocyte chemoattractant ligands for CCR2, a chemokine receptor broadly expressed by monocytes, macrophages and monocyte-derived dendritic cells (Fig. 4E). Based upon the two-fold enrichment of cluster 5 Ccl7+Ccl8+Ccl12+ macrophages in IL31RAKO skin, we hypothesized that the lack of IL-31 signaling in IL31RAKO mice also enables a feedforward loop whereby M2-polarized cutaneous macrophage populations expand in response to HDM by recruiting CCR2+ monocytes (cluster 4, 1.3-fold) as precursors from peripheral blood.
This hypothesis was supported by Monocle analysis of cells in Seurat clusters 0, 1, 4, and 5 (Fig. 4F; see also Fig. S6F). Two component pseudotime projection indicated a developmental relationship between cluster 4 monocytes (concentrated at lower left) and macrophage subsets. Chemokine-expressing cluster 5 macrophages (concentrated at upper right) occupied the most differentiated gene expression states. These states were defined by the M2 macrophage marker Folr2 as well as the chemokines themselves (Ccl7, Ccl8) (Fig. S6G and Data file S1). IL31RAKO macrophages preferentially occupied these differentiated states in the pseudotime trajectory (Fig. 4G), consistent with increased IL-4 and IL-13 driving type 2 macrophage accumulation.
The IL-4/IL-13 receptor, Il4ra, was most highly expressed in cluster 5 macrophages and cluster 9 pDCs, both of which are expanded in IL31RAKO skin (Fig. 4H; see also Fig. S7A–B for Il4ra detection in Folr2+ or Ccl8+ cluster 5 macrophages in HDM dermatitis skin). In addition, Il4ra expression was increased in IL31RAKO cluster 4 monocytes (Fig. 4H and Fig. S7C, right column), cluster 7 Cd207+DCs, and cluster 13 mReg DCs, suggesting another way that IL31RA deficiency may increase responsiveness to type 2 cytokines in the myeloid cell compartment.
Overall, this comparative scRNA-seq analysis of IL31RAKO and WT HDM-treated skin infiltrates revealed a role for IL31RA signaling in controlling myeloid inflammation in allergen-exposed skin, in particular by limiting IL-4/IL-13-responsive M2 macrophage subsets. We suggest that one of these control mechanisms may be through IL-31-dependent signals limiting a chemokine-dependent feedforward loop that otherwise perpetuates expansion and differentiation of cutaneous allergen-induced IL-4-responsive myeloid cells.
IL-31 and HDM activate narrowly-overlapping subsets of TRPV1+ sensory afferents
Our scratching data suggest differential pruritoceptive responses to HDM in the presence and absence of IL-31 (Fig. 1D–E). Therefore, we hypothesized that pathologic expansion of type 2 cutaneous immune cell populations (Th2 cells and M2 macrophages) in Il31- and/or Il31ra-deficiency arises as a consequence of altered sensory neuron-mediated pathways in these animals.
First we mapped dorsal root ganglion (DRG) neuron responsiveness to HDM and IL-31. Prior studies separately reported that both HDM-responsive neurons and IL-31-responsive neurons are capsaicin-responsive, i.e. express calcium channel TRPV1 (2, 29). We previously observed that both HDM dermatitis and overexpression of F2rl1 (which encodes PAR2, a receptor for HDM proteases) increase Il31ra transcription in trigeminal ganglia neurons(20). This suggests overlap between IL-31-responsive and HDM-responsive populations in the non-peptidergic 3 (NP3) subset of TRPV1+ DRG sensory afferent neurons notable for transcription of IL-31 receptor subunits Il31ra and Osmr, F2rl1, and additional pruritogen receptor Mrgprx1 (Fig. 5A, excerpted from Usoskin et al(30)).
Fig. 5. HDM and IL-31 activate partially-overlapping subsets of TRPV1+ sensory afferents.
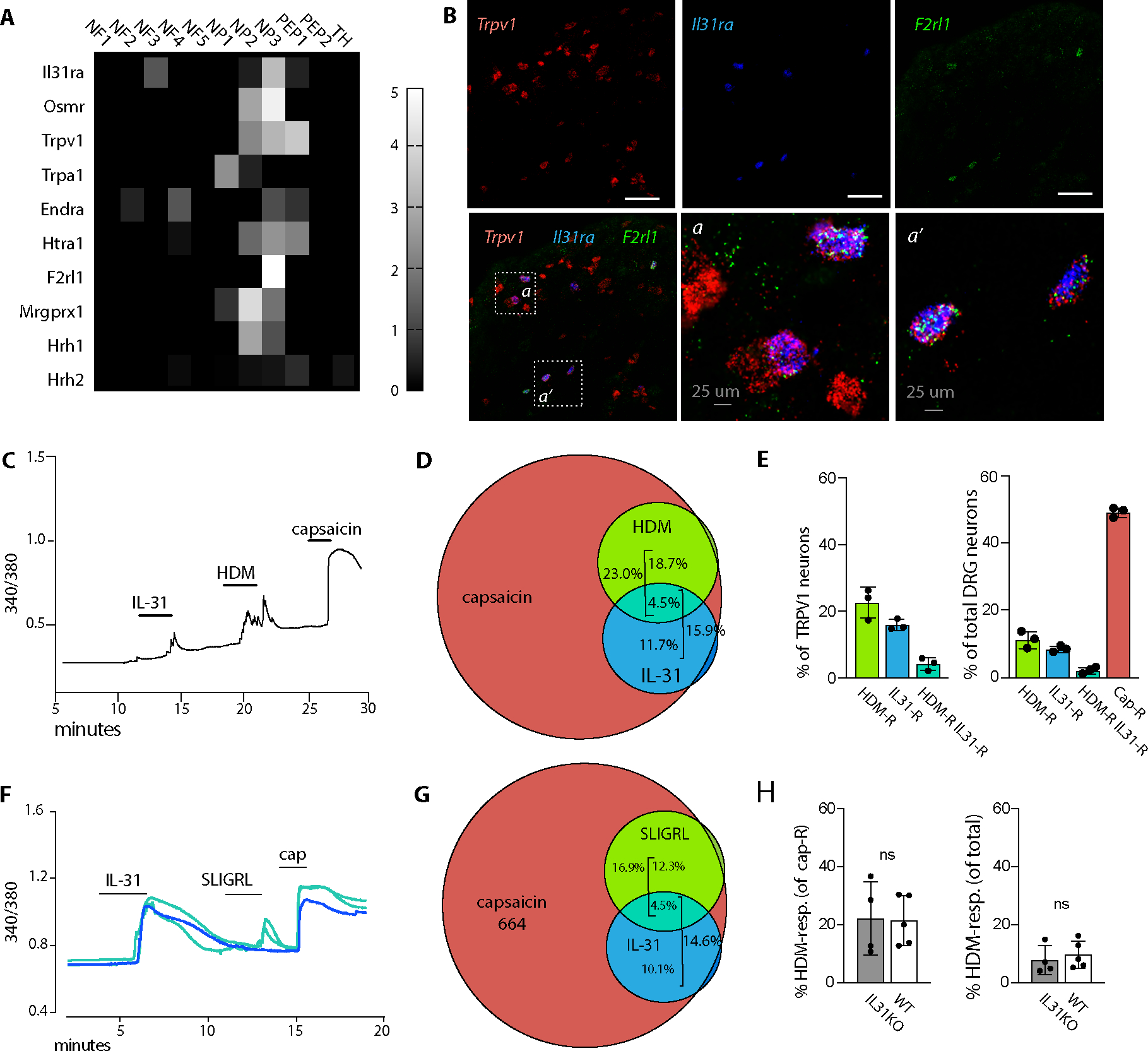
(A) Heat map depicts differential gene expression of select pruritogen receptors in dorsal root ganglia afferent neuron subsets, extracted from Usoskin et al(30). Scale indicates the maximum likelihood estimate of differential expression (log2fold change) in each DRG neuron subset calculated relative to all other DRG subsets, as described in Usoskin et al(30).
(B) Representative images of Trpv1, Il31ra, and F2rl1 in situ hybridization in trigeminal ganglia (TG) tissue sections from untreated animals.
