

Abstract
Withaferin A (WFA), a steroidal lactone, negatively regulates breast cancer growth however, its mechanisms of action remain largely elusive. We found that WFA blocks autophagy flux and lysosomal proteolytic activity in breast cancer cells. WFA increases accumulation of autophagosomes, LC3B-II conversion, expression of autophagy-related proteins and autophagosome/lysosome fusion. Autolysosomes display the characteristics of acidic compartments in WFA-treated cells; however, the protein degradation activity of lysosomes is inhibited. Blockade of autophagic flux reduces the recycling of cellular fuels leading to insufficient substrates for tricarboxylic acid (TCA) cycle and impaired oxidative phosphorylation. WFA decreases expression and phosphorylation of lactate dehydrogenase, the key enzyme that catalyzes pyruvate-to-lactate conversion, reduces adenosine triphosphate levels and increases AMP-activated protein kinase (AMPK) activation. AMPK inhibition abrogates while AMPK activation potentiates WFA’s effect. WFA and 2-deoxy-d-glucose combination elicits synergistic inhibition of breast cancer cells. Genetic knockout of BECN1 and ATG7 fails to rescue cells from WFA treatment; in contrast, addition of methyl pyruvate to supplement TCA cycle protects WFA-treated cells. Together, these results implicate that WFA is a potent lysosomal inhibitor; energetic impairment is required for WFA-induced apoptosis and growth inhibition and combining WFA and 2-DG is a promising therapeutic strategy for breast cancer.
Study shows that WFA mediated blockade of autophagic flux involves inhibition of lysosomal proteolytic activity reducing the recycling of cellular fuels. WFA induces apoptosis and inhibits growth via energetic impairment and WFA and 2-deoxyglucose combination elicits synergistic inhibition.
Introduction
Despite various improved diagnostic techniques and the development of targeted therapeutic approaches, breast cancer is still the second leading cause of cancer-related mortality in women. Ongoing quests to develop more effective, nontoxic and nonendocrine strategies to target breast cancer have now focused on bioactive compounds from natural products and a review of epidemiologic, in vitro and in vivo studies provides robust support for the suitability and efficacy of several bioactive compounds for future clinical development. The root extract of Withania somnifera, exhibiting broad-spectrum pharmacological efficacy, has been successfully used in traditional alternative medicine (1–3) and its therapeutic benefits are attributed to 14 bioactive compounds called withanolides (4,5). Among these withanolides, withaferin A (WFA) is the most abundant and therapeutically effective withanolides with anti-cancer activities (6–8). WFA protects against acetaminophen-induced hepatic toxicity (9) and bromobenzene (an environmental toxin)-induced liver and kidney damage (10). Some clinical trials have investigated the efficacy of W.somnifera extracts (Drug: Sensoril) for bipolar disorder, schizophrenia and generalized anxiety disorder (ClinicalTrials.gov Identifier: NCT00761761, NCT01311180, NCT01793935) and Schlicht and group reported that W.somnifera extracts provided significant benefits in patients with schizophrenia (11). Previous studies from our lab (12) and others (13) have shown that WFA administration decreases breast cancer growth in vitro and in vivo. On one hand WFA has been shown to induce apoptosis (14), effectively inhibit oncogenic transcription factor Stat3 (15), and activate death receptor 5 (DR5) (12) leading to effective inhibition of breast tumors but on the other hand it has also been reported to activate Notch signaling which is involved in growth and progression of breast cancer (16). Thus, developing a complete functional understanding of therapeutic impact of WFA on breast cancer is required to establish surrogate biomarkers and further clinical development of WFA.
