

Abstract
Breast cancer is a serious threat to the health and lives of women. Two novel series of N′-(2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazides and 1-(aryl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)ureas were designed, synthesized and investigated for their anticancer efficacy against the MCF-7 breast cell line. Three compounds of the first series showed potent activity toward MCF-7 with IC50 in the range 8.38–11.67 µM, respectively, as compared to Sorafenib (IC50 = 7.55 µM). N′-(1-butyl-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide inhibited VEGFR-2 with IC50 = 0.33 µM when compared with Sorafenib (IC50 = 0.09 µM). Furthermore, this compound was introduced to PCR assessment, where it increased Bax, caspase 8, caspase 9 and cytochrome C levels by 4.337-, 2.727-, 4.947- and 2.420-fold, respectively, while it decreased levels of Bcl-2, as the anti-apoptotic gene, by 0.359-fold when compared to the untreated control MCF-7. This compound was also arrested in the G2/M phase by 27.07%, compared with 11.31% for the control MCF-7. Furthermore, it induced early and late apoptosis in MCF-7. In addition, a molecular docking study in the VEGFR-2 active site was performed to assess the binding profile for the most active compounds. Moreover, ADME parameters of the targeted compounds were also evaluated.
Keywords: imidazo[2,1-b]thiazole; isatin; MCF-7 cell line; VGEFR-2; PCR; cell cycle; apoptosis; molecular docking; ADME
1. Introduction
Cancer is one of the most lethal diseases, and it is rapidly evolving into one of the world’s most serious health challenges. Furthermore, the unfavorable side effects of classic non-selective chemotherapies, as well as the growth of resistance to existing chemotherapy medications, make the search for more selective novel cancer fighters a top goal. However, it is difficult to design a drug that prevents the production of aberrant cells with no influence on the exerting of normal cells [1,2]. Pathological angiogenesis, on the flip side, occurs not just in the growth of tumors but additionally in a variety of non-neoplastic conditions [3]. Angiogenesis is also suggested to be linked to a variety of disorders, including cancer [4,5]. The inhibition of angiogenesis may decrease the tumor development because it can slow tumor growth without creating significant negative or resistant effects over time [6]. The VEGF (vascular endothelial growth factor) family members are well-known angiogenic agents that govern blood and lymphatic vessel development and balance in both normal and pathological angiogenesis.
VEGF-A assumes a crucial role in directing vascular development and angiogenesis by interacting with VEGFR-2. The nuanced functions of VEGFR-2 in blood vessel development are supported by adaptor proteins. In the realm of cancer, the secretion of VEGF by cancer cells is instrumental in activating the VEGFR-2 pathway in neighboring endothelial cells, thereby participating in the phenomenon of cancer-related angiogenesis. Notably, the activation of VEGFR-2 signaling has been discerned in breast cancer cells [7,8,9].
Consequently, VEGFR has been extensively utilized in cancer therapy [10]. VEGF ligands bind to three different tyrosine kinases (VEGFR-TK): FLT-1 (FLT1), FLT-2 (KDR/FLK1) and FLT-4 (FLT4) [11]. VEGFR-2 is activated by the unique binding of VEGF generated by endothelial cells [12] and many malignancies have VEGFR-2 amplification [13]. Series of VEGFR-2 inhibitors have recently been approved for the management of numerous cancers such as Sorafenib (I) [14], Lenvatinib (II) [15] and Sunitinib (III) [16] with IC50= 90, 1 and 10 nM, respectively, as well as urea-based heterocycles IV (IC50= 1.5 µM) [17] and V (IC50= 6.2 nM) [18] (Figure 1).
Figure 1.

Structure of clinically approved VEGFR-2 inhibitors and some reported anticancer agents (I–IX) with their corresponding IC50 values, in addition to our targeted hybrids 6a–l and 8a–g.
The combinations through the hydrazine group, as a linker between position 3 of isatin (indoline-2,3-dione) and certain heterocycles such as pyrrole, pyridine or quinazoline in compounds VI [19], VII [20] and VIII [21], produce cytotoxic agents against the MCF-7 cancerous cell line with IC50 = 6.25, 6.3 and 2.1 µM, respectively (Figure 1).
A new class of imidazo[2,1-b]thiazoles were developed for anticancer activity and they showed potential activity toward MCF-7 cell line and elicited apoptotic properties such as caspase-9 upregulation; for example, imidazo[2,1-b]thiazole derivatives IX showed IC50 equal to 0.794 µM on the MCF-7 cell line and were further used for comprehensive biological investigations [22] (Figure 1).
As illustrated in Figure 1, isatin in Sunitinib (III) and lead compounds VI–VIII, in addition to the imidazo[2,1-b]thiazole system in the potent anticancer compound IX, are important in the synthesis of potent VEGFR-2 inhibitors and/or MCF-7 anticancer agents. On the other hand, the hydrazine linker in compounds VI–VIII and urea bridge in Sorafenib (I), Lenvatinib (II), IV and V are critical for the same purpose.
In continuation to our research, which deals with the discovery of novel potent anticancer agents [23,24,25,26,27,28], herein we utilized the fragment linking strategy to construct novel anti-proliferative agents for MCF-7 breast cancer therapy and evaluated their effectiveness on VEGFR enzyme inhibition.
The Sorafenib-VEGFR-2 crystal structure showed the role of the pyridine ring of Sorafenib (I) with certain amino acids in the VEGFR-2 hydrophobic pocket [29,30] (Figure 2). However, it is important to understand the SAR of synthetic VEGFR-2 kinase inhibitors [31], which may be a critical factor in determining the efficiency of the agent and its tolerability.
Figure 2.

Docking of Sorafenib (I) with VEGFR-2 binding site and the assumed possible modifications of the targeted structures 6a–l and 8a–g.
The binding mode of VEGFR-2 inhibitors is characterized by a “head” component that binds to the hinge region and possesses crucial H-bond donor and/or acceptor capabilities for interacting with Cys919. Additionally, they have a “linker” spanning three to four chemical bonds with H-bonding moiety to establish the interactions with Asp1046. Furthermore, they have a “tail” segment containing a hydrophobic moiety that occupies the allosteric hydrophobic back pocket [32,33,34].
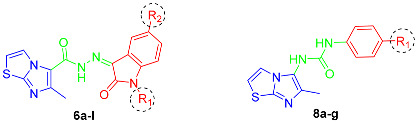
In this context, we developed two sets of N′-(2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazides 6a–l and 1-(aryl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)ureas 8a–g (Figure 1 and Figure 2). Regarding the binding pocket of Sorafenib (I), the pyridine residue was specifically linked with Leu1035, Phe1047, Val848, Val916 and Cys1045 through various forms of interactions in the hinge region, whereas the H interaction was preserved via the urea linker with Asp1046. Furthermore, Leu889 and Glu885 maintained the link between the substituted phenyl ring through distinct interactions (Figure 2). Therefore, the aimed structures 6a–l were designed according to the fragment combination approach, as follows: the 6-methylimidazo[2,1-b]thiazole system replaced the 4-phenoxypyridine moiety in the left side of Sorafenib (I), whereas the isatin moiety with several substitution patterns in position 1 and/or 5 replaced the aryl group of the right side of Sorafenib (I) with a carbohydrazide linker (H-bond acceptor–H-bond donor linker) similar to the urea bridge in Sorafenib (I) in order to obtain an activity similar or higher to that of Sorafenib (I) (Figure 2). We also followed the same approach to design urea derivatives 8a–g (Figure 2). The latter two sets were synthesized and evaluated toward MCF-7 cell cytotoxicity. The most potent compounds were introduced to examine their activity toward VEGFR-2, PCR assessment, apoptosis and cell cycle analysis.
