

Abstract
A receptor-based pharmacophore model describing the binding features required for the multi-kinase inhibition of the target kinases (VEGFR-2, FGFR-1, and BRAF) were constructed and validated. It showed a good overall quality in discriminating between the active and the inactive in a compiled test set compounds with F1 score of 0.502 and Mathew’s correlation coefficient of 0.513. It described the ligand binding to the hinge region Cys or Ala, the glutamate residue of the Glu-Lys αC helix conserved pair, the DFG motif Asp at the activation loop, and the allosteric back pocket next to the ATP binding site. Moreover, excluded volumes were used to define the steric extent of the binding sites. The application of the developed pharmacophore model in virtual screening of an in-house scaffold dataset resulted in the identification of a benzimidazole-based scaffold as a promising hit within the dataset. Compounds 8a-u were designed through structural optimization of the hit benzimidazole-based scaffold through (un)substituted aryl substitution on 2 and 5 positions of the benzimidazole ring. Molecular docking simulations and ADME properties predictions confirmed the promising characteristics of the designed compounds in terms of binding affinity and pharmacokinetic properties, respectively. The designed compounds 8a-u were synthesized, and they demonstrated moderate to potent VEGFR-2 inhibitory activity at 10 µM. Compound 8u exhibited a potent inhibitory activity against the target kinases (VEGFR-2, FGFR-1, and BRAF) with IC50 values of 0.93, 3.74, 0.25 µM, respectively. The benzimidazole derivatives 8a-u were all selected by the NCI (USA) to conduct their anti-proliferation screening. Compounds 8a and 8d resulted in a potent mean growth inhibition % (GI%) of 97.73% and 92.51%, respectively. Whereas compounds 8h, 8j, 8k, 8o, 8q, 8r, and 8u showed a mean GI% > 100% (lethal effect). The most potent compounds on the NCI panel of 60 different cancer cell lines were progressed further to NCI five-dose testing. The benzimidazole derivatives 8a, 8d, 8h, 8j, 8k, 8o, 8q, 8r and 8u exhibited potent anticancer activity on the tested cell lines reaching sub-micromolar range. Moreover, 8u was found to induce cell cycle arrest of MCF-7 cell line at the G2/M phase and accumulating cells at the sub-G1 phase as a result of cell apoptosis.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13065-024-01135-0.
Keywords: VEGFR-2, FGFR-1, BRAF, Multi-kinase, Anti-cancer, CADD, Pharmacophore, Molecular docking
Introduction
Protein kinases are a class of phosphotransferases that play a fundamental role in the regulation of different cellular processes such as cellular survival, growth, proliferation, migration, and apoptosis [1]. More than 30% of cellular proteins are phosphorylated by protein kinases. Protein kinases catalyse the transfer of a gamma phosphate group from ATP to an acceptor amino acid (Tyrosine, serine, or threonine) in a substrate protein [1]. Therefore, protein kinases are categorized mainly into two main categories, the tyrosine kinases such as vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and epidermal growth factor receptor (EGFR) and the serine/threonine kinases such as rapidly accelerated fibrosarcoma (RAF) kinases [2, 3]. In cancer, several protein kinases are dysregulated resulting in the uncontrolled growth, survival, and metastasis of tumour cells [2, 4, 5]. Hence, targeting protein kinases has received a remarkable attention in recent years for the discovery of new targeted chemotherapeutic agents for cancer treatment [6–8]. Based on the fact that cancer is regulated by multiple pathways that can compensate for one another when a single pathway is blocked, targeting multiple kinases is a more efficient strategy than targeting a single kinase [9]. Moreover, multi-kinase inhibition has numerous advantages, such as increasing potency due to its synergistic effect, reducing probable polypharmacy toxicity, avoiding pharmacokinetics incompatibilities, and enhancing selectivity [9, 10].
Angiogenesis, the formation of new blood vessels, plays an essential role in the growth and metastasis of tumour cells [11]. Hence, targeting protein kinases that initiate and sustain the angiogenic process is a prominent approach in cancer treatment [12]. Vascular endothelial growth factor (VEGF) and its receptors (VEGFRs) is a tyrosine kinase system that plays a curial role in angiogenesis both in the physiological as well as pathological conditions [13–15]. In comparison to healthy tissues, VEGFR-2, in particular, is overexpressed in various types of cancer such as malignant melanoma, breast cancer, hepatocellular carcinoma, colon cancer, etc., [16, 17]. In addition, hFGFR family is a group of four isoforms; FGFR-1 to FGFR-4 that are expressed on the cell membrane and participate in various vital physiological and pathological processes, such as proliferation, differentiation, cell migration, survival, as well as angiogenesis [18]. Binding of FGFR to its growth factor (FGF) results in its dimerization and phosphorylation of its intracellular kinase domain resulting in the initiation of a series of downstream signalling pathways. FGFRs overexpression has been reported in different types of solid tumours, for instance, FGFR-1 is amplified in breast and non-small cell lung cancers [19]. Hence, it is believed that FGFRs inhibition by small molecules that competitively bind to the ATP binding pocket is an attractive tactic for the design of novel targeted anticancer agents [20].
Moreover, when the main pro-angiogenic factors VEGF and FGF bind to their target receptors, they result in an activation of the mitogen-activated protein kinase (MAPK) signalling pathway [21]. Downstream signalling of this pathway leads to activation of RAS proteins which in turn causes subsequent activation of RAF kinases [22, 23]. RAF kinases are an intracellular serine/threonine family mediating several transcriptional factors leading to cell growth, survival, and proliferation [22, 23]. Among the RAF family, BRAF is the most sensitive isoform to activation and mutation [24].
The X-ray crystallographic structures of kinases such as VEGFR-2, FGFR-1 and BRAF demonstrated that their kinase domain comprises a smaller N-terminal lobe, larger C-terminal lobe, and an in-between ATP binding region which can be partitioned further into front pocket (front cleft or hinge region), gate area, and back cleft (allosteric back pocket). At the beginning of the C-terminal lobe there is an activation loop (A-loop) that is characterized by a highly conserved aspartate-phenylalanine-glycine (DFG) motif. Based on the 3D orientation of the DFG motif, the A-loop can exist in different conformations resulting in the existence of the protein kinase in its active (DFG-in) or inactive (DFG-out) conformations. In the catalytic cycle, the protein kinase switches between both open and closed conformations [25, 26]. Analysis of the binding modes of the co-crystallized protein kinase inhibitors (PKIs) at their target proteins demonstrated that PKIs can be classified according to their binding modes into six different types [26–30]. Among them, type II inhibitors are regarded as promising ones performing their antagonistic activity on the inactive (DFG-out) conformation accommodating into the hinge region, the gate area and extend further to the less conservative back pocket enhancing their affinity, selectivity, and residence time [31]. For example, sorafenib (I) (Fig. 1) is a VEGFR-2 (PDB ID: 4ASD) and BRAF (PDB ID: 1UWH) inhibitor in which the picolinamide moiety (coloured red in Fig. 1) occupies the front pocket and performs hydrogen bonding interaction with Cys919 (VEGFR-2)/Cys531 (BRAF). The ureido moiety (coloured pink in Fig. 1) extends through the gate area and forms by its NH group a hydrogen bond with the carboxylate group of αC-helix Glu885 (VEGFR-2)/Glu593 (BRAF), furthermore, the oxygen atom of the ureido moiety forms a hydrogen bond with the N–H group of DFG’s Asp1046 (VEGFR-2)/Asp500 (BRAF). In addition, sorafenib (I) extends into the hydrophobic back pocket by a disubstituted phenyl group (coloured blue in Fig. 1) achieving multiple hydrophobic interactions with the surrounding residues [32, 33].
Fig. 1.

Structure of sorafenib (I) in the binding sites of VEGFR-2 (PDB ID: 4ASD) and BRAF (PDB ID: 1UWH)
Benzimidazole is a privileged heterobicyclic scaffold representing the core of several reported targeted chemotherapeutic agents that possess potent protein kinase inhibitory activity [34–37]. Potashman et al., [38] demonstrated the potent VEGFR-2 inhibitory activity and anti-proliferative properties of a series of benzimidazole derivatives. Compound II (PDB ID: 2QU5) (Fig. 2) is a representative for this series showing Ki of 8.7 nM against VEGFR-2. In addition, RAF265 (III) (PDB ID: 5CT7) (Fig. 2) is a potent dual BRAF/VEGFR-2 inhibitor that showed a potent activity against melanoma and colorectal cancer [39, 40]. Dovitinib (IV) (PDB ID: 5AM6) (Fig. 2) is a benzimidazole-based type I multi-kinase inhibitor of VEGFR1-3 (IC50 = 8–13 nM), FGFR1-3 (IC50 = 8–9 nM), and other receptor tyrosine kinases, that showed a potent activity against a wide range of cancers [41–43].
Fig. 2.

Benzimidazole-based multi-kinase inhibitors and their interactions in their targets’ kinase domain
In recent years, significant advancements have been achieved in PKIs exploration, mostly attributed to the utilization of computational techniques [44, 45]. These methods have proven instrumental in delivering valuable insights into diverse protein kinase structures and inhibitors [44, 45]. In computer-aided drug design (CADD), two primary strategies are commonly employed; structure-based drug design (SBDD) and ligand-based drug design (LBDD). These approaches allow researchers to predict and optimize the properties and activities of molecules even before they are synthesized and tested in the laboratory [46].
Due to the continuous resistance development by cancer cells on one hand and the more satisfactory effect and less drawbacks achieved by the concurrent targeting of multiple protein kinases on the other hand [47, 48], there is a continuous demand for developing small molecules that target more than one protein kinase simultaneously (multi-kinase inhibitors).
Encouraged by these facts, the ultimate goal of the current investigation is to extract the common pharmacophoric features required for achieving multi-kinase inhibition of the target kinases; VEGFR-2, FGFR-1, and BRAF. The generated pharmacophore model will be then used to virtually screen a set of in-house synthetically feasible tailored diverse scaffolds to select those satisfy the pharmacophoric features of the target multi-kinase activity. Scaffolds satisfy the constructed pharmacophore model will be structurally optimized to enhance their target kinase binding. Molecular docking will be then used to confirm the ability of the designed derivatives to perform the essential interactions with the three target kinases. Afterwards, the in silico promising derivatives will be synthesized and evaluated for their biochemical inhibitory activity against the target kinases as well as for their cytotoxic activity on several cancer cell lines. Finally, the most potent candidate will be further analyzed for its effect on cell cycle progression and apoptosis induction.
Results and discussion
Molecular modeling study
For the intended study, a common 3D multi-kinase pharmacophore model for type II kinase inhibitors of the target kinases (VEGFR-2, FGFR-1, and BRAF) was constructed using the receptor-based pharmacophore technique. The generated pharmacophore was then validated for its ability to discriminate between active and inactive compounds of the different kinases of interest using a pre-compiled test set of active inhibitors and inactive decoys. Next, the validated pharmacophore was used to virtually screen several in-house datasets with diverse scaffolds and the most promising scaffold was used as a starting point to develop several optimized derivatives with potential synthetic feasibility which should be, by design, multi-kinase inhibitors of the target kinases. Finally, molecular docking simulations were used to investigate the ability of the designed compounds to bind to the active sites of the target kinases accomplishing the key interactions responsible for the kinase inhibitory activity. Promising compounds were passed to the next chemical synthesis step.
Common 3D multi-kinase receptor-based pharmacophore model generation
X-ray crystallographic structures
Several X-ray crystal structures for the target kinases (VEGFR-2, FGFR-1, and BRAF) are available in the protein data bank [49]. For the current work, we are focusing on the design of type II kinase inhibitors due to their reported superiority [20]. Thus, representative structures were selected that are co-crystalized with potent structurally diverse type II kinase inhibitors which bind to the inactive DFG-out kinase conformation occupying the front cleft (hinge region), the gate area and extend beyond the gatekeeper into the hydrophobic allosteric back cleft [50]. Different kinase structures co-crystalized with the same ligand were also preferred. Hence, the X-ray crystallographic structures of VEGFR-2 (PDB ID: 3VHE, 3VNT, and 3VO3), FGFR-1 (PDB ID: 4V01 and 3RHX), and BRAF (PDB ID: 4DBN and 6B8U) were downloaded from the protein data bank [49]. The protein structures were then prepared and aligned using their alpha carbons (See Additional file 1: Section 1: computational studies for further details).
Manual 3D receptor-based pharmacophore models generation
Using the aligned prepared protein structures, several manual 3D pharmacophores were generated to describe the common inhibitors’ interactions. The main common ligand-target interactions involve H-bonding interaction with the hinge region Cys919, Ala564, and Cys532 in VEGFR-2, FGFR-1, and BRAF, respectively, H-bonding with DFG Asp1046, Asp641, and Asp594 in VEGFR-2, FGFR-1, and BRAF, respectively, and H-bonding with αC-helix Glu885, Glu531, and Glu501 in VEGFR-2, FGFR-1, and BRAF, respectively. These interactions were described by hydrogen bond acceptor, donor, and acceptor features, respectively, with their corresponding projected features (Site features). In addition to hydrophobic interactions with the hydrophobic allosteric back pocket in each protein structure which were described by a broader hydrophobic pharmacophoric feature. In addition, excluded volumes were employed to define the binding sites’ steric extent. The different 3D pharmacophores obtained are the result of several combinations of the different pharmacophoric features (in terms of their number and volume) giving a set of 17 combinations (See Additional file 1: Section 1: computational studies for further details).
Pharmacophore model selection and validation
Selection of the best 3D pharmacophore model was carried out with the aid of a compiled test set of 2387 compounds (Table 1) (See Additional file 1: Section 1: computational studies for further details).
Table 1.
Distribution of the test set active inhibitors and inactive decoys for the target kinases VEGFR-2, FGFR-1, and BRAF
| Target kinase | Total test set compounds | Active inhibitors | Inactive decoys |
|---|---|---|---|
| VEGFR-2 | 827 | 26 | 801 |
| FGFR-1 | 620 | 20 | 600 |
| BRAF | 940 | 27 | 913 |
| Total | 2387 | 73 | 2314 |
The 3D pharmacophore ability to discriminate between the test set active and inactive compounds was used to evaluate its quality which was assessed based on its collective results on the whole test set. For each 3D pharmacophore, the total number of true positives TPt, false positives FPt, true negatives TNt, and false negatives FNt were determined from its performance on each kinase test set (See Additional file 1, for further details) and were used in calculating the different assessment metrics; Sensitivity Se, specificity Sp, yield of actives Ya, enrichment E, accuracy Acc, discrimination ratio DR, F1 score F1 and Mathew’s correlation coefficient MCC to evaluate the models’ performance (Table 2 (Metric’s values of the best performing pharmacophore model (Ph4-4) are shown in bold) and see Additional file 1: Section 1: computational studies for further details).
Table 2.