Cells, in normal physiological conditions, undergo macroautophagy (referred to as autophagy hereafter), a self-degradation process where cytosolic components and organelles are recycled using a vacuolar, lysosomal degradation pathway to maintain cellular homeostasis and provide energy in stressed state (17,18). Basal autophagy helps prevent accumulation of damaged proteins and organelles, lower ER stress and reduce the production of reactive oxygen species. Recent studies, using multiple model systems, have shown that cancer cells utilize autophagic process to respond to various environmental stimuli and evade cancer therapy. Autophagy is a complex process involving concerted action of various autophagy-related (ATG) proteins forming a double-membrane bound vacuole known as autophagosome. Following maturation and fusion processes (fusing with endocytic compartments like early and late endosomes), autophagosomes fuse with the lysosomal compartments to form autolysosomes where cargo is degraded to produce energy. Although autophagy is a multistep process, increased number of autophagosome in cytosol is generally used to evaluate autophagic induction but accumulation of autophagosomes can result from either increased formation or decreased turnover of autophagosome. Therefore, it is imperative to examine the entire process of autophagy, known as autophagic flux, to comprehend the involvement and functional impact of autophagy pertaining to anti-cancer regimens.
In this study, we examined the involvement of autophagy in WFA-mediated growth inhibition and apoptosis induction in breast cancer cells. We report that WFA inhibits the autophagic flux in breast cancer cells by abrogating the protein degradation activity of lysosomes without altering the integrity of the other steps of autophagic process such as autophagosome accumulation, LC3B-II conversion and fusion of autophagosome/lysosomes. Blocking autophagic flux can reduce the recycling of cellular fuels resulting in insufficient substrates for tricarboxylic acid (TCA) cycle and impaired oxidative phosphorylation. Indeed, WFA treatment decreases adenosine triphosphate (ATP) levels, activates AMP-activated protein kinase (AMPK) and decreases the expression/phosphorylation of lactate dehydrogenase (LDHA) impacting pyruvate-to-lactate conversion. Blunting autophagosome formation, blocking autophagosome–lysosome fusion or genetic knockout of BECN1 and ATG7 fail to rescue cells from WFA treatment but addition of methyl pyruvate to supplement TCA cycle protects WFA-treated cells. Also, AMPK inhibition interferes with WFA efficacy while AMPK activation potentiates it. In fact, WFA and 2-DG show synergistic inhibition of breast cancer cells indicating that a combined regimen of WFA and 2-DG can be a promising therapeutic strategy. Together, these results implicate that WFA is a potent lysosomal inhibitor and impedes growth and induces apoptosis in breast cancer cells via energetic impairment.
Materials and methods
Ethics statement
Xenograft tumor samples/sections are from our previously published study (12). All animal studies were in accordance with the guidelines of Johns Hopkins University IACUC.
Cell culture and reagents
Breast cancer cell lines MCF7, MDA-MB-231, MDA-MB-468, T47D were obtained from the American Type Culture Collection (HTB-22, HTB-26, HTB-132, HTB-133). SUM149 and SUM159 were procured from Asterand Bioscience (Detroit, MI). These cell lines were authenticated in 2014 and each cell line was of human origin and no inter species contamination was found. Cell lines were maintained according to supplier’s recommendation. Frozen stocks of authenticated cell lines were used. WFA was purchased from Calbiochem. Our preliminary dose-dependent and time-dependent studies showed that 5 µM WFA is an effective dose in breast cancer cells therefore we treated breast cancer cells with 5 µM WFA. Antibodies for MAP1LC3B/LC3B (3868), ATG5 (12994), ATG7 (8558), BECN1 (3495), STK11/LKB1 (3050), cleaved PARP1 (5625), PARP1 (9532), PRKAA1 (2532), phospho-PRKAA1 (Thr172) (2535), LDHA (3582) and phospho-LDHA (Tyr10) (8176) and SQSTM1/p62 (5114) were purchased from Cell Signaling Technology. ACTB/β-actin (A5441) antibody, 3-methyladenine (3-MA; M9281), chloroquine (CQ; C6628), rapamycin (R0395), 2-deoxy-d-glucose (2-DG; D6134), methyl pyruvate (371173) were purchased from Sigma–Aldrich. LysoTracker Red DND-99 (L7528) was purchased from Invitrogen. Bafilomycin (11038) was purchased from Cayman Chemical. DQ™ Green BSA assay (D12050), Earle’s balanced salt solution (EBSS; 14155-063), Alexa Fluor 488 (A-11008) and Alexa Fluor 555 (A-21428) were purchased from Thermo Fisher Scientific.