2. Results and Discussion
2.1. Chemistry
The reaction of 2-chloroethylacetoacetate (1) with 2-aminothiazole (2), in 1,2-dimethoxyethane at 90 °C, yielded the key ester 3. Under reflux circumstances, the latter ester was transformed into acid hydrazide 4 using 99% hydrazine hydrate [29]. Subsequently, compound 4 was reacted with several substituted indoline-2,3-dione derivatives 5a–l to yield the target 6-methyl-N′-(2-oxo-subsituted/unsubstituted-indolin-3-ylidene)imidazo[2,1-b]thiazole-5-carbohydrazides 6a–l (Scheme 1). The 1H NMR data of compounds 6a–d revealed three distinct signals, one around 2.65 ppm for CH3 of imidazo[2,1-b]thiazole and two D2O exchangeable signals around 11.30 and 13.26 ppm for isatin NH and carbohydrazide NH, respectively. Compounds 6e–h with n-propyl substitution in position 1 showed three aliphatic signals of the -CH2-CH2-CH3 group around 0.90, 1.55–1.70 and 3.70 ppm, in addition to a singlet CH3 of imidazo[2,1-b]thiazole around 2.75 ppm. They also showed a D2O exchangeable hydrazide NH signal around 13.20 ppm. Furthermore, butyl-substituted compounds 6i–l were characterized with five aliphatic distinguishing signals arising in the same highlights as prior propyl compounds with an extra multiplet peak of CH2, in addition to the signal of imidazo[2,1-b]thiazole CH3. Concerning 13C NMR, spectra of 6a–l exhibited distinct signal around 16.45 ppm for the CH3 group of imidazothiazole moiety. Moreover, 13C NMR of 6e–h revealed three signals around 11.50, 28.82 and 42.22 ppm for the n-propyl group; on the other hand, compounds 6i–l revealed four separate signals around 13.91, 20.00, 30.30 and 40.80 ppm for n-butyl substitution (Figures S4–S19, Supplementary Data).
Scheme 1.

(i) 1,2-dimethoxyethane, ref., 6 h; (ii) N2H4·H2O/ethanol, ref., 3 h; (iii) EtOH/glacial acetic acid, reflux 4 h.
On the other hand, compound 4 was subjected to a Citrous rearrangement process by its reaction with NaNO2 and HCl to obtain azide 7 [30] which was then refluxed in dioxane with various anilines to yield urea derivatives 8a–g (Scheme 2). The 1H NMR spectra of compounds 8a–g revealed three signals, one around 2.12 ppm for CH3 protons and the remaining two signals around 8.16 ppm and 9.02 ppm for two D2O exchangeable NH protons of the urea linker. Compound 8g was characterized with an additional D2O exchangeable signal of sulphonamide NH2 at 7.17 ppm. The existence of two separate signals around 13.37 ppm and 154.20 ppm for the CH3 and carbonyl groups was seen in 13C NMR spectra of 8a–g. The 1H NMR of compounds 8b and 8c showed the signals of -CH3 and -OCH3 protons, respectively, and the appearance of their carbon signals in 13C NMR (Figures S20–S33, Supplementary Data).
Scheme 2.

(i) NaNO2/HCl/ H2O, stirring 3 h; (ii) dioxane/diff. anilines, reflux 8 h.
2.2. Anticancer Activity
2.2.1. Cytotoxicity of the Designed Compounds 6a–l and 8a–g on MCF-7 and MCF-10A
The anti-cancer potency of 6a–l and 8a–g was investigated towards the MCF-7 breast cell line using MTT assay and their IC50 values were determined, compared to Sorafenib (I) [35]. The majority of the compounds 6a–l and 8a–g have a wide range of activity against the MCF-7 cell line, with IC50 ranging from 23.97 ± 0.67 µM for 6a to 114.61 ± 4.99 µM for 8b comparing with Sorafenib (I) (IC50 = 7.55 ± 0.40 µM) (Table 1). On the other side, compounds 6b, 6i and 6j showed potent anticancer activity toward MCF-7 cell lines, with IC50 = 11.50 ± 0.52, 8.38 ± 0.62 and 11.67 ± 0.52 µM, respectively, as compared to Sorafenib (I) (IC50 = 7.55 ± 0.40 µM) (Table 1).
Table 1.
Anti-proliferative activity of 6a–l and 8a–g toward MCF-7 cell line.
| |||
| Compound | R1 | R2 | IC50 (µM) |
| 6a | H | H | 23.97 ± 0.67 |
| 6b | H | F | 11.50 ± 0.52 |
| 6c | H | Cl | 43.10 ± 3.69 |
| 6d | H | Br | 22.80 ± 1.38 |
| 6e | n -propyl | H | 61.39 ± 3.15 |
| 6f | n -propyl | F | 82.55 ± 2.75 |
| 6g | n -propyl | Cl | 52.47 ± 1.26 |
| 6h | n -propyl | Br | 74.34 ± 2.19 |
| 6i | n - butyl | H | 8.38 ± 0.62 |
| 6j | n - butyl | F | 11.67 ± 0.52 |
| 6k | n - butyl | Cl | 42.72 ± 2.01 |
| 6l | n - butyl | Br | 52.55 ± 2.36 |
| 8a | H | – | 109.68 ± 4.62 |
| 8b | CH3 | – | 114.61 ± 4.99 |
| 8c | OCH3 | – | 106.43 ± 5.32 |
| 8d | F | – | 72.33 ± 4.71 |
| 8e | Cl | – | 35.72 ± 3.32 |
| 8f | Br | – | 31.12 ± 2.47 |
| 8g | SO2NH2 | – | 41.57 ± 1.59 |
| Sorafenib (I) | – | – | 7.55 ± 0.40 |
On the other hand, compounds 6b, 6i and 6j revealed non-cytotoxic activity toward MCF-10A due to its non-tumorigenic origin [36], where their IC50 = 125.26 ± 2.7, 54.63 ± 1.29 and 47.73 ± 0.53 µM, when compared to Sorafenib (IC50 = 22.35 ± 1.29 µM) (Table 2).
Table 2.
IC50 values of 6b, 6i and 6j toward MCF-10A.
| Compound | IC50 (µM) |
|---|---|
| 6b | 125.26 ± 2.7 |
| 6i | 54.63 ± 1.29 |
| 6j | 47.73 ± 0.53 |
| Sorafenib (I) | 22.35 ± 1.29 |
2.2.2. SARs of Compounds 6a–l and 8a–g on MCF-7 Cell Line
SARs assessments of the aforementioned data of compounds 6a–l revealed that the linking between the imidazo[2,1-b]thiazole system and indoline-2,3-dione via the hydrazide bridge showed a wide range of activity as 6c, 6e, 6f, 6g, 6h, 6k and 6l have weak antiproliferative activity, with an IC50 range of 43.10 ± 3.69–82.55 ± 2.75 µM in comparison to Sorafenib (I) (IC50 = 7.55 ± 0.40 µM), while compounds 6a (IC50 = 23.97 ± 0.67 µM) and 6d (IC50 = 22.80 ± 1.38 µM) revealed moderate anti-proliferative activity. Regarding 6b, 6i and 6j, they showed the highest anticancer activity with an IC50 range = 8.38 ± 0.62–11.67 ± 0.52 µM. N′-(1-butyl-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6i) with n-butyl substitution (R = n-butyl) in position 1 of isatin moiety exhibited the highest anticancer activity (IC50 = 8.38 ± 0.62 µM), followed by 6b with fluoro-substitution (R1 = F) in position 5 of isatin moiety, with an IC50 = 11.50 ± 0.52 µM, whereas compound 6j with n-butyl substitution (R1 = n-butyl) and fluoro-substitution (R2 = F) in positions 1 and 5 of isatin moiety, respectively, had an IC50 range = 11.67 ± 0.52 µM (Table 1). From the latter data, the variation in substitutions in positions 1 and 5 of isatin moiety plays an important role in the anticancer activity of hybrids 6a–l; for example, the impact of F is higher than Cl or Br, whereas the effect of n-butyl substitution is higher than that of n-propyl. On the other side, imidazo[2,1-b]thiazoles with arylurea branch compounds 8a–g showed anticancer activities lower than those of 6a–l.
