

Abstract
Cholesterol homeostasis is crucial for cellular and systemic function. The disorder of cholesterol metabolism not only accelerates the onset of cardiovascular disease (CVD) but is also the fundamental cause of other ailments. The regulation of cholesterol metabolism in the human is an extremely complex process. Due to the dynamic balance between cholesterol synthesis, intake, efflux and storage, cholesterol metabolism generally remains secure. Disruption of any of these links is likely to have adverse effects on the body. At present, increasing evidence suggests that abnormal cholesterol metabolism is closely related to various systemic diseases. However, the exact mechanism by which cholesterol metabolism contributes to disease pathogenesis remains unclear, and there are still unknown factors. In this review, we outline the metabolic process of cholesterol in the human body, especially reverse cholesterol transport (RCT). Then, we discuss separately the impact of abnormal cholesterol metabolism on common diseases and potential therapeutic targets for each disease, including CVD, tumors, neurological diseases, and immune system diseases. At the end of this review, we focus on the effect of cholesterol metabolism on eye diseases. In short, we hope to provide more new ideas for the pathogenesis and treatment of diseases from the perspective of cholesterol.
Keywords: cholesterol, cholesterol metabolism, diseases, efflux, reverse cholesterol transport, regulation
At present, increasing evidence suggests that abnormal cholesterol metabolism is closely related to various systemic diseases, including cardiovascular disease, tumors, neurological conditions immune system disorders, and eye diseases. Cholesterol overload caused by abnormal cholesterol metabolism can induce elevated oxidative stress, heightened inflammatory responses, reduced autophagy, and increased apoptosis in cells through various signaling pathways, ultimately accelerating the development of diseases. Therefore, this review will summarize the relationship between cholesterol metabolism and common diseases and aim to provide new perspectives for the pathogenesis of diseases.
1. INTRODUCTION
Cholesterol (C27H46O) was separated from human gallstones over two centuries ago, capturing the attention of scientists and clinicians. Its physiological and pathological significance is undeniable. Like other sterols, cholesterol is predominantly hydrophobic and biosynthesized in mammalian cells. Mainly found on the cell membrane, cholesterol interacts with adjacent lipids, regulating the rigidity, fluidity, and permeability of the bilayer. Additionally, cholesterol can bind to various transmembrane proteins, aiding in maintaining or altering their conformation. Apart from its role in the membrane structure and function, cholesterol generates diverse oxysterols through both enzymatic and nonenzymatic pathways, with some subsequently metabolized into bile acids. 1 Oxidative cleavage of cholesterol side chains produces pregnenolone, a common precursor for all other steroid hormones. These derivatives of cholesterol actively participate in various biological processes. Considering the vital role of cholesterol, disruptions in its metabolism may lead to various congenital human diseases.
Recently, a series of discoveries has contributed to a new understanding of cholesterol metabolism. Cholesterol metabolism in the body comprises four parts: biosynthesis, uptake, efflux, and storage. Disruption in any part of the metabolic process is highly likely to lead to the occurrence of acquired diseases. Currently, significant progress has been made in cardiovascular disease (CVD) research. Researchers have discovered various protective mechanisms to reduce atherosclerosis (AS) and attempted to develop drugs targeting cholesterol metabolism. 2 Nevertheless, the impact of abnormal cholesterol metabolism in the development of other diseases remains unclear. There is increasing evidence suggesting a close relationship between cholesterol metabolism and certain diseases. 3 , 4 , 5 , 6 Especially in eye diseases, for example, studies have shown that genetic variations in lipid metabolism and transport genes—such as liver lipase, cholesterol ester (CE) transferase, apolipoprotein (Apo) E and ATP binding cassette (ABC) transport protein A1—are associated with an increased risk of age‐related macular degeneration (AMD) 7 , 8 , 9 ; and the therapeutic effect of reverse cholesterol transport (RCT) pathway in eye diseases also has become a recent research hotspot 10 ; in addition, the function of cholesterol in the intraocular lens is different from that in other tissues and organs. These make eye disease the focus of this review. Therefore, in this review, we discuss separately the relationship between cholesterol metabolism and common diseases and aim to provide new perspectives for the pathogenesis of diseases.
In this review, we initially describe the metabolic process of cholesterol in the human body, including synthesis, transport, and efflux. Subsequently, we examine separately the impact of abnormal cholesterol metabolism on common diseases and potential therapeutic targets for each disease, including CVD, tumors, neurological conditions, immune system disorders, and eye diseases.
2. CHOLESTEROL METABOLISM IN THE BODY
In this review, we will describe the primary pathways and regulations of various key aspects of cholesterol metabolism, establishing a crucial foundation for understanding the relationship between cholesterol metabolism and diseases in the subsequent text. For more intricate regulatory mechanisms of cholesterol, please refer to the relevant literature on cholesterol regulation.
2.1. Biosynthesis and regulation
Cholesterol is a common lipid molecule essential for normal bodily function. 11 It originates from two pathways: in situ biosynthesis and systemic circulation. For example, in the retina, approximately 28% of cholesterol comes from systemic circulation due to the separation of the blood–retinal barrier, while the remaining cholesterol is synthesized in situ. 12 Here, we discuss the in situ biosynthesis of cholesterol in detail. Cholesterol biosynthesis begins with acetyl‐CoA, which is converted into mevalonate by 3‐hydroxy‐3‐methylglutaryl coenzyme reductase (HMGCR), a rate‐limiting enzyme in cholesterol biosynthesis. Mevalonate is then converted into squalene, subsequently transformed into lanosterol. Cholesterol biosynthesis is then divided into two pathways: the Bloch and Kandutsch–Russell pathways. In the Bloch pathway, lanosterol undergoes five steps to generate desmosterol, and in the Kandutsch–Russell pathway, lanosterol undergoes five steps to generate 7‐dehydrodesmosterol. Finally, 24‐dehydroxysterol reductase (DHCR24) and 7‐dehydroxysterol reductase (DHCR7) catalyze the last step of both pathways to produce cholesterol. 13 , 14
However, cholesterol biosynthesis is a high energy‐consuming process that requires strict regulations. In this section, we briefly review the crucial role of sterol regulatory element‐binding proteins (SREBPs), especially SREBP2, encoded by Srebp2, which enhances the expression of genes related to cholesterol biosynthesis in cholesterol biosynthesis, such as HMGCR. 15 , 16 SREBP2 is initially produced as an endoplasmic reticulum (ER)‐anchored precursor and becomes activated only after translocating from the ER to the Golgi apparatus. 17 However, the activity of SREBP2 is regulated by the cellular cholesterol concentration. When cholesterol concentration is high, SREBP cleavage‐activating protein (SCAP) binds to SREBP2 precursors in the ER membrane and interacts with insulin‐induced gene proteins (INSIGs), leading to the SCAP‐SREBP complex being trapped in the membrane and the blockage of the translocation. 18 , 19 When the concentration is low, INSIGs are degraded by the ubiquitin–proteasome pathway. SREBP2 precursors then move from the ER to the Golgi, releasing the amino‐terminal active domain through the cleavage. 20 In this process, nuclear SREBP undergoes post‐translational modifications. The acetylation of P300 and CBP can enhance the expression of nuclear SREBP, 21 whereas deacetylation of Sirtuin1 (SIRT1) and phosphorylation of AMP‐activated protein kinase (AMPK) can inhibit its expression. 22 , 23 These proteins regulate the transcription of SREBP2 (Figure 1). In addition, mTOR complex 1 (mTORC1) also plays an important role in the regulation of SREBP2. When mTORC1 is inactivated, cell autophagy and endosomal membrane trafficking increase, leading to the rise in cholesterol levels in the ER, which will inhibit SREBP2. By contrast, it reduces cholesterol levels in the ER and activate SRBEP2. 24 A recent study reported that mTORC1 can prevent Lipin1 from entering the nucleus by phosphorylating Lipin1 and promote the expression of SREBP2. 25
FIGURE 1.
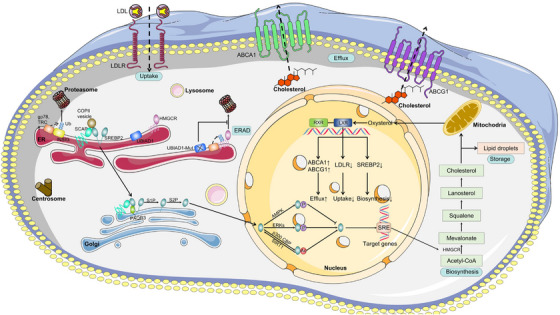
The regulation of cholesterol metabolism in the body. When cholesterol concentration is low, INSIGs are ubiquitylated by gp78 and TRC8, then SCAP interacts with SREBP2 and COPII, allowing the SCAP‐SREBP complex to anchor to the Golgi apparatus via PAQR3, where SREBP2 undergoes proteolytic cleavage of S1P and S2P, entering the nucleus. In the nucleus, SREBP2 interacts with the SRE sequence of promoters of target genes, such as HMGCR, which promotes cholesterol biosynthesis. The generated cholesterol is stored in the form of lipid droplets, and excess cholesterol is excreted out of the cell through the RCT pathway. In the RCT pathway, cholesterol is transported to the mitochondria and activates LXRs. Under normal physiological conditions, the transcription activity of LXRs increases in response to the increase of cholesterol cell level, which decreases the level of cholesterol via multiple pathways. Eventually, excess cholesterol is excreted through the ABCA1 and ABCG. When the concentration is high, SCAP interacts with INSIGs, leading to the blockage of the translocation. HMGCR can also be ubiquitinated and degraded in proteasomes through the ERAD. In addition, when UBIAD1 mutates, it can delay the degradation of HMGCR. COPII, coatomer II; PAQR3, adipoQ receptor 3; S1P, site 1 protease; S2P, site 2 protease; SRE, sterol regulatory element; Ac, acetyl group; P, phosphate group; Ub, ubiquitin; UBIAD1, UbiA prenyltransferase domain‐containing protein‐1; ERAD, ER‐associated degradation; RXR, retinoid X receptor; LXR, liver X receptor; LDL, low‐density lipoprotein; LDLR, low‐density lipoprotein receptor.
2.2. Conversion and transport
As widely known, blood lipids in humans, including cholesterol and triglycerides, are insoluble. Therefore, cholesterol transport in the blood requires association with lipoproteins. In systemic circulation, lipoproteins are classified into five types based on their densities: chylomicrons (CM), very low‐density lipoprotein (VLDL), intermediate‐density lipoprotein (iIDL), low‐density lipoprotein (LDL), and high‐density lipoprotein (HDL), which can convert into each other. Cholesterol in humans comes not only from endogenous biosynthesis but also from external intake. The epithelial cells of the intestine can absorb cholesterol from dietary sources through the Niemann‐Pick type C1‐like 1 protein (NPC1L1), releasing exogenous cholesterol in the form of CM. 26 Subsequently, CM can be transported to the liver through blood circulation and converted into VLDL, the main function of which is to transport endogenous cholesterol. Ultimately, VLDL is transformed into LDL in the blood, transporting cholesterol throughout the body, including the brain and eyes. In this process, low‐density lipoprotein receptor (LDLR), a cell surface glycoprotein, plays a crucial role in peripheral cells' uptake of cholesterol from systemic circulation, and its transcription can be also activated by SREBP2. Numerous studies have shown that VLDL and LDL are risk factors for large‐vessel diseases, such as AS. And they are positively correlated with the risk of diseases. However, HDL is commonly regarded as “good cholesterol,” because it transports excess cholesterol to the liver for metabolism and elimination. 27
The protein components of lipoproteins include ApoA, ApoB, ApoC, ApoD, and ApoE. ApoA1 is the major component of HDL, which exerts a prospective effect by transporting cholesterol from peripheral cells to sites of catabolism and activating lecithin‐cholesterol acyl transferase (LCAT) in the RCT pathway. Studies have shown that ApoA1 can boost cholesterol efflux via the ABCA1. 28 Furthermore, ApoA1 can be upregulated by liver X receptors (LXRs) in a dose‐dependent manner. 29 ApoB is the major component of CM, VLDL, and LDL, which are involved in the lipid delivery. Some data strongly suggest that the risk of CVD is positively correlated with the concentration of ApoB. ApoB has two subtypes, ApoB48 and ApoB100, produced in the intestine and liver respectively. ApoB100 plays an important role in inducing LDL formation and promoting inflammation. 30 And ApoE is responsible for cholesterol transport between cells or tissues, 31 directly regulated by LXRs in macrophages. 32 In blood, ApoE mainly combines with CM and VLDL; however, in the brain and retina, ApoE‐containing lipoproteins have densities similar to those of HDL. 31 It has three isoforms (2, 3, and 4) in the human body. ApoE4 increases the risk of Alzheimer's disease (AD) and reduces the risk of AMD, whereas ApoE2 has the opposite effect, which is secreted by the retinal pigment epithelium (RPE) and increases significantly after exposure to complement in the drusen. 33 , 34 , 35 , 36 This is because ApoE2 has obvious defects in cholesterol transport compared with ApoE4. 37 In addition, ApoE4 can maintain cholesterol homeostasis by regulating cholesterol efflux, preventing the autophagy deficiency and mitochondrial damage caused by complement activation. 37
2.3. Efflux
Almost all cells in the human body produce cholesterol; however, only a few cells can metabolize it. Therefore, to maintain cholesterol homeostasis in cells, excess cholesterol must be expelled, transported to the liver through the systemic circulation, metabolized, and ultimately discharged. This process is also known as the RCT. Cholesterol efflux from cells is an early step of the RCT, in which LXRs and ABCA1/ABCG1 as key regulators play a crucial role (Figure 1).