(C) Representative 340/380 trace from calcium imaging of a Fura-2 loaded lumbar dorsal root ganglion (DRG) neuron double-responsive to IL-31 and HDM. Capsaicin response indicates that the neuron is TRPV1+.
(D) Venn diagram indicating the proportion of WT DRG neurons responding to each of the indicated ligands (capsaicin, HDM, and recombinant IL-31). Capsaicin response marks TRPV1+ neurons.
(E) Quantification of DRG neurons responsive to the indicated ligands. Each dot represents the average % responding cells from at least 4 coverslips from a single animal. Left panel TRPV1+ (capsaicin-responsive), right side all neurons.
(F) Representative 340/380 traces from calcium imaging of Fura-2 loaded lumbar DRG neurons that were monoresponsive to IL-31 (blue) or double-responsive to IL-31 and SLIGRL (turquoise). Capsaicin (cap) responses indicate TRPV1+ neurons.
(G) Venn diagram indicating the proportion of WT DRG neurons responding to each of the indicated ligands (capsaicin, SLIGR, and IL-31).
(H) Quantification of % DRG neurons responsive to HDM in WT (black bars) or IL31KO (open bars). Each dot represents the average % responding cells from at least 4 coverslips from a single animal.
Numbers indicate total responding neurons recorded from at least 4 mice per genotype (D, E, G); and 5 each of WT mice and IL31KO mice (H). Error bars displayed as mean +/−SD.
DRG sensory afferents that co-express Trpv1, Il31ra and F2rl1 were readily identified by in situ hybridization (Fig. 5B). Functional analysis by calcium imaging identified HDM and IL-31 double responsive neurons (Fig. 5C), but they comprised a minority (4.5%) of capsaicin-responsive (TRPV1+) neurons, and just 2.1% of total DRG neurons (Fig. 5D–E). However, substantial populations of TRPV1+ neurons responded solely to IL-31 (11.7%) or HDM (18.7%) (Fig. 5D–E), suggesting that central itch and pain-generating circuits activated by IL-31 and HDM are (for the most part) initiated by different primary afferents.
Analogous experiments using IL-31 and the PAR2 peptide agonist SLIGRL (in place of HDM) yielded consistent results: a high frequency of single-responsive neurons but few double-responsive neurons, where almost all (single- and double-responsive) fell within the capsaicin-responsive cohort (Fig. 5F–G). Collectively, DRG afferents responsive to IL-31 and/or HDM fall predominantly within the capsaicin-responsive set (Fig. 5D–E, G), consistent with prior reports (29, 31). However, there were no pre-existing differences in the frequency of HDM-responsive neurons in WT versus IL31KO (Fig. 5H) to explain differences we observed in HDM dermatitis scratching (Fig. 1D–E) and inflammatory infiltrates (Fig. 2–4).
IL-31 induces neurogenic inflammation suggestive of CGRP-mediated effects on tissue vasculature
We next hypothesized that Il31/Il31ra-dependent limits on HDM-induced expansion of cutaneous type 2 immune cell infiltrates may occur secondary to other functions of sensory neurons, such as neurogenic inflammation. TRPV1+ HDM-responsive neurons can induce neurogenic inflammation characterized by substance P (SP)-mediated vascular leak leading to mast cell degranulation and cDC2 migration to the draining lymph node(29, 32). TRPV1+ IL31RA+ NP3 afferents may also be capable of neurogenic inflammation, as they express multiple neuropeptide transcripts: somatostatin (Sst), neuropeptide b (Nppb), CGRP (Calca) and SP (Tac1) (Fig. 6A, excerpted from Usoskin et al(30)). The pattern of neuropeptide gene expression in this IL31RA+ population therefore aligns with the isolectin B4-reactive subset of paradoxically-named ‘non-peptidergic’ CGRP+ TRPV1+ afferents(33).
Figure 6. IL-31-induced neuronal CGRP release reduces T cell proliferation and IL-13 production.
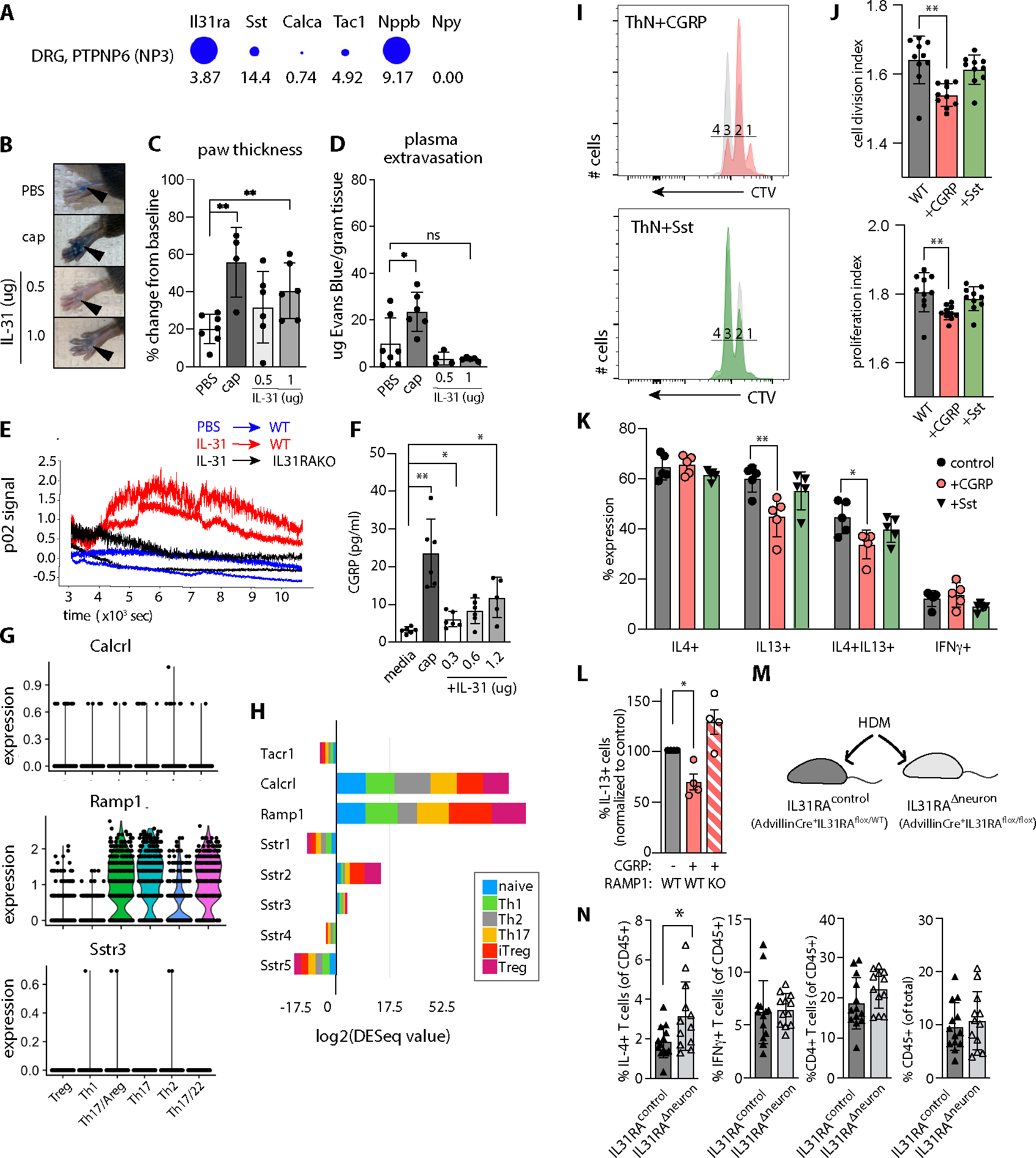
A) Il31ra and neuropeptide expression in NP3 DRG neurons. Diameter indicates proportion of cells expressing the gene, numbers indicate expression relative to other sequenced cells in the dataset. Data extracted from (30, 62) using mousebrain.org visualization tools.
B) Mouse hindpaw images after retro-orbital Evans Blue injection and plantar paw injection with PBS, capsaicin (cap) or recombinant mouse(rm) IL-31. Note: Focal blue injection site staining of stratum corneum is expected.