2.2.3. VGEFR-2 Inhibitory Activity (IC50) for Compounds 6b, 6i and 6j
The VEGFR-2 inhibitory activity for compounds 6b, 6i and 6j was investigated and the results in IC50 computed from the concentration–inhibition response curve compared to Sorafenib (I) are summarized in Table 3. The VEGFR-2 inhibitory effects of 6b, 6i and 6j, respectively, showed good activity, with an IC50s = 3.85 ± 0.15, 0.33 ± 0.01 and 1.51 ± 0.06 µM, respectively, when compared to Sorafenib (I) (IC50 = 0.09 ± 0.004 µM). Compound 6j (IC50 = 1.51 ± 0.06 µM) displayed a significant influence on kinase activity when compared to 6b with IC50 = 3.85 ± 0.15 µM, which indicated the effect of positions 1 and 5 substitutions of isatin moiety. However, compound 6j revealed the highest VEGFR-2 inhibition activity, with IC50 = 0.33 ± 0.01 µM.
Table 3.
In vitro VEGFR-2 inhibitory activity for 6b, 6i and 6j.
| Compounds | R1 | R2 | IC50 (µM) |
|---|---|---|---|
| 6b | H | F | 3.85 ± 0.15 |
| 6i | n -butyl | H | 0.33 ± 0.01 |
| 6j | n -butyl | F | 1.51 ± 0.06 |
| Sorafenib (I) | – | – | 0.09 ± 0.004 |
2.2.4. PCR Assessment for Compound 6i
The gene expression levels of apoptosis genes in MCF-7 cells were measured using RT-PCR to validate the apoptosis-inducing behaviors of the potent lead compound 6i in MCF-7 cells. Compound 6i increased Bax levels by 4.337-fold, caspase 8 levels by 2.727-fold, caspase 9 levels by 4.947-fold and cytochrome C levels by 2.420-fold, while it decreased the level of Bcl-2, as the anti-apoptotic gene, by 0.359-fold when compared to the untreated control MCF-7 (Table 4).
Table 4.
Fold change of apoptosis genes in treated MCF-7 cells with 6i.
| RT-PCR Fold Change | |||||
|---|---|---|---|---|---|
| Bax | Caspase-8 | Caspase-9 | Cytochrome C | Bcl-2 | |
| 6i/MCF-7 | 4.337 | 2.727 | 4.947 | 2.420 | 0.359 |
| Control MCF-7 | 1 | 1 | 1 | 1 | 1 |
The results revealed an inherent apoptotic mechanism including the activation of Bax, cytochrome C, and caspases 3 and 9. As a result, our RT-PCR findings supported prior research by demonstrating that the apoptotic pathway is characterized by the overexpression of proapoptotic genes and the downregulation of anti-apoptotic genes (Figure 3).
Figure 3.

Quantitative RT-PCR results analysis of the apoptosis-related genes, cytochrome, Bax, Caspases 8, 9 and Bcl-2, in 6i-treated MCF-7 cells. Dashed line represents the gene expression for potent 6i and blue line represents untreated control.
2.2.5. Cell Cycle Analysis of Compound 6i on MCF-7
Compound 6i arrested cells in the G2/M phase with %G2/M = 27.07, compared with %G2/M = 11.31 for the control MCF-7 (Table 5 and Figure 4).
Table 5.
Cell cycle of compound 6i in MCF-7.
| %G0–G1 | %S | %G2/M | |
|---|---|---|---|
| 6i/MCF-7 | 53.66 | 19.27 | 27.07 |
| Cont. MCF-7 | 64.73 | 23.96 | 11.31 |
Figure 4.

Cell cycle for cont. MCF-7 (left) and 6i (right).
2.2.6. Apoptotic Assay for Compound 6i
In comparison to the control cells, the apoptotic assay employing Annexin V/PI analysis for compound 6i generated a substantial amount of early and late apoptosis in MCF-7. Compound 6i induced early apoptosis in MCF-7 at rate of 22.05, compared with the control MCF-7 (0.43) (Table 6 and Figure 5). Furthermore, 6i demonstrated late apoptosis at rate 7.61 compared with the control MCF-7 (0.11).
Table 6.
Apoptosis analysis for 6i in MCF-7.
| Total | Early | Late | Necrosis | |
|---|---|---|---|---|
| 6i/MCF-7 | 32.81 | 22.05 | 7.61 | 3.15 |
| Cont. MCF-7 | 1.91 | 0.43 | 0.11 | 1.37 |
Figure 5.

Apoptosis analysis for control MCF-7 (left) and 6i (right).
2.3. Molecular Modeling Study
The computational docking method is used for approximating two fundamental concepts. The main objective is to determine the correct spot and orientation of a particular chemical pose in the binding site of the enzyme. The second concept is the calculation of docking score (the strength of protein-ligand interactions) [37]. In an attempt to explain the cytotoxic action of 6b, 6i and 6j, a docking study in the VEGFR-2 active site was performed. Redocking of sorafenib (I) in the active site of VEGFR-2 (PDB: 4ASD) was nearly in the exact position as the co-crystalized ligand with d. s. = −10.6 kcal/mol) (Figure S1, Supplementary Data).
The docking investigation of 6b, 6i and 6j in the VEGFR-2 active site revealed binding mechanisms that were similar to the lead medicine and they showed docking scores that were higher or comparable to the docked lead. The hot areas were satisfied by targeted compounds, which established H-bonds with Cys919, Glu885 and Asp1046 residues which helped to explain the substantial VEGFR-2 inhibitory effect, as shown in Figure 6.
Figure 6.

(A) 6b in VEGR-2 binding site showed H-bond with Asp1046 and pi-cation interaction with Lys868; (B) compound 6i revealed H-bond with Asp1046, pi-sigma interaction with Cys1045 and pi-cation interaction with Lys868; (C) 6j showed H-interactions with Asp1046 and Lys868 residues (Table S1, Supplementary Data).
As shown in the interactions of 6b, 6i and 6j on the VEGFR-2 active site, they interacted with the same pattern of sorafenib (I) with a hydrogen-bond interaction with amino acid Asp1046 via the linker hydrazide moiety, while imidazothiazole bonded with Val916 and Cys1045 as the pyridine side; on the other hand, phenyl isatin bonded with Leu889 via pi-sigma as the substituted phenyl in the Sorafenib (I) structure (Figure S2, Supplementary Data).
2.4. Computational ADME Study
Three rules, at least, of the following five rules are requested for oral bioavailability: There are no more than five H-bond donors, no more than 500 Da MWt, no more than five log P, only one violation and no more than ten H-bond acceptors, according to Lipinski’s rule of five [38]. The Veber rule predicts that molecules with less than 10 rotatable bonds and at least 140.2 of polar surface area will have appropriate oral bioavailability [39]. ADME parameters of the targeted compounds 6a–l and 8a–g such as the proportion of human intestinal absorption and blood–brain barrier penetration were evaluated using the SWISSADME method. Their BBB penetration was expected to be minimal, which prevents these candidates from passing the BBB or having harmful effects on the central nervous system (Figure S3 and Table S2, Supplementary Data).
3. Materials and Methods
3.1. Chemistry
The specifications of instruments used in the chemistry part are listed in the Supplementary Data, page 42.