LXR, a ligand‐activated nuclear transcription factor, has the opposite effect to SREBP2, activated when cells accumulate excess cholesterol. The two highly homologous LXR proteins in the human body are distributed in different tissues. LXRα is primarily found in the intestine, liver, kidney, fat, and macrophages, while LXRβ is widely distributed in almost all cells. 38 , 39 , 40 In the nucleus, LXR associates with retinoid X receptor (RXR) to form the obligate heterodimer. Upon binding LXR agonists, the heterodimer activates the transcription of target genes, such as ABCA1, ABCG1, SREBP‐1c, and ApoE. 41 In addition, LXR has other functions in the human body. For example, in animal models of diabetes, LXR agonists regulate the metabolism of glucose and reduce blood glucose levels by upregulating the glucose transporter 4 (GLUT4). 42 Furthermore, LXR is one of the key receptors regulating the inflammatory response, 43 which can inhibit the expression of inflammatory factors (such as NF‐κB). 44 LXR also regulates cell apoptosis and autophagy, which will be discussed later. We then discuss the key downstream targets of LXR: ABCA1 and ABCG1. ABCA1 and ABCG1 utilize ATP as an energy source to regulate the transmembrane transport of lipids, sugars and amino acids. ABCA1 is a complete transporter protein widely expressed throughout the body. It consists of two tandem repeats of transmembrane domains, each with six transmembrane segments and a large glycosylated extracellular domain. In the RCT, the main role of ABCA1 is to transfer intracellular cholesterol to lipid‐free ApoA1 when activated by LXR, 45 resulting in the formation of nascent HDL. Subsequently, LCAT induces the formation of mature HDL. 28 Studies have shown that the inactivation or mutation of ABCA1 can significantly reduce the efflux of cellular cholesterol to ApoA1. Therefore, patients with Tangier disease or Abca1−/− mice cannot form mature HDL, and their plasma total cholesterol (TC) is very low, indicating abnormal lipid deposition in various tissues. 46 ABCG1, as a half‐sized transporter, is widely expressed in macrophages and other cell types, except hepatocytes and enterocytes. 47 Studies have shown that mice with Abcg1 deletion show cholesterol accumulation in macrophages and the liver. 47 In contrast, the tissues of ABCG1 overexpression mice were protected from lipid accumulation induced by a high‐fat diet. 48
Cholesterol metabolism is an intricate process that not only depends on the synchronized coordination of various steps but is also regulated by multiple signaling pathways throughout the body. The various diseases we will discuss later are largely a result of disordered cholesterol metabolism, leading to the loss of compensatory mechanisms and thus accelerating disease progression.
3. CHOLESTEROL METABOLISM IN CVDs
Over the past few decades, substantial strides have been achieved in treating CVD, marked by heightened public health awareness, a stronger focus on disease prevention and advancements in percutaneous coronary intervention and stent development. However, CVD persists as the leading global cause of death, 49 driving ongoing research into its pathogenesis. AS is consistently recognized as the linchpin of CVD, serving as the pathophysiological mechanism underlying ischemic heart disease, stroke, peripheral artery disease, and aneurysm formation. Presently, clinical treatment primarily centers on mitigating complications associated with AS using drugs that lower LDL levels, such as statins. 50 Nevertheless, the impact of this multifaceted disease on global mortality underscores the necessity for novel targeted therapies.
To discover new targeted drugs, understanding the pathogenesis of AS is crucial. The formation of foam cells, resulting from the accumulation of atherosclerotic lipoproteins induced by monocyte‐derived macrophages, is a pivotal early sign of AS. In simpler terms, macrophages play a key role in the pathogenesis and development of AS. 51 Normally, cholesterol metabolism in macrophages remains stable. Disruption of metabolism can activate various signaling pathways associated with inflammation and cell apoptosis. Each pathway regulates multiple downstream genes related to inflammation, oxidative stress and cell cycle. This significantly promotes the maintenance and progression of AS. 52 Therefore, we can conclude that the central focus of AS treatment will be reducing cholesterol accumulation.
Because the RCT pathway is presently regarded as the sole mechanism for clearing excess cholesterol in the human body, there has been a growing focus on the RCT research in recent years. Researchers have identified numerous drugs that enhance the pathway, 53 , 54 but the most noteworthy development is the emergence of microRNAs (miRNAs). miRNAs are endogenous noncoding RNA molecules, typically 21–25 nucleotides in length. They generally bind to the 3′ untranslated regions of target genes to repress mRNA translation, regulating various biological processes such as cell differentiation, proliferation, metabolism, and death. 55 According to reports, miRNAs play a role in maintaining cholesterol homeostasis. For example, miR‐33, ‐144, and ‐148 can regulate the RCT pathway by reducing ABCA1 expression. 56 Recently, miRNAs have been regarded as the new target for treating AS due to their important regulatory roles. First, miRNAs can decrease the expression of ABCA1 and inhibit the RCT pathway. Research has shown that ApoE deficient mice treated with miR‐20a/b exhibits reduced ABCA1 expression in the liver and decreased the RCT in vivo. Additionally, miR‐20a/b inhibits HDL formation and promotes AS development. Conversely, miR‐20a/b inhibitors can increase ABCA1 expression and cholesterol efflux, reduce cholesterol content, and inhibit foam cell formation. 57 Similar effects have been observed with other miRNAs, such as miR‐302a, miR‐320b, and miR‐613. 58 , 59 , 60 Second, inhibiting or knocking out miRNAs can alleviate inflammation in macrophages treated with oxidized low‐density lipoproteins (ox‐LDL). Studies have found that inhibiting miR‐652‐3p can reduce foam cell formation and decrease IL‐1β, IL‐6, and TNF‐α in macrophages treated with ox‐LDL, which have proven that inhibiting miR‐652‐3p can regulate lipid metabolism and inflammatory cytokine secretion in macrophages, ultimately alleviating AS. 61 Third, inhibiting or knocking out miRNAs can inhibit oxidative stress in macrophages. For instance, knocking down MiR‐34a‐5p can inhibit cholesterol accumulation and oxidative stress damage. 62 These results may contribute to the development of new therapies related to the RCT‐based treatment strategies. However, the specific mechanism of miRNA is still unclear, and it is uncertain whether their effects on inflammation and oxidative stress are related to cholesterol efflux. In the future, we should also strive to discover other pathways that can eliminate cholesterol accumulation and attempt to discover safer and more effective targets.
4. CHOLESTEROL METABOLISM IN TUMORS
Cholesterol plays a crucial role in the occurrence and development of tumors due to its ability to drive cell proliferation, migration and invasion. Abnormal regulation of cholesterol metabolism in tumor cells mainly involves upregulation of cholesterol synthesis levels, increased cholesterol uptake and abnormal accumulation of metabolic products. These factors contribute to tumor cell proliferation, survival, invasion, metastasis, and enhanced adaptability to the tumor microenvironment, ultimately promoting the occurrence and development of tumors. Some genes related to cholesterol synthesis show increased activity in tumor tissues, leading to the abnormal activation of cholesterol biosynthesis key enzymes, such as HMGCR and squalene monooxygenase (SQLE). 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 which has a significant impact on the development of tumors (Table 1). Next, we will discuss the impact of cholesterol metabolism on tumors, focusing on its effect in prostate cancer (PC) and hepatocellular carcinoma (HCC).
TABLE 1.
The effect of overexpression of cholesterol synthases on tumors.
| Disease | Enzyme | Signal | Functions | References |
|---|---|---|---|---|
| HCC | SQLE | ERK signaling↑ | The growth and migration of HCC↑ | 63 |
| SQLE | TGF‐β/SMAD signaling↑ | The metastasis and chemoresistance of HCC↑ | 64 | |
| PC | HMGCR | The growth of PC↑ | 65 | |
| SQLE | The growth of PC↑ | 66 | ||
| Gastric cancer | HMGCR | Hedgehog/Gli1 signaling↑ | The growth of gastric cancer↑ | 67 |
| Glioblastoma | HMGCR | Hippo signaling↑ | The growth and migration of glioblastoma↑ | 68 |
| Breast cancer | HMGCR | The growth and migration of breast cancer↑ | 69 | |
| Esophageal squamous cell carcinoma | HMGCR | Ras‐ERK signaling↑ | The growth and migration of carcinoma↑ | 70 |
| Lung squamous cell carcinoma | SQLE | ERK signaling↑ | The proliferation and metastasis of carcinoma↑ | 71 |
| Bladder cancer | SQLE | P53 signaling↓ | The proliferation, migration and invasion of bladder cancer↑ | 72 |
| Colorectal cancer | SQLE | MAPK signaling↑ | The proliferation of colorectal cancer↑ | 73 |
Abbreviations: ERK, extracellular signal‐regulated kinase; HCC, hepatocellular carcinoma; MAPK, mitogen‐activated protein kinase; PC, prostate cancer; SMAD, suppressor of mother against decapentaplegic; TGF‐β, transforming growth factor‐β.
4.1. Prostate cancer
PC is the second leading cause of cancer death in men worldwide. 74 Currently, a large number of studies have clearly shown that hypercholesterolemia may be associated with the risk of developing PC. 75 , 76 , 77 A cholesterol‐lowering diet is a favorable factor against the risk of PC. 78 In fact, a positive correlation between cholesterol accumulation in prostate tissue and the presence of PC has been discovered for a long time. 79 Since then, more and more studies have supported the indispensable role of cholesterol in the progression of PC. One hypothesis is that the accumulated cholesterol in PC provides a precursor for androgen synthesis in tumors, which can promote the development of castration‐resistant PC (CRPC). 80 Therefore, the use of statins or enhancement of ABCA1 expression to lower cholesterol levels can reduce the growth of PC by lowering serum or tumor androgen levels. 81 , 82 Meanwhile, studies have shown that cholesterol not only promotes tumor proliferation but also that its metabolite, 27‐hydroxycholesterol (27‐OHC), induces an increase in tumor cell proliferation. 83 Another hypothesis is that an increase in cholesterol levels supports the growth of PC by providing key membrane components such as lipid rafts. 84 , 85 It was also found that epidermal growth factor receptor is a cell membrane binding receptor associated with lipid rafts in PC cells, 86 its activation can lead to the activation of protein kinase B, which promotes the growth of PC. 87 A study has found that LXR agonists induce cell apoptosis and inhibit the growth of tumor cells by activating cholesterol efflux to disrupt lipid rafts and reducing the downregulation of AKT signaling in vitro model. 88
Given that cholesterol accumulation promotes tumor growth, inhibiting the biosynthetic pathway of cholesterol is an attractive direction. In addition to the well‐known statins, such as simvastatin, 89 alternative methods for cholesterol biosynthesis inhibitors are gradually being explored. 2,3‐Oxidosqualene cyclase (OSC), an enzyme that converts 2,3‐monoepoxysqualene to lanosterol, has become a possible new target. Recently a study found that inhibitor of OSC, RO48‐8071 exerts multiple effects. For example, RO48‐8071 can reduce viability of both hormone‐dependent and CRPC cells and induce apoptosis of cells in vitro. 90 The role of SQLE is also worth studying. Previous studies have shown that its expression is associated with poor prognosis. 91 , 92 Recently, Kalogirou et al. 66 showed that high SQLE protein expression is associated with adverse outcomes in PC, which contributes to the growth of PC tumors. They also demonstrated that SQLE inhibition effectively reduces in situ tumor growth without affecting the overall cholesterol metabolism of mice. 66 Moreover, the inhibition of SQLE can effectively suppress lymph node metastasis in PC cells. 93 These findings emphasize that SQLE may be a potential biomarker and therapeutic target in PC patients. In addition to inhibiting key enzymes in the synthesis pathway, blocking cholesterol esterification has become a strategy for cancer treatment. Previously, it has been found that the inhibitor of acyl‐coenzyme A: cholesterol acyltransferase‐1, avasimibe, can significantly reduce the storage of CEs in lipid droplets and increase intracellular free cholesterol levels, leading to cell apoptosis and inhibiting proliferation. 94 Later, Lee et al. 95 further demonstrated that inhibition of cholesterol esterification in mouse models inhibits the development and growth of metastatic PC. The downregulation of the Wnt/β‐catenin pathway after CE depletion was determined, and it was suggested that inhibition of cholesterol esterification could inhibit PC metastasis by impairing Wnt/β‐catenin signaling. 95 In summary, these studies have offered new opportunities for the treatment of PC.
4.2. Hepatocellular carcinoma
HCC is the 6th most common cancer worldwide and the third leading cause of cancer‐related mortality worldwide. 96 , 97 HCC is induced by various diseases, including hepatitis B virus infection, alcoholic liver disease, and metabolic syndrome. 98 Despite progress in the diagnosis and treatment of HCC, the majority of HCC patients still have poor prognosis due to tumor progression or recurrence. 99 Epidemiological studies have shown that cholesterol intake is an independent risk factor for HCC. 100 Cholesterol metabolism disorders are often observed in HCC, and the activation of cholesterol synthesis drives tumor development. 101 , 102 Therefore, reducing cholesterol levels by inhibiting its biosynthesis or promoting its degradation may be an effective strategy for inhibiting the progression of HCC.
How does the cholesterol overload impact the progression of HCC? Numerous studies currently indicate the role of cholesterol in the development of HCC. First, cholesterol overload in the liver can lead to severe cellular redox imbalance, exacerbating various chronic liver diseases. 103 , 104 , 105 Research results have shown that cholesterol overload can lead to antiapoptotic responses and DNA damage. Additionally, redox imbalance can enhance the carcinogenic effect of typical DNA damage inducers such as N‐nitrosodiethylamine. 106 Second, there is also a connection between cholesterol and inflammation, as previously mentioned, NF‐κB pathway is a key link in the inflammatory response, playing an important role in promoting HCC by increasing proliferation and preventing cell apoptosis. 107 Lipopolysaccharides (LPS) are components of Gram‐negative bacterial walls that can activate NF‐κB. In a study aimed at investigating the relationship between inflammation and cholesterol accumulation in HCC cells, the authors found that LPS increases intracellular cholesterol accumulation through the NF‐κB pathway in HCC cells, which in turn promotes LPS/NF‐κB pathway and inflammatory response, indicating the proinflammatory effect of cholesterol in HCC cells. 108 Finally, a recent study also showed that cholesterol is involved in the autophagy process of HCC cells, and demonstrated that cholesterol inhibits the autophagy degradation of receptor tyrosine kinases via Golgi membrane protein 1 to promoting the metastasis of HCC. 109 It is evident that cholesterol plays an important role in the development of HCC, and other potential mechanisms are worth further research.
After clarifying their relationship, research on the treatment of HCC has also emerged. As a key enzyme in the cholesterol synthesis pathway, SQLE still plays an important role in the development of HCC. Research has shown that in the later stages of tumors development, the activation of TGF‐β signals promotes tumor occurrence, metastasis, and chemotherapy resistance. 110 In subtypes of TGF‐β, TGF‐β1 plays a crucial role in fiber formation by activating SMAD2/3 phosphorylation, 111 while SQLE can activate TGF‐β/SMAD signaling pathway and promote the development of HCC. The study by Zhang et al. 64 also demonstrated that chemical inhibitors of SQLE significantly inhibit the development of HCC in vitro and in vivo. In addition, LXR is also a potential therapeutic target, which can inhibit tumor growth through multiple pathways. In a study, the authors have demonstrated that LXR agonists can downregulate forkhead box M1 (FOXM1) in HCC cells, thereby inhibiting the expression of cyclin D1 and cyclin B1, leading to cell cycle and proliferation arrest. 112 Another study also reported that activation of LXR induces the expression of suppressor of cytokine signaling 3, leading to downregulation of cyclin D1 and upregulation of p21 and p27, inhibiting the growth of HCC cells. 113 However, some studies have also pointed out that chronic activation of LXRα promotes HCC, at least in part, by promoting innate immune suppression as a result of accumulation of oxysterols. 114 Therefore, caution is needed when exploring LXR activating drugs for HCC treatment. Finally, due to the core role of SREBP2 in regulating cholesterol homeostasis, its inhibition also exhibits antitumor effects. 115 , 116
It is evident that an excess of cholesterol in tumors can promote the growth and metastasis of tumor cells. Given the substantial impact of cholesterol and its metabolites on tumors, researching the disruption of cholesterol metabolism in cells within the tumor microenvironment will be crucial in the future. This research will offer substantial assistance in antitumor therapy.