The collective assessment metrics of the generated pharmacophore models
| Ph4 no. | Se | Sp | Ya | E | Acc | DR | F1 | MCC |
|---|---|---|---|---|---|---|---|---|
| Ph4-1 | 0.753 | 0.955 | 0.344 | 11.240 | 0.948 | 0.789 | 0.4721 | 0.4875 |
| Ph4-2 | 0.753 | 0.958 | 0.362 | 11.832 | 0.952 | 0.786 | 0.4889 | 0.5017 |
| Ph4-3 | 0.753 | 0.959 | 0.369 | 12.070 | 0.953 | 0.785 | 0.4955 | 0.5073 |
| Ph4-4 | 0.753 | 0.961 | 0.377 | 12.318 | 0.954 | 0.784 | 0.5023 | 0.5131 |
| Ph4-5 | 0.740 | 0.962 | 0.383 | 12.523 | 0.956 | 0.769 | 0.5047 | 0.5128 |
| Ph4-6 | 0.726 | 0.964 | 0.387 | 12.650 | 0.956 | 0.753 | 0.5048 | 0.5106 |
| Ph4-7 | 0.753 | 0.942 | 0.289 | 9.465 | 0.936 | 0.800 | 0.4183 | 0.4422 |
| Ph4-8 | 0.753 | 0.935 | 0.267 | 8.730 | 0.929 | 0.806 | 0.3943 | 0.4220 |
| Ph4-9 | 0.767 | 0.919 | 0.230 | 7.535 | 0.915 | 0.835 | 0.3544 | 0.3908 |
| Ph4-10 | 0.767 | 0.936 | 0.273 | 8.932 | 0.930 | 0.820 | 0.4029 | 0.4318 |
| Ph4-11 | 0.767 | 0.924 | 0.242 | 7.927 | 0.920 | 0.830 | 0.3684 | 0.4027 |
| Ph4-12 | 0.808 | 0.905 | 0.211 | 6.915 | 0.902 | 0.893 | 0.3352 | 0.3822 |
| Ph4-13 | 0.849 | 0.773 | 0.105 | 3.448 | 0.775 | 1.099 | 0.1876 | 0.2486 |
| Ph4-14 | 0.849 | 0.824 | 0.132 | 4.323 | 0.825 | 1.031 | 0.2288 | 0.2918 |
| Ph4-15 | 0.849 | 0.861 | 0.161 | 5.279 | 0.860 | 0.987 | 0.2713 | 0.3328 |
| Ph4-16 | 0.479 | 0.952 | 0.240 | 7.839 | 0.938 | 0.504 | 0.3196 | 0.3100 |
| Ph4-17 | 0.425 | 0.986 | 0.484 | 15.838 | 0.969 | 0.431 | 0.4526 | 0.4375 |
(Metric’s values of the best performing pharmacophore model (Ph4-4) are shown in bold)
As can be seen in Table 2, Ph4-16 and Ph4-17 showed low sensitivity (0.479 and 0.425, respectively) meaning that they yielded a low number of true positives, however, they showed good specificity (0.952 and 0.986, respectively) and so could discard decoys and correctly consider them as inactive compounds, so these two models are biased towards inactive compounds and that is reflected in their low F1 score and MCC (0.3196 and 0.3100, respectively, for Ph4-16 and 0.4526 and 0.4375, respectively, for Ph4-17) (Table 2 and Fig. 3). On the contrary, models Ph4-12 to Ph4-15 showed good sensitivity (0.808, 0.849, 0.849 and 0.849, respectively) meaning that they yielded a high number of true positives, however, they showed low specificity (0.905, 0.773, 0.824 and 0.861, respectively) and so could not discard decoys properly and predict large number of decoys as active compounds, so these models are biased towards active compounds and this is reflected in their low MCC (0.3822, 0.2486, 0.2918 and 0.3328, respectively) (Table 2 and Fig. 3). Models Ph4-1 to Ph4-10 showed a balance between sensitivity and specificity with a sensitivity range of (0.726–0.767) and a specificity range of (0.919–0.964) indicating that the models are not biased towards either of actives or decoys (Table 2 and Fig. 3).
Fig. 3.

3D pharmacophore models’ performance represented by their F1 score F1 and Mathew’s correlation coefficient MCC values
Model Ph4-4 (Fig. 4) was selected as the best model because it showed the best overall performance on the test set. Ph4-4 selected 146 hits out of 2387 compounds of which 55 compounds were true positives (Se = 0.753) and it assigned 2241 compounds as inactive compounds from which 2223 are true negatives (Sp = 0.961). It showed a yield of actives Ya of 0.377 and an enrichment value E of 12.318 proving the success of the pharmacophore model in improving the selection process of active compounds via the virtual screening technique versus random methods. Moreover, Ph4-4 model had an accuracy Acc of 0.954 emphasizing that it can accurately identify active compounds while dismissing the inactive ones. Lastly, it had a discrimination ratio DR of 0.784 which shows that this model has a high prediction potential for discriminating between the active and the inactive compounds. Moreover, Ph4-4 showed F1 score of 0.5023 and the highest MCC of 0.5131 indicating its good overall quality (Table 2 and Fig. 3).
Fig. 4.

The selected pharmacophore model (Ph4-4) (distances in Å)
Figure 4 shows the selected 3D pharmacophore model, Ph4-4, its pharmacophoric features, and inter-feature distances (in Å) in 3D space. Ph4-4 consists of 4 main features [F1-F4]. Feature 1 (F1:Acc), a hydrogen bond acceptor, where ligands bind to the hinge region Cys or Ala, and the direction of this hydrogen bond acceptor lone pair is indicated by its projected site point feature (F5:Acc2). Feature 2 (F2:Don), a hydrogen bond donor, describing the feature required for binding to the glutamate residue of the Glu-Lys αC helix conserved pair and its projected site point feature (F6:Don2) indicates the direction of the hydrogen bond donor hydrogen. Feature 3 (F3:Acc), a hydrogen bond acceptor mapping where the ligands bind to the DFG motif Asp at the activation loop, in addition to its projected site point feature (F7:Acc2) indicating the direction of the hydrogen bond acceptor lone pair. Finally, the broadest feature (F4:Hyd) where ligands’ hydrophobic moieties occupy the allosteric back pocket next to the ATP binding site. Moreover, thirty excluded volumes (Not shown in Fig. 4 for clarity) were also added to this pharmacophore to define the steric extent of the binding sites and to restrict the highly flexible compounds (if any) to bind in the desired conformations to the binding site, simulating the actual binding scenario.
Virtual screening
The pharmacophore model exhibited the best performance on the test set (Best discrimination between actives and inactive compounds), Ph4-4, was then used to screen an in-house dataset of diverse scaffolds. The benzimidazole scaffold 8a was selected by Ph4-4 as the most promising hit with the least RMSD from the assigned pharmacophore model features’ centroids (RMSD = 0.979Å) (Fig. 5).
Fig. 5.

A The selected promising scaffold 8a by Ph4-4. B The promising scaffold 8a mapped onto Ph4-4 with RMSD of 0.979Å
Hit optimization
The promising hit scaffold was then used as a starting point to develop several optimized derivatives 8a-u with potential chemical synthesis feasibility and probable good binding affinity which should be, by design, multi-kinase type II inhibitors for the target kinases (Fig. 6).
Fig. 6.

Suggested chemically feasible optimized derivatives 8a-u from the selected promising scaffold 8a
The design strategy took into consideration that the benzimidazole core would occupy the gate area of the target protein kinases and the imidazole moiety would be involved in hydrogen bonding with the key amino acids Glu885 and Asp1046 in VEGFR-2, Glu531 and Asp641 in FGFR-1, as well as Glu501 and Asp594 in BRAF. Thus, the 5-position of the benzimidazole moiety was substituted with different aryl groups which are decorated with hydroxy and methoxy groups at different positions to be involved in hydrogen bonding with the key amino acid Cys919 (VEGFR-2), Ala564 (FGFR-1), and Cys532 (BRAF) at the hinge region. Moreover, (un)substituted aryl groups were introduced at 2-position of the benzimidazole scaffold to occupy the allosteric hydrophobic back pocket to engage in hydrophobic interactions with the surrounding amino acid (Fig. 6).
Molecular docking simulation
Molecular docking is a well stablished technique for the investigation of the binding mode and binding affinity of drug-like molecules in their proposed biological targets [51–55]. In the current study, to confirm and to study the binding characteristics of the designed compounds in the binding sites of the target kinases VEGFR-2, FGFR-1 and BRAF, molecular docking studies were performed using Molecular Operating Environment (MOE, 2022.02) software. The X-ray crystallographic structures of VEGFR-2 (PDB ID: 4ASD), FGFR-1 (PDB ID: 4V01) and BRAF (PDB ID: 5CT7) in their DGF-out inactive conformation were downloaded from the Protein Data Bank (PDB) [32, 39, 49, 56]. The downloaded protein structures are co-crystalized with a type II PK inhibitor, sorafenib (I), ponatinib, and RAF265, respectively. Molecular docking setup was initially validated by self-docking of the co-crystalized ligands in the binding sites of their corresponding target kinases. These simulations successfully reproduced the binding pattern of the co-crystalized ligands in the target binding sites, VEGFR-2, FGFR-1 and BRAF, with energy scores of − 15.18, − 17.00 and − 15.82 kcal/mol, respectively, and with an RMSD of 0.470, 0.398 and 0.419 Å, respectively, between the docked poses and the co-crystalized ligands (For further details see Additional file 1). Additionally, the docking poses reproduced all the key interactions achieved by the co-crystallized ligands with the binding site hot spots in VEGFR-2 (Glu885, Cys919 and Asp1046), FGFR-1 (Glu531, Ala564 and Asp641), and BRAF (Glu501, Cys532 and Asp594). The validation step results indicated the suitability of the used molecular docking protocol for the molecular docking study of the target compounds 8a-u in the binding sites of VEGFR-2, FGFR-1 and BRAF.
The docked compounds showed analogous binding patterns in the target kinases with predicted docking energy score ranges of − 14.89 to − 12.73 kcal/mol in VEGFR-2, − 14.18 to − 11.62 kcal/mol in FGFR-1, and − 13.65 to − 11.49 kcal/mol in BRAF, in comparison to the co-crystalized ligands docking score of − 15.19, − 17.00, and − 15.82 kcal/mol, respectively (See Additional file 1: Section 1: computational studies for further details).
The docked compounds showed promising binding patterns in VEGFR-2, FGFR-1 and BRAF interacting with the key amino acids in their binding pocket. The benzimidazole ring fits in the gate area stabilized via hydrogen bond interactions. By its imidazole ring, it interacts with the side chain carboxylate of Glu885, Glu531 and Glu501 of the αC helix in VEGFR-2, FGFR-1, and BRAF, respectively, and/or with Asp1046, Asp594, and Asp641 of the conserved DFG motif in VEGFR-2, FGFR-1, and BRAF, respectively.
The 2-phenyl substitution of the benzimidazole ring is directed towards the allosteric back pocket forming hydrophobic interactions with the hydrophobic side chains of the amino acids lining the pocket, Ile888, Leu889, Ile892, Val899, Leu1019, Ile1025, and Ile1044 amino acids of VEGFR-2, Ala512, Val513, Met534, Met535, Ile538, Ile545, Leu614, Leu634, Ile639, Ala640, and Phe642 of FGFR-1, and Val504, Leu505, Ile527, Leu565, Leu567, Ile572, and Ile573 of BRAF.
The substituted benzylidene-hydrazide moiety is accommodated in the hinge region interacting in most of the target compounds through hydrogen bonding with Cys919, Ala564, and Cys532 of VEGFR-2, FGFR-1, and BRAF, respectively. Additionally, it is involved in hydrophobic interactions with the hydrophobic side chains of the amino acids in the hinge region, Leu840, Val848, Ala866, Val899, Val916, Phe918, and Leu1035 amino acids of VEGFR-2, Leu484, Val492, Ala512, Val559, Val561, Leu630, and Phe642 of FGFR-1, and Ile463, Val471, Ala481, Leu514, Trp531, Phe583, Phe595, and Leu597 of BRAF (Figs. 7, 8, 9 and see Additional file 1: Section 1: computational studies for further details).
Fig. 7.
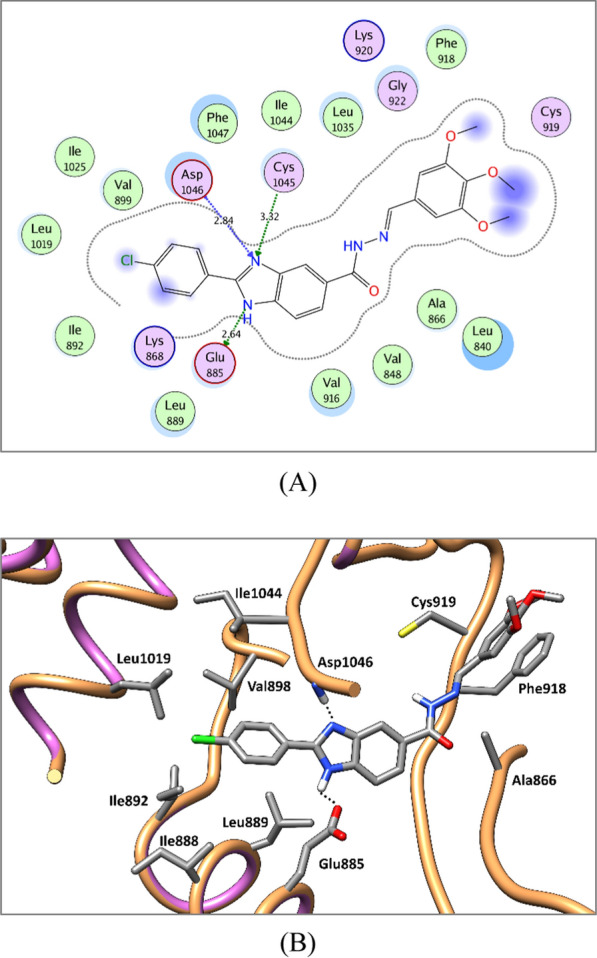
2D diagram (A) and 3D representation (B) of compound 8u showing its interaction with the VEGFR-2 active site
Fig. 8.

2D diagram (A) and 3D representation (B) of compound 8u showing its interaction with the FGFR-1 active site
Fig. 9.
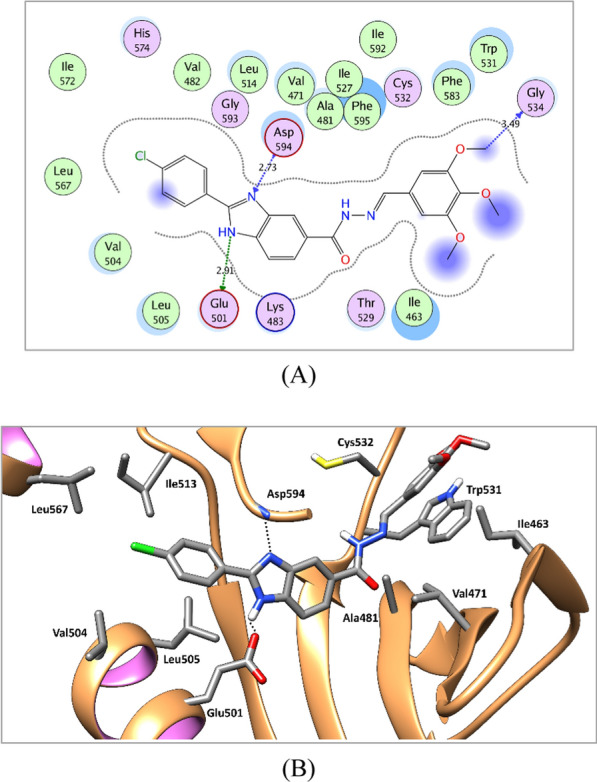
2D diagram (A) and 3D representation (B) of compound 8u showing its interaction with the BRAF active site
ADME properties prediction
SwissADME online web tool [57–59] was used to predict the physicochemical and AMDE properties of the target compounds 8a-u. SwissADME showed that the newly synthesized compounds possess promising predicted physiochemical and pharmacokinetic properties.
All target compounds possess promising predicted physicochemical properties and moderate predicted aqueous solubility. Moreover, they complied with Lipinski’s rule of 5 indicating that they are predicted to be orally bioavailable, and they possess a predicted SwissADME bioavailability score of 0.55 (See Additional file 1: Section 1: computational studies for further details).