3.1.1. Preparation of Carbohydrazide 4
A mixture of 2-aminothiazole (1) (10.0 g, 100 mmol) and ethyl 2-chloroacetoacetate (2) (16.5 g, 100 mmol) was refluxed for 6 h in 1,2-dimethoxyethane (50 mL). After evaporation of the solvent, off-white precipitate was formed and then recrystallized from EtOH to afford compound 3. The latter ester 3 (2.10 g, 10 mmol) was treated with hydrazine hydrate (99%, 2 mL) for 4 h in refluxed EtOH (50 mL). The solvent was evaporated to obtain white precipitate, which then recrystallized from EtOH to give carbohydrazide 4 [40].
3.1.2. Synthesis of Hybrids 6a–l
A mixture of carbohydrazide 4 (0.196 g, 1.0 mmol) and indoline-2,3-dione derivatives 5a–l (1.0 mmol) in EtOH (30 mL) and AcOH (0.3 mL) was refluxed for 4 h. Crystallization of the resulting solid from EtOH/DMF gave compounds 6a–l, respectively.
6-Methyl-N′-(2-oxoindolin-3-ylidene)imidazo[2,1-b]thiazole-5-carbohydrazide (6a)
Yield = 68%; m.p. = >300 °C; IR: 1697 (>C=O), 3263 (>N-H), 3444 (>N-H); 1H NMR: 2.65 (s, 3H, -CH3), 6.92 (d, J = 8.1 Hz, 1H, Ar-H), 7.07 (d, J = 8.1 Hz, 1H, Ar-H), 7.31–7.36 (m, 1H, Ar-H), 7.41 (d, J = 4.5 Hz, 1H, Ar-H), 7.53 (d, J = 7.5 Hz, 1H, Ar-H), 8.21 (d, J = 4.5 Hz, 1H, Ar-H), 11.11 (s, 1H, >N-H), 13.26 (s, 1H, >N-H); 13C NMR: 16.88 (-CH3), 111.73, 115.41, 117.66, 120.61, 121.22, 121.74, 132.04, 137.52, 142.82, 148.80, 152.63, 157.08 (>C=O), 163.16 (>C=O); Anal. for C15H11N5O2S (325.35): calcd.: C, 55.38; H, 3.41; N, 21.53; found: C, 55.50; H, 3.47; N, 21.37.
N′-(5-Fluoro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6b)
Yield = 74%; m.p. = >300 °C; IR: 1710 (>C=O), 3294 (>N-H), 3420 (>N-H); 1H NMR: 2.65 (s, 3H, -CH3), 6.93 (q, J = 4.0 Hz, 1H, Ar-H), 7.15–7.19 (m, 1H, Ar-H), 7.33 (dd, J = 8.1 Hz, 1H, Ar-H), 7.42 (d, J = 4.1 Hz, 1H, Ar-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 11.14 (s, 1H, >N-H), 13.28 (s, 1H, >N-H); 13C NMR: 16.89 (-CH3), 40.73, 108.34, 112.85, 115.54 (2C), 117.59, 118.29, 121.75 (2C), 139.05, 149.06, 152.81, 157.01 (>C=O), 163.29 (>C=O); Anal. for C15H10FN5O2S (343.34): calcd.: C, 52.47; H, 2.94; N, 20.40; found: C, 52.21; H, 2.99; N, 20.53.
N′-(5-Chloro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6c)
Yield = 67%; m.p. = 292–293 °C; IR: 1705 (>C=O), 3124 (>N-H), 3367 (>N-H); 1H NMR: 2.68 (s, 3H, -CH3), 6.95 (d, J = 8.5 Hz, 1H, Ar-H), 7.39 (d, J = 4.0 Hz, 1H, Ar-H), 7.48 (d, J = 4.0 Hz, 1H, Ar-H), 7.51 (m, 1H, Ar-H), 8.26 (d, J = 4.4 Hz, 1H, Ar-H),11.38 (s, 1H, >N-H), 13.26 (s, 1H, >N-H); 13C NMR: 16.91 (-CH3), 113.25, 115.53, 117.56, 120.67, 121.75, 122.31, 127.42, 131.39, 136.48, 141.44, 149.03, 152.80, 156.92 (>C=O), 162.94 (>C=O); Anal. for C15H10ClN5O2S (359.79): calcd.: C, 50.08; H, 2.80; N, 19.47; found: C, 49.91; H, 3.01; N, 19.50.
N′-(5-Bromo-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6d)
Yield = 66%; m.p. = 296–297 °C; IR: 1705 (>C=O), 3116 (>N-H), 3383 (>N-H); 1H NMR: 2.62 (s, 3H, -CH3), 6.85 (s, 1H, Ar-H), 7.42–7.46 (m, 2H, Ar-Hs), 7.56 (s, 1H, Ar-H), 8.20 (s, 1H, Ar-H), 11.33 (s, 1H, >N-H), 13.20 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.07 (2C), 115.59, 115.76 (2C), 118.58, 118.98, 1120.82, 136.62, 137.29, 144.84, 154.29, 156.98 (C=O), 158.87 (C=O); Anal. for C15H10BrN5O2S (404.24): calcd.: C, 44.57; H, 2.49; N, 17.33; found: C, 44.30; H, 2.36; N, 17.37.
6-Methyl-N′-(2-oxo-1-propylindolin-3-ylidene)imidazo[2,1-b]thiazole-5-carbohydrazide (6e)
Yield = 51%; m.p. = 256–257 °C; IR: 1702 (>C=O), 3236 (>N-H); 1H NMR: 0.89 (t, J = 7.5 Hz, 3H, -CH3), 1.62–1.69 (m, 2H, >CH2), 2.67 (s, 3H, -CH3), 3.71 (t, J = 7.5 Hz, 2H, >CH2), 7.12–7.19 (m, 2H, Ar-Hs), 7.41–7.44 (m, 2H, Ar-Hs), 7.59 (d, J = 8.1 Hz, 1H, Ar-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 13.21 (s, 1H, >N-H); 13C NMR: 11.55 (-CH3), 17.03 (-CH3), 28.89 (>CH2), 41.65 (>CH2), 112.80, 113.01, 115.65, 121.82, 123.48, 134.33, 135.65, 142.20, 148.27, 152.71, 156.25, 161.10 (>C=O); Anal. for C18H17N5O2S (367.43): calcd.: C, 58.84; H, 4.66; N, 19.06; found: C, 59.07; H, 4.59; N, 19.20.
N′-(5-Fluoro-2-oxo-1-propylindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6f)
Yield = 66%; m.p. = 243–244 °C; IR: 1689 (>C=O), 3224 (>N-H); 1H NMR: 0.92 (t, J = 7.5 Hz, 3H, -CH3), 1.64–1.70 (m, 2H, >CH2), 2.72 (s, 3H, -CH3), 3.76 (t, J = 7.5 Hz, 2H, >CH2), 7.26–7.32 (m, 2H, Ar-Hs), 7.43–7.45 (m, 1H, Ar-H), 7.48 (d, J = 4.4 Hz, 1H, Ar-H), 8.38 (s, 1H, Ar-H), 13.21 (s, 1H, >N-H); 13C NMR: 11.51 (-CH3), 17.20 (-CH3), 29.90 (>CH2), 41.71 (>CH2), 113.21, 113.29, 114.50, 123.18, 125.58, 132.10, 134.23, 143.60, 149.55, 153.79, 156.63, 162.12 (>C=O); Anal. for C18H16FN5O2S (385.42): calcd.: C, 56.09; H, 4.18; N; 18.17; found: C, 55.82; H, 4.21; N; 18.30.