5. CHOLESTEROL METABOLISM IN NEUROLOGICAL DISEASES
The brain is the organ with the highest cholesterol content, accounting for approximately 25% of total body cholesterol. 117 Unesterified cholesterol is the main sterol in the adult brain, along with small amounts of deamination sterols and CEs. Cholesterol is not only a crucial structural component of cell membranes and myelin, but also necessary for synaptic and dendritic formation. 118 , 119 The consumption of cholesterol in neurons can impair the exocytosis, neuronal activity and neurotransmission of synaptic vesicles, leading to dendritic spines and synaptic degeneration. 120 , 121 , 122 Therefore, cholesterol is crucial for neuronal physiology, and cholesterol metabolism may be related to some central nervous system (CNS) diseases (Table 2), such as AD and Parkinson's disease (PD). 123 , 124 , 125 , 126 , 127 , 128
TABLE 2.
The relationship between common neurological disorders and cholesterol metabolism.
| Disease | Experimental model | Experimental aim | Conclusion | References |
|---|---|---|---|---|
| AD | AD mice | The effect of LXR agonist TO901317 on the cognitive function of AD mice. | TO901317 reduces the production of Aβ in the brain by promoting cholesterol efflux. | 123 |
| AD mice | The effect of increased cholesterol abundance on neurodegeneration. | The increase in cholesterol content in neurons in the brain may help induce and/or worsen AD. | 124 | |
| The effect of AD on the ability of ABCA1‐mediated cholesterol efflux. | In AD patients, ability of ABCA1‐mediated cholesterol efflux decreases. | 125 | ||
| The contribution of ApoE4 and astrocytes to amyloidosis in AD. | ApoE4 astrocytes induce amyloidosis through cholesterol oversupply. | 126 | ||
| PD | PD mice | The relationship between hypercholesterolemia and dopaminergic neurodegeneration. | Hypercholesterolemia can lead to oxidative stress and mitochondrial dysfunction, inducing dopaminergic neurotoxicity. | 127 |
| Levels of cholesterol metabolites in cerebrospinal fluid of PD patients. | 7α‐hydroxycholesterol levels are positively correlated with depression in PD patients. | 128 |
Abbreviations: Aβ, amyloid proteins‐β; AD, Alzheimer's disease; PD, Parkinson's disease.
5.1. Alzheimer's disease
AD is the main cause of dementia in the elderly. The pathological features of AD include the aggregation of Aβ and neurofibrillary tangles, twisted fibers of tau. So far, the amyloid hypothesis has dominated research on AD. However, the repeated failures of clinical trials have challenged our narrow understanding of the pathogenesis of AD, indicating that Aβ may be a prominent pathological feature, but not the only pathogenic factor, and has sparked extensive investigation into the potential mechanisms of AD. 129 , 130 An increasing amount of data indicates a close correlation between brain cholesterol deficiency and the pathogenesis of AD, forming a new cholesterol hypothesis. 131 , 132 More and more studies also show that AD patients have lower brain cholesterol levels, reduced cholesterol synthesis and trafficking. 133 , 134 In addition, due to the presence of the blood–brain barrier, although some studies have shown that people with high plasma cholesterol levels are more prone to AD, 135 , 136 we cannot easily conclude that high plasma cholesterol is a risk factor for AD, and further research is needed to elucidate the relationship between cholesterol and AD.
At present, changes in cholesterol metabolism have always been considered a key factor in AD's development. 137 Most of the cholesterol in the brain is catalyzed by the cytochrome P450 46A1 (CYP46A1) to 27‐OHC and 24‐S‐hydroxycholesterol (24‐OHC). Research has shown that 27‐OHC is a risk factor for the development and exacerbation of AD pathology by disrupting cholesterol metabolism and increasing Aβ in the brain, while 24‐OHC can become a protective factor for AD. 138 Therefore, the exploration of CYP46A1 has become a hot topic in the treatment of AD. At present, CYP46A1 has been studied in different models. For example, impaired learning and defective hippocampal long‐term potentiation were observed in the hippocampus of Cyp46a1 knockout mice. 139 Studies on mice overexpressing CYP46A1 have also found that increased CYP46A1 activity may improve memory. 140 A recent study showed that activating CYP46A1 improves animal performance in hippocampal dependent MWM and situational fear regulation tests. Improved behavior is accompanied by an increase in 24‐OHC brain content, neuronal cholesterol turnover, and specific presynaptic and postsynaptic proteins. At the same time, an increase in astrocyte response and a decrease in microglial activation were observed. 141 Therefore, we can believe that increasing the level of 24‐OHC in the brain and the activity of CYP46A1 may be an effective strategy for treating AD. In addition to CYP46A1, statins are also widely used due to their anti‐inflammatory, antioxidant, and antithrombotic properties, which can reduce the risk of AD events and dementia. However, observational studies and randomized controlled trials of AD have shown conflicting results, 142 , 143 which also reflect the complexity of the relationship between cholesterol and cognitive decline in the brain. Therefore, further research on the metabolism of cholesterol in the brain may be crucial for understanding how changes in blood cholesterol affect cognitive and neurodegenerative processes.
5.2. Parkinson's disease
PD is the second most common neurodegenerative disease globally, characterized by various motor and nonmotor symptoms, including static tremor, rigidity, motor delay, and gait/posture instability. 144 As widely known, α‐synuclein aggregation and neuroinflammation are the two main pathological features of PD. 145 , 146 However, the role of cholesterol in PD has not been appropriately addressed. Some studies have shown that cholesterol plays a role in dopaminergic neurodegeneration, and further evaluation is needed to understand its role in PD.
Although the role of cholesterol in PD is still unknown, epidemiological studies have shown that statins have the lowest risk of PD. They can act as antioxidants and anti‐inflammatory agents to reduce the formation of reactive oxygen species (ROS) and exert neuroprotective effects through various mechanisms, such as inhibiting proinflammatory cytokines, inhibiting microglial cell activation, inhibiting oxidative stress, and reducing α‐syn aggregation. 147 Although additional randomized controlled trials and observational studies are needed to confirm this conclusion, it also supports the important role of statins. For example, simvastatin can inhibit the activation of NF‐κB pathway, reduce proinflammatory cytokines, restore striatal dopamine (DA) levels, and improve motor function in PD patients. 148 However, there is still controversy regarding the use of statins. In a prospective study on the relationship between statin use and PD, statin use may be associated with a higher risk of PD, while higher TC may be associated with a lower risk. 149 These data are inconsistent with the hypothesis that statins can prevent PD. Some studies even suggest that the use of statins may have adverse effects on baseline nigrostriatal DA degradation and long‐term outcomes in PD patients. 150 In the future, we should enhance our understanding of the pathogenesis of PD and clarify the relationship between the disease and statin therapy.
The above content informs us that the metabolism of cholesterol in the brain is a complex process. There is an urgent need to conduct in‐depth research on the specific role of cholesterol metabolism in neurodegenerative diseases. With the deepening of research, effective intervention measures targeting cholesterol and its metabolites also have important clinical significance for delaying the development of diseases and finding new therapeutic targets.
6. CHOLESTEROL METABOLISM IN IMMUNE SYSTEM DISEASES
Diseases of the immune system are mainly caused by the accumulation of a large number of immune cells and immunoglobulins generated by an excessive immune response, which damages normal tissues, causing pathological changes and functional damage. Their occurrence is closely related to the activation of inflammatory reactions. However, abnormal cholesterol metabolism often leads to an enhanced immune response, which seems to suggest that cholesterol also plays an important role in the pathogenesis of immune system diseases. At present, there is currently very little research on cholesterol metabolism and the immune system. Therefore, in this review, we discuss systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), attempting to explore the role of cholesterol in the development of these diseases.
6.1. Systemic lupus erythematosus
SLE is a common and potentially fatal autoimmune disease characterized by multiple organ damage associated with autoantibodies, including the kidneys, cardiovascular system, neurological system and skin, primarily affecting women of childbearing age. 151 In the past few years, we have made some interesting progress in understanding the pathogenesis of SLE, including metabolic disorders, signaling and biochemical defects in immune cells, as well as impaired perception and repair of DNA damage. 152 Among these factors, dyslipidemia is a common feature of patients with SLE, and associated with accelerated AS and increased risk of CVD. 153 Recent studies have shown that the ability of cholesterol efflux is impaired to varying degrees in SLE patients, 154 which provides new insights into the pathogenesis of SLE. The study found that both ABCA1/ABCG1‐mediated cholesterol efflux is reduced, which may have a significant impact on the formation of foam cells and the inflammatory activation of macrophages. 155 And they believed that the impairment is related to the dysfunction of HDL. This view has also been verified in another study. They found in vitro that resveratrol can enhance the cholesterol outflow of macrophages through ABCA1 and ABCG1 pathways, effectively resisting the impact of SLE on AS. 156 Therefore, we may conclude that the occurrence of SLE is also related to the deposition of cholesterol. At the same time, drug treatment for SLE is gradually advancing, including statins. 157 With the progress of confirmatory trials, its therapeutic effect is worth looking forward to.
6.2. Rheumatoid arthritis
RA is also an autoimmune disease in which the immune system mistakenly attacks the host's body systems. Its clinical features typically manifest as chronic noninfectious synovial inflammation, cartilage and bone destruction. At present, there are no effective drugs for RA, and only disease‐relieving antirheumatic drugs and biological agents can be used to alleviate symptoms. However, these drugs can also bring serious side effects during therapy. 158 Patients with active RA often have the clinical characteristics of the lipid paradox, with lower levels of TC, HDL‐C, and LDL‐C, leading to a higher risk of cardiovascular incidence rate and RA mortality. 159 The reason for this phenomenon is that, in RA patients, a large amount of cholesterol accumulates in macrophages. Studies have shown that chronic inflammation contributes to the accumulation of lipids in the synovium. 160 So, how can we change this paradox? Some studies have showed that the suppression of LXRα agonism can improve lipid metabolism and inflammatory process in RA. 161 Although this contradicts our traditional understanding, 162 it will also help develop new drugs for treating RA with dyslipidemia.
In summary, given the complexity of immune system diseases, we can only offer a rough overview of the role of cholesterol in their pathogenesis and provide clues for future research. We aim to make more progress in the future to further identify potential therapeutic targets.
7. CHOLESTEROL METABOLISM IN EYE DISEASES
At present, there is limited research on the metabolism of cholesterol in the eye. As a common lipid present in the retina, we also have little understanding of its role in retinal function. As both the brain and retina belong to the CNS and share many similarities, numerous studies compare the two. Similar to the brain, due to the blood–retinal barrier, cholesterol in the retina primarily comes from in situ synthesis, and its metabolism is strictly regulated by various mechanisms. In this review, we will discuss not only retinal diseases like AMD and diabetic retinopathy (DR) but also several other common eye conditions, including cataracts, corneal diseases and dry eye syndrome (DES), which are the primary focus of our review (Table 3). 163 — 174
TABLE 3.
The relationship between common eye diseases and cholesterol metabolism.
| Disease | Experimental model | Experimental aim | Conclusion | References |
|---|---|---|---|---|
| AMD | The mice of targeted deletion of macrophage Abca1 and Abcg1 | The effect of deletion of Abca1 and Abcg1 on cholesterol homeostasis. | The lack of Abca1 and Abcg1 will lead to cholesterol accumulation and trigger early AMD. | 163 |
| The potential function of ABCA1 in the eye. | Impaired activity of the ABCA1‐mediated lipid efflux pathway may contribute to the progression of AMD. | 164 | ||
| Human iPSC‐derived RPE cells of Abca1 deletion | The identification of conditions for reducing the expression and function of ABCA1 in RPE. | The deletion of Abca1 leads to intracellular lipid accumulation and promotes the development of AMD. | 165 | |
| The mice of Tspo knockout | The impact of Tspo knockout on RPE in mice. | The lack of TSPO in mice leads to the defect of cholesterol efflux in RPE cells and induces the onset of AMD. | 166 | |
| Cataract | Male and female Sprague–Dawley rats | The mechanism of cataract induced by E2012. | The reduction of cholesterol in lens has been proved to be a prerequisite for cataract induced by E2012. | 167 |
| Male Sprague–Dawley rats | The relationship between lens opacity and cholesterol biosynthesis inhibition. | The inhibition of cholesterol biosynthesis is one of the reasons for lens turbidity. | 168 | |
| The knock‐in mouse model with G589S mutation in Lss | The roles of LSS in cataractogenesis. | The down‐regulation of cholesterol biosynthesis may lead to the lens development defect during cataract. | 169 | |
| The Shumiya cataract rat | The mechanism of cataract formation. | Cholesterol in the lens may be important for maintaining normal lens homeostasis. | 170 | |
| Corneal diseases | To understand abnormal cholesterol accumulation in cornea. | LCAT, ApoD, ApoA1 and ABCA1 are essential to prevent cholesterol accumulation and maintain vision. | 171 | |
| DES | The potential correlation between DES and statin or dyslipidemia | History of statin use or dyslipidemia is related to the increased probability of diagnosis of DES. | 172 | |
| The heterozygous Ubiad1 G184R knock‐in mice | To study how UBIAD1 promotes the occurrence of SCD. | The Ubiad1 mutation associated with SCD impairs the transport of ER to Golgi apparatus, finally leads to accumulation of cholesterol in the cornea. | 173 | |
| DR | The mice of ablation of Cyp46a1 | The role of CYP46A1 in retina and retinal vessels. | The ablation of Cyp46a1 leads to the increase of cholesterol in the retina and induce some manifestations of early DR. | 174 |
Abbreviations: AMD, age‐related macular degeneration; DES, dry eye syndrome; DR, diabetic retinopathy; LSS, lanosterol synthase; SCD, Schnyder corneal dystrophy; TSPO, 18‐kDa translocator protein.
7.1. Age‐related macular degeneration
7.1.1. The relationship of AMD and cholesterol metabolism
AMD is the leading cause of blindness worldwide and has a significant impact on quality of life. 175 About 80% of AMD patients are diagnosed with early dry subtypes, characterized by the formation of drusen under RPE, thickening of the Bruch membrane, and accumulation of lipofuscin in RPE. 176 A study in 2010 showed that over 40% of the volume of drusen is composed of lipid‐containing particles. 177 Similarly, Rodriguez et al. 178 , 179 found that 7‐ketocholesterol (7KCh), the main component of ox‐LDL, is mainly located in the outer layer of the retina. Therefore, we believe that the occurrence and development of AMD are closely related to cholesterol metabolism.