Furthermore, as shown in SwissADME Boiled-Egg chart (Fig. 10), all target compounds showed high predicted GIT absorption with no predicted blood brain barrier (BBB) permeation and so devoid of CNS side effects. Moreover, Fig. 10 shows that all compounds are not p-glycoprotein (P-gp) substrates.
Fig. 10.

SwissADME BOILED-Egg chart for the designed compounds 8a-u
In summary, the designed benzimidazole derivatives 8a-u are predicted to be promising type II-like multi-kinase inhibitors in terms of binding affinity and pharmacokinetic properties and can be progressed further to chemical synthesis and biological evaluation.
Chemistry
As can be seen in Fig. 11, the target 2,5-disubstituted benzimidazole derivatives 8a-u were synthesized by the reaction of the benzaldehyde derivatives 1a-c with sodium metabisulfite to give the corresponding intermediates 2a-c which were condensed with 3,4-diaminobenzoic acid (3) to give the 2-aryl-benzimidzole-5-carboxylic acids 4a-c [34]. Esterification of 4 was then carried out to afford the corresponding ethyl esters 5a-c [60]. Hydrazinolysis of the benzimidazole esters 5a-c was carried out to yield the benzimidazole acid hydrazides 6a-c [61] which was followed by the reaction with different hydroxy and methoxybenzaldehyde derivatives 7a-g to afford the target benzimidazoles 8a-u in excellent yield.
Fig. 11.

Schematic pathway for the synthesis of the target 2,5-disubstituted benzimidazoles 8a-u
Biology
Biochemical assay
VEGFR-2 inhibitory activity
All the target 2,5-disubstituted benzimidazoles 8a-u were screened for their inhibitory activity on VEGFR-2 at 10 µM concentration and the % of inhibition was depicted in Table 3 using sorafenib (I) as a reference standard.
Table 3.
VEGFR-2 inhibitory activity of the synthesized 2,5-disubstituted benzimidazole derivatives 8a-u at 10 µM in reference to sorafenib (I)
 | |||
|---|---|---|---|
| ID | R1 | R2 | % Inhibition |
| 8a | H | 2-OH | 43.03 ± 2.32 |
| 8b | H | 3-OH | 23.82 ± 1.47 |
| 8c | H | 3-OMe | 18.52 ± 0.95 |
| 8d | H | 2-OH, 3-OMe | 53.80 ± 2.62 |
| 8e | H | 3-OH, 4-OMe | 36.36 ± 1.91 |
| 8f | H | 2,5-OMe | 58.29 ± 2.98 |
| 8g | H | 3,4,5-OMe | 47.91 ± 3.53 |
| 8h | OMe | 2-OH | 62.88 ± 3.89 |
| 8i | OMe | 3-OH | 45.16 ± 2.22 |
| 8j | OMe | 3-OMe | 15.67 ± 1.41 |
| 8k | OMe | 2-OH, 3-OMe | 69.50 ± 4.37 |
| 8l | OMe | 3-OH, 4-OMe | 50.62 ± 5.29 |
| 8m | OMe | 2,5-OMe | 6.67 ± 0.08 |
| 8n | OMe | 3,4,5-OMe | 20.81 ± 1.78 |
| 8o | Cl | 2-OH | 22.30 ± 1.95 |
| 8p | Cl | 3-OH | 22.32 ± 0.86 |
| 8q | Cl | 3-OMe | 21.49 ± 1.54 |
| 8r | Cl | 2-OH, 3-OMe | 25.71 ± 1.87 |
| 8s | Cl | 3-OH, 4-OMe | 26.72 ± 1.83 |
| 8t | Cl | 2,5-OMe | 33.99 ± 2.57 |
| 8u | Cl | 3,4,5-OMe | 80.0 ± 3.98 |
| Sorafenib (I) | – | – | 99 ± 0.50 |
Mean % inhibition (duplicate test) at a single dose (10 μM). Data are represented as mean value ± SD
The designed and synthesized 2,5-disubstituted benzimidazole 8a-u inhibited VEGFR-2 to variable extent. In series 8a-g, the 2,5-disubstituted benzimidazoles 8a, 8d, 8f and 8g displayed moderate inhibition of VEGFR-2 at 10 µM with inhibition % range of 43.03% to 58.29%. The substitution on the phenyl group at 5-position of the benzimidazole greatly affects the inhibition %. Compound 8a exhibiting 2-hydroxyphenyl moiety showed inhibition % of 43.03%, whereas further introduction of a methoxy group at 3-position to yield compound 8d resulted in increasing the potency (inhibition % = 53.80%). On the contrary, the 3-hydroxyphenyl 8b, 3-methoxyphenyl 8c, 3-hydroxy,4-methoxyphenyl 8e derivatives displayed weak potency against VEGFR-2 with inhibition % of 23.82%, 18.52% and 36.36%, respectively. Compound 8f exhibiting 2,5-dimethoxyphenyl group showed the most promising inhibition % of 58.29%, while introduction of 3,4,5-trimethoxyphenyl moiety in 8g resulted in a slight decrease in the potency (inhibition % = 47.91%).
In series 8h-n, compounds 8h and 8k displayed potent VEGFR-2 inhibition % of 62.88% and 69.50%, respectively. Meanwhile, their regioisomers 8i and 8l, respectively, demonstrated a decrease in the potency (inhibition % of 45.16% and 50.62%, respectively). Compounds 8j, 8m, and 8n with 3-methoxyphenyl, 2,5-dimethoxyphenyl and 3,4,5-trimethoxyphenyl groups, respectively, displayed weak VEGFR-2 inhibitory activity with inhibition % of 15.67%, 6.67%, and 20.81%, respectively.
In series 8o-u, which exhibit 4-chlorophenyl group at the 2 position, compound 8u incorporating 3,4,5-trimethoxyphenyl group displayed a promising VEGFR-2 inhibitory activity with an inhibition % of 80%, whereas replacement of the 3,4,5-trimethoxyphenyl moiety with hydroxy or methoxyphenyl groups at different positions resulted in a weak inhibitory activity on VEGFR-2 with (inhibition % range of 21.49—33.99%) in compounds 8o-t.
Multi-kinase inhibitory activity of 8u
In reference to the potent activity of 8u on VEGFR-2 (Table 3), it was further investigated for its inhibitory activity on VEGFR-2, FGFR-1 and BRAF at different concentrations and its IC50 values are presented in Table 4.
Table 4.
IC50 (µM) values of compound 8u on VEGFR-2, FGFR-1 and BRAF in reference to sorafenib (I)
 | ||||
|---|---|---|---|---|
| IC50 (µM) | ||||
| VEGFR-2 | FGFR-1 | BRAF | ||
| 8u | 0.93 ± 0.10 | 3.74 ± 0.34 | 0.25 ± 0.03 | |
| Sorafenib | 0.10 ± 0.01 | 0.58 ± 0.10 [62] | 0.02 ± 0.002 | |
The disubstituted benzimidazole derivative 8u showed an interesting potent multi-kinase inhibitory activity on BRAF (IC50 = 0.25 µM), followed by VEGFR-2 (IC50 = 0.93 µM) and FGFR-1 (IC50 = 3.74 µM) in reference to sorafenib (I) which demonstrated IC50 = 0.02, 0.10 and 0.58 µM against BRAF, VEGFR-2 and FGFR-1, respectively.
Antiproliferative activity
Antiproliferative activity on NCI cancer cell lines at 10 µM concentration
All the target 2,5-disubstituted benzimidazoles 8a-u were screened by NCI (USA) for their ability to supress the growth of NCI 60 cancer cell lines at 10 micromolar concentration and the results were depicted in Table 5.
Table 5.
In vitro growth inhibition % (GI %) of target 2,5-diarylbenzimidazole 8a-u against NCI panel of 60 tumor cell lines at 10 µM concentration
| Cell name | GI% | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8a | 8b | 8c | 8d | 8e | 8f | 8g | 8h | 8i | 8j | 8k | 8l | 8m | 8n | 8o | 8p | 8q | 8r | 8s | 8t | 8u | |
| Leukemia | |||||||||||||||||||||
| CCRF-CEM | 88.16 | 54.37 | 44.52 | La | 35.93 | 80.48 | 61.96 | 94.27 | 80.82 | 35.00 | L | 89.42 | 45.90 | 38.76 | L | 71.92 | 46.97 | L | L | 56.12 | L |
| HL-60(TB) | 95.58 | 14.38 | 38.07 | 90.03 | 24.68 | 36.28 | 17.05 | 82.91 | 55.15 | 30.37 | L | 52.20 | 18.79 | – | 47.15 | – | 50.70 | L | 45.32 | 12.89 | 67.85 |
| K-562 | 78.58 | 21.43 | 41.67 | 60.91 | 26.24 | 63.00 | 30.39 | 99.48 | 60.56 | 88.06 | L | 48.52 | 22.75 | 23.67 | 85.46 | 49.86 | 98.12 | L | 50.30 | 52.90 | L |
| MOLT-4 | 89.54 | 31.49 | 35.43 | 65.79 | 36.14 | 49.49 | 21.20 | 88.52 | 78.97 | 59.41 | L | 78.57 | 35.76 | 22.13 | 60.79 | 68.62 | 71.63 | L | 83.69 | 58.90 | 96.04 |
| RPMI-8226 | 88.59 | 9.59 | 81.63 | 98.47 | 23.38 | 81.35 | 86.93 | 96.71 | 24.77 | L | L | 32.41 | 76.82 | 44.49 | 88.15 | 56.71 | L | L | L | 62.25 | L |
| SR | 92.31 | 36.75 | 36.30 | 77.01 | 44.12 | 36.30 | 17.59 | 85.9 | 81.89 | 92.65 | L | 72.20 | 28.49 | 26.93 | 75.20 | 26.42 | L | L | 77.15 | 31.36 | L |
| Non-small cell lung cancer | |||||||||||||||||||||
| A549/ATCC | L | –b | 20.15 | 79.76 | – | 17.21 | 29.27 | L | 11.15 | L | 76.72 | 5.94 | 13.69 | – | 86.99 | 7.53 | L | L | 7.66 | 8.76 | 70.84 |
| EKVX | 96.49 | 13.81 | 41.83 | 87.59 | 19.18 | 22.69 | 12.24 | 92.19 | 23.40 | L | 98.06 | 12.95 | 10.58 | – | 67.24 | 7.59 | L | L | 37.02 | 42.46 | 90.75 |
| HOP-62 | L | 19.26 | 15.41 | ndc | 21.42 | nd | nd | L | 41.98 | L | L | 55.53 | 19.45 | 14.62 | ndc | 32.45 | L | L | nd | 18.43 | L |
| HOP-92 | L | – | – | 68.37 | 6.30 | 7.10 | – | L | 27.01 | L | 70.36 | 17.43 | – | – | L | 6.79 | L | L | – | 36.64 | L |
| NCI-H226 | 93.32 | – | 19.33 | L | 11.44 | 49.54 | 50.35 | L | 7.75 | 98.27 | 82.17 | 24.51 | – | 22.33 | 88.60 | 30.08 | L | L | 37.63 | 35.69 | L |
| NCI-H23 | 83.12 | – | 9.68 | L | – | 46.67 | 49.43 | 94.63 | 6.56 | L | L | 23.97 | – | 10.75 | L | 56.65 | L | L | 29.22 | 33.93 | L |
| NCI-H322M | 94.14 | – | 6.09 | 88.01 | – | 12.31 | 15.67 | 96.71 | 18.26 | L | 95.89 | 24.04 | – | – | 70.15 | – | L | L | 18.15 | 28.86 | 96.14 |
| NCI-H460 | L | – | 13.08 | 86.38 | – | 15.82 | 56.31 | L | 5.05 | L | L | 14.98 | – | – | L | 56.63 | L | L | 63.79 | 17.46 | L |
| NCI-H522 | L | 38.06 | 25.96 | L | 29.39 | 37.77 | 50.69 | L | 43.41 | 89.70 | L | 38.23 | 21.28 | 96.2 | L | 55.77 | L | L | 30.91 | 40.86 | L |
| Colon cancer | |||||||||||||||||||||
| COLO 205 | L | – | 32.62 | nd | 35.42 | nd | nd | L | 32.41 | L | L | 76.84 | – | 33.71 | nd | 34.57 | L | L | nd | 18.27 | L |
| HCC-2998 | 73.20 | – | – | 98.55 | – | 8.92 | 44.26 | 99.64 | 14.86 | 88.46 | L | 15.01 | – | – | 77.99 | 15.94 | 79.58 | L | 86.84 | 14.58 | L |
| HCT-116 | L | – | 12.67 | 97.43 | 15.03 | 52.52 | 41.62 | L | 42.79 | L | L | 43.83 | 22.29 | 23.4 | 98.39 | 37.67 | L | L | 69.55 | 43.08 | L |
| HCT-15 | 94.00 | – | 27.47 | 71.57 | – | 65.20 | 38.39 | L | 5.17 | 79.23 | 78.11 | – | 13.68 | – | L | – | 63.00 | 95.20 | 23.27 | 58.25 | 44.27 |