N′-(5-Chloro-2-oxo-1-propylindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6g)
Yield = 61%; m.p. = 254–255 °C; IR: 1693 (>C=O), 3236 (>N-H); 1H NMR: E/Z, 0.83, 0.87 (2t, J = 7.0 Hz, J = 7.5 Hz, 3H, -CH3), 1.54–1.63 (2m, 2H, >CH2), 2.52, 2.68 (2s, 3H, -CH3), 3.66, 3.72 (2t, J = 7.0 Hz, J = 7.5 Hz, 2H, >CH2), 7.18, 7.26 (2d, J = 8.5 Hz, J = 9.0 Hz, 1H, Ar-H), 7.39–7.45 (2d, J = 4.0 Hz, J = 4.4 Hz, 1H, Ar-H), 7.48–7.55 (m, 2H, Ar-Hs), 8.18, 8.25 (2s, 1H, Ar-H), 11.46, 13.20 (2s, 1H, >N-H); 13C NMR: 11.43 (-CH3), 16.30 (-CH3), 28.40 (>CH2), 43.51 (>CH2), 112.56, 113.65, 118.58, 124.29, 124.52, 132.18, 134.23, 144.64, 149.05, 154.59, 155.83, 162.23(>C=O); Anal. for C18H16ClN5O2S (401.87): calcd.: C, 53.80; H, 4.01; N, 17.43; found: C, 53.53; H, 3.84; N, 17.57.
N′-(5-Bromo-2-oxo-1-propylindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6h)
Yield = 50%; m.p. = 261–262 °C; IR: 1724 (>C=O), 3232 (>N-H); 1H NMR: E/Z, 0.83, 0.86 (2t, J = 7.0 Hz, J = 7.5 Hz, 3H, -CH3), 1.54–1.63 (2m, 2H, >CH2), 2.36, 2.66 (2s, 3H, -CH3), 3.65, 3.69 (2t, J = 7.0 Hz, J = 7.5 Hz, 2H, >CH2), 7.12, 7.18 (2d, J = 8.5 Hz, J = 9.0 Hz, 1H, Ar-H), 7.38, 7.44 (2d, J = 4.0 Hz, J = 4.5 Hz, 1H, Ar-H), 7.57–7.64 (m, 2H, Ar-Hs), 8.06, 8.23 (2d, J = 4.0 Hz, J = 5.0 Hz, 1H, ArH), 11.14, 13.60 (2s, D2O exchangeable, 1H, >N-H); 13C NMR: 11.58 (-CH3), 17.00 (-CH3), 28.87 (>CH2), 41.60 (>CH2), 112.75, 115.47, 115.60, 121.78, 123.28, 134.10, 135.43, 142.50, 149.17, 152.89, 156.92, 161.04 (>C=O); Anal. for C18H16BrN5O2S (446.32): calcd.: C, 48.44; H, 3.61; N, 15.69; found: C, 48.56; H, 3.53; N, 15.71.
N′-(1-Butyl-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6i)
Yield = 45%; m.p. = 250–251 °C; IR: 1670 (>C=O), 3240 (>N-H); 1H NMR: 0.92 (t, J = 7.2 Hz, 3H, -CH3), 1.35–1.39 (m, 2H, >CH2), 1.60–1.66 (m, 2H, >CH2), 2.73 (s, 3H, -CH3), 3.79 (t, J = 7.3 Hz, 2H, >CH2), 7.17–7.24 (m, 2H, Ar-Hs), 7.45–7.49 (m, 2H, Ar-Hs), 7.64 (d, J= 7.2 Hz, 1H, Ar-H), 8.34 (s, 1H, Ar-H): 13C NMR: 14.02 (-CH3), 16.97 (-CH3), 20.04 (>CH2), 29.64 (>CH2), 39.24 (>CH2), 4077, 42.23, 110.65, 115.48, 117.63, 120.02, 121.06, 121.76, 123.66, 132.02, 136.63, 143.38, 148.91, 161.32 (>C=O); Anal. for C19H19N5O2S (381.45): calcd.: C, 59.83; H, 5.02; N, 18.36; found: C, 59.67; H, 4.85; N, 18.39.
N′-(1-Butyl-5-fluoro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6j)
Yield = 50%; m.p. = 237–238 °C; IR: 1670 (>C=O), 3244 (>N-H); 1H NMR: 0.86 (t, J = 7.0 Hz, 3H, -CH3), 1.26–1.33 (m, 2H, >CH2), 1.53–1.59 (m, 2H, >CH2), 2.66 (s, 3H, -CH3), 3.71 (t, J = 7.5 Hz, 2H, >CH2), 7.17–7.20 (m, 1H, Ar-H), 7.23–7.27 (m, 1H, Ar-H), 7.37 (d, J = 8.1 Hz, 1H, Ar-H), 7.44 (d, J = 4.0 Hz, 1H, Ar-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 13.20 (s, 1H, >N-H); 13C NMR: 14.00 (-CH3), 16.97 (-CH3), 20.01 (>CH2), 29.56 (>CH2), 39.74 (>CH2), 108.16, 108.36, 111.94, 115.59, 117.56, 118.07, 121.43, 136.13, 139.60, 149.14, 152.88, 156.97, 158.35, 160.26, 161.35 (>C=O); Anal. for C19H18FN5O2S (399.44): calcd.: C, 57.13; H, 4.54; N, 17.53; found: C, 57.26; H, 4.57; N, 17.80.
N′-(1-Butyl-5-chloro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6k)
Yield = 67%; m.p. = 224–225 °C; IR: 1693 (>C=O), 3224 (>N-H); 1H NMR: 0.85 (t, J = 7.5 Hz, 3H, -CH3), 1.27–1.32 (m, 2H, >CH2), 1.55–1.58 (m, 2H, >CH2), 2.68 (s, 3H, -CH3), 3.74 (t, J = 7.5 Hz, 2H, >CH2), 7.24 (d, J = 8.1 Hz, 1H, Ar-H), 7.46–7.49 (m, 2H, Ar-Hs), 7.56 (s, 1H, Ar-H), 8.25 (d, J = 4.0 Hz, 1H, Ar-H), 13.19 (s, 1H, >N-H); 13C NMR: 14.02 (-CH3), 15.99 (-CH3), 20.00 (>CH2), 30.56 (>CH2), 40.52 (>CH2), 43.69, 44.96, 118.70, 121.10, 122.18, 125.52, 126.31, 127.96, 130.24, 138.18, 144.56, 149.16, 157.09, 161.58 (>C=O); Anal. for C19H18ClN5O2S (415.90): calcd.: C, 54.87; H, 4.36; N, 16.84; found: C, 55.06; H, 4.38; N, 17.00.
N′-(5-Romo-1-butyl-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6l)
Yield = 39%; m.p. = 237–238 °C; IR: 1693 (>C=O), 3228 (>N-H); 1H NMR: 0.86 (t, J = 7.1 Hz, 3H, -CH3), 1.27–1.33 (m, 2H, >CH2), 1.53–1.57 (m, 2H, >CH2), 2.66 (s, 3H, -CH3), 3.71 (t, J = 7.5 Hz, 2H, >CH2), 7.16 (s, 1H, Ar-H), 7.44 (d, J = 5.0 Hz, 1H, Ar-H), 7.58 (s, J = 9.5 Hz, 1H, Ar-H), 7.63 (s, 1H, Ar-H), 8.22 (s, 1H, Ar-H), 13.14 (s, 1H, >N-H); 13C NMR: 14.00 (-CH3), 16.99 (-CH3), 20.00 (>CH2), 29.56 (>CH2), 40.52 (>CH2), 40.69, 40.76, 112.72, 115.48, 115.61, 117.55, 121.78, 123.30, 134.13, 142.42, 149.18, 152.90, 156.93, 160.97 (>C=O); Anal. for C19H18BrN5O2S (460.35): calcd.: C, 49.57; H, 3.94; N, 15.21; found: C, 49.52; H, 4.08; N, 15.34.