Currently, many studies have shown that 7KCh has a significant impact on the pathogenesis of AMD (Figure 2). First, concerning autophagy, P62, one of the targets of autophagic degradation, represents the autophagic flux of cells. Some studies have indicated that the level of P62 protein increases in 7KCh‐treated RPE cells, indicating that 7KCh can inhibit autophagy. 180 Regarding the mechanism, we suspect that under conditions of high oxygen consumption and accumulation of lipid peroxidation products, oxidative stress is triggered, damaging mitochondria in RPE cells, in turn, increasing oxidative stress. Finally, some oxidation products polymerize, and lipofuscin accumulates in the lysosome of the cell, affecting the lysosomal phagocytosis of autophagy and leading to a decrease in the autophagic flux of RPE cells (Figure 2A). 181 , 182 , 183 Second, in terms of inflammation, 7KCh can induce the expression of inflammatory cytokines, such as IL‐6, IL‐1β, and TNFα, mainly by activating the NF‐κB pathway. 44 , 184 , 185 NF‐κB, as a transcription factor, is critical in mediating the expression of cytokines and other inflammatory markers. Huang et al. 185 showed that inhibiting the NF‐κB pathway suppresses 7KCh‐induced inflammation and provides significant protection against 7KCh‐induced cell death. In addition, other signaling pathways, such as extracelluar signal‐regulated kinase (ERK), can also cause the increases of inflammation. 180 Simultaneously, with a decrease in autophagy, the accumulation of ROS can induce the activation of NOD‐like receptor protein 3 (NLRP3) inflammatory bodies in human ARPE‐19 cells, 186 , 187 leading to a further enhancement of the cellular inflammatory response (Figure 2B). 188 Third, in terms of apoptosis, 7KCh is also associated with apoptosis. Previous studies have shown that in ARPE‐19 and rat neuro‐retinal R28 cell cultures, 7KCh significantly increases the activities of caspase‐3, ‐8, and ‐12 that promote apoptosis. 189 , 190 , 191 In addition, a study showed that the injection of 7KCh into the rat retina can induce apoptosis of photoreceptors and RPE cells, leading to the detachment of outer microvilli (Figure 2C). 180 Therefore, we conclude that 7KCh can affect cell viability and normal physiological functions by regulating autophagy, inflammation, and apoptosis, ultimately accelerating AMD progression. In addition, there is some evidence that 7KCh can also affect cell adhesion, migration, and angiogenesis (Figure 2D), which requires further exploration. 192
FIGURE 2.
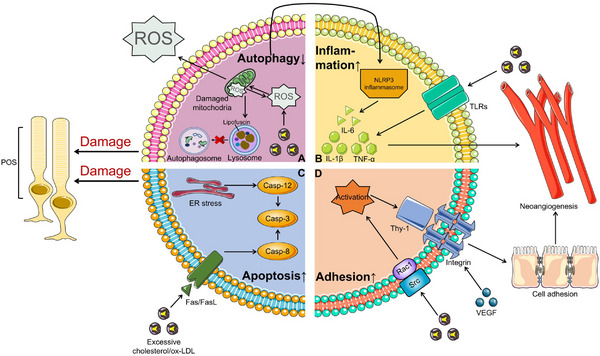
Effects of the accumulation of excess cholesterol/ox‐LDL in AMD. Excessive cholesterol is easily oxidized to ox‐LDL in the retina, which is the main component of 7KCh and widely affects multiple cellular processes. (A) First, under the condition of excessive lipid accumulation, oxidative stress can be triggered, leading to the mitochondrial damage in RPE cells, thereby increasing oxidative stress. Finally, some oxidation products polymerize, and lipofuscin accumulates in the lysosomes of cells, affecting the phagocytosis of lysosomes against autophagy and resulting in the impact on autophagy in the POS. In addition, decreased autophagy and mitochondrial damage can activate NLRP3 inflammasomes and further release inflammatory factors. (B) Second, excessive cholesterol activates NF‐κB pathway by binding to TLRs, increasing the expression of inflammatory factors, such as IL‐6, IL‐1β, and TNF‐α. (C) Third, the activation of Fas/FasL signals and ER under stress can increase the activities of Caspase‐3, ‐8, and ‐12 that promote apoptosis, leading to the increase of apoptosis of POS. (D) Fourth, the activation of Rac1 and Src by 7KCh leads to VEGF‐dependent expression of Thy‐1 and induces cell adhesion and migration, resulting to vascular rupture and angiogenesis with the increased expression of inflammatory factors. TLRs, Toll‐like receptors; VEGF, vascular endothelial growth factor; FasL, Fas receptor‐Fas ligand; POS, outer segment photoreceptor; NLRP3, NOD‐like receptor protein 3.
7.1.2. Role of cholesterol metabolism regulation
After understanding the impact of excessive cholesterol on REC, potential treatment options have also emerged. Therapeutic goals can be achieved by reducing cholesterol biosynthesis and increasing cholesterol efflux. To achieve reduced cholesterol biosynthesis, it is necessary to inhibit the key rate‐limiting enzymes in the synthesis pathway. Although studies have shown that statins do not increase or reduce the risk of AMD, 193 , 194 a recent study has demonstrated that simvastatin can inhibit the biosynthesis of retinal cholesterol, which is valuable for future clinical research on statins as potential treatment methods for AMD. 195 Additionally, DHCR24 inhibitors can also reduce the TC content in the retina.
However, the cholesterol efflux pathway is worth exploring due to its complex nature. In the retina, cholesterol efflux from cells is largely dependent on ABCA1/ABCG1. Before cholesterol is expelled from cells, it needs to be transported to the mitochondria and metabolized into steroids through the cytochrome P450 enzyme in the mitochondria. This process involves a complex of proteins, including the steroidogenic acute regulatory protein (StAR), 18‐kDa translocator protein (TSPO), voltage‐dependent anion channel (VDAC) and possibly the adenine nucleotide channel. STAR is divided into 15 different members and 6 subfamilies; STARD1 and STARD3 in the STARD1 subfamily are considered to play important roles in lipid transport and metabolism. 196 STARD1 is a mitochondrial protein that regulates steroid production by transferring cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane and converting it into pregnenolone via the cytochrome P450 enzyme. 197 Moreover, Borthwick and coworkers 198 showed that STARD3 in ARPE‐19 cells can enhance cholesterol efflux, which is located on the membrane of the late endosome and transfers cholesterol from the late endosome to the ER and mitochondria. 199 Almarhoun et al. 200 found that the overexpression of STARD3 in ARPE‐19 cells can enhance cholesterol efflux in RPE cells by upregulating the expression of ABCA1 and reducing the accumulation of ox‐LDL, which ultimately inhibits oxidative stress and inflammation induced by ox‐LDL. Regarding TSPO, as a mitochondrial protein, it has the same effect as STARD1. TSPO combines with other proteins, such as STARD1 and VDAC, forming a multiprotein complex responsible for cholesterol transport. 201 Some studies have shown that the activation of TSPO in mice leads to an increase in cholesterol efflux from RPE cells, a decrease in the levels of lipids (cholesterol, triglycerides, and phospholipids) in the retina, an increase in the expression of cholesterol‐related genes (LXR, ABCA1/ABCG1, and ApoE) and the inhibition of inflammation. 166 , 202 , 203 , 204 Simultaneously, TSPO can regulate the activity of STARD1, 205 but the relationship between TSPO and STARD3 remains unknown. We previously reported that LXR plays a crucial role in cholesterol efflux by activating ABCA1 and ABCG1 to increase cholesterol efflux and inhibit inflammation. Through further investigation, we found that the activation of LXR can reduce the apoptosis of RPE cells induced by oxysterol by maintaining mitochondrial membrane stasis, which jointly activates mitochondrial autophagy via the mTOR/p62 pathway to promote mitochondrial metabolism. 206 In addition to the above regulatory proteins, other molecular signals that increase the expression of ABCA1/ ABCG1 can also promote cholesterol efflux from cells and reduce the accumulation of ox‐LDL, delaying disease progression.
7.2. Corneal diseases
Like AMD, corneal diseases are also caused by the accumulation of cholesterol. Maintaining normal vision relies on corneal clarity. Excessive cholesterol accumulation in the corneal stroma can cause opacity, thus affecting clarity. In some cases, corneal transplantation is necessary to restore normal vision. In the cornea, collagen and proteoglycans form a dense connective tissue called the corneal stroma, which contains corneal cells. The outer and inner surfaces of the corneal stroma are covered by multiple layers of corneal epithelium and a single layer of corneal endothelial cells, respectively. Our findings indicate that ABCA1, ApoA1, ApoD, ApoE, and LCAT are present in the cornea, keratocytes, epithelial cells and endothelial cells. 171 , 207 Certain inherited diseases, including Tangier disease, familial LCAT deficiency, and familial ApoA1 deficiency, result in varying degrees of corneal opacity due to corneal cholesterol accumulation. 208 , 209 , 210
In addition to mutations in RCT‐related genes, the mutation of UbiA prenyltransferase domain‐containing protein‐1 (UBIAD1) also leads to cholesterol accumulation. 211 UBIAD1 is a member of the UbiA superfamily of integral membrane prenyltransferases. In animals, UBIAD1 can catalyze generation of menaquinone‐4 by transferring the 20‐carbon geranylgeranyl moiety from geranylgeranyl pyrophosphate (GGpp). Under normal conditions, to prevent the excessive accumulation of cholesterol, sterols can induce HMGCR to combine with ER‐anchored INSIG‐1 and INSIG‐2, then some ubiquitin ligases ubiquitin HMGCR together with other cofactors, leading to HMGCR degradation in the proteasome, which is called the ER‐associated degradation (ERAD) of reductase. 212 , 213 , 214 , 215 , 216 , 217 However, sterol also induces the binding of reductase to UBIAD1, inhibiting the ubiquitination step of ERAD reductase and allowing continued mevalonate production. 218 Concurrently, the accumulation of GGpp in the ER membrane induces the release of UBIAD1 from the reductase, alleviating the inhibition of ERAD and facilitating the transport of UBIAD1 from the ER to the Golgi. 219 It is precisely due to the above regulatory mechanisms that cholesterol in the corneal stroma can always maintain a stable level for a long time. However, when Ubiad1 is mutated, the transfer of UBIAD1 mediated by GGpp is inhibited, leading to the inhibition of ERAD and the accumulation of cholesterol, eventually resulting in corneal opacity, also known as Schnyder corneal dystrophy (SCD) (Figure 1). 219 Currently, we are seeking drugs that accelerate the degradation of UBIAD1 related to SCD, trigger the transport of variants from ER to Golgi apparatus or prevent their interaction with reductase, which may prevent cholesterol accumulation and corneal opacity related to SCD.
7.3. Cataract
7.3.1. The relationship of cataract and cholesterol metabolism
Unlike AMD and corneal diseases, cataracts seem to rely on high concentrations of cholesterol. 220 To achieve clear vision, we depend on the lens to refract light onto the retina. Therefore, lens transparency is crucial for clear vision. A recent study has found that the long‐term inhibition of cholesterol biosynthesis in the lens reduces the rate of fiber proliferation and differentiation, increasing the risk of lens opacity. 168 This suggests that cholesterol is beneficial for the lens.
The lens is different from the retina, it is an avascular structure, which means not all nutrients in the blood can easily penetrate it. Dietary cholesterol does not affect the cholesterol content in the lens. 221 In addition, the cholesterol content of the aqueous humor is extremely low, which is negligible compared with that of plasma. Therefore, De Vries et al. 222 concluded that cholesterol accumulation in lens depends on in situ synthesis. This perspective implies that cholesterol is valuable for the lens. So why is cholesterol beneficial for the lens?
Cholesterol plays a crucial in maintaining lens transparency. First, cholesterol is necessary for the formation of cytoplasmic membranes during lens epithelial cell (LEC) differentiation. 223 Cholesterol in the cell membrane can form a highly hydrophobic barrier between cells to reduce light scattering and maintain lens transparency by maintaining an appropriate volume and water content in lens fibers. 224 Second, the high cholesterol content in the membranes maintains a low oxygen partial pressure. When the cholesterol content of the cells is high, the oxygen permeability coefficient of the membrane is lower than that of the water layer of the same thickness, which is crucial for lens transparency. 225 , 226 Thus, a high cholesterol level is beneficial for the lens. Once cholesterol biosynthesis is affected, lens transparency cannot be guaranteed, and the risk of cataracts will increase. For example, approximately 20% of patients with Smith Lemli Opitz syndrome who are deficient in DHCR7 have cataract 227 ; the missense mutation of lanosterol synthase (LSS, cyclinase in cholesterol biosynthesis) causes cataract in pedigree analysis of close relatives of Caucasian descent; cataracts occur in approximately 30% of patients with mevalonate aciduria caused by mevalonate kinase deficiency. 228 Therefore, maintaining a high cholesterol content is crucial for the normal function of the lens.
7.3.2. Role of cholesterol metabolism regulation
Given the crucial role of cholesterol in the lens, the metabolic regulation of cholesterol within the lens has become a focus of research. At present, studies have found that LSS, as a key rate‐limiting enzyme in cholesterol synthesis, plays a crucial role in cataractogenesis. 229 The rs2968 polymorphism of Lss gene is associated with karyotype risk of age‐related cataracts in the Chinese population. 230 However, as a causal gene of cataracts, the role of Lss in the lens remains largely unknown. Now, we summarize the role of LSS based on existing research findings. First, LSS, as a key rate‐limiting enzyme in the cholesterol synthesis pathway, plays a critical role in maintaining high cholesterol content in the lens. 170 Second, LSS plays an important role in lens development. Zhao et al. 169 found that blockage of primary fiber differentiation and delay of secondary fiber differentiation, as well as incomplete karyolysis, occur in mice with a homozygous LssG589S/G589S mutation. Simultaneously, the polarity of the lens fiber cells from top to bottom is lost, potentially leading to a disorder in lens fiber differentiation. 169 In addition, LSS can promote the synthesis of lanosterol in the biosynthetic pathway, which decomposes large protein aggregates into small protein aggregates to reverse protein aggregation in cataracts. 231 Third, LSS also have an effect on oxidative stress. Oxidative stress regulates various cellular processes related to cell survival, such as proliferation, differentiation, aging, and death, 170 which is the major cause of age‐related ophthalmopathy. 232 , 233 In a recent study, researchers increased oxidative stress in cells by exposing LECs to UV‐B and found that LSS reduces UV‐B‐induced apoptosis and exerts an antioxidant effects in the early stages of cell damage. 229 In another study, Ishida et al. 170 analyzed that Lss deficiency may lead to the reduction of antioxidant gene expression. Therefore, LSS is highly anticipated due to its important functions. However, due to its complex physiological properties, it has not yet been applied to clinical practice, and further exploration is needed in the future.