C) Percent change in paw thickness immediately prior to and 15 minutes after PBS, capsaicin, or rmIL-31 intradermal paw injection. Mixed-effects model testing p=0.0282 (*), pairwise post-test probabilities n.s.
D) Quantification of Evans Blue dye in paw tissue harvested 15 minutes after PBS, cap, or rmIL-31 intradermal injection. Mixed-effects model testing p=0.0123 (*), post-test probabilities nonsignificant.
E) Quantification of local blood flow measured by Oxyflow probe applied to mouse paw, immediately after injection of IL-31 or PBS.
F) CGRP concentration in TRG neuron culture media after 30 minute incubation with capsaicin or rmIL-31. Representative of 3 experiments. Welch’s one-way ANOVA 0.0006; post-test probabilities as indicated.
G) Neuropeptide receptor expression in HDM-treated skin CD4+ T cell subsets. Tacr1 not detected.
H) Neuropeptide receptor expression in in vitro-polarized CD4+ T cell subsets, data extracted from ThExpress(37).
I) CellTrace Violet (CTV) dilution histograms as a measure of mouse lymph node CD4+ T cell proliferation in culture. Colored histograms depict T cells cultured in the presence of neuropeptides as indicated (CGRP orange, Sst green), overlaid on paired controls (grey). Numbers indicate cell division gates where 1 = undivided cells.
J) Cell division index (upper panel) and proliferation index (lower panel) of CTV-labeled CD4+ T cells cultured +/−neuropeptides as indicated. Representative of 3 experiments. Wilcoxon matched-pairs signed rank test for division index p=0.0003, proliferation index p=0.0037).
K) Effector cytokine expression in in vitro-differentiated CD4+ T cells cultured +/− neuropeptides: CGRP (orange bars), somatostatin (green bars), or no neuropeptide (control, grey bars). IL-4+ (left panel, n.s.), IL-13+ (left center; one-way Welch’s ANOVA p=0.0019, pairwise post-test significance as indicated), IL-4+IL-13+ (right center; one-way Welch’s ANOVA p=0.0042, pairwise post-test significance as indicated), or IFNγ+ (right, n.s.).
L) Percent IL-13+ CD4+ T cells in in vitro-differentiated WT or RAMP1KO T cellscultured in the presence or absence of CGRP. Brown-Forsythe one-sided ANOVA, p=0.0108 (*).
M) Advillin-Cre+.Il31raflox/flox (Il31raΔneuron) mice exhibit conditional ablation of Il31ra in Advillin+ cells (sensory and sympathetic neurons). Advillin-Cre+.Il31raflox/WT littermates were used as controls for HDM dermatitis (N).
N) Flow cytometry-based quantification of skin cell populations from HDM-treated Advillin-Cre+IL31raflox/WT (=IL31raflox/WT) and Advillin+IL31raflox/flox (=IL31Δneuron) mouse dorsal skin: IFN-γ+ CD4+ T cells, IL-13+ CD4+ T cells, total CD4+ T cells, CD45+ cells. Representative of 2 experiments, n≥5/group for HDM-treated animals. Significance determined by unpaired 2-tailed student’s t-test. See Fig. S3 for gating strategy.
Data points reflect biological replicates. Error bars displayed as mean +/−SD. For all experiments, ns = p>0.05, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
Intradermal injection of recombinant IL-31 (rIL31, Fig. 6B) provoked paw swelling (Fig. 6C) but not extravasation of Evans Blue labeled plasma albumin (Fig. 6D). These results suggest that IL-31 can selectively boost local blood flow (vasodilatation, an indication of CGRP activity) without increasing vascular permeability (an indication of SP activity). Intradermal rIL31 injection also induced a rapid increase in doppler p02 signal, a metric for blood flow, in an IL31RA-dependent manner (Fig. 6E). Together, these findings suggest that IL-31 injection is sufficient to trigger neurogenic inflammation via peripheral sensory neuron release of CGRP but not SP (34, 35). Moreover, IL-31 treatment of short-term primary mouse trigeminal ganglion (TRG) neuron cultures induced a dose-dependent release of CGRP at concentrations comparable to previous studies (Fig. 6F)(36). Thus, IL-31 can directly trigger CGRP release from sensory neurons.
CD4+ T cells express neuropeptide receptors and respond to CGRP by decreasing proliferation and cytokine production
To determine whether CD4+ T cells could be a direct target of neuropeptides, we examined their neuropeptide receptor gene expression. In our HDM-treated skin CD45+ scRNA-seq dataset, we detected expression of Sstr3 and CGRP coreceptors Ramp1 and Calca (Fig. 6G), but not other somatostatin receptors or the SP receptor Tacr1 (not shown). These in vivo data correspond to neuropeptide receptor gene expression in in vitro-differentiated CD4+ T cell subsets, where Sstr2 was additionally detected (Fig. 6H; excerpted from ThExpress(37)).
Neuropeptide receptor expression patterns indicated that CGRP and/or Sst may signal directly to CD4+ T cells in HDM-treated skin. Consistent with prior studies (38–40), CGRP supplementation reduced T cell proliferation in a CD4+ T cell culture system that favors Th2 cell differentiation (41, 42), when measured either by cell division index (average number of cell divisions that a cell in the original population has undergone) or cell proliferation index (total number of divisions divided by the number of dividing cells) (Fig. 6I–J). In addition, CGRP decreased the proportion of IL-13-producing CD4+ T cells (Fig. 6K), an effect that was lost in T cells from mice lacking the CGRP receptor subunit Ramp1 (Fig. 6L). Somatostatin had no appreciable effects on proliferation or cytokine production.
Sensory neurons mediate IL-31-dependent regulation of allergic skin inflammation
We returned to the HDM model to validate the in vivo disease relevance of our working model, which posits that IL-31-responsive neurons are responsible for CGRP-mediated blunting of CD4+ effector T cell functions in skin. First, we ablated the broad class of TRPV1+ sensory neurons with resiniferatoxin (RTX), which should remove both a TRPV1+SP+ subset known to enhance inflammation (29) plus the TRPV1+IL31RA+ CGRP+ subset that may reduce effector CD4+ T cell responses. In our topical HDM model, the net result of RTX-mediated TRPV1 afferent ablation was marked reduction in cytokine-producing skin CD4+ T cells (Fig. S8A). This result demonstrates that the TRPV1+SP+ neuron-mast cell pathway dominates the TRPV1+CGRP+ neuron-T cell pathways described here. Yet, as these two subsets of TRPV1+ neurons are distinct(30), and IL31RA+ cells comprise only a minor fraction of all TRPV1+ neurons, this result does not rule out a separate pathway mediated by IL-31/IL31RA.
To isolate the pathway specifically dependent on IL-31-responsive neurons (rather than all TRPV1+ neurons), we generated a conditional mouse strain whose sensory neurons lack Il31ra: Advillin-Cre+.Il31raflox/flox (denoted as Il31raΔneuron, Fig. 6M and S8B–E). In the HDM dermatitis model, Il31raΔneuron mouse skin accumulated significantly more type 2 cytokine-producing CD4+ T cells than Advillin-Cre+IL31raflox/WT littermate controls (Fig. 6N, left panel). Based upon the consistent increase of skin type 2 CD4+ T cells in IL31KO, IL31RAKO, and now also Il31raΔneuron dermatitis skin, we conclude that this IL-31-mediated pathway requires IL31RA+ sensory neurons.
Discussion:
Here, we demonstrate that IL-31 activates sensory neuron-mediated pathways of dual purpose in allergic dermatitis: to induce itch, and to stimulate neurogenic inflammation via CGRP release. Our experiments in sensory neuron-specific Il31ra mutant mice confirm the requirement for IL31RA+ sensory neurons in limiting cutaneous type 2 cell populations in this allergic dermatitis model. The capacity of IL-31 to incite CGRP release from sensory afferents is surprising and relevant to understanding the exacerbated inflammatory phenotypes we and others have observed in IL31KO or IL31RAKO in various type 2 inflammatory disease models (5, 7–9). We found that CGRP acts directly on CD4+ T cells to limit their proliferation and IL-13 cytokine production. Conversely, absence of IL-31-activated neuroimmune feedback in IL31KO/IL31RAKO mice resulted in excessive recruitment and/or expansion of type 2 cytokine-producing CD4+ T cells and IL-4/13-responsive myeloid cells in a dermatitis model. Thus, the net effect of IL-31 and CGRP on type 2 immune cell populations is immunoregulatory rather than pro-inflammatory. Other cytokines with the capacity to communicate directly or indirectly with both cutaneous inflammatory cells and sensory neurons, including the epithelial alarmin TSLP and IL-4, enhance both cutaneous inflammation and pruritus (43, 44). IL-31, on the other hand, enhances pruritoception but limits allergic inflammation.