3.1.3. Synthesis of Urea Derivatives 8a–g
Compound 4 was stirred for 3 h in a solution of NaNO2 with HCl to form the 6-methylimidazo[2,1-b]thiazole-5-carbonyl azide [41]. Azide 7 (0.21 g, 1 mmol) was refluxed in 1,4-dioxane (40 mL) with different anilines (1.0 mmol) for 8 h. Crystallization from EtOH for the formed solid gave the targeted urea derivatives 8a–g, respectively.
1-(6-Methylimidazo[2,1-b]thiazol-5-yl)-3-phenylurea (8a)
Yield = 54%; m.p. = 287–288 °C; IR: 1639 (>C=O), 3263 (>N-H), 3290 (>N-H); 1H NMR: 2.09 (s, 3H, -CH3), 6.92 (t, J = 7.5 Hz, 1H, Ar-H), 7.08 (d, J = 4.5 Hz, 1H, Ar-H), 7.22 (t, J = 7.4 Hz, 1H, Ar-H), 7.42 (d, J = 7.8 Hz, 2H, Ar-Hs), 7.53 (d, J = 5.0 Hz, 1H, Ar-H), 8.09 (s, 1H, NH), 8.93 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.05, 118.63, 118.98 (3C), 122.48, 129.21 (2C), 137.23, 140.25, 144.81, 154.18 (>C=O); Anal. for C13H12N4OS (272.33): calcd.: C, 57.34; H, 4.44; N, 20.57; found: C, 57.17; H, 4.26; N, 20.79.
1-(6-Methylimidazo[2,1-b]thiazol-5-yl)-3-(p-tolyl)urea (8b)
Yield = 47%; m.p. = 282–283 °C; IR: 1701 (>C=O), 3267 (>N-H), 3360 (>N-H); 1H NMR: 2.14 (s, 3H, -CH3), 2.25 (s, 3H, -CH3), 7.08 (d, J = 8.0 Hz, 2H, Ar-Hs), 7.13 (d, J = 4.4 Hz, 1H, Ar-H), 7.36 (d, J = 7.8 Hz, 2H, Ar-Hs), 7.57 (d, J = 4.4 Hz, 1H, Ar-H), 8.09 (s, 1H, >N-H), 8.83 (s, 1H, >N-H); 13C NMR: 13.35 (-CH3), 20.87 (-CH3), 112.05, 118.70, 118.98, 119.13 (2C), 129.60 (2C), 131.32, 137.19, 137.65, 144.79, 154.20 (>C=O); Anal. for C14H14N4OS (286.35): calcd.: C, 58.72; H, 4.93; N, 19.57; found: C, 58.66; H, 5.10; N, 19.59.
1-(4-Methoxyphenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8c)
Yield = 50%; m.p. = 284–285 °C; IR: 1639 (>C=O), 3140 (>N-H), 3278 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 3.66 (s, 3H, -O-CH3), 6.80 (d, J = 8.5 Hz, 2H, ArH), 7.08 (d, J = 4.5 Hz, 1H, Ar-H), 7.32 (d, J = 8.5 Hz, 2H, Ar-Hs), 7.52 (d, J = 5.0 Hz, 1H, Ar-H), 8.04 (s, 1H, >N-H), 8.74 (s, 1H, >N-H); 13C NMR: 13.37 (-CH3), 55.67 (-O-CH3), 111.98, 114.37 (2C), 118.83, 120.6 (2C), 133.29, 137.13 144.73, 144.73, 154.37, 155.05 (>C=O); Anal. for C14H14N4O2S (302.35): calcd.: C, 55.62; H, 4.67; N, 18.53; found: C, 55.35; H, 4.69; N, 18.45.
1-(4-Fluorophenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8d)
Yield = 56%; m.p. = 281–282 °C; IR (KBr): 1635 (>C=O), 3163 (>N-H), 3278 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 7.04–7.10 (m, 3H, Ar-Hs), 7.42–7.44 (m, 2H, Ar-Hs), 7.54 (d, J = 5.0 Hz, 1H, Ar-H), 8.12 (s, 1H, >N’-H), 8.98 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.07, 115.59 (2C), 115.76 (2C), 118.58, 118.98, 120.82, 136.62, 137.29, 144.84, 154.29 (>C=O); Anal. for C13H11FN4OS (290.32): calcd.: C, 53.78; H, 3.82; N, 19.30; found: C, 53.80; H, 4.02; N, 19.53.
1-(4-Chlorophenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8e)
Yield = 48%; m.p. = 285–286 °C; IR: 1708 (>C=O), 3267 (>N-H), 3360 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 7.09 (d, J = 4.0 Hz, 1H, Ar-H), 7.26 (d, J = 9.0 Hz, 2H, Ar-Hs), 7.45 (d, J = 8.5 Hz, 1H, Ar-H), 7.54 (d, J = 4.5 Hz, 1H, Ar-H), 8.19 (s, 1H, >N-H), 9.12 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.10, 118.46, 118.99, 120.56 (2C), 126.00, 129.03 (2C), 137.35, 139.32, 144.89, 154.13 (>C=O); Anal. for C13H11ClN4OS (306.77): calcd.: C, 50.90; H, 3.61; N, 18.26; found: C, 50.73; H, 3.84; N, 18.39.
1-(4-Bromophenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8f)
Yield = 50%; m.p. = 288–289 °C; IR: 1709 (>C=O), 3263 (>N-H), 3360 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 7.09 (d, J = 4.5 Hz, 1H, Ar-H), 7.37–7.42 (m, 4H, Ar-Hs), 7.55 (d, J = 4.5 Hz, 1H, Ar-H), 8.27 (s, 1H, >N-H), 9.20 (s, 1H, >N-H); 13C NMR: 13.37 (-CH3), 112.09, 113.87, 118.48, 119.00, 120.96 (2C), 131.92 (2C), 137.30, 139.79, 144.87, 154.12 (>C=O); Anal. for:C13H11BrN4OS (351.22): calcd.: C, 44.46; H, 3.16; N, 15.95; found: C, 44.68; H, 3.28; N, 16.17.
4-(3-(6-Methylimidazo[2,1-b]thiazol-5-yl)ureido)benzenesulfonamide (8g)
Yield = 56%; m.p. = 283–284 °C; IR: 1708 (>C=O), 3120 (>N-H), 3275 (>N-H); 1H NMR: 2.09 (s, 3H, -CH3), 7.09 (d, J = 4.5 Hz, 1H, Ar-H), 7.17 (s, 2H, -NH2), 7.58 (d, J = 9.0 Hz, 1H, Ar-H), 7.57 (t, J = 8.5 Hz, 3H, Ar-Hs), 7.68 (d, J = 8.5 Hz, 2H, Ar-Hs), 8.26 (s, 1H, >N-H), 9.35 (s, 1H, >N-H); 13C NMR: 13.37 (-CH3), 112.18, 118.26, 118.32 (2C), 119.01, 127.21 (2C), 137.39, 137.55, 143.42, 144.98, 153.99 (>C=O); Anal. for C13H13N5O3S2 (351.40); calcd.: C, 44.43; H, 3.73; N, 19.93; found: C, 44.25; H, 3.56; N, 20.11.
3.2. Anticancer Activity
3.2.1. MTT Assay
The developed compounds were evaluated in vitro using the MTT cell viability assay following the published techniques [42,43] (Supplementary Data, page 42).
3.2.2. VEGFR-2 Kinase Activity
This assay was performed using the methodology described below. (Supplementary Data, page 42) [44,45].
3.2.3. PCR Assay
The gene expression levels of apoptosis genes were evaluated in MCF-7 cells using the following approaches in the Supplementary Data, page 43.