7.4. Dry eye syndrome
DES is a disorder of tear secretion that causes eye discomfort, eye fatigue and visual impairment due to insufficient or excessive evaporation of tears. 234 The tear film is composed of lipid, aqueous, and mucin layers, among which the lipid layer protects the eye from environmental pressure. 235 The meibomian gland, the main component of the tear film lipid layer, is responsible for the secretion of lipids, including cholesterol and CEs. Dysfunction of the Meibomian gland is a common ocular ailment that results in DES by causing local dyslipidemia. Hence, systemic dyslipidemia may be associated with the pathogenesis of DES. 236 A recent study investigated the relationship between dyslipidemia and DES in a middle‐aged Korean population. They defined dyslipidemia as TC > 240 mg/dL, HDL‐C < 40 mg/dL, LDL‐C > 160 mg/dL, or triglycerides > 200 mg/dL. Moreover, they discovered that the odds ratio for DES in men with dyslipidemia was 1.29 (95% confidence interval, 0.97−1.71) compared with men without DES. However, no significant correlation between female dyslipidemia and DES was observed, suggesting a potential association between dyslipidemia and DES prevalence in Korean men, but not women. 236 In another study, Aldaas et al. 172 examined the relationship between statin use and DES. They found that the local use of statins is beneficial for the treatment of dry eyes related to blepharitis, but the risks of DES for low‐intensity, medium‐intensity and high‐intensity statins in the treatment of blepharitis are similar, which suggests that the effect of statins may not be dose‐dependent, or the correlation between statins and DES may be false. 172 Therefore, additional research is needed to clarify the association between dyslipidemia and DES.
7.5. Diabetic retinopathy
DR is a serious complication of diabetes, characterized by retinal ischemia and neurodegeneration, eventually leading to vision loss. 237 According to the development of DR, it is be classified into nonproliferative diabetes retinopathy (NPDR) and proliferative diabetes retinopathy (PDR). NPDR is characterized by changes in the retinal vascular system, such as increased permeability. Untreated, these changes induce ischemia, leading to neovascularization, commonly referred to as PDR. Opreanu et al. 238 previously demonstrated that abnormal retinal lipid metabolism can induce retinal degeneration by activating chronic inflammation. Recent studies indicate that dyslipidemia and lipid peroxidation are significant contributors to DR pathogenesis. 239 , 240 These findings suggest that regulating related metabolism could delay or prevent vision loss attributed to DR. Consequently, enhancing RCT is advantageous for preventing and treating DR. LXR, a crucial receptor in the RCT pathway, its decreased signal transduction is a hallmark feature of type 2 diabetes, which can lead to retinal cholesterol metabolism disorder and increased production of proinflammatory cytokines. 241 A study demonstrated that LXR is involved not only in regulating lipid metabolism but also in the insulin signaling pathway, influencing glucose metabolism. 242 Given the effectiveness of LXR, it may emerge as a potential target for future DR treatment.
While our current grasp of cholesterol metabolism in the eye is not exhaustive, we believe eye diseases are closely linked to abnormal cholesterol metabolism. Simultaneously, we have identified numerous potential therapeutic targets and summarized them in this review (Table 4), 243 , 244 , 245 with the aim of fostering a more comprehensive understanding of disease prevention and treatment in the future.
TABLE 4.
The functions and effects of potential therapeutic targets for eye diseases.
| Disease | Signals | Functions | Roles | References |
|---|---|---|---|---|
| AMD | Statins |
HMGCR↓ IL‐6/8↓ |
Cholesterol synthesis↓ Inflammation↓ |
243 |
| The inhibitors of miRNA | ABCA1↑ | Cholesterol efflux↑ | 244 | |
| AMPK | ABCA1/ABCG1↑ | Cholesterol efflux↑ | 245 | |
| Oxysterols/LXR agonists |
ABCA1/ABCG1↑ The mTOR/p62 pathway↑ The NF‐κB pathway↓ |
Cholesterol efflux↑ Autophagy↑ Inflammation↓ |
44 , 206 | |
| Gypenosides |
ABCA1/ABCG1↑ CYP46A1↑ The NF‐κB pathway↓ The ox‐LDL accumulation↓ |
Cholesterol efflux↑ Inflammation↓ Antioxidant capacity↑ |
184 | |
| STARD3 |
ABCA1↑ IL‐1β↓ TNF‐α↓ The ox‐LDL accumulation↓ |
Cholesterol efflux↑ Inflammation↓ Antioxidant capacity↑ |
200 | |
| TSPO |
ABCA1/ABCG1↑ CYP46A1↑ IL‐1β↓ TNF‐α↓ The ox‐LDL accumulation↓ Lipogenesis↓ |
Cholesterol efflux↑ Inflammation↓ Antioxidant capacity↑ Cholesterol synthesis↓ |
166 , 202 , 204 – | |
| Cataract | LSS |
Lipogenesis↑ Lenosterol↑ |
Cholesterol synthesis↑ Protein aggregation↓ |
169 , 170 |
| DR | LXR agonists |
ABCA1/ABCG1↑ GLUT4↑ |
Cholesterol efflux↑ Blood glucose↓ |
242 |
8. CONCLUSIONS AND PROSPECTS
Widely distributed throughout the human body, cholesterol plays a crucial role in various physiological processes. For example, in the brain, cholesterol synthesized by oligodendrocytes and Schwann cells mainly exists in the form of myelin sheath, a vital component for the nervous system's normal functioning. 246 A well‐regulated cholesterol metabolism is also crucial for maintaining the body's normal functioning and keeping cholesterol levels stable. Once cholesterol synthesis or efflux is impaired, it may lead to the occurrence of diseases. Our studies reveal that defects in cholesterol metabolism are closely linked to various diseases, including CVD, tumors, neurological disorders, immune system issues, and eye diseases. Excessive accumulation of cholesterol often promotes the progression of diseases and even plays a role as a “marker.” Cholesterol overload can induce elevated oxidative stress, heightened inflammatory responses, reduced autophagy, and increased apoptosis in cells through various signaling pathways, ultimately accelerating the development of diseases. However, high levels of cholesterol within the lens are beneficial for maintaining lens transparency, and inhibiting cholesterol synthesis often leads to lens opacity, resulting in cataracts. It is important to note that in elderly individuals, surpassing the cholesterol solubility threshold can result in the formation of cholesterol crystals, causing pathological manifestations. 247
Although the exact mechanism by which cholesterol metabolism plays a role in the pathogenesis of diseases is not yet clear, reducing cholesterol accumulation undoubtedly contributes to the treatment of diseases. On the one hand, we can employ inhibitors to decrease cholesterol production by targeting key enzymes in the cholesterol synthesis pathway, like statins, widely utilized in clinical practice for their antioxidant and anti‐inflammatory properties. Nevertheless, further efforts are required to minimize the side effects of statins and discover more effective and safe medications for modifying cholesterol metabolism. On the other hand, mounting evidence indicates the significance of the RCT pathway in cholesterol metabolism. Activating this pathway to enhance cholesterol efflux offers a potential avenue for clinical practice. Specific receptors in the RCT pathway, like LXRs, have emerged as potential therapeutic targets. Currently, the development of selective LXR receptor agonists that do not affect liver and plasma triglyceride levels has become a research focus. In the near future, LXR agonists could evolve into tailored therapeutic targets for addressing cholesterol metabolism disorders.
However, some unresolved issues persist concerning cholesterol metabolism. Due to barriers, cholesterol in the brain and retina primarily results from in situ synthesis. It remains uncertain if cholesterol in these regions is regulated by the entire body, posing a challenge in explaining the relationship between plasma cholesterol levels and disease risk. Additionally, the influence of cholesterol metabolites on cholesterol homeostasis merits further investigation, including the role of 24‐OHC and CYP46A1 in diseases and their effect on cholesterol turnover rate.
AUTHOR CONTRIBUTIONS
J. G. collected literature and drafted the manuscript. S. C., Y. Z., J. L., and L. J. drew the figures and tables. L. H. and K. Y. reviewed and made significant revisions to the manuscript. Y. Y. and X. C. finalized the manuscript and contributed to funding acquisition, validation, and resources. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
Thanks to all authors for their hard work and helpful comments.
Guo J, Chen S, Zhang Y, et al. Cholesterol metabolism: physiological regulation and diseases. MedComm. 2024;5:e476. 10.1002/mco2.476
Contributor Information
Yibo Yu, Email: yuyibo@zju.edu.cn.
Xiangjun Chen, Email: chenxiangjun@zju.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Luu W, Sharpe LJ, Capell‐Hattam I, Gelissen IC, Brown AJ. Oxysterols: old tale, new twists. Annu Rev Pharmacol Toxicol. 2016;56:447‐467. [DOI] [PubMed] [Google Scholar]
- 2. Okuhira K. [Challenges in drug development targeting anti‐atherosclerotic proteins]. Yakugaku Zasshi. 2020;140(2):153‐157. [DOI] [PubMed] [Google Scholar]
- 3. Pikuleva IA, Curcio CA. Cholesterol in the retina: the best is yet to come. Prog Retin Eye Res. 2014;41:64‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6(4):254‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meng Y, Wang Q, Lyu Z. Cholesterol metabolism and tumor. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2021;50(1):23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Courtney R, Landreth GE. LXR regulation of brain cholesterol: from development to disease. Trends Endocrinol Metab. 2016;27(6):404‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conley YP, Thalamuthu A, Jakobsdottir J, et al. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age‐related maculopathy. Hum Mol Genet. 2005;14(14):1991‐2002. [DOI] [PubMed] [Google Scholar]
- 8. Neale BM, Fagerness J, Reynolds R, et al. Genome‐wide association study of advanced age‐related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci USA. 2010;107(16):7395‐7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W, Stambolian D, Edwards AO, et al. Genetic variants near TIMP3 and high‐density lipoprotein‐associated loci influence susceptibility to age‐related macular degeneration. Proc Natl Acad Sci USA. 2010;107(16):7401‐7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang X, Wang K, Zhu L, Wang Q. Reverse cholesterol transport pathway and cholesterol efflux in diabetic retinopathy. J Diabetes Res. 2021;2021:8746114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cardoso D, Perucha E. Cholesterol metabolism: a new molecular switch to control inflammation. Clin Sci (Lond). 2021;135(11):1389‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983;22(2):79‐131. [DOI] [PubMed] [Google Scholar]
- 13. Mitsche MA, McDonald JG, Hobbs HH, Cohen JC. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell‐type specific pathways. Elife. 2015;4:e07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hubler Z, Friedrich RM, Sax JL, et al. Modulation of lanosterol synthase drives 24,25‐epoxysterol synthesis and oligodendrocyte formation. Cell Chem Biol. 2021;28(6):866‐875. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Horton JD, Shah NA, Warrington JA, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003;100(21):12027‐12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Y, Song Y, Zhao M, et al. A novel role for CRTC2 in hepatic cholesterol synthesis through SREBP‐2. Hepatology. 2017;66(2):481‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tao R, Xiong X, DePinho RA, Deng CX, Dong XC. Hepatic SREBP‐2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J Lipid Res. 2013;54(10):2745‐2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang T, Espenshade PJ, Wright ME, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG‐1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489‐500. [DOI] [PubMed] [Google Scholar]
- 19. Yabe D, Brown MS, Goldstein JL. Insig‐2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element‐binding proteins. Proc Natl Acad Sci USA. 2002;99(20):12753‐12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vergnes L, Chin RG, de Aguiar Vallim T, et al. SREBP‐2‐deficient and hypomorphic mice reveal roles for SREBP‐2 in embryonic development and SREBP‐1c expression. J Lipid Res. 2016;57(3):410‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giandomenico V, Simonsson M, Grönroos E, Ericsson J. Coactivator‐dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol. 2003;23(7):2587‐2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodgers JT, Puigserver P. Fasting‐dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA. 2007;104(31):12861‐12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet‐induced insulin‐resistant mice. Cell Metab. 2011;13(4):376‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eid W, Dauner K, Courtney KC, et al. mTORC1 activates SREBP‐2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci USA. 2017;114(30):7999‐8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altmann SW, Davis HR, Jr. , Zhu LJ, et al. Niemann‐Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303(5661):1201‐1204. [DOI] [PubMed] [Google Scholar]
- 27. Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. Jama. 2009;302(18):1993‐2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saito H, Dhanasekaran P, Nguyen D, et al. Alpha‐helix formation is required for high affinity binding of human apolipoprotein A‐I to lipids. J Biol Chem. 2004;279(20):20974‐20981. [DOI] [PubMed] [Google Scholar]
- 29. Hong YF, Kim H, Kim HS, Park WJ, Kim JY, Chung DK. Lactobacillus acidophilus K301 inhibits atherogenesis via induction of 24 (S), 25‐epoxycholesterol‐mediated ABCA1 and ABCG1 production and cholesterol efflux in macrophages. PLoS One. 2016;11(4):e0154302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choudhary M, Tayyari F, Handa JT, Malek G. Characterization and identification of measurable endpoints in a mouse model featuring age‐related retinal pathologies: a platform to test therapies. Lab Invest. 2022;102(10):1132‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saadane A, Petrov A, Mast N, et al. Mechanisms that minimize retinal impact of apolipoprotein E absence. J Lipid Res. 2018;59(12):2368‐2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laffitte BA, Repa JJ, Joseph SB, et al. LXRs control lipid‐inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci U S A. 2001;98(2):507‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. La Cunza N, Tan LX, Thamban T, et al. Mitochondria‐dependent phase separation of disease‐relevant proteins drives pathological features of age‐related macular degeneration. JCI Insight. 2021;6(9):e142254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Malek G, Li CM, Guidry C, Medeiros NE, Curcio CA. Apolipoprotein B in cholesterol‐containing drusen and basal deposits of human eyes with age‐related maculopathy. Am J Pathol. 2003;162(2):413‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mayeux R, Stern Y, Ottman R, et al. The apolipoprotein epsilon 4 allele in patients with Alzheimer's disease. Ann Neurol. 1993;34(5):752‐754. [DOI] [PubMed] [Google Scholar]