Our sequencing data demonstrated that IL31RA+ sensory afferents express transcripts for several neuropeptides and have peptidergic activity in vivo (Fig. 6A,C,E). IL-31 is sufficient for CGRP release from primary neurons in vitro and cutaneous vascular changes attributable to CGRP (arteriolar vasodilatation (34)) in vivo. CGRP also inhibits proliferation of lymphocytes (38) including Th2-polarized CD4+ cells(39), as we observed. However, its directional effects on CD4+ T cell and innate lymphoid cell cytokine production appear to differ by inflammatory context in vivo(39), where CGRP receptor subunit Ramp1 is also expressed by non-lymphocyte populations including vascular endothelial cells(46). Here, in chronic HDM dermatitis, our in vivo and in vitro data support a model whereby IL-31 induces sensory afferents to release CGRP, whose downstream effects include direct signaling in CD4+ T cells to limit their proliferation and production of the type 2 cytokine IL-13. While our experiments in sensory neuron lineage-specific Il31ra conditional mutant mice revealed a necessary contribution of IL-31-responsive IL31RA+ neurons to limiting type 2 inflammatory responses, we cannot rule out additional contributions from myeloid populations or other cell types that express IL31RA/OSMRβ. Future experiments will be needed to investigate the direct and indirect consequences of CGRP/RAMP1 signaling to T cells in vitro and in vivo in HDM dermatitis and other inflammatory skin disease models.
Others have described HDM-responsive TRPV1+ sensory neurons that promote allergic inflammation via SP acting on mast cells and dendritic cells(29, 32). Consistent with this, our experiments in RTX-treated mice reveal that TRPV1+ neuron pathways also promote type 2 CD4+ T cell accumulation (Fig. S8A). However, our additional data from sensory neuron-specific IL31RA mutant mice (Fig. 6M–N) revealed that the TRPV1+SP+ sensory neurons that respond to HDM by secreting SP to activate mast cells(29) are different from the TRPV1+IL31RA+CGRP+ neurons described here, which respond to IL-31 by secreting CGRP to modulate CD4+ T cell activity. Hence IL31RA marks a distinct - and counterbalancing - subset of TRPV1+ sensory neurons that constrain cutaneous allergic inflammation.
ScRNA-seq also uncovered an Il31ra-dependent expansion of skin-infiltrating myeloid cells, with disproportionate increases in highly phagocytic and chemokine-producing macrophage subsets (25) (47–49). Although these macrophage subsets (clusters 0, 1 and 5) did not express transcripts for the canonical markers of either M1 or M2 fates, M2 macrophages with similar gene signatures (Sepp1, Mrc1, Cd163, Cbr2, Folr2) have been described in the setting of atherosclerosis (25) and in the oxazolone model of dermatitis (47–49). Il4ra was expressed in multiple macrophage populations in HDM dermatitis skin, including those that expand preferentially in the absence of IL-31/IL31RA signaling. In addition, skin-infiltrating monocytes expressed more Il4ra in IL31RAKO mice. This pattern of expression suggests that an IL-4 and/or IL-13-mediated feedforward loop may preferentially promote monocyte recruitment and differentiation into type 2 macrophages in IL31KO/IL31RAKO skin. Overall, the macrophage differentiation trajectory appears to be fueled by a combination of increased IL-4/IL-13 production by cutaneous T cells and increased responsiveness of myeloid cells to these cytokines.
Independently-derived Il31-deficient mice also exhibited decreased scratching in contact dermatitis models(5). Although no significant alterations in serum cytokine production were reported, IFN-γ and TNF-α production trended toward increases in those Il31-deficient mice, consistent with results presented here. Differences in the magnitude of our findings likely reflect the acute versus chronic nature of cutaneous allergen exposure and sensitivity differences between cytokine detection techniques. Here, cytokine detection by intracellular antibody staining for flow cytometry allowed us to evaluate CD4+ T cell cytokine production at both single cell and population perspectives.
How the IL-31/IL31RA pathway described here contributes to disease processes in atopic dermatitis, lichen amyloidosis, prurigo nodularis, psoriasis, and other conditions associated with IL-31 remains to be determined(4, 50, 51). Given the translational relevance of these conclusions to clinical dermatology, we note similar results from anti-IL31RA (Nemolizumab) clinical trials for atopic dermatitis (3, 52, 53). Nemolizumab-treated AD patients experienced rapid improvement in pruritus but delayed improvements in dermatitis metrics, paradoxical dermatitis flares (19–21% vs 13% placebo in phase IIB(54); 23–24% vs 21% placebo in phase III(3)), and dose-dependent asthma exacerbations (total 14 patients in Phase IIB (54)). The phase III study also uncovered “abnormal cytokine” levels (7% treatment vs 0% placebo), and elevated serum levels of the IL-4-responsive epithelial cytokine TARC, with an ~3-fold mean increase from baseline in all Nemolizumab-treated AD patients (3, 53). Overall impressive clinical improvement in Nemolizumab-treated AD patients despite these increased inflammatory markers in a sizable subgroup suggests functional dominance of IL31RA+ pruritoceptive pathways in this disease. We interpret these clinical data as consistent with the complex phenotypic profile of our Il31-deficient animals, in which pruritus and dermatitis pathways are similarly (and pathologically) decoupled.
Materials and Methods:
Study Design
The objective of this study was to elucidate the effects of IL-31 signaling on the intersecting processes of cutaneous inflammation and itch, which are both active during allergic and atopic skin inflammation in genetically-predisposed individuals. To do so, we employed an established mouse model for allergic dermatitis induced by serial exposure to house dust mite, a common environmental allergen. As readouts for Il31- and Il31ra-dependent differences in cutaneous inflammation, we used a combination of flow cytometry and scRNA-seq of skin infiltrates. To detect alterations in sensory neuron responses, we performed spontaneous behavior assays, dorsal root ganglion in situ hybridization, calcium imaging, and primary neuron culture assays. Details of these experimental methods are delineated in this section.
Animal studies and institutional ethics approval
WT and KO animals in our colony were age- and sex-matched, then randomly assigned to control vs treatment groups. For all experiments, we used mixed cohorts of male and female mice (aged 8–12 weeks for in vivo experiments; aged 4.5–6 weeks for in vitro T cell culture assays). In age-matching, a maximum tolerance of 2 weeks age difference was considered acceptable. A power calculation was used to determine sample sizes needed for in vivo dermatitis models. Groups of 5–6 mice/HDM or OVA treatment group were used, providing <10% chance of improper rejection of the null hypothesis (no difference in the mean frequency of infiltrating inflammatory cells in any two sets of mice), assuming the difference to be measured is (at minimum) a doubling of the number of rare inflammatory immune cell subsets from 1% to 2% with a standard error of 0.5% in each group. These assumptions are consistent with our own observations and with published data on dermatitis models.
Euthanasia procedures and experimental protocols were approved by the University of California, San Francisco IACUC committee (protocols AN183584–01B and AN183265–01E), and performed in accordance with University of California, San Francisco IACUC Committee guidelines and regulations.
IL31KO transgene design and transgenic animal screening
The IL31tm1e(EUCOMM)Wtsi transgene allele interrupts the endogenous Il31 locus (mm10 ch5:123480419–123482049, minus strand) at position 123481232; it was generated by the European Mutant Mouse Consortium (EUCOMM) according to the insertional mutagenesis scheme described by Skarnes et al(15). The IL31tm1e(EUCOMM)Wtsi transgene contains a neomycin-selection cassette flanked by FRT sites, and paired loxp sites flanking Il31 exon 2. When excised by serial genetic crosses to Flp-recombinase- and β-actin-Cre recombinase-expressing mouse strains, the resulting IL31null allele causes a frameshift mutation and generation of a nonsense transcript.