3.2.4. Cell Cycle Analysis and Apoptosis
These were performed using the approach described in the Supplementary Data, page 44 [46].
3.3. Docking Protocol
Docking studies were performed using AutoDockVina 1.5.7 (Supplementary Data, page 44).
3.4. In Silico Predictive ADME Study
The ADME study was performed using the SWISSADME server [47] (Supplementary Data, page 45).
4. Conclusions
Three derivatives, 6b, 6i and 6j, of isatin–imidazo[2,1-b]thiazole hybrids with a carbohydrazide function as a linker showed significant anti-cancerous activity using the MTT assay, with the most active candidates being submitted for further investigation to test their invitro VEGFR-2 inhibitory efficacy compared to Sorafenib as a reference drug. The potent candidate 6i was then subjected to a PCR study to measure Bax, Bcl2, caspase 8, caspase 9 and cytochrome C to understand the potency of the most active candidate 6i on malignant cells. Ultimately, for the full investigation regarding its activity, 6i was utilized for the cell cycle and apoptosis. It additionally arrested the cell cycle in the G2/M phase and induced apoptosis at rates higher than a standard drug. A docking protocol and ADME study were also performed and our candidates had the same binding mode of Sorafenib through interactions with the amino acid residues Lys868, Cys1045, Glu885 and Asp1046. As a consequence, 6i was specified as a promising lead for additional research to design efficient anti-cancer agents.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph17020216/s1, Figures S1 and S2 and Table S1 (Molecular docking); Figure S3 and Table S2 (Virtual ADME assessment); Figures S4–S19 (1H NMR and 13C NMR spectra of 6a–l); Figures S20–S33 (1H NMR and 13C NMR spectra of 8a–g).
Author Contributions
Conceptualization, methodology, software, validation, formal analysis, investigation, resources, data curation, writing—original draft preparation, M.K.E. and M.S.E.; writing—review and editing, visualization, M.K.E., M.S.E. and N.A.A.; supervision, H.A.A.-A.; project administration, N.A.A.; funding acquisition, N.A.A. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary material.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R403), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Pincheira J. Invited Review Cell proliferation and cancer. Histol. Histopathol. 1998;13:1197–1214. doi: 10.14670/HH-13.1197. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 4.Karamysheva A.F. Mechanisms of angiogenesis. Biochemistry. 2008;73:751–762. doi: 10.1134/S0006297908070031. [DOI] [PubMed] [Google Scholar]
- 5.Kerbel R.S. Tumor angiogenesis: Past, present and the near future. Carcinogenesis. 2000;21:505–515. doi: 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- 6.Traxler P. Tyrosine kinases as targets in cancer therapy—Successes and failures. Expert Opin. Ther. Targets. 2003;7:215–234. doi: 10.1517/14728222.7.2.215. [DOI] [PubMed] [Google Scholar]
- 7.Guo S., Colbert L.S., Fuller M., Zhang Y., Gonzalez-Perez R.R. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim. Biophys. Acta Rev. Cancer. 2010;1806:108–121. doi: 10.1016/j.bbcan.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bando H., Weich H.A., Brokelmann M., Horiguchi S., Funata N., Ogawa T., Toi M. Association between intratumoral free and total VEGF, soluble VEGFR-1, VEGFR-2 and prognosis in breast cancer. Br. J. Cancer. 2005;92:553–561. doi: 10.1038/sj.bjc.6602374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X., Zhou W. The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front. Endocrinol. 2015;6:159. doi: 10.3389/fendo.2015.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiau J.P., Wu C.C., Chang S.J., Pan M.R., Liu W., Ou-Yang F., Chen F.M., Hou M.F., Shih S.L., Luo C.W. FAK regulates VEGFR2 expression and promotes angiogenesis in triple-negative breast cancer. Biomedicines. 2021;9:1789. doi: 10.3390/biomedicines9121789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes K., Roberts O.L., Thomas A.M., Cross M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Narayanan J., Tamilanban T., Kumar P.S., Guru A., Muthupandian S., Kathiravan M.K., Arockiaraj J. Role and mechanistic actions of protein kinase inhibitors as an effective drug target for cancer and COVID. Arch. Microbiol. 2023;205:238. doi: 10.1007/s00203-023-03559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Musumeci F., Radi M., Brullo C., Schenone S. Vascular endothelial growth factor (VEGF) receptors: Drugs and new inhibitors. J. Med. Chem. 2012;55:10797–10822. doi: 10.1021/jm301085w. [DOI] [PubMed] [Google Scholar]
- 14.El Hadi S.R.A., Lasheen D.S., Soliman D.H., Elrazaz E.Z., Abouzid K.A.M. Scaffold hopping and redesign approaches for quinazoline based urea derivatives as potent VEGFR-2 inhibitors. Bioorg. Chem. 2020;101:103961. doi: 10.1016/j.bioorg.2020.103961. [DOI] [PubMed] [Google Scholar]
- 15.Fodor D., Jung I., Turdean S., Satala C., Gurzu S. Angiogenesis of hepatocellular carcinoma: An immunohistochemistry study. World J. Hepatol. 2019;11:294–304. doi: 10.4254/wjh.v11.i3.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng F.W., Liu D.K., Zhang Q.W., Xu Y.G., Shi L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012–2016) Expert Opin. Ther. Pat. 2017;27:987–1004. doi: 10.1080/13543776.2017.1344215. [DOI] [PubMed] [Google Scholar]
- 17.Oguro Y., Miyamoto N., Okada K., Takagi T., Iwata H., Awazu Y., Miki H., Hori A., Kamiyama K., Imamura S. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo[3,2-d] pyrimidine derivatives: Novel VEGFR2 kinase inhibitors binding to inactive kinase conformation. Bioorg. Med. Chem. 2010;18:7260–7273. doi: 10.1016/j.bmc.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Aziz M.A., Serya R.A.T., Lasheen D.S., Abdel-Aziz A.K., Esmat A., Mansour A.M., Singab A.N., Abouzid K. Discovery of Potent VEGFR-2 Inhibitors based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci. Rep. 2016;6:24460. doi: 10.1038/srep24460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dweedar H.E., Mahrous H., Ibrahim H.S., Abdel-Aziz H.A. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014;78:275–280. doi: 10.1016/j.ejmech.2014.03.058. [DOI] [PubMed] [Google Scholar]
- 20.Eldehna W.M., Altoukhy A., Mahrous H., Abdel-Aziz H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015;90:684–694. doi: 10.1016/j.ejmech.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Fares M., Eldehna W.M., Abou-Seri S.M., Abdel-Aziz H.A., Aly M.H., Tolba M.F. Design, synthesis and in Vitro antiproliferative activity of novel isatin-quinazoline hybrids. Arch. Pharm. 2015;348:144–154. doi: 10.1002/ardp.201400337. [DOI] [PubMed] [Google Scholar]
- 22.Kamal A., Dastagiri D., Ramaiah M.J., Reddy J.S., Bharathi E.V., Srinivas C., Pushpavalli S.N., Pal D., Pal-Bhadra M. Synthesis of imidazothiazole-chalcone derivatives as anticancer and apoptosis inducing agents. ChemMedChem. 2010;5:1937–1947. doi: 10.1002/cmdc.201000346. [DOI] [PubMed] [Google Scholar]