| HT29 | L | 16.35 | 33.92 | 84.54 | 95.82 | 14.69 | 47.26 | L | 74.50 | L | L | L | 25.35 | nd | L | 19.88 | L | L | L | 20.28 | L |
| KM12 | 84.00 | 19.80 | 25.98 | L | 21.86 | 30.49 | 36.44 | L | 38.72 | L | L | 34.46 | 11.66 | 13.78 | 85.44 | 62.74 | L | L | 68.41 | 23.33 | L |
| SW-620 | 77.39 | – | – | 70.84 | – | 30.32 | 37.93 | L | 6.72 | L | 98.83 | 21.77 | – | – | L | 6.31 | L | L | 69.62 | 7.98 | L |
| CNS cancer | |||||||||||||||||||||
| SF-268 | L | 16.82 | 15.05 | 99.03 | 16.00 | 36.62 | 31.99 | L | 37.35 | 84.67 | L | 40.44 | 9.02 | 21 | L | 24.23 | L | L | 72.71 | 43.02 | L |
| SF-295 | L | – | 18.50 | 83.16 | 10.86 | 31.38 | 54.12 | L | 21.96 | 167.36 | 96.39 | 50.95 | 19.91 | 29.8 | L | 11.79 | L | L | 84.96 | 51.90 | L |
| SF-539 | L | 13.68 | 28.61 | L | 32.21 | 36.88 | 49.79 | L | 53.27 | 34.15 | L | 42.58 | 32.57 | 24.35 | L | 32.42 | L | L | 47.21 | 45.33 | L |
| SNB-19 | 95.02 | – | – | 90.36 | – | 21.40 | 25.17 | L | 15.85 | L | L | 16.06 | – | 36.2 | L | nd | L | L | 39.72 | 45.69 | L |
| SNB-75 | L | 6.56 | 11.00 | 99.21 | – | – | – | 95.58 | 17.83 | L | 90.95 | 25.75 | – | – | L | 7.89 | L | L | 6.41 | 27.17 | L |
| U251 | L | – | 14.27 | 99.86 | 14.62 | 20.71 | 42.32 | L | 32.10 | L | 97.28 | 44.79 | 9.71 | 15.28 | L | 75.53 | L | L | 75.23 | 25.71 | L |
| Melanoma | |||||||||||||||||||||
| LOX IMVI | 88.38 | 7.43 | – | 92.90 | – | 62.46 | 56.54 | 98.7 | 26.07 | L | L | 37.90 | 18.79 | 7.53 | L | 14.74 | L | L | 44.94 | 34.64 | L |
| MALME-3 M | 99.32 | 39.71 | 47.13 | L | 78.42 | 40.49 | 73.63 | L | L | L | L | 87.59 | 18.33 | 76.79 | L | 58.88 | L | L | L | 18.37 | L |
| M14 | 81.80 | – | 8.28 | L | 19.74 | 34.40 | 43.27 | L | 39.14 | 70.41 | 90.97 | 63.49 | – | 40.28 | 90.88 | 25.37 | L | L | 42.14 | 30.86 | L |
| MDA-MB-435 | 63.10 | 5.45 | 18.89 | 74.61 | 30.39 | 73.65 | 48.54 | L | 63.84 | 76.54 | 76.03 | 26.54 | – | 24.97 | L | 15.16 | 98.89 | L | 89.90 | 14.33 | L |
| SK-MEL-2 | 60.21 | – | 19.50 | 34.31 | 8.51 | 16.71 | 21.79 | nd | 37.55 | 80.99 | 39.06 | 25.77 | 11.11 | nd | L | 23.97 | L | L | 76.06 | – | L |
| SK-MEL-28 | 82.67 | – | 20.43 | 96.19 | 28.87 | 29.06 | 40.41 | L | 52.90 | 64.23 | L | 52.45 | 5.70 | 31.45 | 78.18 | 15.97 | L | L | 70.75 | 14.62 | L |
| SK-MEL-5 | 85.73 | 10.91 | 39.03 | L | 39.52 | 67.75 | 89.35 | L | 40.51 | L | L | 55.34 | 22.29 | 42.62 | L | 69.47 | L | L | 53.32 | 68.78 | L |
| UACC-257 | 88.80 | – | 14.61 | L | 13.76 | – | 54.30 | 85.05 | 47.38 | 57.67 | L | 52.86 | 14.32 | 25.57 | 42.37 | – | L | L | 24.65 | – | L |
| UACC-62 | 75.88 | – | 14.66 | 99.46 | 18.69 | 35.47 | 70.39 | L | 50.84 | 58.87 | 98.01 | 47.57 | 6.39 | 68.48 | 95.30 | 19.57 | L | L | 37.51 | 24.28 | L |
| Ovarian cancer | |||||||||||||||||||||
| IGROV1 | 82.83 | 12.96 | 10.09 | 79.92 | 21.96 | 10.98 | 16.96 | L | 11.20 | L | 84.81 | 20.28 | – | – | 84.28 | 5.71 | L | L | – | 7.82 | 92.38 |
| OVCAR-3 | L | 11.09 | 7.38 | L | 7.25 | 12.52 | 29.45 | L | 8.04 | L | L | 37.61 | – | – | L | – | L | L | 47.55 | 10.78 | L |
| OVCAR-4 | 94.42 | 12.43 | 20.07 | L | – | 37.84 | 21.95 | L | 27.06 | L | 86.01 | 9.88 | 8.31 | – | L | 15.65 | L | L | 20.81 | 38.89 | L |
| OVCAR-5 | 77.04 | – | – | 63.24 | – | – | – | L | – | 91.17 | 69.48 | – | – | – | 55.93 | – | L | L | – | – | 37.36 |
| OVCAR-8 | L | 10.32 | 24.52 | L | 25.20 | 52.57 | 48.68 | L | 38.12 | L | L | 28.08 | 23.61 | 15.01 | L | 19.01 | L | L | 43.17 | 17.17 | L |
| NCI/ADR-RES | 63.15 | – | 6.59 | 47.95 | – | 24.51 | – | 63.2 | – | 31.99 | 28.06 | – | – | – | 56.35 | – | 23.04 | 49.30 | – | 11.23 | 24.32 |
| SK-OV-3 | L | 25.63 | 63.95 | nd | 56.26 | nd | nd | L | 53.95 | 68.31 | L | – | – | 20.23 | nd | 36.21 | L | L | nd | 15.10 | L |
| Renal cancer | |||||||||||||||||||||
| 786–0 | 91.76 | 5.43 | 21.32 | 86.04 | 16.58 | 18.59 | 27.80 | L | 23.41 | L | L | 18.10 | 29.04 | – | L | 22.17 | L | L | 52.04 | 26.55 | L |
| A498 | – | – | – | – | – | – | – | 54.5 | – | – | – | – | – | – | 53.91 | 10.17 | L | L | – | 56.16 | L |
| ACHN | L | – | – | 83.27 | – | 36.51 | 33.83 | L | – | L | 87.70 | – | 8.59 | 9.17 | 94.16 | – | L | L | – | 53.09 | 69.94 |
| CAKI-1 | L | 9.41 | 10.00 | 91.59 | 17.64 | 41.47 | 49.73 | L | 5.16 | 85.29 | 84.62 | – | – | 23.76 | L | – | L | L | 18.49 | 38.36 | 97.13 |
| RXF 393 | L | – | 29.22 | 85.95 | – | 49.96 | 30.81 | L | 14.49 | L | L | 37.22 | 15.21 | 20.09 | L | 31.57 | L | L | 69.12 | 96.54 | L |
| SN12C | nd | nd | nd | 91.53 | nd | 35.21 | 32.21 | L | nd | nd | nd | 26.96 | nd | 9.66 | 59.42 | nd | nd | nd | 31.33 | nd | nd |
| TK-10 | 65.04 | – | – | 50.64 | – | 18.99 | 6.66 | L | – | L | 40.00 | – | – | – | 95.02 | – | L | L | – | 10.93 | 62.64 |
| UO-31 | 86.22 | 21.59 | 30.63 | 98.53 | 38.19 | 54.04 | 45.55 | L | 10.49 | 98.91 | 83.20 | 9.13 | 9.12 | 21.84 | L | 14.89 | 81.69 | L | 8.70 | 31.95 | 41.11 |
| Prostate cancer | |||||||||||||||||||||
| PC-3 | 90.74 | 11.75 | 24.64 | 82.18 | 11.47 | 55.54 | 37.87 | L | 34.39 | L | 87.92 | 51.22 | 13.61 | 12.94 | 78.51 | 48.79 | L | L | 56.49 | 58.53 | L |
| DU-145 | L | – | 18.43 | 79.68 | – | 18.43 | 17.07 | L | 13.98 | L | 85.12 | 27.88 | – | – | L | – | L | L | 16.20 | 51.99 | L |
| Breast cancer | |||||||||||||||||||||
| MCF7 | 96.94 | 43.15 | 50.96 | 98.08 | 70.45 | 54.14 | 85.99 | L | 88.20 | L | L | 77.80 | 20.64 | 73.09 | 93.74 | 72.33 | L | L | 84.87 | 48.06 | L |
| MDA-MB-231/ATCC | 88.16 | 14.06 | – | 94.36 | – | 38.00 | 30.48 | 98.72 | 22.38 | L | 97.65 | 26.37 | 14.33 | 7.37 | L | 45.17 | L | L | 75.12 | 89.71 | L |
| HS 578 T | L | – | 19.99 | 92.89 | 6.89 | 49.91 | 51.04 | L | 52.61 | L | 89.32 | 46.23 | – | 37.73 | L | 27.07 | L | L | 86.87 | 48.77 | L |
| BT-549 | L | – | – | 96.59 | – | 38.19 | 35.43 | L | 32.71 | 29.81 | L | 10.40 | 17.16 | – | 60.58 | – | 6.97 | L | 42.90 | 25.73 | L |
| T-47D | 93.65 | 50.90 | 69.11 | nd | 39.30 | nd | nd | L | 86.79 | L | 94.97 | 52.49 | 40.60 | 38.08 | nd | 79.76 | L | L | nd | 61.77 | L |
| MDA-MB-468 | 99.81 | 18.93 | 22.71 | L | 7.65 | 24.73 | 39.16 | L | 54.60 | L | L | 44.35 | 16.21 | – | L | 65.77 | L | L | 88.69 | 30.39 | L |
| Mean growth inhibition % | 97.73 | 8.49 | 20.74 | 92.51 | 16.75 | 33.74 | 36 | L | 33.08 | L | L | 34.39 | 10.90 | 13.64 | L | 25.43 | L | L | 49.49 | 33.49 | L |
aGI% > 100; bGI% < 5%; cnot detected
The substitution pattern on the 2 and 5 phenyl moieties has a great influence on the growth inhibitory activity of the synthesized benzimidazoles 8a-u on cancer cell lines (Table 5, Fig. 12). In the 2-phenylbenzimidazole series 8a-g, incorporation of 2-hydroxyphenyl and 2-hydroxy, 3-methoxyphenyl moieties at the 5-position in 8a and 8d, respectively, resulted in a potent mean growth inhibition % (GI%) of 97.73% and 92.51%, respectively, with a broad spectrum antiproliferative activity against the different NCI sub-panels. Isomeric shifting of the hydroxy group to the 3-position in 8b and 8e, respectively, decreased the mean GI% to 8.49% and 16.75%, respectively. On the other side, the 3-methoxyphenyl 8c, 2,5-dimethoxyphenyl 8f and 3,4,5-trimethoxyphenyl 8g derivatives showed moderate mean GI% of 20.74%, 33.74% and 36.00%, respectively.
Fig. 12.

Structure activity relationship diagram showing the effect of the substitution pattern of 2- and 5-phenyl moieties of 8a-u on the antiproliferative activity
In series 8h-n which incorporates 2-(4-methoxyphenyl)benzimidazole moiety, the 2-hydroxyphenyl derivatives 8h and 8k showed mean GI% > 100% (lethal effect) on the tested cell lines. On the contrary, the 3-hydroxyphenyl congeners 8i and 8l demonstrated moderate mean GI% of 33.08 and 34.39%, respectively. Replacement of 3-hydroxyphenyl group in 8i with 3-methoxyphenyl moiety in 8j increased the inhibitory activity (mean GI% of 33.08% versus 100% (lethal effect), respectively). However, the 2,5-dimethoxyphenyl derivative 8m and the 3,4,5-trimethoxyphenyl derivative 8n demonstrated weak mean GI% of 10.90 and 13.64%, respectively.
Replacement of the 2-(4-methoxyphenyl)benzimidazole moiety in series 8h-n with 2-(4-chlorophenyl)benzimidazole moiety in series 8o-u, showed the same pattern of mean GI%. The benzimidazole derivative incorporating 2-hydroxyphenyl, 3-methoxyphenyl and 2-hydroxy, 3-methoxyphenyl 8o, 8q and 8r, respectively showed a mean GI% > 100% (lethal effect). On the other hand, the derivatives 8p, 8s and 8t demonstrated moderate mean GI% of 25.43, 49.49, and 33.49%, respectively. Furthermore, the 5-(3,4,5-trimethoxyphenyl)benzimidazole derivative 8u showed a mean GI% > 100% (lethal effect) on the tested cell lines.
Antiproliferative activity on NCI cancer cell lines at five different concentrations
The 2,5-diaryl benzimidazole derivatives 8a, 8d, 8h, 8j, 8k, 8o, 8q, 8r, and 8u were selected by NCI to be further assayed for their growth inhibitory activity at five dose level and their GI50 results were depicted in Table 6. The selected 2,5-diaryl benzimidazoles displayed potent inhibitory activity against the tested cell lines with GI50 up to 0.12 µM. Close examination showed that 8d displayed the most potent GI50 against most of the cell lines with mean GI50 of 2.62 µM. Meanwhile compounds 8a, 8h, 8j, 8k, 8o, 8q, 8r and 8u demonstrated potent inhibitory activity against most of the tested cell lines with mean GI50 range of 2.98 to 7.98 µM.
Table 6.