C57Bl/6 frozen embryos containing the IL31tm1a(EUCOMM)Wtsi transgene were obtained from EUCOMM via the European Mutant Mouse Archive (EMMA). Founder pups were generated in the UCSF transgenesis core facility by transgenic embryo implantation into pseudopregnant females. Pups were screened for WT Il31 and mutant Il31tm1a(EUCOMM)Wtsi transgene alleles with the following PCR primer pairs:
IL31 tm1a(EUCOMM)Wtsi CGAGAAGTTCCTATTCCGAAGT and GGAGGTTATCTGTGTCTCCATC
IL31WT allele GCAGCCAAAGATGTCCTAGT and TAGCCATCTGCAACCTGTTC
IL31null CGAGAAGTTCCTATTCCGAAGT and ACTTACTCAAGGTTATGCAGCT
IL31RAflox transgene design and transgenic animal crosses to generate IL31RAΔneuron mice
The IL31RAtm1a(EUCOMM)Hmgu transgene allele interrupts the endogenous Il31ra locus (GRC m38 ch13: 112649439–112717266 bp, minus strand) at position 112685666; it was generated by the European Mutant Mouse Consortium (EUCOMM) according to the insertional mutagenesis scheme described by Skarnes et al(15). The IL31RAtm1a(EUCOMM)Hmgu transgene contains a neomycin-selection cassette flanked by FRT sites, and paired loxp sites flanking Il31ra exon 6. When excised by serial genetic crosses to Flp-recombinase- and the sensory neuron lineage-specific Cre-recombinase-expressing mouse strain Advillin-Cre, the resulting IL31RAflox allele is excised, causing a frameshift mutation and generation of a nonsense transcript (IL31RAΔneuron) in Advillin-Cre-expressing cells and their progeny. The following primer pair detects both IL31RA WT (240 bp product) and IL31RAflox (388 bp product) alleles: F primer GGA ACT TAC TGT AGA GCA GGC, R primer ACA GAT TGC CCA AAG TCC AG.
In vitro-differentiated primary mouse CD4 T cell cultures
Bulk Th1, ThN, and Th2-polarized CD4 T cell cultures were generated by in vitro-differentiation of purified IL31KO or WT lymph node T cells, as previously described(10, 55). Recombinant mouse neuropeptides (1 uM CGRP or 0.1 uM Sst, Sigma) were added as indicated.
CellTrace Violet Proliferation Assay
Purified lymph node CD4 T cells were labeled with CellTrace Violet (CTV) per the manufacturer’s instructions (Thermo), then cultured for 36 hours, as above. CTV signal was detected by flow cytometry (BD Fortessa), as described below. Cell Division Index and Proliferation Index were calculated using Flojo Proliferation Platform tools.
Chronic topical HDM sensitization
Sensitization with HDM ointment (Biostir AD, active ingredient Dermatophagoides farinae) from Biostir, Inc. (Osaka, Japan) was performed per published protocol(18, 20). In brief, shaved skin was treated twice-weekly with 4% SDS in PBS solution followed by Biostir ointment application after 2 hours. For behavior and serum cytokine experiments cheek skin was sensitized for 8 weeks, analysis in week 9. For flow cytometry and histology, dorsal neck skin was sensitized 4 weeks then harvested in week 5. Toenails were clipped weekly from week 3 onward to prevent scratching-induced skin injury.
Behavior monitoring and analysis
Scratching bouts per 30-minute window of videorecording were tabulated by a blinded observer, following a standard protocol validated by Lamotte(17). For HDM-treated animals, spontaneous scratching bouts were tabulated in week 5 and week 9, and at least 2 days following the previous HDM treatment, as previously described(20).
Skin digestion
Dorsal neck skin sections were minced then digested 1.5h in RPMI media (Sigma) supplemented with type XI collagenase and bovine deoxyribouclease I (Sigma). Enzymatic activity was quenched with fetal bovine serum (FBS, Omega Scientific) prior to mechanical dissociation by GentleMACS (Miltenyi), then filtered through 70 μm nylon fabric mesh.
Ex vivo activation of skin T cells
When indicated, cells were resuspended in DMEM media (Sigma) containing FBS, L-glutamine and Sodium Pyruvate (Sigma) and cultured for 4 hours at 37°C, 10% CO2 in the presence of phorbol 12-myristate 13-acetate, ionomycin, and Brefeldin A (Sigma).
Flow cytometry
Cells were stained with DAPI or fixable viability dye eFluor-780 (eBioscience) and fluorophore-conjugated anti-mouse antibodies to cell surface antigens. For intracellular cytokine staining, cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences), then permeabilized in eBioscience transcription factor staining kit buffer (Thermo). Samples were collected on an LSRII Fortessa (Beckton-Dickinson) cytometers equipped with FACS Diva software. Flow cytometry data analysis was performed with Flowjo 10 software (Treestar).
Antibodies for flow cytometry included: BUV395 or BV711-conjugated anti-mouse CD45 (BD, clone 30-F11); APC or AlexaFluor488-conjugated anti-mouse CD11b (eBiosciences; M1/70); PE-Cf594-conjugated anti-mouse CD3ζ (Biolegend, 145–2C11); PE-Cy7-conjugated anti-mouse CD4 (Biolegend, RM4–5); BV605-conjugated anti-mouse CD8α (Biolegend, 53–6.7); BV605-conjugated anti-mouse γδTCR (Biolegend, GL3), V450-conjugated anti-mouse Ly6C/G (BD Pharmingen; Gr-1); FITC or PE-conjugated anti-mouse SiglecF (BD Biosciences; E50–2440); PECy7-conjugated anti-mouse CD11c (Biolegend;N418); PerCP-Cy5.5-conjugated anti-mouse I-A/I-E (BD Pharmingen; M5/114.15.2); BV605-conjugated anti-mouse CD117 (c-Kit; BD Pharmingen; 2B8); BV785-conjugated CD90.2 (Biolegend; 30-H12); APC-conjugated anti-mouse IL-4 (eBiosciences; 11B11), PE-eFluor610-conjugated anti-mouse IL-13 (eBiosciences; eBio13A); eFluor450-conjugated anti-mouse IFN-γ (eBiosciences; XMG1.2); Alexa Fluor 488-conjugated anti-mouse IL-17A (eBiosciences; eBio17B7); PE-conjugated anti-mouse IL-31 (Biolegend; W17037B).
IgE and IL-4 ELISAs
For serum IgE quantification, MaxiSorp 96-well plates (Nunc) were coated with anti-mouse IgE antibody (R35–72, BD) and blocked with 1% BSA in PBS. Serum samples (1:25 dilution) and a serially-titrated mouse IgE standard (C48–2, BD) were incubated for 2h. After washing in PSBT, plates were incubated with biotin-conjugated anti-mouse IgE secondary antibody (R35–118, BD), washed, and incubated with HRP Streptavidin (Biolegend). For serum IL-4 quantification, plates were coated with anti-mouse IL-4 antibody (11B11, Biolegend), blocked as above. Serum samples (1:4 dilution) and a serially-titrated mouse IL-4 standard (Peprotech) were added and incubated for 2h. After washing as above, plates were incubated biotin-conjugated anti-mouse IL-4 secondary antibody (BVD6–24G2, BD), washed again, then incubated with HRP Streptavidin (Biolegend). After final washes, plates were developed with TMB substrate (Invitrogen), stopped with 2N H2SO4, and read at 450nm on a microplate reader (Molecular Devices).
Skin histology
Paraffin embedding, tissue sectioning and H&E staining of skin was performed by the UCSF Mouse Pathology core. Images were captured with a 10x objectives on a Zeiss AxioImager M2 microscope.