- 23.Elsawi A.E., Elbadawi M.M., Nocentini A., Almahli H., Giovannuzzi S., Shaldam M., Salem R., Ibrahim T.M., Abdel-Aziz H.A., Supuran C.T., et al. 1,5-diaryl-1,2,4-triazole ureas as new SLC-0111 analogues endowed with dual carbonic anhydrase and VEGFR-2 inhibitory activities. J. Med. Chem. 2023;66:10. doi: 10.1021/acs.jmedchem.3c00721. [DOI] [PubMed] [Google Scholar]
- 24.Fakhry M.M., Mattar A.A., Alsulaimany M., Al-Olayan E.M., Al-Rashood S.T., Abdel-Aziz H.A. New Thiazolyl-Pyrazoline Derivatives as Potential Dual EGFR/HER2 Inhibitors: Design, Synthesis, Anticancer Activity Evaluation and In Silico Study. Molecules. 2023;28:7455. doi: 10.3390/molecules28217455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdelsalam E.A., Abd El-Hafeez A.A., Eldehna W.M., El Hassab M.A., Marzouk H.M.M., Elaasser M.M., Abou Taleb N.A., Amin K.M., Abdel-Aziz H.A., Ghosh P., et al. Discovery of novel thiazolyl-pyrazolines as dual EGFR and VEGFR-2 inhibitors endowed with in vitro antitumor activity towards non-small lung cancer. J. Enzyme Inhib. Med. Chem. 2022;37:2265–2282. doi: 10.1080/14756366.2022.2104841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elewa M.A.F., Eldehna W.M., Hamdan A.M.E., Abd El-kawi S.H., El-Kalaawy A.M., Majrashi T.A., Barghash R.F., Abdel-Aziz H.A., Hashem K.S., Al-Gayyar M.M.H. WRH-2412 alleviates the progression of hepatocellular carcinoma through regulation of TGF-β/β-catenin/α-SMA pathway. J. Enzyme Inhib. Med. Chem. 2023;38:2185761. doi: 10.1080/14756366.2023.2185761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eldehna W.M., Mohammed E.E., Al-Ansary G.H., Berrino E., Elbadawi M.M., Ibrahim T.M., Jaballah M.Y., Al-Rashood S.T., Binjubair F.A., Celik M., et al. Design and synthesis of 6-arylpyridine-tethered sulfonamides as novel selective inhibitors of carbonic anhydrase IX with promising antitumor features toward the human colorectal cancer. Eur. J. Med. Chem. 2023;258:115538. doi: 10.1016/j.ejmech.2023.115538. [DOI] [PubMed] [Google Scholar]
- 28.El-Atawy M.A., Alshaye N.A., Elrubi N., Hamed E.A., Omar A.Z. Pyrimidines-Based Heterocyclic Compounds: Synthesis, Cytoxicity Evaluation and Molecular Docking. Molecules. 2022;27:4912. doi: 10.3390/molecules27154912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dietrich J., Hulme C., Hurley L.H. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: A structural analysis of the binding interactions of Gleevec ®, Nexavar®, and BIRB-796. Bioorg. Med. Chem. 2010;18:5738–5748. doi: 10.1016/j.bmc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- 30.Xie Q.Q., Xie H.Z., Ren J.X., Li L.L., Yang S.Y. Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2. J. Mol. Graph. Model. 2009;27:751–758. doi: 10.1016/j.jmgm.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Sobhy M.K., Mowafy S., Lasheen D.S., Farag N.A., Abouzid K.A.M. 3D-QSAR pharmacophore modelling, virtual screening and docking studies for lead discovery of a novel scaffold for VEGFR 2 inhibitors: Design, synthesis and biological evaluation. Bioorg. Chem. 2019;89:102988. doi: 10.1016/j.bioorg.2019.102988. [DOI] [PubMed] [Google Scholar]
- 32.Abdel-Mohsen H.T., Abdullaziz M.A., El Kerdawy A.M., Ragab F.A.F., Flanagan K.J., Mahmoud A.E.E., Ali M.M., Diwani H.I.E., Senge M.O. Targeting receptor tyrosine kinase VEGFR-2 in hepatocellular cancer: Rational design, synthesis and biological evaluation of 1,2-disubstituted benzimidazoles. Molecules. 2020;25:770. doi: 10.3390/molecules25040770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyde C.A.C., Giese A., Stuttfeld E., Abram Saliba J., Villemagne D., Schleier T., Binz H.K., Ballmer-Hofer K. Targeting Extracellular Domains D4 and D7 of Vascular Endothelial Growth Factor Receptor 2 Reveals Allosteric Receptor Regulatory Sites. Mol. Cell Biol. 2012;32:3802–3813. doi: 10.1128/MCB.06787-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Modi S.J., Kulkarni V.M. Exploration of structural requirements for the inhibition of VEGFR-2 tyrosine kinase: Binding site analysis of type II, ‘DFG-out’ inhibitors. J. Biomol. Struct. Dyn. 2022;40:5712–5727. doi: 10.1080/07391102.2021.1872417. [DOI] [PubMed] [Google Scholar]
- 35.El-Adl K., Ibrahim M.K., Khedr F., Abulkhair H.S., Eissa I.H. Design, synthesis, docking, and anticancer evaluations of phthalazines as VEGFR-2 inhibitors. Arch. Pharm. 2022;355:e2100278. doi: 10.1002/ardp.202100278. [DOI] [PubMed] [Google Scholar]
- 36.Liu S., Lin Y.C. Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17β. Breast J. 2004;10:514–521. doi: 10.1111/j.1075-122X.2004.21410.x. [DOI] [PubMed] [Google Scholar]
- 37.Li J., Fu A., Zhang L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. 2019;11:320–328. doi: 10.1007/s12539-019-00327-w. [DOI] [PubMed] [Google Scholar]
- 38.Lipinski C.A., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 39.Duchowicz P.R., Talevi A., Bellera C., Bruno-Blanch L.E., Castro E.A. Application of descriptors based on Lipinski’s rules in the QSPR study of aqueous solubilities. Bioorg. Med. Chem. 2007;15:3711–3719. doi: 10.1016/j.bmc.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 40.Samala G., Devi P.B., Saxena S., Meda N., Yogeeswari P., Sriram D. Design, synthesis and biological evaluation of imidazo[2,1-b]thiazole and benzo[d]imidazo[2,1-b]thiazole derivatives as Mycobacterium tuberculosis pantothenate synthetase inhibitors. Bioorganic Med. Chem. 2016;24:1298–1307. doi: 10.1016/j.bmc.2016.01.059. [DOI] [PubMed] [Google Scholar]
- 41.Cesur N., Cesur Z., Guner H. Fused Heterocycles: Synthesis of Some New Imidazothiazoles. Heterocycl. Commun. 2002;8:433–438. doi: 10.1515/HC.2002.8.5.433. [DOI] [Google Scholar]
- 42.Slater T.F., Sawyer B., Sträuli U., van de Loosdrecht A.A., Beelen R.H.J., Ossenkoppele G.J. Studies on succinate-tetrazolium reductase systems. III. Points of coupling of four different tetrazolium salts III. Points of coupling of four different tetrazolium salts. J. Immunol. Methods. 1994;77:311–320. [Google Scholar]
- 43.van de Loosdrecht A.A., Beelen R.H.J., Ossenkoppele G.J., Broekhoven M.G., Langenhuijsen M.M.A.C. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods. 1994;174:311–320. doi: 10.1016/0022-1759(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 44.Fontanella C., Ongaro E., Bolzonello S., Guardascione M., Fasola G., Aprile G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann. Transl. Med. 2014;2:123. doi: 10.3978/j.issn.2305-5839.2014.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma K., Suresh P.S., Mullangi R., Srinivas N.R. Quantitation of VEGFR2 (vascular endothelial growth factor receptor) inhibitors—Review of assay methodologies and perspectives. Biomed. Chromatogr. 2015;29:803–834. doi: 10.1002/bmc.3370. [DOI] [PubMed] [Google Scholar]
- 46.Darzynkiewicz Z. Critical aspects in analysis of cellular DNA content. Curr. Protoc. Cytom. 2011;7:7.2.1–7.2.8. doi: 10.1002/0471142956.cy0702s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article and supplementary material.