GI50 of compounds 8a, 8d, 8h, 8j, 8k, 8o, 8q, 8r, and 8u on NCI cancer cell lines
| Cell name | GI50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 8a | 8d | 8h | 8j | 8k | 8o | 8q | 8r | 8u | |
| Leukemia | |||||||||
| CCRF-CEM | 0.26 | 0.30 | 0.92 | 7.91 | 1.05 | > 38.8 | 1.95 | 0.43 | 6.27 |
| HL-60(TB) | 3.08 | 2.42 | 2.09 | 36.5 | 7.66 | 0.31 | 19.5 | 1.95 | 3.90 |
| K-562 | 3.39 | 2.04 | 1.36 | 6.92 | 3.74 | 0.15 | 3.14 | 0.49 | 1.96 |
| MOLT-4 | 1.62 | 2.06 | 1.70 | 22.6 | 3.80 | 0.28 | 5.96 | 0.63 | 3.58 |
| RPMI-8226 | 1.19 | 1.86 | 1.39 | 4.14 | 2.80 | > 38.8 | 1.47 | 2.22 | 3.21 |
| SR | 1.68 | 1.80 | 1.17 | 10.6 | 2.06 | 0.12 | 4.15 | 0.50 | 3.76 |
| Non-small cell lung cancer | |||||||||
| A549/ATCC | 3.41 | 3.39 | 3.03 | 3.05 | 9.04 | 1.80 | 3.22 | 3.68 | 7.55 |
| EKVX | 1.85 | 1.86 | 3.24 | 2.76 | 2.30 | 9.31 | 3.52 | 4.56 | 14.80 |
| HOP-62 | 1.47 | 1.65 | 5.09 | 2.70 | 3.11 | 0.84 | 1.92 | 1.04 | 4.68 |
| HOP-92 | nda | 1.58 | 2.83 | nd | nd | > 38.8 | nd | 1.25 | 5.95 |
| NCI-H226 | 4.02 | 0.53 | 6.94 | 2.13 | 3.14 | 4.33 | 2.57 | 1.65 | 1.31 |
| NCI-H23 | 3.31 | 1.87 | 2.06 | 2.95 | 2.14 | 0.31 | 2.20 | 1.15 | 4.72 |
| NCI-H322M | 2.13 | 1.41 | 4.23 | 3.03 | 3.04 | 1.38 | 3.57 | 1.59 | 8.29 |
| NCI-H460 | 2.37 | 2.63 | 1.87 | 1.85 | 3.27 | 0.25 | 3.34 | 1.35 | 3.19 |
| NCI-H522 | 1.87 | 1.97 | 1.54 | 3.30 | 2.98 | 0.26 | 2.26 | 1.17 | 4.90 |
| Colon cancer | |||||||||
| COLO 205 | 1.97 | 2.34 | 2.90 | 4.30 | 3.94 | 0.89 | 4.02 | 2.33 | 2.84 |
| HCC-2998 | 3.91 | 1.79 | 4.43 | 3.52 | 2.33 | 1.38 | 4.42 | 1.48 | 11.10 |
| HCT-116 | 0.88 | 1.84 | 4.93 | 2.99 | 2.90 | 1.08 | 3.06 | 1.19 | 3.26 |
| HCT-15 | 1.07 | 2.07 | 1.25 | 3.92 | 3.35 | 0.20 | 5.52 | 2.72 | 73.0 |
| HT29 | 2.63 | 3.41 | nd | 2.80 | 3.67 | 1.08 | 2.38 | 4.17 | 2.98 |
| KM12 | 3.72 | 2.58 | 4.24 | 4.34 | 2.45 | 1.24 | 4.42 | 1.66 | 4.50 |
| SW-620 | 3.48 | 3.77 | 1.75 | 3.10 | 4.35 | 0.33 | 2.93 | 1.32 | 4.40 |
| CNS cancer | |||||||||
| SF-268 | 2.78 | 1.76 | 0.92 | 3.28 | 2.16 | 0.84 | 2.18 | 1.21 | 4.40 |
| SF-295 | 2.70 | 2.93 | 1.26 | 2.93 | 2.48 | 1.60 | 2.17 | 2.20 | 4.18 |
| SF-539 | 2.74 | 1.31 | 2.05 | 11.5 | 2.80 | 0.37 | 7.14 | 1.07 | 2.02 |
| SNB-19 | 4.16 | 2.41 | 1.76 | 3.18 | 3.33 | 1.12 | 2.17 | 1.31 | 3.42 |
| SNB-75 | 2.20 | nd | 0.39 | 1.78 | 3.05 | 0.74 | 5.69 | nd | 1.28 |
| U251 | 2.11 | 0.76 | 1.64 | 2.18 | 2.17 | 0.83 | 2.03 | 1.50 | 2.59 |
| Melanoma | |||||||||
| LOX IMVI | 2.00 | 1.34 | 2.23 | 1.98 | 2.34 | 0.43 | 1.82 | 1.18 | 4.67 |
| MALME-3 M | 1.88 | 1.37 | 1.77 | 2.45 | 1.09 | 1.03 | 1.54 | 1.36 | 1.12 |
| M14 | 0.92 | 1.57 | 3.65 | 3.42 | 2.81 | 1.81 | 2.15 | 2.07 | 3.77 |
| MDA-MB-435 | 5.77 | 4.69 | 1.95 | 6.25 | 6.20 | 0.53 | 3.90 | 1.81 | 4.13 |
| SK-MEL-2 | 3.19 | 1.64 | 5.17 | 10.6 | 6.30 | 4.60 | 3.07 | nd | 11.0 |
| SK-MEL-28 | 4.62 | 1.78 | 2.96 | 3.93 | 3.32 | 1.85 | 2.15 | 1.62 | 1.83 |
| SK-MEL-5 | 3.75 | 1.40 | 1.73 | 3.45 | 2.86 | 0.51 | 4.90 | 1.34 | 2.83 |
| UACC-257 | 4.00 | 2.34 | 6.32 | 11.0 | 3.43 | 7.35 | 4.80 | 2.35 | 4.67 |
| UACC-62 | 3.66 | 1.22 | 2.53 | 6.50 | 2.78 | 1.69 | 4.92 | 1.23 | 1.24 |
| Ovarian cancer | |||||||||
| IGROV1 | 3.90 | 2.34 | 2.26 | 3.49 | 3.52 | 0.95 | 3.01 | 1.25 | 11.0 |
| OVCAR-3 | 1.94 | 0.42 | 1.98 | 2.05 | 1.25 | 0.19 | 3.24 | 0.56 | 2.28 |
| OVCAR-4 | 2.24 | 1.85 | 2.24 | 2.01 | 3.26 | 1.25 | 3.07 | 1.41 | 2.35 |
| OVCAR-5 | 8.50 | 2.77 | 6.55 | 4.69 | 7.72 | 2.12 | 5.35 | 2.17 | 15.6 |
| OVCAR-8 | 2.49 | 1.62 | 1.97 | 1.92 | 2.71 | 0.98 | 2.69 | 1.58 | 3.87 |
| NCI/ADR-RES | 3.51 | 4.65 | 6.74 | 6.35 | > 100 | 1.15 | 94.8 | > 100 | nd |
| SK-OV-3 | 2.62 | 2.56 | 7.30 | 5.39 | 4.31 | 4.53 | 2.02 | 2.08 | 15.0 |
| Renal cancer | |||||||||
| 786–0 | 3.84 | 2.47 | nd | 3.84 | 3.69 | 0.83 | 1.85 | 1.49 | 4.88 |
| A498 | 16.90 | 36.6 | 9.21 | 21.5 | > 100 | 8.10 | 15.3 | 12.1 | 26.4 |
| ACHN | 3.28 | 2.28 | 3.02 | 4.25 | 4.19 | 0.81 | 4.21 | 2.92 | 7.93 |
| CAKI-1 | 3.48 | 2.68 | 1.78 | 2.81 | 3.64 | 0.22 | 3.84 | 2.31 | 11.2 |
| RXF 393 | 2.37 | 3.21 | 6.29 | 2.26 | 2.58 | 0.79 | 2.06 | 1.41 | 2.22 |
| SN12C | 4.67 | 2.08 | 4.99 | 2.78 | 3.81 | 3.41 | 2.22 | 1.72 | 3.22 |
| TK-10 | 4.53 | 4.78 | 6.19 | 12.6 | > 100 | 5.08 | 23.9 | 6.46 | 28.8 |
| UO-31 | 0.60 | 2.34 | 1.70 | 1.78 | 3.34 | 0.31 | 3.46 | 2.07 | > 100 |
| Prostate cancer | |||||||||
| PC-3 | nd | 2.71 | 1.77 | nd | nd | > 38.8 | nd | 1.62 | 5.73 |
| DU-145 | 2.75 | 3.05 | 2.33 | 3.28 | 5.06 | 1.36 | 2.04 | 1.99 | 6.77 |
| Breast cancer | |||||||||
| MCF7 | 2.63 | 1.06 | 1.67 | 2.50 | 1.72 | 0.16 | 2.57 | 0.66 | 0.66 |
| MDA-MB-231/ATCC | 2.03 | 2.41 | 4.48 | 2.43 | 2.80 | 1.41 | 1.89 | 1.82 | 3.39 |
| HS 578 T | 2.49 | 0.62 | 0.94 | 1.90 | 2.22 | 0.26 | 2.71 | 0.70 | 2.58 |
| BT-549 | 1.80 | 1.52 | 6.99 | 5.47 | 2.85 | 3.57 | 10.1 | 1.36 | 2.69 |
| T-47D | 3.38 | 1.93 | 2.60 | 2.62 | 3.48 | 1.48 | 1.95 | 0.60 | 3.48 |
| MDA-MB-468 | 3.45 | 0.89 | 2.98 | 3.06 | 1.95 | 1.51 | 2.97 | 1.44 | 3.27 |
| Mean GI50 | 3.00 | 2.62 | 2.98 | 5.24 | 7.70 | 5.44 | 5.40 | 3.39 | 7.98 |
aNot detected
Cell cycle analysis
Encouraged by the potent multi-kinase inhibitory activity of the 2,5-diaryl benzimidazole derivative 8u as well as its potent and broad spectrum of antiproliferative activity, it was selected as a representative to be examined for its influence on the cell cycle progression of MCF-7 cell line at its GI50 concentration by flow cytometry analysis using propidium iodide (PI) stain. Comparison with breast cancer MCF-7 cells treated with DMSO as control was carried out, and the results were presented in Fig. 13 and Table 7. The % of MCF-7 cells that was accumulated in the G1 phase showed a decrease from 56.30% to 45.32% after treatment with 8u. On the other side, the accumulated % of cells in the G2/M phase increased from 20.60% to 25.57% (Table 7). These results emphasized that 8u arrests the MCF-7 cell line at the G2/M phase. Furthermore, an increase in the percent of cells accumulated in the sub-G1 phase, from 2.64% in the control to 4.40% in the treated cells was noticed as a result of cell apoptosis.
Fig. 13.

Effect of compound 8u on the phases of cell cycle of MCF-7 cells
Table 7.
Effect of compounds 8u on the phases of cell cycle of MCF-7 cells
| Comp. | %G0/G1 | %S | %G2/M | %Sub-G1 |
|---|---|---|---|---|
| Control | 56.30 | 23.10 | 20.60 | 2.64 |
| 8u | 45.32 | 29.11 | 25.57 | 4.40 |
Apoptosis assay
The capability of 8u to enhance apoptosis of MCF-7 cell line at its GI50 concentration was explored (Fig. 14). The presented results showed that the % of cells in the early apoptosis and late apoptosis phase increased from 0.25% and 2.36% to 1.46 and 4.54%, respectively, after treatment with 8u, which indicates that 8u induces cell apoptosis in MCF-7 cell line.
Fig. 14.

Effect of 8u on the percentage of annexin V-FITC-positive staining in MCF-7 cells. The four quadrants identified as: Q2-3, viable; Q2-4, early apoptotic; Q2-2, late apoptotic; Q2-1, necrotic
Conclusion
Several receptor-based pharmacophore models describing the binding features required for the multi-kinase inhibition of the target kinases (VEGFR-2, FGFR-1, and BRAF) were constructed based on the experimental binding mode and binding interactions of several inhibitors for these target kinases. Using a compiled test set of 73 active inhibitors for the target kinases as well as 2314 inactive decoys, (Ph4-4) was selected as the best model showing F1 score of 0.5023 and Mathew’s correlation coefficient of 0.5131 indicating its good overall quality in discriminating between the active and the inactive compounds. Virtual screening of an in-house dataset of diverse scaffolds using the selected pharmacophore model yielded a benzimidazole-based scaffold as a promising hit among the dataset compounds with RMSD of 0.979Å. Structural optimization of the hit benzimidazole-based scaffold through (un)substituted aryl substitution on 2 and 5 positions of the benzimidazole ring produced compounds 8a-u. Based on molecular docking simulations and ADME properties predictions, the optimization products were predicted to be promising type II-like multi-kinase inhibitors in terms of binding affinity and pharmacokinetic properties and can be progressed further to chemical synthesis and biological evaluation.
The designed compounds 8a-u were synthesized, and they were tested for their VEGFR-2 inhibitory activity at 10 µM concentration. The benzimidazole derivatives 8h, 8k, and 8u showed potent VEGFR-2 inhibition % of 62.88, 69.50, and 80.00%, respectively. The benzimidazole derivative 8u exhibited a potent inhibitory activity against the target kinases (VEGFR-2, FGFR-1, and BRAF) with IC50 values of 0.93, 3.74 and 0.25 µM, respectively.
Simultaneously, compounds 8a-u were examined at 10 µM for their antiproliferative efficacy at NCI (USA). Compounds 8a, 8d, 8h, 8j, 8k, 8o, 8q, 8r and 8u demonstrated potent activity with GI% > 90% and were further selected to be tested at the five dose assay. It is obvious that incorporation of 2-hydroxyphenyl group in 8a, 8h, 8o or 2-hydroxy, 3-methoxyphenyl group in 8d, 8k, 8r is favourable (mean GI50 = 2.62–7.70 µM). Meanwhile, incorporation of 3-methoxyphenyl group is favourable at the five position of 2-(4-methoxyphenylbenzimidazole) 8j and 2-(4-chlorophenylbenzimidazole) 8q (mean GI50 = 5.24 and 5.40 µM, respectively). Interestingly, the introduction of 3,4,5-trimethoxyphenyl derivative is promising only in 2-(4-chlorophenylbenzimidazole) 8u (mean GI50 = 7.98 µM). Encouraged by the potent activity of 8u on both the biochemical and cellular assays, it was further assessed for its effect on cell cycle and apoptosis of MCF-7 cell line. Interestingly, 8u was found to induce cell cycle arrest in MCF-7 cell line at the G2/M phase and accumulating cells at the sub-G1 phase as a result of cell apoptosis.
Experimental
Molecular modeling study
The molecular modeling study was carried out using Molecular Operating Environment software (MOE 2022.02) according to the following steps:
Common 3D multi-kinase receptor-based pharmacophore model generation
Retrieving X-ray crystallographic structures
The X-ray crystallographic structures of VEGFR-2 (PDB ID: 3VHE, 3VNT, and 3VO3), FGFR-1 (PDB ID: 4V01 and 3RHX), and BRAF (PDB ID: 4DBN and 6B8U) were downloaded from the protein data bank [49]. MOE was used to prepare the retrieved protein structures (For further details see Additional file 1: Section 1: computational studies). Finally, correctness of ligands’ structures and reported ligand interactions at the active site were further checked after the protonation step. The different prepared protein structures of VEGFR-2, FGFR-1 and BRAF were aligned and superposed using Align protocol in MOE using protein structures’ αCs. Consequently, the co-crystalized ligands were aligned in their bioactive conformation in the binding sites of the different protein structures.
Manual 3D receptor-based pharmacophore models generation
Using the pharmacophore query editor in MOE, the aligned co-crystalized ligands were used to generate several manual 3D pharmacophores based on their common interactions with the target kinases’ binding sites in the different protein structures. The assigned pharmacophoric features include recognition, shape, and projected site point features representing the main common features responsible for the inhibitors’ binding to the kinase domain hotspots in the different proteins. Moreover, several excluded volumes (with different volumes and number) were included to define the steric extent of the binding sites (See Additional file 1: Section 1: computational studies for further details).
Pharmacophore model selection and validation
For pharmacophore model selection and validation, for each kinase of the target kinases, a test set was constructed from active inhibitors and inactive decoys with type II-kinase-inhibitor-like structures (Based on their visual inspection). The test set compounds for each kinase were compiled from the Directory of Useful Decoys-Enhanced (DUD-E) [63] and/or DEKOIS 2.0 [64] databases. A large decoys/actives ratio (≈30) was maintained to mimic the natural ratio in the chemical space between the active and inactive compounds. Table 1 shows the distribution of the actives and decoys for each target kinase (See Additional file 1, Section 1: computational studies for the structure of the active inhibitors used in the test set for each target kinase).
Conformational search was then carried out for the test set compounds using Stochastic search in MOE which generates conformations by randomly rotating all bonds (including ring bonds) and randomly inverting tetrahedral centres followed by an all-atom energy minimization.
The generated conformers were virtually screened using the different manually generated pharmacophore models to test their ability to discriminate between the active and inactive compounds in the compiled test sets. MOE pharmacophore search-algorithm begins with prefiltration of the conformers database by calculating the conformer similarity to the pharmacophore model with respect to feature type and distance; followed by a more computationally expensive alignment of the conformer atoms to the query feature points minimizing their deviation from each other using root mean square deviation (RMSD) as the fitness criteria for the alignment quality.
The 3D pharmacophore ability in discriminating between the test set active and inactive compounds was assessed based on its collective results on the whole test set. For each 3D pharmacophore, the total number of true positives TPt, false positives FPt, true negatives TNt, and false negatives FNt were determined (see Additional file 1, Section: computational studies). To assess the performance of the different generated pharmacophores, a set of assessment metrics were used to select and validate the best one. These metrics include sensitivity Se, specificity Sp, yield of actives Ya, enrichment E, accuracy Acc, discrimination ratio DR, F1 score and Mathew’s correlation coefficient MCC to evaluate the models’ performance (For further details see Additional file 1, Sect. 1: computational studies) [65].
Virtual screening
The pharmacophore model exhibited the best performance on the test set in discriminating between actives and inactive compounds was used to screen an in-house dataset of diverse scaffolds after performing conformational search on the in-house dataset using the same protocol used for the test set compounds (Vide supra).
Hit optimization
The promising scaffold was then used as a starting point to develop several optimized derivatives with potential synthetic feasibility which should be, by design, multi-kinase type II inhibitors for the target kinases.
Molecular docking study
Finally, molecular docking was used to investigate the ability of the designed compounds to bind to the binding sites of the target kinases accomplishing the key interactions responsible for the kinase inhibitory activity.
The X-ray crystallographic structure of VEGFR-2, FGFR-1, and BRAF co-crystallized with Type II kinase inhibitors (PDB ID: 4ASD, 4V01 and 5CT7, respectively) were downloaded from the protein data bank [32, 39, 49, 56] and were utilized to perform the molecular docking study. Details of the molecular docking procedures are discussed in the Additional file 1, Section 1: computational studies.
ADME properties prediction
SwissADME online web tool [57–59] was used to predict the physicochemical and AMDE properties of the target compounds 8a-u (See Additional file 1, Section 1: computational studies for further details).