- 36. Klaver CC, Kliffen M, van Duijn CM, et al. Genetic association of apolipoprotein E with age‐related macular degeneration. Am J Hum Genet. 1998;63(1):200‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fan D, Qiu S, Overton CD, et al. Impaired secretion of apolipoprotein E2 from macrophages. J Biol Chem. 2007;282(18):13746‐13753. [DOI] [PubMed] [Google Scholar]
- 38. Apfel R, Benbrook D, Lernhardt E, Ortiz MA, Salbert G, Pfahl M. A novel orphan receptor specific for a subset of thyroid hormone‐responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol Cell Biol. 1994;14(10):7025‐7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Auboeuf D, Rieusset J, Fajas L, et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator‐activated receptors and liver X receptor‐alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46(8):1319‐1327. [DOI] [PubMed] [Google Scholar]
- 40. Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9(9):1033‐1045. [DOI] [PubMed] [Google Scholar]
- 41. Edwards PA, Kennedy MA, Mak PA. LXRs; oxysterol‐activated nuclear receptors that regulate genes controlling lipid homeostasis. Vascul Pharmacol. 2002;38(4):249‐256. [DOI] [PubMed] [Google Scholar]
- 42. Laffitte BA, Chao LC, Li J, et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci U S A. 2003;100(9):5419‐5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hazra S, Rasheed A, Bhatwadekar A, et al. Liver X receptor modulates diabetic retinopathy outcome in a mouse model of streptozotocin‐induced diabetes. Diabetes. 2012;61(12):3270‐3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. El‐Darzi N, Mast N, Buchner DA, et al. Low‐dose anti‐HIV drug efavirenz mitigates retinal vascular lesions in a mouse model of Alzheimer's disease. Front Pharmacol. 2022;13:902254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oram JF, Vaughan AM. ATP‐binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006;99(10):1031‐1043. [DOI] [PubMed] [Google Scholar]
- 46. Orsó E, Broccardo C, Kaminski WE, et al. Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1‐deficient mice. Nat Genet. 2000;24(2):192‐196. [DOI] [PubMed] [Google Scholar]
- 47. Kennedy MA, Barrera GC, Nakamura K, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1(2):121‐131. [DOI] [PubMed] [Google Scholar]
- 48. Vaughan AM, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid‐depleted apolipoproteins. J Biol Chem. 2005;280(34):30150‐30157. [DOI] [PubMed] [Google Scholar]
- 49. Virani SS, Alonso A, Benjamin EJ, et al. Heart disease and stroke statistics‐2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139‐e596. [DOI] [PubMed] [Google Scholar]
- 50. Almeida SO, Budoff M. Effect of statins on atherosclerotic plaque. Trends Cardiovasc Med. 2019;29(8):451‐455. [DOI] [PubMed] [Google Scholar]
- 51. Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104(4):503‐516. [DOI] [PubMed] [Google Scholar]
- 52. Varghese JF, Patel R, Yadav UCS. Sterol regulatory element binding protein (SREBP)‐1 mediates oxidized low‐density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell Signal. 2019;53:316‐326. [DOI] [PubMed] [Google Scholar]
- 53. He XW, Yu D, Li WL, et al. Anti‐atherosclerotic potential of baicalin mediated by promoting cholesterol efflux from macrophages via the PPARgamma‐LXRalpha‐ABCA1/ABCG1 pathway. Biomed Pharmacother. 2016;83:257‐264. [DOI] [PubMed] [Google Scholar]
- 54. Gui YZ, Yan H, Gao F, Xi C, Li HH, Wang YP. Betulin attenuates atherosclerosis in apoE(‐/‐) mice by up‐regulating ABCA1 and ABCG1. Acta Pharmacol Sin. 2016;37(10):1337‐1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brümmer A, Hausser J. MicroRNA binding sites in the coding region of mRNAs: extending the repertoire of post‐transcriptional gene regulation. Bioessays. 2014;36(6):617‐626. [DOI] [PubMed] [Google Scholar]
- 56. Baldan A, de Aguiar Vallim TQ. miRNAs and high‐density lipoprotein metabolism. Biochim Biophys Acta. 2016;1861(12 Pt B):2053‐2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liang B, Wang X, Song X, et al. MicroRNA‐20a/b regulates cholesterol efflux through post‐transcriptional repression of ATP‐binding cassette transporter A1. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(9):929‐938. [DOI] [PubMed] [Google Scholar]
- 58. Meiler S, Baumer Y, Toulmin E, Seng K, Boisvert WA. MicroRNA 302a is a novel modulator of cholesterol homeostasis and atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(2):323‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu X, Yang B, Yang H, et al. MicroRNA‐320b modulates cholesterol efflux and atherosclerosis. J Atheroscler Thromb. 2022;29(2):200‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao R, Feng J, He G. miR‐613 regulates cholesterol efflux by targeting LXRalpha and ABCA1 in PPARgamma activated THP‐1 macrophages. Biochem Biophys Res Commun. 2014;448(3):329‐334. [DOI] [PubMed] [Google Scholar]
- 61. Liu H, Zuo C, Cao L, Yang N, Jiang T. Inhibition of miR‐652‐3p regulates lipid metabolism and inflammatory cytokine secretion of macrophages to alleviate atherosclerosis by improving TP53 expression. Mediators Inflamm. 2022;2022:9655097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kong J, Liu L, Song L, Zhao R, Feng Y. MicroRNA miR‐34a‐5p inhibition restrains oxidative stress injury of macrophages by targeting MDM4. Vascular. 2023;31(3):608‐618. [DOI] [PubMed] [Google Scholar]
- 63. Sui Z, Zhou J, Cheng Z, Lu P. Squalene epoxidase (SQLE) promotes the growth and migration of the hepatocellular carcinoma cells. Tumour Biol. 2015;36(8):6173‐6179. [DOI] [PubMed] [Google Scholar]
- 64. Zhang Z, Wu W, Jiao H, et al. Squalene epoxidase promotes hepatocellular carcinoma development by activating STRAP transcription and TGF‐beta/SMAD signalling. Br J Pharmacol. 2023;180(12):1562‐1581. [DOI] [PubMed] [Google Scholar]
- 65. Ashida S, Kawada C, Inoue K. Stromal regulation of prostate cancer cell growth by mevalonate pathway enzymes HMGCS1 and HMGCR. Oncol Lett. 2017;14(6):6533‐6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kalogirou C, Linxweiler J, Schmucker P, et al. MiR‐205‐driven downregulation of cholesterol biosynthesis through SQLE‐inhibition identifies therapeutic vulnerability in aggressive prostate cancer. Nat Commun. 2021;12(1):5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is up‐regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene. 2016;587(1):42‐47. [DOI] [PubMed] [Google Scholar]
- 68. Qiu Z, Yuan W, Chen T, et al. HMGCR positively regulated the growth and migration of glioblastoma cells. Gene. 2016;576(1 Pt 1):22‐27. [DOI] [PubMed] [Google Scholar]
- 69. Singh R, Yadav V, Kumar S, Saini N. MicroRNA‐195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci Rep. 2015;5:17454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhong C, Fan L, Yao F, Shi J, Fang W, Zhao H. HMGCR is necessary for the tumorigenecity of esophageal squamous cell carcinoma and is regulated by Myc. Tumour Biol. 2014;35(5):4123‐4129. [DOI] [PubMed] [Google Scholar]
- 71. Ge H, Zhao Y, Shi X, et al. Squalene epoxidase promotes the proliferation and metastasis of lung squamous cell carcinoma cells though extracellular signal‐regulated kinase signaling. Thorac Cancer. 2019;10(3):428‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zou F, Chen W, Song T, et al. SQLE Knockdown inhibits bladder cancer progression by regulating the PTEN/AKT/GSK3β signaling pathway through P53. Cancer Cell Int. 2023;23(1):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. He L, Li H, Pan C, et al. Squalene epoxidase promotes colorectal cancer cell proliferation through accumulating calcitriol and activating CYP24A1‐mediated MAPK signaling. Cancer Commun (Lond). 2021;41(8):726‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang L, Lu B, He M, Wang Y, Wang Z, Du L. Prostate cancer incidence and mortality: global status and temporal trends in 89 countries from 2000 to 2019. Front Public Health. 2022;10:811044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Farwell WR, D'Avolio LW, Scranton RE, Lawler EV, Gaziano JM. Statins and prostate cancer diagnosis and grade in a veterans population. J Natl Cancer Inst. 2011;103(11):885‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kitahara CM, Berrington de González A, Freedman ND, et al. Total cholesterol and cancer risk in a large prospective study in Korea. J Clin Oncol. 2011;29(12):1592‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tulloch‐Reid MK, McFarlane‐Anderson N, Bennett FI, Aiken WD, Jackson MD. Effects of cholesterol, C‐reactive protein, and interleukin‐6 on prostate cancer risk in a population of African ancestry. Cancer Causes Control. 2017;28(11):1313‐1321. [DOI] [PubMed] [Google Scholar]
- 78. Masko EM, Allott EH, Freedland SJ. The relationship between nutrition and prostate cancer: is more always better? Eur Urol. 2013;63(5):810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schaffner CP. Prostatic cholesterol metabolism: regulation and alteration. Prog Clin Biol Res. 1981;75a:279‐324. [PubMed] [Google Scholar]
- 80. El‐Kenawi A, Dominguez‐Viqueira W, Liu M, et al. Macrophage‐derived cholesterol contributes to therapeutic resistance in prostate cancer. Cancer Res. 2021;81(21):5477‐5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pan T, Lin SC, Lee YC, et al. Statins reduce castration‐induced bone marrow adiposity and prostate cancer progression in bone. Oncogene. 2021;40(27):4592‐4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ossoli A, Giorgio E, Cetti F, et al. HDL‐mediated reduction of cholesterol content inhibits the proliferation of prostate cancer cells induced by LDL: Role of ABCA1 and proteasome inhibition. Biofactors. 2022;48(3):707‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Raza S, Meyer M, Goodyear C, Hammer KDP, Guo B, Ghribi O. The cholesterol metabolite 27‐hydroxycholesterol stimulates cell proliferation via ERbeta in prostate cancer cells. Cancer Cell Int. 2017;17:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115(4):959‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Freeman MR, Cinar B, Kim J, et al. Transit of hormonal and EGF receptor‐dependent signals through cholesterol‐rich membranes. Steroids. 2007;72(2):210‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol‐rich lipid rafts mediate akt‐regulated survival in prostate cancer cells. Cancer Res. 2002;62(8):2227‐2231. [PubMed] [Google Scholar]
- 87. Toren P, Zoubeidi A. Targeting the PI3K/Akt pathway in prostate cancer: challenges and opportunities (review). Int J Oncol. 2014;45(5):1793‐1801. [DOI] [PubMed] [Google Scholar]
- 88. Pommier AJ, Alves G, Viennois E, et al. Liver X Receptor activation downregulates AKT survival signaling in lipid rafts and induces apoptosis of prostate cancer cells. Oncogene. 2010;29(18):2712‐2723. [DOI] [PubMed] [Google Scholar]
- 89. Mak B, Lin HM, Duong T, et al. Modulation of plasma lipidomic profiles in metastatic castration‐resistant prostate cancer by simvastatin. Cancers (Basel). 2022;14(19):4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liang Y, Mafuvadze B, Aebi JD, Hyder SM. Cholesterol biosynthesis inhibitor RO 48–8071 suppresses growth of hormone‐dependent and castration‐resistant prostate cancer cells. Onco Targets Ther. 2016;9:3223‐3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stopsack KH, Gerke TA, Sinnott JA, et al. Cholesterol metabolism and prostate cancer lethality. Cancer Res. 2016;76(16):4785‐4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stopsack KH, Gerke TA, Andrén O, et al. Cholesterol uptake and regulation in high‐grade and lethal prostate cancers. Carcinogenesis. 2017;38(8):806‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Xu Z, Huang L, Dai T, et al. SQLE mediates metabolic reprogramming to promote LN metastasis in castration‐resistant prostate cancer. Onco Targets Ther. 2021;14:4285‐4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee SS, Li J, Tai JN, Ratliff TL, Park K, Cheng JX. Avasimibe encapsulated in human serum albumin blocks cholesterol esterification for selective cancer treatment. ACS Nano. 2015;9(3):2420‐2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee HJ, Li J, Vickman RE, et al. Cholesterol esterification inhibition suppresses prostate cancer metastasis by impairing the Wnt/beta‐catenin pathway. Mol Cancer Res. 2018;16(6):974‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. [DOI] [PubMed] [Google Scholar]
- 97. McGlynn KA, Petrick JL, El‐Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73(Suppl 1):4‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118‐1127. [DOI] [PubMed] [Google Scholar]
- 99. Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology. 2007;72(Suppl 1):30‐44. [DOI] [PubMed] [Google Scholar]
- 100. Ioannou GN, Morrow OB, Connole ML, Lee SP. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology. 2009;50(1):175‐184. [DOI] [PubMed] [Google Scholar]
- 101. Peng L, Yan Q, Chen Z, et al. Research progress on the role of cholesterol in hepatocellular carcinoma. Eur J Pharmacol. 2023;938:175410. [DOI] [PubMed] [Google Scholar]
- 102. Wang Y, Wang J, Li X, et al. N(1)‐methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat Commun. 2021;12(1):6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nuño‐Lámbarri N, Domínguez‐Pérez M, Baulies‐Domenech A, et al. Liver cholesterol overload aggravates obstructive cholestasis by inducing oxidative stress and premature death in mice. Oxid Med Cell Longev. 2016;2016:9895176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Domínguez‐Pérez M, Nuño‐Lámbarri N, Clavijo‐Cornejo D, et al. Hepatocyte growth factor reduces free cholesterol‐mediated lipotoxicity in primary hepatocytes by countering oxidative stress. Oxid Med Cell Longev. 2016;2016:7960386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gomez‐Quiroz LE, Seo D, Lee YH, et al. Loss of c‐Met signaling sensitizes hepatocytes to lipotoxicity and induces cholestatic liver damage by aggravating oxidative stress. Toxicology. 2016;361‐362:39‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Enríquez‐Cortina C, Bello‐Monroy O, Rosales‐Cruz P, et al. Cholesterol overload in the liver aggravates oxidative stress‐mediated DNA damage and accelerates hepatocarcinogenesis. Oncotarget. 2017;8(61):104136‐104148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Dapito DH, Mencin A, Gwak GY, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21(4):504‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. He M, Zhang W, Dong Y, et al. Pro‐inflammation NF‐κB signaling triggers a positive feedback via enhancing cholesterol accumulation in liver cancer cells. J Exp Clin Cancer Res. 2017;36(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shao WQ, Zhu WW, Luo MJ, et al. Cholesterol suppresses GOLM1‐dependent selective autophagy of RTKs in hepatocellular carcinoma. Cell Rep. 2022;39(3):110712. [DOI] [PubMed] [Google Scholar]