Clonogenicity and cell viability assay
Anchorage-dependent clonogenicity assay (19) was conducted in the presence of WFA. Colonies containing >50 normal appearing cells were counted and pictures were taken using a digital camera. Viability was examined using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Thermo Fisher Scientific, M6494) following the manufacturer’s instructions.
Transmission electron microscopy, immunofluorescence and confocal imaging
Breast cancer cells were treated with WFA or vehicle control for 3 or 6 h, specimens were embedded in EPON, sectioned, stained and examined with an H7600 transmission electron microscope (Hitachi, Tokyo, Japan). Cells were fixed and immunofluorescently stained (20) followed by imaging using a Zeiss LSM510 Meta (Zeiss, Dublin, CA) laser scanning confocal system configured to a Zeiss Axioplan 2 upright microscope (Zeiss).
Immunoblotting
Cells were scraped in modified RIPA buffer to prepare whole cell lysates and subjected to immunoblotting (12,21). For RNA isolation and RT-PCR (22,23), total cellular RNA was extracted using the TRIzol Reagent (ThermoFisher Scientific, 15596026) and RT-PCR was performed using specific sense and antisense PCR primers.
Breast tumorigenesis assay
MDA-MB-231 cells xenografts from our previously published study (12) were used to determine the effect of WFA on autophagy. All animal studies were in accordance with the guidelines of Johns Hopkins University IACUC.
Apoptosis, acridine orange staining
Cells were cultured on chamber slides, treated with WFA followed by Hoechst 33342 staining (Thermo Fisher Scientific, H1399) according to the manufacturer’s protocol. Breast cancer cells were cultured on six-well plates and treated with vehicle or 2.5 µM or 5 µM WFA for 24 h followed by incubation with acridine orange (Sigma–Aldrich, A9231) and were examined with confocal microscope (24).
ATG7 and BECN1 knockout with CRISPR/Cas9
Digested and purified LentiCRISPRv2 plasmid [lentiCRISPRv2 was a gift from Feng Zhang (Addgene, 52961)] was incubated with phosphorylated, annealed oligos for ATG7 and BECN1 in a ligation reaction, transformed into Stbl3 bacteria (ThermoFisher Scientific, C7373-03). lentiCRISPR with inserted sequences were cotransfected into HEK293T cells with packinging plasmids. MCF7 cells were transfected twice and selected for a week. MCF7 cells were examined for the ATG7 and BECN1 knockout using immunoblot analyses.
ATP assay
Cells were treated with vehicle or WFA for 1, 3, 6, 12 or 24 h, lysed in cell lysis solution (Perkin Elmer, 6016739), substrate solution was added and ATP levels were detected by measuring luminescence. ATP levels were measured using the ATPlite luminescence assay system (Perkin Elmer, 6016739) according to the manufacturer’s instructions.
Survival analysis
We assessed the correlation between AMPK and LDHA expression and survival using an independent multicentre gene expression database established as described previously (PMID: 23836010). All samples were measured using Affymetrix HGU133A or HUG133 plus 2 gene chips. We performed Cox proportional hazards regression analysis to correlate expression and relapse-free survival and Kaplan–Meier plots were drawn for visualization.
Statistical analysis
All experiments were performed multiple times using independent biological replicates. Statistical analysis was performed using Microsoft Excel software. Significant differences were analyzed using the Student’s t-test and two-tailed distribution. Results were considered to be statistically significant if P <0.05. Results were expressed as mean ± SE between triplicate experiments performed thrice.