RNA preparation and qPCR
For whole-skin or trigeminal ganglia mRNA expression assays, RNA was extracted using the RNeasy Fibrous Tissue kit (Qiagen). For bulk CD4 T cell or bone marrow RNA extraction, 106 cells/sample were processed following the miRNeasy micro kit (Qiagen). After RNA quantification, cDNA was synthesized per Invitrogen Superscript III kit protocol (Thermo). Il31, Il4, Il13 and Ifng cDNA abundance in CD4 T cells was quantified by qPCR using published Taqman primer/probe sets(1, 56). Il31, Il31ra, and Osmr cDNA abundance in skin or trigeminal ganglia was quantified by SYBR Green qPCR and the following primers: Il31 (F GGATGTCAGCAGACGAATCAA, R TGACTTTCTCCAGATGTGCTATG), Il31ra (F AAGACTCGTGGGTTTCCTTTAG, R GTAAACAGGTCTGCAACCTCA), Osmr (F CCATCTGGGTGGAGAATTATAG, R CCATCTGGGTGGAGAATTATAG). Il31ra cDNA abundance in AdvillinCre+.IL31RAflox/flox and littermate control animals was validated using Taqman primers/probe designed to amplify Il31ra exon 6–7 (Thermo).
RNA preparation for scRNA-seq
HDM-treated dorsal neck skin from 4 pooled IL31RAKO and 4 pooled WT C57Bl/6 mice was digested and prepared for flow cytometry as above. The live (DAPI-negative) CD45+ cell fraction of each sample was sorted using a Moflo XDP (Beckman Coulter) directly into ice-cold 0.5% BSA in PBS, and immediately processed through the Chromium Single Cell 3′ v2 Library Kit (10X Genomics) per the manufacturer’s protocol by the UCSF Genomics Core Facility of the UCSF Institute for Human Genetics. Each channel was loaded with 30,000 cells per sample. The cells were then partitioned into Gel Beads in Emulsion in the instrument, where cell lysis and barcoded reverse transcription of RNA occurred, followed by amplification, shearing, and 5′ adaptor and sample index attachment. Libraries were sequenced on an Illumina HiSeq 4000. Single Cell 3′ libraries used standard Illumina sequencing primers for both sequencing and index reads and were run using paired-end sequencing with single indexing where Read 1 is 26 cycles and Read 2 is 98 cycles.
scRNA-seq Analysis
Initial post-processing and quality control including single-cell transcript alignment to mouse genome GRC38/mm10 were also performed by the UCSF Genomics Core Facility, using the 10X Cell Ranger package (Cell Ranger Version 3.1, 10X Genomics).
R package Seurat (version 3.2.2, Satija Lab(57)) was used for single cell transcriptome analysis. Expression matrices for WT and IL31RAKO were combined using the Seurat merge command. The SCTransform normalization method was used to control for between-sample sequencing depth variation. Cells were removed according to the following thresholds: <500 genes/cell or >5000 genes per cell, >20000 UMIs/cell, 10% mitochondrial content, <0.01% hemoglobin content. Among the cells retained, the effects of mitochondrial and ribosomal content were regressed out prior to clustering. Clustering was performed using the default Seurat parameters: clusters are identified using Shared Nearest Neighbors method then optimized using the original Louvain algorithm. Genes were excluded from the final dataset if they were expressed in fewer than 3 cells, resulting in a total of 15,592 genes. For differential expression (DEG) analysis between clusters, genes were detected if they are expressed in at least 10% of cells in a cluster with a log fold change of at least 0.25. For DEG analysis between comparison groups, genes were detected if expressed in at least 5% of cells in the group with a log fold change of at least 0.25. Monocle 3 pseudotime trajectory calculations were performed on the Seurat object, as previously described(58).
Myeloid cell clusters identified by inspection of the initial Seurat cluster DEGs were selected to generate a Myeloid Seurat object using Seurat’s subset command, and re-clustering was performed using SCTransform parameters as above. These DEGs were used as cell marker genes to assign myeloid cluster identities (Fig. S5). Neutrophils and a cluster containing predominantly T cell-myeloid doublets (identified by co-expression of genes encoding subunits and Cd209/DC-SIGN) were removed from subsequent analyses. For comparative analysis within macrophages or within dendritic cells, as depicted in Fig. 3D–E, additional Seurat objects were generated from the Myeloid Seurat object, and cluster markers genes for these new Seurat objects were identified using the same computational workflow and DEG detection parameters described above.
RNAscope in situ hybridization
In situ hybridization was performed on flash-frozen sections of trigeminal ganglia and dorsal neck skin using the RNAscope Fluorescent Multiplex in situ hybridization kit (ACD Biosciences), per manufacturer’s instructions. RNAscope probes (ACD Biosciences) included: IL31RA (C1, C2 and C3); OMSR (C3); TRPV1 (C1, C2 and C3); TRPA1 (C2); IL4RA (C1 and C2); F2RL1 (C1, C2 and C3); MRC1 (C1); FOLR2 (C2); CHIL3 (C2); CCL8 (C2) with Alexa-488, Atto-550 and Atto-647 dyes.
Ex vivo calcium imaging of DRG neurons
Lumbar spinal cord DRG from 6–10 week-old WT and IL31KO mice were collected, enzymatically-digested with collagenase II (Worthington), dispase II (Sigma) and papain (Worthington), then mechanically triturated as previously described(59). The resultant single cell suspension was plated on poly-D-lysine/laminin-coated coverslips (Corning Biocoat), incubated at 37 °C in 95% humidity (95%) and 5% CO2, and imaged within 16–36 h.
DRG cells were loaded with Fura-2-AM and 0.01% Pluronic F-127 (Invitrogen), then mounted in the open chamber of a Nikon Eclipse TE300 microscope for imaging. Cells were sequentially challenged by serial addition of the following agonists prepared in calcium Ringer’s solution: HDM extract (5 ng/ml, D. farinae, Stallergenes Greer) or SLIGRL (100 μM; R&D Systems), rmIL-31 (300 nM; Peprotech), and capsaicin (1 μM; Sigma), followed by KCl (50 mM; Sigma) to distinguish neurons from non-neural cells.
Fura-2-AM fluorescence was measured at 340 nm and 380 nm excitation and 530 nm emission. COOL LED PE300 was used as the excitation source. Images were acquired using a Nikon TE300 microscope fitted with a Hamamatsu ORCA-ER camera and MetaFluor v7.6.9 software (Molecular Devices). Images were analyzed using a custom journal in MetaMorph v7.6.9 software (Molecular Devices). Increased 340/380 ratio ≥0.2 over baseline counted as a positive response to agonist.
Blood flow measurements
An Oxyflo Pro™ (OX-OXYFLO) sensor was used to measure blood flow, per manufacturer instructions. Mice were weighed and anesthetized with an intraperitoneal injection of Avertin (Sigma) prepared in sterile water. Anesthesia was assessed by pinching hindpaws until mice showed no sign of reflex withdrawal. Mice were immobilized by taping to a solid surface and a miniature Oxyflo skin probe (MSP300NX) was taped to the right hindpaw. Recording started immediately after the Oxyflow probe was secured and continued until a stable baseline was established. Mice were then injected with 10 ul of either saline (vehicle control) or 500 ug or 1000 ug rmIL-31(Peprotech) on the plantar surface of the right hindpaw. The probe was immediately re-stabilized to limit motion artifact. Perfusion data (pO2) was collected at 10 Hz for a minimum of 15 minutes per animal, then extracted as a csv file for analysis in LabChart Pro Software. Raw data collected was in blood perfusion units (BPU); this was corrected per manufacturer instructions to account for backscatter of the laser light, which was the percentage of laser light remitted from the tissue relative to the total amount of laser light incident to the tissue. The corrected BPU signal for each animal was normalized to its 30-second baseline to optimize inter-animal comparisons. Timestamps for probe placement and injections were recorded in LabChart Pro Software.