- 110. Hu HH, Chen DQ, Wang YN, et al. New insights into TGF‐β/Smad signaling in tissue fibrosis. Chem Biol Interact. 2018;292:76‐83. [DOI] [PubMed] [Google Scholar]
- 111. Chen L, Yang T, Lu DW, et al. Central role of dysregulation of TGF‐β/Smad in CKD progression and potential targets of its treatment. Biomed Pharmacother. 2018;101:670‐681. [DOI] [PubMed] [Google Scholar]
- 112. Hu C, Liu D, Zhang Y, et al. LXRalpha‐mediated downregulation of FOXM1 suppresses the proliferation of hepatocellular carcinoma cells. Oncogene. 2014;33(22):2888‐2897. [DOI] [PubMed] [Google Scholar]
- 113. Xiong H, Zhang Y, Chen S, et al. Induction of SOCS3 by liver X receptor suppresses the proliferation of hepatocellular carcinoma cells. Oncotarget. 2017;8(38):64083‐64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Xie Y, Sun R, Gao L, et al. Chronic activation of LXRalpha sensitizes mice to hepatocellular carcinoma. Hepatol Commun. 2022;6(5):1123‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Fan M, Chen Z, Shao W, et al. SREBP2 inhibitor betulin sensitizes hepatocellular carcinoma to lenvatinib by inhibiting the mTOR/IL‐1β pathway. Acta Biochim Biophys Sin (Shanghai). 2023;55(9):1479‐1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yue X, Kong Y, Zhang Y, et al. SREBF2‐STARD4 axis confers sorafenib resistance in hepatocellular carcinoma by regulating mitochondrial cholesterol homeostasis. Cancer Sci. 2023;114(2):477‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375‐1397. [DOI] [PubMed] [Google Scholar]
- 118. Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol‐induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29(2):190‐201. [DOI] [PubMed] [Google Scholar]
- 119. Fester L, Zhou L, Bütow A, et al. Cholesterol‐promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus. 2009;19(8):692‐705. [DOI] [PubMed] [Google Scholar]
- 120. Linetti A, Fratangeli A, Taverna E, et al. Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci. 2010;123(Pt 4):595‐605. [DOI] [PubMed] [Google Scholar]
- 121. Liu Q, Trotter J, Zhang J, et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age‐dependent synapse loss and neurodegeneration. J Neurosci. 2010;30(50):17068‐17078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu Q, Zerbinatti CV, Zhang J, et al. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56(1):66‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Luo Y, Tan X, Zhang X, Li Y, Huang J, Deng Y. Effect of liver X receptor agonist TO901317 on cognitive function in APP/PS1 double transgenic mice with Alzheimer's disease and the underlying mechanism. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2022;47(10):1324‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Djelti F, Braudeau J, Hudry E, et al. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer's disease. Brain. 2015;138(Pt 8):2383‐2398. [DOI] [PubMed] [Google Scholar]
- 125. Yassine HN, Feng Q, Chiang J, et al. ABCA1‐mediated cholesterol efflux capacity to cerebrospinal fluid is reduced in patients with mild cognitive impairment and Alzheimer's disease. J Am Heart Assoc. 2016;5(2):e002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lee SI, Jeong W, Lim H, et al. APOE4‐carrying human astrocytes oversupply cholesterol to promote neuronal lipid raft expansion and Abeta generation. Stem Cell Reports. 2021;16(9):2128‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Paul R, Choudhury A, Kumar S, Giri A, Sandhir R, Borah A. Cholesterol contributes to dopamine‐neuronal loss in MPTP mouse model of Parkinson's disease: Involvement of mitochondrial dysfunctions and oxidative stress. PLoS One. 2017;12(2):e0171285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Griffiths WJ, Abdel‐Khalik J, Moore SF, et al. The cerebrospinal fluid profile of cholesterol metabolites in Parkinson's Disease and their association with disease state and clinical features. Front Aging Neurosci. 2021;13:685594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rayaprolu S, Higginbotham L, Bagchi P, et al. Systems‐based proteomics to resolve the biology of Alzheimer's disease beyond amyloid and tau. Neuropsychopharmacology. 2021;46(1):98‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sun BL, Chen Y, Fan DY, Zhu C, Zeng F, Wang YJ. Critical thinking on amyloid‐beta‐targeted therapy: challenges and perspectives. Sci China Life Sci. 2021;64(6):926‐937. [DOI] [PubMed] [Google Scholar]
- 131. Bai X, Mai M, Yao K, et al. The role of DHCR24 in the pathogenesis of AD: re‐cognition of the relationship between cholesterol and AD pathogenesis. Acta Neuropathol Commun. 2022;10(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Borràs C, Mercer A, Sirisi S, et al. HDL‐like‐Mediated Cell Cholesterol Trafficking in the Central Nervous System and Alzheimer's Disease Pathogenesis. Int J Mol Sci. 2022;23(16):9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mai M, Guo X, Huang Y, et al. DHCR24 knockdown induces tau hyperphosphorylation at Thr181, Ser199, Ser262, and Ser396 sites via activation of the lipid raft‐dependent Ras/MEK/ERK signaling pathway in C8D1A astrocytes. Mol Neurobiol. 2022;59(9):5856‐5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhang WB, Huang Y, Guo XR, Zhang MQ, Yuan XS, Zu HB. DHCR24 reverses Alzheimer's disease‐related pathology and cognitive impairment via increasing hippocampal cholesterol levels in 5xFAD mice. Acta Neuropathol Commun. 2023;11(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kivipelto M, Solomon A. Cholesterol as a risk factor for Alzheimer's disease ‐ epidemiological evidence. Acta Neurol Scand Suppl. 2006;185:50‐57. [DOI] [PubMed] [Google Scholar]
- 136. Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014;71(2):195‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Loera‐Valencia R, Vazquez‐Juarez E, Muñoz A, et al. High levels of 27‐hydroxycholesterol results in synaptic plasticity alterations in the hippocampus. Sci Rep. 2021;11(1):3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wang T, Cui S, Hao L, et al. Regulation of Th17/Treg balance by 27‐hydroxycholesterol and 24S‐hydroxycholesterol correlates with learning and memory ability in mice. Int J Mol Sci. 2022;23(8):4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ramirez DM, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24‐hydroxylase in the mouse brain. J Comp Neurol. 2008;507(5):1676‐1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Maioli S, Båvner A, Ali Z, et al. Is it possible to improve memory function by upregulation of the cholesterol 24S‐hydroxylase (CYP46A1) in the brain? PLoS One. 2013;8(7):e68534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Petrov AM, Lam M, Mast N, et al. CYP46A1 activation by efavirenz leads to behavioral improvement without significant changes in amyloid plaque load in the brain of 5XFAD mice. Neurotherapeutics. 2019;16(3):710‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Szwast SJ, Hendrie HC, Lane KA, et al. Association of statin use with cognitive decline in elderly African Americans. Neurology. 2007;69(19):1873‐1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62(7):1047‐1051. [DOI] [PubMed] [Google Scholar]
- 144. Tolosa E, Garrido A, Scholz SW, Poewe W. Challenges in the diagnosis of Parkinson's disease. Lancet Neurol. 2021;20(5):385‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dirkx MF, Zach H, Bloem BR, Hallett M, Helmich RC. The nature of postural tremor in Parkinson disease. Neurology. 2018;90(13):e1095‐e1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Rana AQ, Siddiqui I, Mosabbir AA, Qureshi AR, Fattah A, Awan N. Is action tremor in Parkinson's disease related to resting tremor? Neurol Res. 2014;36(2):107‐111. [DOI] [PubMed] [Google Scholar]
- 147. Yan J, Qiao L, Tian J, et al. Effect of statins on Parkinson's disease: a systematic review and meta‐analysis. Medicine (Baltimore). 2019;98(12):e14852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Carroll CB, Wyse RKH. Simvastatin as a potential disease‐modifying therapy for patients with Parkinson's disease: rationale for clinical trial, and current progress. J Parkinsons Dis. 2017;7(4):545‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Huang X, Alonso A, Guo X, et al. Statins, plasma cholesterol, and risk of Parkinson's disease: a prospective study. Mov Disord. 2015;30(4):552‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Jeong SH, Lee HS, Chung SJ, et al. Effects of statins on dopamine loss and prognosis in Parkinson's disease. Brain. 2021;144(10):3191‐3200. [DOI] [PubMed] [Google Scholar]
- 151. Mohamed A, Chen Y, Wu H, Liao J, Cheng B, Lu Q. Therapeutic advances in the treatment of SLE. Int Immunopharmacol. 2019;72:218‐223. [DOI] [PubMed] [Google Scholar]
- 152. Lo MS, Tsokos GC. Recent developments in systemic lupus erythematosus pathogenesis and applications for therapy. Curr Opin Rheumatol. 2018;30(2):222‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. López‐Pedrera C, Aguirre M, Barbarroja N, Cuadrado MJ. Accelerated atherosclerosis in systemic lupus erythematosus: role of proinflammatory cytokines and therapeutic approaches. J Biomed Biotechnol. 2010;2010:607084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Quevedo‐Abeledo JC, Sánchez‐Pérez H, Tejera‐Segura B, et al. Differences in capacity of high‐density lipoprotein cholesterol efflux between patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res (Hoboken). 2021;73(11):1590‐1596. [DOI] [PubMed] [Google Scholar]
- 155. Ronda N, Favari E, Borghi MO, et al. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. 2014;73(3):609‐615. [DOI] [PubMed] [Google Scholar]
- 156. Voloshyna I, Teboul I, Littlefield MJ, et al. Resveratrol counters systemic lupus erythematosus‐associated atherogenicity by normalizing cholesterol efflux. Exp Biol Med (Maywood). 2016;241(14):1611‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ruiz‐Limon P, Barbarroja N, Perez‐Sanchez C, et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: effects of in vivo statin treatment. Ann Rheum Dis. 2015;74(7):1450‐1458. [DOI] [PubMed] [Google Scholar]
- 158. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Amezaga Urruela M, Suarez‐Almazor ME. Lipid paradox in rheumatoid arthritis: changes with rheumatoid arthritis therapies. Curr Rheumatol Rep. 2012;14(5):428‐437. [DOI] [PubMed] [Google Scholar]
- 160. Pérez‐Baos S, Barrasa JI, Gratal P, et al. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: understanding the lipid paradox associated with rheumatoid arthritis. Br J Pharmacol. 2017;174(18):3018‐3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Xie Y, Feng SL, Mai CT, et al. Suppression of up‐regulated LXRα by silybin ameliorates experimental rheumatoid arthritis and abnormal lipid metabolism. Phytomedicine. 2021;80:153339. [DOI] [PubMed] [Google Scholar]
- 162. Dragoljevic D, Lee MKS, Pernes G, et al. Administration of an LXR agonist promotes atherosclerotic lesion remodelling in murine inflammatory arthritis. Clin Transl Immunol. 2023;12(4):e1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ban N, Lee TJ, Sene A, et al. Impaired monocyte cholesterol clearance initiates age‐related retinal degeneration and vision loss. JCI Insight. 2018;3(17):e120824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Storti F, Raphael G, Griesser V, et al. Regulated efflux of photoreceptor outer segment‐derived cholesterol by human RPE cells. Exp Eye Res. 2017;165:65‐77. [DOI] [PubMed] [Google Scholar]
- 165. Peters F, Ebner LJA, Atac D, et al. Regulation of ABCA1 by AMD‐associated genetic variants and hypoxia in iPSC‐RPE. Int J Mol Sci. 2022;23(6):3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Alamri A, Biswas L, Watson DG, Shu X. Deletion of TSPO resulted in change of metabolomic profile in retinal pigment epithelial cells. Int J Mol Sci. 2019;20(6):1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Nakano‐Ito K, Fujikawa Y, Hihara T, et al. E2012‐induced cataract and its predictive biomarkers. Toxicol Sci. 2014;137(1):249‐258. [DOI] [PubMed] [Google Scholar]
- 168. Miyashita T, Senshu M, Ibi K, et al. Evaluation of lens opacity due to inhibition of cholesterol biosynthesis using rat lens explant cultures. Toxicology. 2022;465:153064. [DOI] [PubMed] [Google Scholar]
- 169. Zhao M, Mei T, Shang B, et al. Defect of LSS disrupts lens development in cataractogenesis. Front Cell Dev Biol. 2021;9:788422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ishida H, Shibata T, Nakamura Y, et al. Identification of Differential gene expression pattern in lens epithelial cells derived from cataractous and noncataractous lenses of Shumiya cataract rat. Biomed Res Int. 2020;2020:7319590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Flores R, Jin X, Chang J, et al. LCAT, ApoD, and ApoA1 expression and review of cholesterol deposition in the cornea. Biomolecules. 2019;9(12):785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Aldaas KM, Ismail OM, Hakim J, et al. Association of dry eye disease with dyslipidemia and statin use. Am J Ophthalmol. 2020;218:54‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Jiang SY, Tang JJ, Xiao X, et al. Schnyder corneal dystrophy‐associated UBIAD1 mutations cause corneal cholesterol accumulation by stabilizing HMG‐CoA reductase. PLoS Genet. 2019;15(7):e1008289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Saadane A, Mast N, Trichonas G, et al. Retinal vascular abnormalities and microglia activation in mice with deficiency in cytochrome P450 46A1‐mediated cholesterol removal. Am J Pathol. 2019;189(2):405‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Vujosevic S, Alovisi C, Chakravarthy U. Epidemiology of geographic atrophy and its precursor features of intermediate age‐related macular degeneration. Acta Ophthalmol. 2023;101(8):839‐856. [DOI] [PubMed] [Google Scholar]
- 176. Liu K, Xie B. Today and future of age‐related macular degeneration. ISRN Ophthalmol. 2012;2012:480212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Wang L, Clark ME, Crossman DK, et al. Abundant lipid and protein components of drusen. PLoS One. 2010;5(4):e10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Rodriguez IR, Alam S, Lee JW. Cytotoxicity of oxidized low‐density lipoprotein in cultured RPE cells is dependent on the formation of 7‐ketocholesterol. Invest Ophthalmol Vis Sci. 2004;45(8):2830‐2837. [DOI] [PubMed] [Google Scholar]