Chemistry
General remarks
Chemicals, reagents and solvents were purchased from commercial suppliers. Chemical reactions were followed up by TLC using aluminium plates precoated with silica gel 60 F245 (Merck). Uncorrected melting points were measured on a Stuart SMP30 melting point apparatus. Spectral and elemental analyses of 8a-u were recorded in the laboratory central services, National Research Centre, Cairo, Egypt and faculty of pharmacy, Cairo University. IR spectra (4000–400 cm−1) were detected on a Jasco FT/IR 300 E Fourier transform infrared spectrophotometer. 1HNMR and 13CNMR (DMSO-d6) were measured at 500 (125) MHz and 400 (100) MHz on Bruker instruments.
General procedure for the synthesis of N'-(substitutedbenzylidene)-2-(substituted)phenyl-1H-benzo[d]imidazole-5-carbohydrazide 8a-u
A mixture of 2-phenyl-1H-benzo[d]imidazole-5-carbohydrazides 6a-c (0.50 mmol) and benzaldehyde derivatives 7a-g (0.50 mmol) were reacted in EtOH (20 mL) under reflux in the presence of glacial acetic acid (1 mL) for 2h. The solution was poured into ice–water and neutralized with few drops of NH4OH solution. The precipitated crude products 8a-u were filtered and were crystalized from EtOH to give the pure 2,5-diaryl benzimidazoles 8a-u.
N'-(2-Hydroxybenzylidene)-2-phenyl-1H-benzo[d]imidazole-5-carbohydrazide (8a)
Pale brown powder; yield = 84%; mp 287–289 °C; IR (KBr) ṽ 3221, 3059, 2936, 2859, 1663, 1620, 1574, 1489 cm−1; 1H NMR (500 MHz; DMSO-d6) δH 6.91–6.98 (m, 2H), 7.30 (t, 3J = 7.5 Hz, 1H), 7.39 (dt, 3J = 8.0 Hz, 4J = 1.5 Hz, 1H), 7.54 (d, 3J = 7.0 Hz, 1H), 7.58 (t, 3J = 7.5 Hz, 2H), 7.68 (dd, 3J = 8.0 Hz, 4J = 1.0 Hz, 1H), 7.73 (d, 3J = 8.5 Hz, 1H), 7.86 (d, 3J = 8.5 Hz, 1H), 8.22 (d, 3J = 7.5 Hz, 1H), 8.27 (s, 1H), 8.67 (s, 1H), 8.99 (s, 1H), 11.46 (s, 1H), 12.20 ppm (s, 1H); 13C NMR (125 MHz; DMSO-d6) δC 116.40, 116.49, 118.14, 118.66, 119.26, 119.54, 122.13, 126.72, 128.98, 129.58, 129.70, 130.34, 130.86, 131.17, 133.15, 148.10, 153.42, 157.50, 158.62, 162.78, 163.27 ppm; Anal. Calcd for C21H16N4O2: C, 70.77; H, 4.53; N, 15.72. Found: C, 70.45; H, 4.88; N, 15.85.
N'-(3-Hydroxybenzylidene)-2-phenyl-1H-benzo[d]imidazole-5-carbohydrazide (8b)
Pale brown powder; yield = 89%; mp 308–310 °C; 1H NMR (400 MHz; DMSO-d6) δH 6.83 (d, 3J = 6.8 Hz, 1H), 7.11 (d, 3J = 7.2 Hz, 1H), 7.22 (s, 1H), 7.25 (t, 3J = 8.0 Hz, 1H), 7.51–7.60 (m, 3H), 7.70 (d, 3J = 8.4 Hz, 1H), 7.82 (d, 3J = 8.0 Hz, 1H), 8.21 (d, 3J = 7.2 Hz, 3H), 8.41 (s, 1H), 9.62 (s, 1H), 11.83 (s, 1H), 13.27 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 112.69, 117.40, 118.83, 122.25, 126.78, 127.42, 129.15, 129.67, 129.97, 130.48, 135.82, 147.49, 153.39, 157.72, 163.65 ppm; Anal. Calcd for C21H16N4O2: C, 70.77; H, 4.53; N, 15.72. Found: C, 70.90; H, 4.75; N, 15.50.
N'-(3-Methoxybenzylidene)-2-phenyl-1H-benzo[d]imidazole-5-carbohydrazide (8c)
Pale brown powder; yield = 82%; mp 163–165 °C; IR (KBr) ṽ 3159, 3075, 3005, 2974, 2940, 1663, 1604, 1570, 1462 cm−1; 1H NMR (500 MHz; DMSO-d6) δH 3.80 (s, 3H), 7.00 (d, 3J = 7.0 Hz, 1H), 7.29–7.31 (m, 1H), 7.37 (t, 3J = 7.5 Hz, 1H), 7.40–7.45 (m, 1H), 7.53 (d, 3J = 7.0 Hz, 1H), 7.58 (t, 3J = 7.0 Hz, 2H), 7.71 (br., 1H), 7.83 (d, 3J = 7.5 Hz, 1H), 8.22 (d, 3J = 7.0 Hz, 2H), 8.48 (s, 1H), 8.68 (s, 1H), 11.93 (s, 1H), 13.25 ppm (br., 1H);13C NMR (125 MHz; DMSO-d6) δC 55.11, 111.17, 112.53, 116.05, 117.44, 119.97, 121.18, 126.69, 127.29, 128.97, 129.69, 129.86, 129.94, 130.27, 135.16, 135.93, 147.18, 153.34, 159.55, 161.25, 163.70 ppm; Anal. Calcd for C22H18N4O2: C, 71.34; H, 4.90; N, 15.13. Found: C, 71.00; H, 5.21; N, 15.35.
N'-(2-hydroxy-3-methoxybenzylidene)-2-phenyl-1H-benzo[d]imidazole-5-carbohydrazide (8d)
Pale brown powder; yield = 77%; mp 188–190 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.81 (s, 3H), 6.86 (t, 3J = 8.0 Hz, 1H), 7.03 (d, 3J = 8.0 Hz, 1H), 7.14 (d, 3J = 7.6 Hz, 1H), 7.51–7.60 (m, 4H), 7.72 (d, 3J = 8.4 Hz, 1H), 7.85 (d, 3J = 8.4 Hz, 1H), 8.22 (d, 3J = 7.2 Hz, 2H), 8.26 (s, 1H), 8.68 (s, 1H), 11.17 (s, 1H), 12.15 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.86, 113.82, 118.97, 119.08, 121.10, 122.25, 126.76, 126.81, 129.13, 129.63, 130.49, 147.28, 148.00, 148.03, 153.51, 163.35 ppm; Anal. Calcd for C22H18N4O3: C, 68.38; H, 4.70; N, 14.50. Found: C, 68.09; H, 4.97; N, 14.32.
N'-(3-Hydroxy-4-methoxybenzylidene)-2-phenyl-1H-benzo[d]imidazole-5-carbohydrazide (8e)
Pale brown powder; yield = 72%; mp 267–269 °C; 1H NMR (500 MHz; DMSO-d6) δH 3.80 (s, 3H), 6.97 (d, 3J = 8.5 Hz, 1H), 7.06 (d, 3J = 7.5 Hz, 1H), 7.29 (s, 1H), 7.53 (t like, 3J = 7.5 Hz, 1H), 7.58 (t like, 3J = 7.0 Hz, 2H), 7.68 (br., 1H), 7.80 (d, 3J = 7.0 Hz, 1H), 8.21 (d, 3J = 7.5 Hz, 2H), 8.34 (s, 1H), 8.51 (s, 1H), 9.34 (s, 1H), 11.72 (s, 1H), 13.22 ppm (br., 1H); 13C NMR (125 MHz; DMSO-d6) δC 55.58, 111.94, 112.41, 120.11, 126.68, 127.39, 128.98, 129.71, 130.27, 146.88, 147.51, 149.68, 153.26, 160.52, 163.38 ppm; Anal. Calcd for C22H18N4O3: C, 68.38; H, 4.70; N, 14.50. Found: C, 68.72; H, 4.34; N, 14.77.
N'-(2,5-Dimethoxybenzylidene)-2-phenyl-1H-benzo[d]imidazole-5-carbohydrazide (8f)
Pale brown powder; yield = 75%; mp 249–251 °C; IR (KBr) ṽ 3256, 3051, 2967, 2928, 1663, 1624, 1601, 1562, 1493, 1454 cm−1; 1H NMR (500 MHz; DMSO-d6) δH 3.75 (s, 3H), 3.82 (s, 3H), 6.99 (dd, 3J = 8.5 Hz, 4J = 2.5 Hz, 1H), 7.05 (d, 3J = 9.0 Hz, 1H), 7.41 (s, 1H), 7.53 (t like, 3J = 7.5 Hz, 1H), 7.58 (t like, 3J = 7.5 Hz, 2H), 7.70 (d, 3J = 8.0 Hz, 1H), 7.85 (d, 3J = 8.5 Hz, 1H), 8.21 (d, 3J = 7.0 Hz, 2H), 8.24 (br., 1H), 8.83 (s, 1H), 11.94 (s, 1H), 13.19 ppm (br., 1H); 13C NMR (125 MHz; DMSO-d6) δC 55.41, 56.23, 109.28, 113.40, 117.40, 122.15, 123.18, 126.68, 127.31, 128.97, 129.59, 130.30, 142.66, 152.24, 153.27, 163.41 ppm; Anal. Calcd for C23H20N4O3: C, 68.99; H, 5.03; N, 13.99. Found: C, 68.71; H, 5.24; N, 14.34.
2-Phenyl-N'-(3,4,5-trimethoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8g)
Pale brown powder; yield = 80%; mp 178–180 °C; 1H NMR (500 MHz; DMSO-d6) δH 3.70 (s, 3H), 3.84 (s, 6H), 7.04 (s, 2H), 7.53 (t like, 3J = 7.0 Hz, 1H), 7.58 (t, 3J = 7.5 Hz, 2H), 7.71 (s, 1H), 7.82 (d, 3J = 7.5 Hz, 1H), 8.21 (d like, 3J = 7.0 Hz, 3H), 8.43 (s, 1H), 11.91 (s, 1H), 13.24 ppm (s, 1H); 13C NMR (125 MHz; DMSO-d6) δC 55.92, 60.07, 104.33, 122.19, 126.66, 127.38, 127.38, 127.44, 128.97, 129.69, 129.99, 130.26, 139.23, 147.39, 153.17, 163.68 ppm; Anal. Calcd for C24H22N4O4: C, 66.97; H, 5.15; N, 13.02. Found: C, 66.72; H, 5.37; N, 13.29.
N'-(2-Hydroxybenzylidene)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carbohydrazide (8h)
Pale brown powder; yield = 81%; mp 242–244 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.85 (s, 3H), 6.93 (t, 3J = 8.4 Hz, 2H), 7.14 (d, 3J = 9.2 Hz, 2H), 7.30 (t, 3J = 7.6 Hz, 1H), 7.53 (d, 3J = 7.2 Hz, 1H), 7.67 (d, 3J = 8.4 Hz, 1H), 7.82 (d, 3J = 8.4 Hz, 1H), 8.15 (d, 3J = 8.8 Hz, 2H), 8.20 (br., 1H), 8.66 (s, 1H), 11.44 (s, 1H), 12.14 (s, 1H), 13.15 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.42, 114.52, 116.44, 118.71, 119.34, 121.89, 121.95, 126.41, 128.44, 129.70, 131.24, 147.98, 153.50, 157.50, 161.11, 163.27 ppm; Anal. Calcd for C22H18N4O3: C, 68.38; H, 4.70; N, 14.50. Found C, 68.61; H, 4.95; N, 14.32.
N'-(3-Hydroxybenzylidene)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carbohydrazide (8i)
Pale brown powder; yield = 90%; mp 298–300 °C; IR (KBr) ṽ 3210, 3063, 2993, 2940, 2893, 1663, 1613, 1582, 1543 cm−1; 1H NMR (400 MHz; DMSO-d6) δH 3.85 (s, 3H), 6.83 (d, 3J = 8.0 Hz, 1H), 7.10 (d, 3J = 7.2 Hz, 1H), 7.14 (d, 3J = 9.2 Hz, 2H), 7.21 (s, 1H), 7.25 (t, 3J = 7.6 Hz, 1H), 7.65 (d, 3J = 8.4 Hz, 1H), 7.79 (d, 3J = 8.4 Hz, 1H), 8.15 (d, 3J = 8.8 Hz, 3H), 8.40 (s, 1H), 9.62 (s, 1H), 11.80 (s, 1H), 13.10 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.42, 112.62, 114.52, 117.34, 118.77, 121.94, 122.10, 127.08, 128.40, 129.92, 135.82, 147.36, 153.47, 157.70, 161.05, 163.63 ppm; Anal. Calcd for C22H18N4O3: C, 68.38; H, 4.70; N, 14.50. Found: C, 68.14; H, 4.91; N, 14.88.
N'-(3-Methoxybenzylidene)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carbohydrazide (8j)
Pale brown powder; yield = 90%; mp 203–205 °C; IR (KBr) ṽ 3159, 3075, 3028, 3009, 2971, 1663, 1612, 1566, 1493 cm−1; 1H NMR (400 MHz; DMSO-d6) δH 3.80 (s, 3H), 3.84 (s, 3H), 7.00 (d, 3J = 7.6 Hz, 1H), 7.14 (d, 3J = 8.4 Hz, 2H), 7.30 (br., 2H), 7.37 (t, 3J = 8.0 Hz, 1H), 7.66 (d, 3J = 8.4 Hz, 1H), 7.81 (d, 3J = 8.4 Hz, 1H), 8.16 (d, 3J = 8.8 Hz, 2H), 8.19 (s, 1H), 8.48 (s, 1H), 11.90 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.17, 55.40, 111.15, 114.50, 116.09, 120.01, 121.99, 127.05, 128.40, 129.95, 135.97, 147.15, 153.42, 159.57, 161.07, 163.66 ppm; Anal. Calcd for C23H20N4O3: C, 68.99; H, 5.03; N, 13.99. Found: C, 68.67; H, 5.31; N, 14.23.
N'-(2-Hydroxy-3-methoxybenzylidene)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carbohydrazide (8k)
Pale brown powder; yield = 82%; mp 192–194 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.81 (s, 3H), 3.85 (s, 3H), 6.87 (t, 3J = 8.0 Hz, 1H), 7.03 (d, 3J = 8.0 Hz, 1H), 7.13 (d, 3J = 6.4 Hz, 1H), 7.14 (d, 3J = 8.8 Hz, 2H), 7.67 (d, 3J = 8.4 Hz, 1H), 7.82 (d, 3J = 8.4 Hz, 1H), 8.16 (d, 3J = 8.8 Hz, 2H), 8.20 (s, 1H), 8.67 (s, 1H), 11.18 (s, 1H), 12.13 (s, 1H), 13.32 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.47, 55.88, 113.83, 114.58, 118.98, 119.10, 121.12, 121.96, 122.02, 126.49, 128.51, 147.28, 148.01, 153.60, 161.17, 163.39 ppm; Anal. Calcd for C23H20N4O4: C, 66.34; H, 4.84; N, 13.45. Found: C, 66.67; H, 4.98; N, 13.17.