Evans Blue extravasation and paw swelling quantification
Evans Blue (EB) solution was prepared fresh by dissolving Evans Blue (Sigma) at 12.5 mg/ml in sterile saline on the day of experiment. Animals were anaesthetized by Avertin injection as above. Baseline thickness of each hindpaw was measured by caliper prior to injection of any compound, then retro-orbital injection of 100 ul EB solution was performed. Although intravascular distribution of EB is rapid (within seconds of retro-orbital injection), mice were rested for 5 minutes to ensure EB distribution throughout the body. Mice were then injected with 10 ul of either vehicle or compound as above. After 15 minutes, thickness of both ipsilateral (treated) and contralateral (untreated) hindpaws was re-measured by caliper. Cervical dislocation was performed and a 4×4 mm segment of plantar hindpaw skin surrounding the injection site of both ipsilateral and contralateral sides was immediately dissected. Skin was placed in a 1.5 ml eppendorf tube and dessicated on a 50°C heat block for 24–48 hours. Dessicated tissue was weighed and pulverized to facilitate EB extraction. Tissue was then submerged in 100 ul dimethylformamide and transferred to a 55°C heat block for 48h incubation to extract EB. Absorbance of the supernatant was measured at 620 nm using a Nanodrop spectrophotometer. Dimethylformamide was used as a blank. Weight of EB per sample was calculated based upon a standard curve of serially-diluted fresh EB standard solution, and normalized to mg tissue and to extraction volume. These procedures were all performed for both ipsilateral and contralateral hindpaw for each animal. Weight of EB/g of tissue was calculated for ipsilateral (treated) side and normalized to the contralateral (untreated) side of the same animal.
TRPV1 sensory neuron ablation with resiniferatoxin was performed as previously described(60). Hot plate testing was performed as a functional assay for TRPV1 sensory neuron ablation, as previously described(61). Lack of behavioral reaction (no licking or wiping paws) on/before a cutoff 40 seconds confirmed successful RTX treatment.
Primary mouse sensory neuron culture and CGRP ELISA detection
Trigeminal ganglia were collected and digested with 1 mg/ml of Collagenase/Dispase (Millipore) in a 37°C shaking incubator at 150 rpm for 60 min. Cell suspensions were then serially triturated through decreasing diameter syringe needles (18G, 22G, 25G). The resultant cell suspension was filtered through a 70 μm cell strainer, pelleted by centrifugation, and resuspended in neurobasal media (Gibco). The cell suspension was then mixed with 15% Percoll in 1x HBSS (Sigma), centrifuged at 800 g for 20 min without brake, and resuspended in complete neurobasal media supplemented with N2, B27, and Glutamax (Gibco), 25ng/ml each of human β-NGF/BDNF/GDNF (Peprotech), and 10μM of arabinosylcytosine (Sigma).
Cells in complete neurobasal media were subsequently seeded at a density of 6 × 105 cells on a 13 mm plastic coverslip (Nunc Thermanox) precoated with 0.1% polyethyleneimine (PEI) and 300 μg/ml of Matrigel, and placed in a 24-well plate. After culture for 5–6 days at 37°C and 5% CO2, media was gently removed and replaced with 200 ul/well of neurobasal media containing: no additives (vehicle control), 1 μM capsaicin (Sigma), 300 nM, 600 nM or 1200 nM murine IL-31 (Peprotech), then incubated at 37°C for one hour. Supernatants was collected and assayed for CGRP concentration with the CGRP EIA kit (Cayman Chemical) according to manufacturer instructions.
Statistical testing and analysis
Data were analyzed using Prism 8 software (Graphpad) by comparison of means testing using unpaired two-tailed student’s t-tests when only two groups were compared, or by one-way Welch’s ANOVA when results of an intervention on two or more groups were examined and the data was not assumed to be normally distributed. A Brown-Forsythe ANOVA was used to test for equality of variance where data in one or more groups was skewed, as in Figs. 2B, 6L, S8E, S2B. Mixed effects model testing was applied when repeated measures were performed in matched groups, Figs. 6C–D. A two-tailed Wilcoxon matched-pairs signed rank test was used to determine significance between matched T cell culture samples +/− neuropeptide (Fig. 6J). Where Welch’s ANOVA, Brown-Forsythe ANOVA, or Wilcoxon signed-rank tests were employed, significance symbols indicate significant differences in post-hoc pairwise multiple comparisons testing. All error bars indicate mean ± standard deviation. All data points reflect unique biological replicates; no technical replicates or repeated measurements were performed except as indicated (Fig. 6C–D). For experiments where data collection required manual counting (behavior analysis, cell counting in in situ hybridization slides), scoring was performed by a lab member blinded to sample identity, genotype, and intervention/control.
Supplementary Material
Figure S1. Transgene schematic and serial crosses to generate Il31null allele
Figure S2. Supplementary data to accompany Figure 3
Figure S3. Gating strategy for cutaneous CD45+ cell subsets
Figure S4. Gating strategy for CD4 T cell intracellular cytokine detection
Figure S5. Cell Marker genes for the 13 myeloid clusters
Figure S6. Supplementary data to accompany Figures 4–5
Figure S7. RNAscope in situ hybridization of HDM skin sections demonstrates Il4ra expression in c5 macrophages and c4 monocytes.
Figure S8. Supplementary data regarding RTX-treated and IL31RAΔneuron mice.
Data file S1. Top 100 DEGs by Pseudotime, as defined by Monocle 2 analysis
Data file S2. Raw data file
Supplementary Methods
Acknowledgments:
We acknowledge Nico Ghilardi and Shannon Turley of Genentech (IL31RAKO mice), Biolegend (early access to anti-mouse IL-31 monoclonal antibody), the UCSF Flow Cytometry and Genomics CoLabs, and additional members of the Ansel, Fassett, and Basbaum laboratories.
Funding:
This work was supported by grants from the Dermatology Foundation (Career Development Award to M.S.F.), the Sandler Foundation (to K.M.A.) and the NIH (NIAMS K08AR074556 to M.S.F., NHLBI R01HL109102 to K.M.A., and NINDS R35NS097306 to A.I.B.). We also acknowledge UCSF Parnassus Flow Cytometry CoLab funding from NIH P30 DK63720.
Footnotes
Competing interests: Authors declare that they have no competing interests.
This manuscript has been accepted for publication in Science Immunology. This version has not undergone final editing. Please refer to the complete version of record at www.scienceimmunology.org. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Data and materials availability: All data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials. Raw sequencing files have been deposited at NCBI, GEO accession number GSE236744. ES cells containing the Il31 targeted allele are available from EUCOMM via the European Mutant Mouse Archive/INFRAFRONTIER. ES cells containing the Il31ra targeted allele are available from EUCOMM via the MRC Harwell Institute. IL31RAKO mice were obtained by MTA from Genentech and cannot be distributed by the authors. All other materials are available directly from commercial suppliers.