- 179. Rodriguez IR, Clark ME, Lee JW, Curcio CA. 7‐ketocholesterol accumulates in ocular tissues as a consequence of aging and is present in high levels in drusen. Exp Eye Res. 2014;128:151‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Yang C, Xie L, Gu Q, Qiu Q, Wu X, Yin L. 7‐Ketocholesterol disturbs RPE cells phagocytosis of the outer segment of photoreceptor and induces inflammation through ERK signaling pathway. Exp Eye Res. 2019;189:107849. [DOI] [PubMed] [Google Scholar]
- 181. Bergmann M, Schütt F, Holz FG, Kopitz J. Inhibition of the ATP‐driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2‐E may contribute to the pathogenesis of age‐related macular degeneration. Faseb j. 2004;18(3):562‐564. [DOI] [PubMed] [Google Scholar]
- 182. Vives‐Bauza C, Anand M, Shiraz AK, et al. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283(36):24770‐24780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hammer M, Richter S, Guehrs KH, Schweitzer D. Retinal pigment epithelium cell damage by A2‐E and its photo‐derivatives. Mol Vis. 2006;12:1348‐1354. [PubMed] [Google Scholar]
- 184. Biswas L, Zeng Z, Graham A, Shu X. Gypenosides mediate cholesterol efflux and suppress oxidized LDL induced inflammation in retinal pigment epithelium cells. Exp Eye Res. 2020;191:107931. [DOI] [PubMed] [Google Scholar]
- 185. Huang JD, Amaral J, Lee JW, Rodriguez IR. 7‐Ketocholesterol‐induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS One. 2014;9(7):e100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE‐19 cells–implications for age‐related macular degeneration (AMD). Immunol Lett. 2012;147(1‐2):29‐33. [DOI] [PubMed] [Google Scholar]
- 187. Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Luthra S, Fardin B, Dong J, et al. Activation of caspase‐8 and caspase‐12 pathways by 7‐ketocholesterol in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2006;47(12):5569‐5575. [DOI] [PubMed] [Google Scholar]
- 190. Neekhra A, Luthra S, Chwa M, et al. Caspase‐8, ‐12, and ‐3 activation by 7‐ketocholesterol in retinal neurosensory cells. Invest Ophthalmol Vis Sci. 2007;48(3):1362‐1367. [DOI] [PubMed] [Google Scholar]
- 191. Luthra S, Dong J, Gramajo AL, et al. 7‐Ketocholesterol activates caspases‐3/7, ‐8, and ‐12 in human microvascular endothelial cells in vitro. Microvasc Res. 2008;75(3):343‐350. [DOI] [PubMed] [Google Scholar]
- 192. Wang H, Han X, Kunz E, Hartnett ME. Thy‐1 regulates VEGF‐mediated choroidal endothelial cell activation and migration: implications in neovascular age‐related macular degeneration. Invest Ophthalmol Vis Sci. 2016;57(13):5525‐5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Gehlbach P, Li T, Hatef E. Statins for age‐related macular degeneration. Cochrane Database Syst Rev. 2016;2016(8):Cd006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Memarzadeh E, Heidari‐Soureshjani S. The relationship between statin and risk of age‐related macular degeneration: a systematic review and meta‐analysis. J Ophthalmol. 2022;2022:8564818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Mast N, Bederman IR, Pikuleva IA. Retinal cholesterol content is reduced in simvastatin‐treated mice due to inhibited local biosynthesis albeit increased uptake of serum cholesterol. Drug Metab Dispos. 2018;46(11):1528‐1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Alpy F, Tomasetto C. Give lipids a START: the StAR‐related lipid transfer (START) domain in mammals. J Cell Sci. 2005;118(Pt 13):2791‐2801. [DOI] [PubMed] [Google Scholar]
- 197. Clark BJ, Soo SC, Caron KM, Ikeda Y, Parker KL, Stocco DM. Hormonal and developmental regulation of the steroidogenic acute regulatory protein. Mol Endocrinol. 1995;9(10):1346‐1355. [DOI] [PubMed] [Google Scholar]
- 198. Taylor JM, Borthwick F, Bartholomew C, Graham A. Overexpression of steroidogenic acute regulatory protein increases macrophage cholesterol efflux to apolipoprotein AI. Cardiovasc Res. 2010;86(3):526‐534. [DOI] [PubMed] [Google Scholar]
- 199. Wilhelm LP, Wendling C, Védie B, et al. STARD3 mediates endoplasmic reticulum‐to‐endosome cholesterol transport at membrane contact sites. Embo J. 2017;36(10):1412‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Almarhoun M, Biswas L, Alhasani RH, et al. Overexpression of STARD3 attenuates oxidized LDL‐induced oxidative stress and inflammation in retinal pigment epithelial cells. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866(7):158927. [DOI] [PubMed] [Google Scholar]
- 201. Liu J, Rone MB, Papadopoulos V. Protein‐protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J Biol Chem. 2006;281(50):38879‐38893. [DOI] [PubMed] [Google Scholar]
- 202. Biswas L, Farhan F, Reilly J, Bartholomew C, Shu X. TSPO ligands promote cholesterol efflux and suppress oxidative stress and inflammation in choroidal endothelial cells. Int J Mol Sci. 2018;19(12):3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Biswas L, Ibrahim KS, Li X, et al. Effect of a TSPO ligand on retinal pigment epithelial cholesterol homeostasis in high‐fat fed mice, implication for age‐related macular degeneration. Exp Eye Res. 2021;208:108625. [DOI] [PubMed] [Google Scholar]
- 204. Farhan F, Almarhoun M, Wong A, et al. Deletion of TSPO causes dysregulation of cholesterol metabolism in mouse retina. Cells. 2021;10(11):3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Rone MB, Fan J, Papadopoulos V. Cholesterol transport in steroid biosynthesis: role of protein‐protein interactions and implications in disease states. Biochim Biophys Acta. 2009;1791(7):646‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Xie L, Gu Q, Wu X, Yin L. Activation of LXRs reduces oxysterol lipotoxicity in RPE cells by promoting mitochondrial function. Nutrients. 2022;14(12):2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Ashraf F, Cogan DG, Kruth HS. Apolipoprotein A‐I and B distribution in the human cornea. Invest Ophthalmol Vis Sci. 1993;34(13):3574‐3578. [PubMed] [Google Scholar]
- 208. Barchiesi BJ, Eckel RH, Ellis PP. The cornea and disorders of lipid metabolism. Surv Ophthalmol. 1991;36(1):1‐22. [DOI] [PubMed] [Google Scholar]
- 209. Winder AF, Borysiewicz LK. Corneal opacification and familial disorders affecting plasma high‐density lipoprotein. Birth Defects Orig Artic Ser. 1982;18(6):433‐440. [PubMed] [Google Scholar]
- 210. Bron AJ. Corneal changes in the dislipoproteinaemias. Cornea. 1989;8(2):135‐140. [PubMed] [Google Scholar]
- 211. Schumacher MM, Jun DJ, Johnson BM, DeBose‐Boyd RA. UbiA prenyltransferase domain‐containing protein‐1 modulates HMG‐CoA reductase degradation to coordinate synthesis of sterol and nonsterol isoprenoids. J Biol Chem. 2018;293(1):312‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Cao J, Wang J, Qi W, et al. Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG‐CoA reductase. Cell Metab. 2007;6(2):115‐128. [DOI] [PubMed] [Google Scholar]
- 213. Jiang LY, Jiang W, Tian N, et al. Ring finger protein 145 (RNF145) is a ubiquitin ligase for sterol‐induced degradation of HMG‐CoA reductase. J Biol Chem. 2018;293(11):4047‐4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Sever N, Yang T, Brown MS, Goldstein JL, DeBose‐Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig‐1 to its sterol‐sensing domain. Mol Cell. 2003;11(1):25‐33. [DOI] [PubMed] [Google Scholar]
- 215. Song BL, Sever N, DeBose‐Boyd RA. Gp78, a membrane‐anchored ubiquitin ligase, associates with Insig‐1 and couples sterol‐regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19(6):829‐840. [DOI] [PubMed] [Google Scholar]
- 216. Tsai YC, Leichner GS, Pearce MM, et al. Differential regulation of HMG‐CoA reductase and Insig‐1 by enzymes of the ubiquitin‐proteasome system. Mol Biol Cell. 2012;23(23):4484‐4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Menzies SA, Volkmar N, van den Boomen DJ, et al. The sterol‐responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG‐CoA reductase together with gp78 and Hrd1. Elife. 2018;7:e40009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Schumacher MM, Jun DJ, Jo Y, Seemann J, DeBose‐Boyd RA. Geranylgeranyl‐regulated transport of the prenyltransferase UBIAD1 between membranes of the ER and Golgi. J Lipid Res. 2016;57(7):1286‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Jun DJ, Schumacher MM, Hwang S, Kinch LN, Grishin NV, DeBose‐Boyd RA. Schnyder corneal dystrophy‐associated UBIAD1 is defective in MK‐4 synthesis and resists autophagy‐mediated degradation. J Lipid Res. 2020;61(5):746‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Shin S, Zhou H, He C, et al. Qki activates Srebp2‐mediated cholesterol biosynthesis for maintenance of eye lens transparency. Nat Commun. 2021;12(1):3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Widomska J, Subczynski WK. Why is very high cholesterol content beneficial for the eye lens but negative for other organs? Nutrients. 2019;11(5):1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. De Vries AC, Vermeer MA, Bredman JJ, Bär PR, Cohen LH. Cholesterol content of the rat lens is lowered by administration of simvastatin, but not by pravastatin. Exp Eye Res. 1993;56(4):393‐399. [DOI] [PubMed] [Google Scholar]
- 223. Cenedella RJ, Kuszak JR, Al‐Ghoul KJ, Qin S, Sexton PS. Discordant expression of the sterol pathway in lens underlies simvastatin‐induced cataracts in Chbb: Thom rats. J Lipid Res. 2003;44(1):198‐211. [DOI] [PubMed] [Google Scholar]
- 224. Enkavi G, Javanainen M, Kulig W, Róg T, Vattulainen I. Multiscale simulations of biological membranes: the challenge to understand biological phenomena in a living substance. Chem Rev. 2019;119(9):5607‐5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Mainali L, Raguz M, O'Brien WJ, Subczynski WK. Properties of membranes derived from the total lipids extracted from the human lens cortex and nucleus. Biochim Biophys Acta. 2013;1828(6):1432‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Plesnar E, Szczelina R, Subczynski WK, Pasenkiewicz‐Gierula M. Is the cholesterol bilayer domain a barrier to oxygen transport into the eye lens? Biochim Biophys Acta Biomembr. 2018;1860(2):434‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Goodwin H, Brooks BP, Porter FD. Acute postnatal cataract formation in Smith‐Lemli‐Opitz syndrome. Am J Med Genet A. 2008;146a(2):208‐211. [DOI] [PubMed] [Google Scholar]
- 228. Hübner C, Hoffmann GF, Charpentier C, et al. Decreased plasma ubiquinone‐10 concentration in patients with mevalonate kinase deficiency. Pediatr Res. 1993;34(2):129‐133. [DOI] [PubMed] [Google Scholar]
- 229. Hua H, Yang T, Huang L, et al. Protective effects of lanosterol synthase up‐regulation in UV‐B‐induced oxidative stress. Front Pharmacol. 2019;10:947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Zou X, Wang H, Zhou D, et al. The polymorphism rs2968 of LSS gene confers susceptibility to age‐related cataract. DNA Cell Biol. 2020;39(11):1970‐1975. [DOI] [PubMed] [Google Scholar]
- 231. Chen XJ, Hu LD, Yao K, Yan YB. Lanosterol and 25‐hydroxycholesterol dissociate crystallin aggregates isolated from cataractous human lens via different mechanisms. Biochem Biophys Res Commun. 2018;506(4):868‐873. [DOI] [PubMed] [Google Scholar]
- 232. Chang JR, Koo E, Agrón E, et al. Risk factors associated with incident cataracts and cataract surgery in the age‐related eye disease study (AREDS): AREDS report number 32. Ophthalmology. 2011;118(11):2113‐2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. McCarty CA, Nanjan MB, Taylor HR. Attributable risk estimates for cataract to prioritize medical and public health action. Invest Ophthalmol Vis Sci. 2000;41(12):3720‐3725. [PubMed] [Google Scholar]
- 234. Borchman D, Ramasubramanian A, Foulks GN. Human meibum cholesteryl and wax ester variability with age, sex, and meibomian gland dysfunction. Invest Ophthalmol Vis Sci. 2019;60(6):2286‐2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Goto E, Dogru M, Fukagawa K, et al. Successful tear lipid layer treatment for refractory dry eye in office workers by low‐dose lipid application on the full‐length eyelid margin. Am J Ophthalmol. 2006;142(2):264‐270. [DOI] [PubMed] [Google Scholar]
- 236. Choi HR, Lee JH, Lee HK, Song JS, Kim HC. Association between dyslipidemia and dry eye syndrome among the korean middle‐aged population. Cornea. 2020;39(2):161‐167. [DOI] [PubMed] [Google Scholar]
- 237. Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. Jama. 2004;291(3):335‐342. [DOI] [PubMed] [Google Scholar]
- 238. Opreanu M, Tikhonenko M, Bozack S, et al. The unconventional role of acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes. 2011;60(9):2370‐2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Aldebasi YH, Mohieldein AH, Almansour YS, Almutairi BL. Dyslipidemia and lipid peroxidation of Saudi type 2 diabetics with proliferative retinopathy. Saudi Med J. 2013;34(6):616‐622. [PubMed] [Google Scholar]
- 240. Chung YR, Park SW, Choi SY, et al. Association of statin use and hypertriglyceridemia with diabetic macular edema in patients with type 2 diabetes and diabetic retinopathy. Cardiovasc Diabetol. 2017;16(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Hammer SS, Vieira CP, McFarland D, et al. Fasting and fasting‐mimicking treatment activate SIRT1/LXRα and alleviate diabetes‐induced systemic and microvascular dysfunction. Diabetologia. 2021;64(7):1674‐1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Pettersson AM, Stenson BM, Lorente‐Cebrián S, et al. LXR is a negative regulator of glucose uptake in human adipocytes. Diabetologia. 2013;56(9):2044‐2054. [DOI] [PubMed] [Google Scholar]
- 243. Tian B, Al‐Moujahed A, Bouzika P, et al. Atorvastatin promotes phagocytosis and attenuates pro‐inflammatory response in human retinal pigment epithelial cells. Sci Rep. 2017;7(1):2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Gnanaguru G, Wagschal A, Oh J, et al. Targeting of miR‐33 ameliorates phenotypes linked to age‐related macular degeneration. Mol Ther. 2021;29(7):2281‐2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Kim YK, Hong HK, Yoo HS, Park SP, Park KH. AICAR upregulates ABCA1/ABCG1 expression in the retinal pigment epithelium and reduces Bruch's membrane lipid deposit in ApoE deficient mice. Exp Eye Res. 2021;213:108854. [DOI] [PubMed] [Google Scholar]
- 246. Hussain G, Wang J, Rasul A, et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019;18(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Preston Mason R, Tulenko TN, Jacob RF. Direct evidence for cholesterol crystalline domains in biological membranes: role in human pathobiology. Biochim Biophys Acta. 2003;1610(2):198‐207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.