N'-(3-Hydroxy-4-methoxybenzylidene)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carbohydrazide (8l)
Pale brown powder; yield = 82%; mp > 250 °C [66]; 1H NMR (400 MHz; DMSO-d6) δH 3.80 (s, 3H), 3.85 (s, 3H), 6.97 (d, 3J = 8.4 Hz, 1H), 7.06 (d, 3J = 8.0 Hz, 1H), 7.14 (d, 3J = 8.8 Hz, 2H), 7.28 (s, 1H), 7.65 (d, 3J = 8.4 Hz, 1H), 7.78 (d, 3J = 8.4 Hz, 1H), 8.15 (d, 3J = 8.4 Hz, 2H), 8.16 (s, 1H), 8.33 (s, 1H), 9.30 (s, 1H), 11.68 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.50, 55.66, 111.96, 112.43, 114.61, 120.32, 121.92, 122.05, 127.42, 128.52, 146.93, 147.61, 149.80, 153.42, 161.19, 163.54 ppm; Anal. Calcd for C23H20N4O4: C, 66.34; H, 4.84; N, 13.45. Found: C, 66.02; H, 4.71; N, 13.21.
N'-(2,5-Dimethoxybenzylidene)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carbohydrazide (8m)
Pale brown powder; yield = 80%; mp 181–183 °C; IR (KBr) ṽ 3206, 3159, 3059, 3005, 2940, 1643, 1613, 1555, 1493, 1466 cm−1; 1H NMR (400 MHz; DMSO-d6) δH 3.76 (s, 3H), 3.82 (s, 3H), 3.85 (s, 3H), 7.00 (dd, 3J = 9.2 Hz, 4J = 2.8 Hz, 1H), 7.06 (d, 3J = 9.2 Hz, 1H), 7.14 (d, 3J = 8.8 Hz, 2H), 7.40 (s, 1H), 7.64 (d, 3J = 8.4 Hz, 1H), 7.80 (d, 3J = 8.4 Hz, 1H), 8.15 (d, 3J = 8.8 Hz, 2H), 8.18 (s, 1H), 8.82 (br., 1H), 11.88 (s, 1H), 13.14 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.42, 55.46, 56.26, 109.25, 113.42, 114.52, 117.48, 122.03, 123.17, 127.03, 128.42, 142.63, 152.27, 153.30, 153.43, 161.08, 163.54 ppm; Anal. Calcd for C24H22N4O4: C, 66.97; H, 5.15; N, 13.02. Found: C, 66.70; H, 5.44; N, 13.30.
2-(4-Methoxyphenyl)-N'-(3,4,5-trimethoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8n)
Pale brown powder; yield = 87%; mp 290–292 °C [66]; IR (KBr) ṽ 3198, 3163, 3079, 2944, 1636, 1613, 1578, 1559, 1504, 1454 cm−1; 1H NMR (400 MHz; DMSO-d6) δH 3.71 (s, 3H), 3.84 (s, 3H), 3.85 (s, 6H), 7.03 (s, 2H), 7.14 (d, 3J = 8.8 Hz, 2H), 7.66 (d, 3J = 8.4 Hz, 1H), 7.80 (d, 3J = 8.4 Hz, 1H), 8.15 (d, 3J = 8.8 Hz, 2H), 8.17 (s, 1H), 8.43 (s, 1H), 11.87 (s, 1H), 13.27 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.47, 56.00, 60.19, 104.33, 114.59, 121.84, 122.12, 127.23, 128.50, 130.09, 139.19, 147.41, 153.26, 153.42, 161.19, 163.76 ppm; Anal. Calcd for C25H24N4O5: C, 65.21; H, 5.25; N, 12.17. Found: C, 65.01; H, 5.55; N, 12.47.
2-(4-Chlorophenyl)-N'-(2-hydroxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8o)
Pale brown powder; yield = 70%; mp 287–289 °C; IR (KBr) ṽ 3206, 3063, 1667, 1624, 1555, 1478, 1447 cm−1; 1H NMR (400 MHz; DMSO-d6) δH 6.91–6.95 (m, 2H), 7.30 (t like, 3J = 7.2 Hz, 1H), 7.53 (d, 3J = 6.8 Hz, 1H), 7.66 (d, 3J = 8.4 Hz, 2H), 7.73 (br., 1H), 7.85 (d, 3J = 8.0 Hz, 1H), 8.22 (d, 3J = 8.4 Hz, 2H), 8.32 (br., 1H), 8.66 (s, 1H), 11.43 (s, 1H), 12.16 (s, 1H), 13.31 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 111.83, 116.45, 118.71, 119.33, 122.57, 126.82, 128.45, 128.55, 129.19, 129.71, 131.26, 135.08, 148.06, 152.42, 157.53, 163.23 ppm; Anal. Calcd for C21H15ClN4O2: C, 64.54; H, 3.87; N, 14.34. Found: C, 64.78; H, 3.57; N, 14.51.
2-(4-Chlorophenyl)-N'-(3-hydroxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8p)
Pale brown powder; yield = 75%; mp 313–315 °C; 1H NMR (400 MHz; DMSO-d6) δH 6.83 (d, 3J = 8.0 Hz, 1H), 7.10 (d, 3J = 7.2 Hz, 1H), 7.22 (s, 1H), 7.25 (t, 3J = 8.0 Hz, 1H), 7.66 (d, 3J = 8.4 Hz, 2H), 7.70 (d, 3J = 8.4 Hz, 1H), 7.82 (d, 3J = 8.4 Hz, 1H), 8.22 (d, 3J = 8.8 Hz, 2H), 8.40 (s, 1H), 9.62 (s, 1H), 11.83 (s, 1H), 11.94 (br., 1H), 13.28 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 112.62, 117.35, 118.78, 122.43, 127.56, 128.45, 128.58, 129.20, 129.90, 135.07, 135.78, 147.44, 152.27, 157.70, 163.50 ppm; Anal. Calcd for C21H15ClN4O2: C, 64.54; H, 3.87; N, 14.34. Found: C, 64.19; H, 3.95; N, 14.55.
2-(4-Chlorophenyl)-N'-(3-methoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8q)
Pale brown powder; yield = 78%; mp 171–173 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.81 (s, 3H), 7.01 (d, 3J = 7.6 Hz, 1H), 7.30 (s, 2H), 7.37 (t, 3J = 7.6 Hz, 1H), 7.66 (d, 3J = 8.8 Hz, 2H), 7.71 (d, 3J = 8.4 Hz, 1H), 7.84 (d, 3J = 8.0 Hz, 1H), 8.22 (d, 3J = 8.8 Hz, 3H), 8.47 (s, 1H), 11.91 (s, 1H), 13.78 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.17, 111.17, 116.13, 120.04, 122.47, 127.57, 128.40, 128.48, 129.20, 129.95, 135.15, 135.94, 147.29, 152.22, 159.57, 163.57 ppm; Anal. Calcd for C22H17ClN4O2: C, 65.27; H, 4.23; N, 13.84. Found: C, 65.49; H, 4.50; N, 13.63.
2-(4-Chlorophenyl)-N'-(2-hydroxy-3-methoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8r)
Pale brown powder; yield = 87%; mp 187–190 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.81 (s, 3H), 6.86 (t, 3J = 7.6 Hz, 1H), 7.03 (d, 3J = 7.6 Hz, 1H), 7.14 (d, 3J = 7.6 Hz, 1H), 7.69 (d, 3J = 8.8 Hz, 2H), 7.75 (d, 3J = 8.4 Hz, 1H), 7.90 (d, 3J = 8.4 Hz, 1H), 8.23 (d, 3J = 8.4 Hz, 2H), 8.27 (s, 1H), 8.69 (s, 1H), 11.14 (br., 1H), 12.19 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.82, 113.79, 114.61, 115.27, 118.92, 119.02, 121.01, 122.89, 127.45, 128.73, 129.30, 135.67, 138.17, 140.30, 147.24, 147.95, 148.10, 151.98, 162.98 ppm; Anal. Calcd for C22H17ClN4O3: C, 62.79; H, 4.07; N, 13.31. Found: C, 62.98; H, 4.32; N, 13.66.
2-(4-Chlorophenyl)-N'-(3-hydroxy-4-methoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8s)
Pale brown powder; yield = 87%; mp 298–300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.81 (s, 3H), 6.97 (d, 3J = 8.0 Hz, 1H), 7.06 (d, 3J = 7.6 Hz, 1H), 7.29 (s, 1H), 7.67 (t like, 3J = 8.8 Hz, 3H), 7.81 (d, 3J = 8.4 Hz, 1H), 8.22 (d, 3J = 8.8 Hz, 3H), 8.34 (s, 1H), 9.29 (s, 1H), 11.70 (s, 1H), 13.24 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.61, 111.92, 112.36, 120.25, 122.38, 127.37, 127.77, 128.47, 128.57, 129.22, 135.09, 146.91, 147.60, 149.75, 152.23, 163.36 ppm; Anal. Calcd for C22H17ClN4O3: C, 62.79; H, 4.07; N, 13.31. Found: C, 62.51; H, 4.19; N, 13.42.
2-(4-Chlorophenyl)-N'-(2,5-dimethoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8t)
Pale brown powder; yield = 91%; mp 190–192 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.75 (s, 3H), 3.82 (s, 3H), 6.99 (dd, 3J = 8.8 Hz, 4J = 2.8 Hz, 1H), 7.05 (d, 3J = 9.2 Hz, 1H), 7.41 (s, 1H), 7.66 (d, 3J = 8.4 Hz, 2H), 7.69 (d, 3J = 8.8 Hz, 1H), 7.85 (d, 3J = 8.4 Hz, 1H), 8.22 (d, 3J = 8.8 Hz, 2H), 8.24 (d, 4J = 2.0 Hz, 1H), 8.83 (s, 1H), 11.92 (s, 1H), 13.39 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 56.06, 56.24, 109.24, 113.39, 117.48, 122.44, 123.13, 127.52, 128.46, 129.19, 135.10, 142.73, 152.22, 152.27, 153.28, 163.40 ppm; Anal. Calcd for C23H19ClN4O3: C, 63.52; H, 4.40; N, 12.88. Found: C, 63.70; H, 4.72; N, 12.51.
2-(4-Chlorophenyl)-N'-(3,4,5-trimethoxybenzylidene)-1H-benzo[d]imidazole-5-carbohydrazide (8u)
Pale brown powder; yield = 86%; mp 300–302 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.71 (s, 3H), 3.84 (s, 6H), 7.04 (s, 2H), 7.73 (d, 3J = 8.4 Hz, 2H), 7.78 (d, 3J = 8.4 Hz, 1H), 7.90 (d, 3J = 8.4 Hz, 1H), 8.21–8.25 (m, 3H), 8.44 (s, 1H), 11.94 ppm (s, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.95, 60.12, 104.27, 114.51, 114.97, 123.31, 126.68, 128.61, 128.89, 129.41, 129.94, 136.06, 139.19, 147.64, 151.60, 153.20, 163.15 ppm; Anal. Calcd for C24H21ClN4O4: C, 62.00; H, 4.55; N, 12.05. Found C, 62.32; H, 4.40; N, 12.28.
Biology
Screening of the inhibitory activity of 2,5-disubstituted benzimidazoles 8a-u on VEGFR-2
The VEGFR-2 inhibitory activity of 8a-u was investigated at 10 µM using VEGFR-2 assay kit (BPS Biosciences—San Diego—CA—US) according to the protocol provided by the manufacturer [68] and the % of inhibition was determined (For further details see Additional file 1: section II: practical results) [67].
Screening of the inhibitory activity of 8u on diverse kinases
The inhibitory activity of 8u was investigated at different concentrations using VEGFR-2, FGFR-1 and BRAF assay kits (BPS Biosciences—San Diego—CA—US) according to the protocol provided by the manufacturer (For further details see Additional file 1: section II: practical results).
Cell cycle analysis assay
The diaryl benzimidazole 8u was applied at its GI50 concentration to MCF-7 cancer cells. After the cells were handled as previously described [69], the distribution of the cells at each stage of the cell cycle was analysed. (For further details see Additional file 1: section II: practical results).
Apoptosis assay
As previously reported, the Annexin V-FITC apoptosis detection kit (Abcam Inc., Cambridge, UK) in conjunction with two fluorescent channels flow cytometry was used to identify the populations of apoptosis and necrosis cells [69]. (For further details see Additional file 1: section II: practical results).
Supplementary Information
Additional file 1. Section I: Computational Studies: 1. Common 3D multi-kinase receptor-based pharmacophore model generation, 2. Pharmacophore model selection and validation, 3. Molecular docking simulation, 4. ADME properties prediction. Section II: Practical Results: 1. NMR Spectra of 2,5-disubstituted benzimidazole 8a-u, 2. Biochemical kinase assay procedure, 3. Dose response curves of 8u on VEGFR-2, FGFR-1 and BRAF, 4. Screening of cytotoxic activity against a panel of sixty human tumor cell lines, 5. Dose response curves of the 2,5-diaryl benzimidazole conjugates on NCI cancer cell lines, 6. Analysis of cell cycle distribution, 7. Apoptosis assay.
Acknowledgements
This paper is based upon work supported by Science, Technology & Innovation Funding Authority (STDF) under grant ID 37225. The authors are grateful to the National Cancer Institute (NCI), Bethesda, Maryland, USA for testing compounds 8a-u for their anticancer activity.
Abbreviations
- Å
Angstrom
- Acc
Acceptor feature
- Acc
Accuracy
- ADME
Absorption, distribution, metabolism, and excretion
- ATP
Adenosine triphosphate
- BBB
Blood brain barrier
- BRAF
B-rapidly accelerated fibrosarcoma
- CADD
Computer-aided drug design
- CNS
Central nervous system
- DMSO
Dimethyl sulfoxide
- DR
Discrimination ratio
- DUD-E
Directory of useful decoys-enhanced
- E
Enrichment
- EGFR
Epidermal Growth Factor Receptor
- FGF
Fibroblast Growth Factor
- FGFR-1
Fibroblast growth factor receptor-1
- FNt
Total false negatives
- FPt
Total false positives
- GIT
Gastrointestinal tract
- GI
Growth inhibition
- hFGFR
Human fibroblast growth factor receptor
- Hyd
Hydrophobic feature
- IC50
Half maximal inhibitory concentration
- LBDD
Ligand-based drug design
- MAPK
Mitogen activated protein kinases
- MCC
Mathew’s correlation coefficient
- MHz
Megahertz
- µM
Micromolar
- MOE
Molecular operating environment
- NCI
National Cancer Institute
- NMR
Nuclear magnetic resonance
- nd
Not detected
- PDB
Protein Data Bank
- P-gp
Protein glycoprotein
- Ph4
Pharmacophore
- PKIs
Protein kinase inhibitors
- ppm
Parts per million
- PI
Propidium iodide
- RAF
Rapidly accelerated fibrosarcoma
- RAS
Reticular activating system
- RMSD
Root mean square deviation
- SAR
Structure activity relationship
- SBDD
Structure-based drug design
- SD
Standard deviation
- Se
Sensitivity
- Sp
Specificity
- 3D
Three dimensional
- TLC
Thin layer chromatography
- TPt
Total true positives
- TNt
Total true negatives
- 2D
Two dimensional
- USA
United States of America
- VEGF
Vascular endothelial growth factor
- VEGFR-2
Vascular endothelial growth factor receptor-2
- Ya
Yield of actives
Author contributions
H. T. A. Participated in suggesting the research point, performing the organic synthesis and the structure elucidation of the target compounds, analysis of the biological results, writing, revising, and finalizing the manuscript; M. A. I. Participated in performing the computational studies of the manuscript; A. M. N. Participated in performing the biochemical screening of the target compounds on protein kinases; A. M. El K. Participated in suggesting the research point, performing the computational studies of the manuscript, analysing the obtained results, writing, revising, and finalizing the manuscript.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). Open access funding provided by Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Availability of data and materials
Supporting materials word file is available in the online version of this article.
Declarations
Ethics approval and consent to participate
Egyptian National Research Centre Medical Research Ethics Committee (Approval number 315062023).