References
- 1.S. R. Dillon et al. , Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 5, 752–760 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Cevikbas F et al. , A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J. Allergy Clin. Immunol. 133, 448–460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kabashima K, Matsumura T, Komazaki H, Kawashima M, Nemolizumab JPSG, Trial of Nemolizumab and Topical Agents for Atopic Dermatitis with Pruritus. N. Engl. J. Med. 383, 141–150 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Stander S et al. , Trial of Nemolizumab in Moderate-to-Severe Prurigo Nodularis. N. Engl. J. Med. 382, 706–716 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Takamori A et al. , IL-31 is crucial for induction of pruritus, but not inflammation, in contact hypersensitivity. Sci. Rep. 8, 6639 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arai I et al. , Repeated administration of IL-31 upregulates IL-31 receptor A (IL-31RA) in dorsal root ganglia and causes severe itch-associated scratching behaviour in mice. Exp. Dermatol. 24, 75–78 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Perrigoue JG, Zaph C, Guild K, Du Y, Artis D, IL-31-IL-31R interactions limit the magnitude of Th2 cytokine-dependent immunity and inflammation following intestinal helminth infection. J. Immunol. 182, 6088–6094 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrigoue JG et al. , IL-31-IL-31R interactions negatively regulate type 2 inflammation in the lung. J. Exp. Med. 204, 481–487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neuper T et al. , IL-31 transgenic mice show reduced allergen-induced lung inflammation. Eur. J. Immunol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bilsborough J, Mudri S, Chadwick E, Harder B, Dillon SR, IL-31 receptor (IL-31RA) knockout mice exhibit elevated responsiveness to oncostatin M. J. Immunol. 185, 6023–6030 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Yosipovitch G, Berger T, Fassett MS, Neuroimmune interactions in chronic itch of atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 34, 239–250 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mollanazar NK, Smith PK, Yosipovitch G, Mediators of Chronic Pruritus in Atopic Dermatitis: Getting the Itch Out? Clin. Rev. Allergy Immunol. 51, 263–292 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Bilsborough J et al. , IL-31 is associated with cutaneous lymphocyte antigen-positive skin homing T cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 117, 418–425 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Sonkoly E et al. , IL-31: a new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 117, 411–417 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Skarnes WC et al. , A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stott B et al. , Human IL-31 is induced by IL-4 and promotes TH2-driven inflammation. J. Allergy Clin. Immunol. 132, 446–454 e445 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Shimada SG, LaMotte RH, Behavioral differentiation between itch and pain in mouse. Pain 139, 681–687 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto M et al. , A novel atopic dermatitis model induced by topical application with dermatophagoides farinae extract in NC/Nga mice. Allergol Int 56, 139–148 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M et al. , Contribution of itch-associated scratch behavior to the development of skin lesions in Dermatophagoides farinae-induced dermatitis model in NC/Nga mice. Arch. Dermatol. Res. 301, 739–746 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Braz JM et al. , Genetic priming of sensory neurons in mice that overexpress PAR2 enhances allergen responsiveness. Proc. Natl. Acad. Sci. U. S. A. 118, e2021386118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haspeslagh E, Debeuf N, Hammad H, Lambrecht BN, Murine Models of Allergic Asthma. Methods Mol. Biol. 1559, 121–136 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Maier B et al. , A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanemaru K et al. , Clec10a regulates mite-induced dermatitis. Sci Immunol 4, eaax6908 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA, Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front. Immunol. 5, 402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chinetti-Gbaguidi G, Staels B, Macrophage polarization in metabolic disorders: functions and regulation. Curr. Opin. Lipidol. 22, 365–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakarov S et al. , Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, (2019). [DOI] [PubMed] [Google Scholar]
- 27.Kolter J et al. , A Subset of Skin Macrophages Contributes to the Surveillance and Regeneration of Local Nerves. Immunity 50, 1482–1497 e1487 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Kolter J, Kierdorf K, Henneke P, Origin and Differentiation of Nerve-Associated Macrophages. Journal of immunology (Baltimore, Md. : 1950) 204, 271–279 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Serhan N et al. , House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat. Immunol. 20, 1435–1443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usoskin D et al. , Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Buhl T et al. , Protease-Activated Receptor-2 Regulates Neuro-Epidermal Communication in Atopic Dermatitis. Front. Immunol. 11, 1740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perner C et al. , Substance P Release by Sensory Neurons Triggers Dendritic Cell Migration and Initiates the Type-2 Immune Response to Allergens. Immunity 53, 1063–1077 e1067 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price TJ, Flores CM, Critical evaluation of the colocalization between calcitonin gene-related peptide, substance P, transient receptor potential vanilloid subfamily type 1 immunoreactivities, and isolectin B4 binding in primary afferent neurons of the rat and mouse. J Pain 8, 263–272 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grant AD, Gerard NP, Brain SD, Evidence of a role for NK1 and CGRP receptors in mediating neurogenic vasodilatation in the mouse ear. Br J Pharmacol 135, 356–362 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YJ, Granstein RD, Roles of calcitonin gene-related peptide in the skin, and other physiological and pathophysiological functions. Brain Behav Immun Health 18, 100361 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinho-Ribeiro FA et al. , Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173, 1083–1097 e1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stubbington MJ et al. , An atlas of mouse CD4(+) T cell transcriptomes. Biol Direct 10, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umeda Y, Takamiya M, Yoshizaki H, Arisawa M, Inhibition of mitogen-stimulated T lymphocyte proliferation by calcitonin gene-related peptide. Biochem Biophys Res Commun 154, 227–235 (1988). [DOI] [PubMed] [Google Scholar]
- 39.Nagashima H et al. , Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity 51, 682–695 e686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Millet I, Bottomly K, Vignery A, Calcitonin gene-related peptide inhibits interleukin 2 production by murine T lymphocytes. J Biol Chem 267, 21052–21057 (1992). [PubMed] [Google Scholar]
- 41.Fassett MS, Pua HH, Simpson LJ, Steiner DF, Ansel KM, Identification of Functionally Relevant microRNAs in the Regulation of Allergic Inflammation. Methods Mol. Biol. 1799, 341–351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pua HH et al. , MicroRNAs 24 and 27 Suppress Allergic Inflammation and Target a Network of Regulators of T Helper 2 Cell-Associated Cytokine Production. Immunity 44, 821–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oetjen LK et al. , Sensory Neurons Co-opt Classical Immune Signaling Pathways to Mediate Chronic Itch. Cell 171, 217–228 e213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson SR et al. , The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 155, 285–295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kempkes C, Buddenkotte J, Cevikbas F, Buhl T, Steinhoff M, in Itch: Mechanisms and Treatment, Carstens E, Akiyama T, Eds. (CRC Press/Taylor & Francis, Boca Raton (FL: ), 2014), chap. 11. [Google Scholar]
- 46.Ding W et al. , Regulation of Cutaneous Immunity In Vivo by Calcitonin Gene-Related Peptide Signaling through Endothelial Cells. J Immunol 208, 633–641 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamoutounour S et al. , Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 39, 925–938 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Cheng JB et al. , Transcriptional Programming of Normal and Inflamed Human Epidermis at Single-Cell Resolution. Cell Rep. 25, 871–883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y et al. , Single-Cell Profiling Reveals Divergent, Globally Patterned Immune Responses in Murine Skin Inflammation. iScience 23, 101582 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsoi LC et al. , Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat Commun 8, 15382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dousset L et al. , A Th2 cytokine interleukin-31 signature in a case of sporadic lichen amyloidosis. Acta Derm. Venereol. 95, 223–224 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Kabashima K et al. , Nemolizumab in patients with moderate-to-severe atopic dermatitis: Randomized, phase II, long-term extension study. J. Allergy Clin. Immunol. 142, 1121–1130 e1127 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Ruzicka T et al. , Anti-Interleukin-31 Receptor A Antibody for Atopic Dermatitis. N. Engl. J. Med. 376, 826–835 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Silverberg JI et al. , Phase 2B randomized study of nemolizumab in adults with moderate-to-severe atopic dermatitis and severe pruritus. J. Allergy Clin. Immunol. 145, 173–182 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Gagnon JD et al. , miR-15/16 Restrain Memory T Cell Differentiation, Cell Cycle, and Survival. Cell Rep. 28, 2169–2181 e2164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson LJ et al. , A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat. Immunol. 15, 1162–1170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stuart T et al. , Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao J et al. , The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sadeghi M et al. , Structure-Activity Studies Reveal the Molecular Basis for GABAB-Receptor Mediated Inhibition of High Voltage-Activated Calcium Channels by alpha-Conotoxin Vc1.1. ACS Chem. Biol. 13, 1577–1587 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Sandor K, Helyes Z, Elekes K, Szolcsanyi J, Involvement of capsaicin-sensitive afferents and the Transient Receptor Potential Vanilloid 1 Receptor in xylene-induced nocifensive behaviour and inflammation in the mouse. Neurosci. Lett. 451, 204–207 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Mulder GB, Pritchett K, Rodent analgesiometry: the hot plate, tail flick and Von Frey hairs. Contemp Top Lab Anim Sci 43, 54–55 (2004). [PubMed] [Google Scholar]
- 62.Zeisel A et al. , Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014 e1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Transgene schematic and serial crosses to generate Il31null allele
Figure S2. Supplementary data to accompany Figure 3
Figure S3. Gating strategy for cutaneous CD45+ cell subsets
Figure S4. Gating strategy for CD4 T cell intracellular cytokine detection
Figure S5. Cell Marker genes for the 13 myeloid clusters
Figure S6. Supplementary data to accompany Figures 4–5
Figure S7. RNAscope in situ hybridization of HDM skin sections demonstrates Il4ra expression in c5 macrophages and c4 monocytes.
Figure S8. Supplementary data regarding RTX-treated and IL31RAΔneuron mice.
Data file S1. Top 100 DEGs by Pseudotime, as defined by Monocle 2 analysis
Data file S2. Raw data file
Supplementary Methods