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cheng HC, Qi RZ, Paudel H, Zhu HJ. Regulation and function of protein kinases and phosphatases. Enzyme Res. 2011;2011:794089. doi: 10.4061/2011/794089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ardito F, Giuliani M, Perrone D, Troiano G, Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review) Int J Mol Med. 2017;40(2):271–280. doi: 10.3892/ijmm.2017.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roskoski R., Jr A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1–23. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul MK, Mukhopadhyay AK. Tyrosine kinase—role and significance in cancer. Int J Med Sci. 2004;1(2):101–115. doi: 10.7150/ijms.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17(1):48. doi: 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cicenas J, Zalyte E, Bairoch A, Gaudet P. Kinases and cancer. Cancers (Basel). 2018;10(3):63. 10.3390/cancers10030063. [DOI] [PMC free article] [PubMed]
- 8.Parang K, Sun G. Protein kinase inhibitors drug discovery. In: Gad SX, editor. Drug discovery handbook. John Wiley & Sons, Ltd; 2005. p. 1191–1257.
- 9.Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J Clin Oncol. 2011;2(2):80–93. doi: 10.5306/wjco.v2.i2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garuti L, Roberti M, Bottegoni G. Multi-kinase inhibitors. Curr Med Chem. 2015;22(6):695–712. doi: 10.2174/0929867321666141216125528. [DOI] [PubMed] [Google Scholar]
- 11.Griffioen AW. Angiogenesis. In: Schwab M, editor. Encyclopedia of cancer. Springer, Berlin Heidelberg: Berlin, Heidelberg; 2011. pp. 185–186. [Google Scholar]
- 12.Liu Z-L, Chen H-H, Zheng L-L, Sun L-P, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8(1):198. doi: 10.1038/s41392-023-01460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal. 2016;10(4):347–354. doi: 10.1007/s12079-016-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 15.Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov. 2019;2:100009. doi: 10.1016/j.medidd.2019.100009. [DOI] [Google Scholar]
- 16.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 17.Muto J, Shirabe K, Sugimachi K, Maehara Y. Review of angiogenesis in hepatocellular carcinoma. Hepatol Res. 2015;45(1):1–9. doi: 10.1111/hepr.12310. [DOI] [PubMed] [Google Scholar]
- 18.Dai S, Zhou Z, Chen Z, Xu G, Chen Y. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells. 2019;8(6):614. 10.3390/cells8060614. [DOI] [PMC free article] [PubMed]
- 19.Krook MA, Reeser JW, Ernst G, Barker H, Wilberding M, Li G, Chen HZ, Roychowdhury S. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer. 2021;124(5):880–892. doi: 10.1038/s41416-020-01157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eldehna WM, El Kerdawy AM, Al-Ansary GH, Al-Rashood ST, Ali MM, Mahmoud AE. Type IIA–Type IIB protein tyrosine kinase inhibitors hybridization as an efficient approach for potent multikinase inhibitor development: design, synthesis, anti-proliferative activity, multikinase inhibitory activity and molecular modeling of novel indolinone-based ureides and amides. Eur J Med Chem. 2019;163:37–53. doi: 10.1016/j.ejmech.2018.11.061. [DOI] [PubMed] [Google Scholar]
- 21.Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, Rusu A, Irimie A, Atanasov AG, Slaby O, et al. A comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers (Basel) 2019 doi: 10.3390/cancers11101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rusconi P, Caiola E, Broggini M. RAS/RAF/MEK inhibitors in oncology. Curr Med Chem. 2012;19(8):1164–1176. doi: 10.2174/092986712799320510. [DOI] [PubMed] [Google Scholar]
- 23.Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G. Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta. 2007;1773(8):1196–1212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Nassan HB. Recent progress in the identification of BRAF inhibitors as anti-cancer agents. Eur J Med Chem. 2014;72:170–205. doi: 10.1016/j.ejmech.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 25.Rahman R, Ung PM, Schlessinger A. KinaMetrix: a web resource to investigate kinase conformations and inhibitor space. Nucleic Acids Res. 2019;47(D1):D361–D366. doi: 10.1093/nar/gky916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roskoski R., Jr Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res. 2019;144:19–50. doi: 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Kooistra AJ, Kanev GK, van Linden OP, Leurs R, de Esch IJ, de Graaf C. KLIFS: a structural kinase-ligand interaction database. Nucleic Acids Res. 2016;44(D1):D365–371. doi: 10.1093/nar/gkv1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamoto K, Ikemori-Kawada M, Jestel A, von Konig K, Funahashi Y, Matsushima T, Tsuruoka A, Inoue A, Matsui J. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med Chem Lett. 2015;6(1):89–94. doi: 10.1021/ml500394m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roskoski R., Jr Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48. doi: 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Roskoski R., Jr Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res. 2020;152:104609. doi: 10.1016/j.phrs.2019.104609. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS. Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery? ACS Chem Biol. 2014;9(6):1230–1241. doi: 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci U S A. 2012;109(45):18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 34.Abdel-Mohsen HT, Abdullaziz MA, Kerdawy AME, Ragab FAF, Flanagan KJ, Mahmoud AEE, Ali MM, Diwani HIE, Senge MO. Targeting receptor tyrosine kinase VEGFR-2 in hepatocellular cancer: rational design, synthesis and biological evaluation of 1,2-disubstituted benzimidazoles. Molecules. 2020;25(4):770. doi: 10.3390/molecules25040770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdullaziz MA, Abdel-Mohsen HT, El Kerdawy AM, Ragab FAF, Ali MM, Abu-Bakr SM, Girgis AS, El Diwani HI. Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors. Eur J Med Chem. 2017;136:315–329. doi: 10.1016/j.ejmech.2017.04.068. [DOI] [PubMed] [Google Scholar]
- 36.Ali IH, Abdel-Mohsen HT, Mounier MM, Abo-Elfadl MT, El Kerdawy AM, Ghannam IAY. Design, synthesis and anticancer activity of novel 2-arylbenzimidazole/2-thiopyrimidines and 2-thioquinazolin-4(3H)-ones conjugates as targeted RAF and VEGFR-2 kinases inhibitors. Bioorg Chem. 2022;126:105883. doi: 10.1016/j.bioorg.2022.105883. [DOI] [PubMed] [Google Scholar]
- 37.Abdel-Mohsen H, El Kerdawy A. Design, synthesis, molecular docking studies and in silico prediction of ADME properties of new 5-nitrobenzimidazole/thiopyrimidine hybrids as anti-angiogenic agents targeting hepatocellular carcinoma. Egypt J Chem. 2023;0(0):0–0. doi: 10.21608/ejchem.2023.212212.7998. [DOI] [Google Scholar]
- 38.Potashman MH, Bready J, Coxon A, DeMelfi TM, Jr, DiPietro L, Doerr N, Elbaum D, Estrada J, Gallant P, Germain J, et al. Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors. J Med Chem. 2007;50(18):4351–4373. doi: 10.1021/jm070034i. [DOI] [PubMed] [Google Scholar]
- 39.Williams TE, Subramanian S, Verhagen J, McBride CM, Costales A, Sung L, Antonios-McCrea W, McKenna M, Louie AK, Ramurthy S, et al. Discovery of RAF265: a potent mut-B-RAF inhibitor for the treatment of metastatic melanoma. ACS Med Chem Lett. 2015;6(9):961–965. doi: 10.1021/ml500526p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian S, Costales A, Williams TE, Levine B, McBride CM, Poon D, Amiri P, Renhowe PA, Shafer CM, Stuart D, et al. Design and synthesis of orally bioavailable benzimidazole reverse amides as pan RAF kinase inhibitors. ACS Med Chem Lett. 2014;5(9):989–992. doi: 10.1021/ml5002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porta C, Giglione P, Liguigli W, Paglino C. Dovitinib (CHIR258, TKI258): structure, development and preclinical and clinical activity. Future Oncol. 2015;11(1):39–50. doi: 10.2217/fon.14.208. [DOI] [PubMed] [Google Scholar]
- 42.Bunney TD, Wan S, Thiyagarajan N, Sutto L, Williams SV, Ashford P, Koss H, Knowles MA, Gervasio FL, Coveney PV, et al. The effect of mutations on drug sensitivity and kinase activity of fibroblast growth factor receptors: a combined experimental and theoretical study. EBioMedicine. 2015;2(3):194–204. doi: 10.1016/j.ebiom.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105(7):2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 44.Gagic Z, Ruzic D, Djokovic N, Djikic T, Nikolic K. In silico methods for design of kinase inhibitors as anticancer drugs. Front Chem. 2019;7:873. doi: 10.3389/fchem.2019.00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agafonov RV, Wilson C, Kern D. Evolution and intelligent design in drug development. Front Mol Biosci. 2015;2:27. doi: 10.3389/fmolb.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y-f, Fu L-w. Mechanisms of acquired resistance to tyrosine kinase inhibitors. Acta Pharm Sin B. 2011;1(4):197–207. doi: 10.1016/j.apsb.2011.10.007. [DOI] [Google Scholar]
- 48.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6(3):1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Protein Data Bank. https://www.rcsb.org/.
- 50.Eldehna WM, Maklad RM, Almahli H, Al-Warhi T, Elkaeed EB, Abourehab MAS, Abdel-Aziz HA, El Kerdawy AM. Identification of 3-(piperazinylmethyl)benzofuran derivatives as novel type II CDK2 inhibitors: design, synthesis, biological evaluation, and in silico insights. J Enzyme Inhib Med Chem. 2022;37(1):1227–1240. doi: 10.1080/14756366.2022.2062337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ezugwu JA, Okoro UC, Ezeokonkwo MA, Hariprasad KS, Rudrapal M, Ugwu DI, Gogoi N, Chetia D, Celik I, Ekoh OC. Design, synthesis, molecular docking, molecular dynamics and in vivo antimalarial activity of new dipeptide-sulfonamides. ChemistrySelect. 2022;7(5):e202103908. doi: 10.1002/slct.202103908. [DOI] [Google Scholar]
- 52.Othman IMM, Mahross MH, Gad-Elkareem MAM, Rudrapal M, Gogoi N, Chetia D, Aouadi K, Snoussi M, Kadri A. Toward a treatment of antibacterial and antifungal infections: design, synthesis and in vitro activity of novel arylhydrazothiazolylsulfonamides analogues and their insight of DFT, docking and molecular dynamic simulations. J Mol Struct. 2021;1243:130862. doi: 10.1016/j.molstruc.2021.130862. [DOI] [Google Scholar]
- 53.Rudrapal M, Eltayeb WA, Rakshit G, El-Arabey AA, Khan J, Aldosari SM, Alshehri B, Abdalla M. Dual synergistic inhibition of COX and LOX by potential chemicals from Indian daily spices investigated through detailed computational studies. Sci Rep. 2023;13(1):8656. doi: 10.1038/s41598-023-35161-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudrapal M, Gogoi N, Chetia D, Khan J, Banwas S, Alshehri B, Alaidarous MA, Laddha UD, Khairnar SJ, Walode SG. Repurposing of phytomedicine-derived bioactive compounds with promising anti-SARS-CoV-2 potential: molecular docking, MD simulation and drug-likeness/ADMET studies. Saudi J Biol Sci. 2022;29(4):2432–2446. doi: 10.1016/j.sjbs.2021.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudrapal M, Issahaku AR, Agoni C, Bendale AR, Nagar A, Soliman MES, Lokwani D. In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J Biomol Struct Dyn. 2022;40(20):10437–10453. doi: 10.1080/07391102.2021.1944909. [DOI] [PubMed] [Google Scholar]
- 56.Tucker JA, Klein T, Breed J, Breeze AL, Overman R, Phillips C, Norman RA. Structural insights into FGFR kinase isoform selectivity: diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure. 2014;22(12):1764–1774. doi: 10.1016/j.str.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 57.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daina A, Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.SwissADME. http://www.swissadme.ch/index.php.
- 60.Yoon YK, Ali MA, Wei AC, Choon TS, Ismail R. Synthesis and evaluation of antimycobacterial activity of new benzimidazole aminoesters. Eur J Med Chem. 2015;93:614–624. doi: 10.1016/j.ejmech.2013.06.025. [DOI] [PubMed] [Google Scholar]
- 61.Acar Cevik U, Saglik BN, Osmaniye D, Levent S, Kaya Cavusoglu B, Karaduman AB, Atlid O, Atli Eklioglu O, Kaplancikli ZA. Synthesis, anticancer evaluation and molecular docking studies of new benzimidazole- 1,3,4-oxadiazole derivatives as human topoisomerase types I poison. J Enzyme Inhib Med Chem. 2020;35(1):1657–1673. doi: 10.1080/14756366.2020.1806831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravi S, Singal AK. Regorafenib: an evidence-based review of its potential in patients with advanced liver cancer. Core Evid. 2014;9:81–87. doi: 10.2147/CE.S48626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DUD-E. http://dude.docking.org/targets.
- 64.DEKOIS 2.0. www.dekois.com.
- 65.El Kerdawy AM, Osman AA, Zaater MA. Receptor-based pharmacophore modeling, virtual screening, and molecular docking studies for the discovery of novel GSK-3beta inhibitors. J Mol Model. 2019;25(6):171. doi: 10.1007/s00894-019-4032-5. [DOI] [PubMed] [Google Scholar]
- 66.Azizian H, Pedrood K, Moazzam A, Valizadeh Y, Khavaninzadeh K, Zamani A, Mohammadi-Khanaposhtani M, Mojtabavi S, Faramarzi MA, Hosseini S, et al. Docking study, molecular dynamic, synthesis, anti-alpha-glucosidase assessment, and ADMET prediction of new benzimidazole-Schiff base derivatives. Sci Rep. 2022;12(1):14870. doi: 10.1038/s41598-022-18896-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abdel-Mohsen HT, Girgis AS, Mahmoud AEE, Ali MM, El Diwani HI. New 2,4-disubstituted-2-thiopyrimidines as VEGFR-2 inhibitors: design, synthesis, and biological evaluation. Arch Pharm (Weinheim) 2019;352(11):e1900089. doi: 10.1002/ardp.201900089. [DOI] [PubMed] [Google Scholar]
- 68.Abd El-Karim SS, Syam YM, El Kerdawy AM, Abdel-Mohsen HT, Rational design and synthesis of novel quinazolinone Nacetohydrazides as type II multi-kinase inhibitors and potential anticancer agents. Bioorg Chem 2024;142:106920. 10.1016/j.bioorg.2023.106920. [DOI] [PubMed]
- 69.Abdel-Mohsen HT, El Kerdawy AM, Omar MA, Petreni A, Allam RM, El Diwani HI, Supuran CT. Application of the dual-tail approach for the design and synthesis of novel thiopyrimidine-benzenesulfonamide hybrids as selective carbonic anhydrase inhibitors. Eur J Med Chem. 2022;228:114004. doi: 10.1016/j.ejmech.2021.114004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Section I: Computational Studies: 1. Common 3D multi-kinase receptor-based pharmacophore model generation, 2. Pharmacophore model selection and validation, 3. Molecular docking simulation, 4. ADME properties prediction. Section II: Practical Results: 1. NMR Spectra of 2,5-disubstituted benzimidazole 8a-u, 2. Biochemical kinase assay procedure, 3. Dose response curves of 8u on VEGFR-2, FGFR-1 and BRAF, 4. Screening of cytotoxic activity against a panel of sixty human tumor cell lines, 5. Dose response curves of the 2,5-diaryl benzimidazole conjugates on NCI cancer cell lines, 6. Analysis of cell cycle distribution, 7. Apoptosis assay.
Data Availability Statement
Supporting materials word file is available in the online version of this article.