Results
WFA inhibits breast cancer growth and increases intracellular autophagosomes and autophagy markers
WFA exerted a significant decrease in anchorage-dependent clonogenicity and cell viability in a concentration dependent manner and more effective inhibition was observed with 5–12.5 μM WFA treatment. Breast cancer is a heterogeneous disease and gene expression profiling has revealed five molecular subtypes (25) of clinical implications therefore WFA-mediated growth inhibition was corroborated in breast cancer cells spanning multiple subtypes including luminal A (MCF7, T47D), luminal B (BT474), basal (MDA-MB-468, SUM149), claudin-low (MDA-MB-231, SUM159), HER2 (SKBR3) (Supplementary Figures S1A and B, S2A–D, S3A–D, S4A–C, available at Carcinogenesis Online). WFA-mediated inhibition of breast cancer growth was associated with increased apoptotic cell death (Supplementary Figure S5, available at Carcinogenesis Online). Next, we examined the ability of WFA to induce autophagy in breast cancer cells. Using transmission electron microscopy (TEM), we observed ultrastructural changes in cells treated with 5 μM WFA within 3 and 6 h post-treatment. WFA-treated cells acquired significantly higher number (3- to 10-fold increase) of autophagic vacuoles in comparison to control cells (Figure 1A). Formation of autophagosomes is accompanied with a marked redistribution of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 α) from the cytosol to autophagosomes. Confocal microscopy detected increased LC3B puncta formation in WFA-treated MCF7, MDA-MB-231, MDA-MB-468, SUM149 and SUM159 cells while a diffuse, cytoplasmic green fluorescence was observed in control cells (Figure 1B). LC3B-I to LC3B-II conversion is an important sign of autophagy activity (26) and WFA treatment resulted in increased time-dependent accumulation of LC3B-II (Figure 1C). These in vitro observations were corroborated using MDA-MB-231 xenograft-tumors and immunoblotting of tumor lysates from WFA-treated mice showed increased expression of LC3B-II in comparison to tumors from vehicle-treated mice (Figure 1D). Immunohistochemical analysis also showed increased expression of LC3B-II in breast tumors from WFA-treated mice while tumors from vehicle-treated mice exhibited low LC3B-II staining (Figure 1F). In addition to augmented LC3B-conversion, mRNA level and protein expression of other autophagy-related (ATG) markers, such as ATG5, ATG7 and BECN1 were also increased upon WFA treatment. Some ATG proteins (such as ATG5) showed a sustained increase in expression while others (e.g. Beclin) showed a transient increase in expression in response to WFA. Of note, all ATG proteins exhibited increased expression upon 6-h WFA treatment (Figure 1E and G). Augmented LC3B-II puncta and expression levels correlate either with an increase in autophagosomes synthesis or with a decrease in autophagosomes turnover resulting from delayed trafficking to the lysosomes and decreased fusion with lysosomes (27). To discriminate between these two scenarios in WFA-treated cells, we utilized a plasmid encoding membrane-localized red fluorescent protein (mRFP)-EGFP-LC3B (tandem fluorescent-tagged LC3B [tfLC3B]) (28). This probe results in both green and red fluorescence and is used to identify autophagosomes (green flurescent protein [GFP]-positive and red flurescent protein [RFP]-positive; merged as yellow) and autolysosomes (GFP-negative and RFP-positive; merged as red) because enhanced green fluorescent protein (EGFP) fluorescence is quenched in acidic compartments whereas mRFP is more stable in low pH environment. Breast cancer cells were transfected with tfLC3B, treated with WFA and distribution of mRFP-EGFP-LC3B fusion protein was analyzed. MDA-MB-231 and MCF7 cells exhibited an increase in both red and yellow punctate fluorescence upon WFA treatment (Figure 1H) indicating the WFA does not impair the clearance of autophagosomes.
Figure 1.

WFA induces autophagosome accumulation, LC3B conversion and other autophagy related proteins. (A) MCF7 and MDA-MB-231 cells were treated with 5 µM WFA for 3 and 6 h as indicated and visualized under an electron microscope. Scale bars: 2 µm. Pictures are shown with ~×7400 magnifications. Double-membrane autophagosomes were counted in randomly selected ~100 cells. The number of autophagosomes was counted from randomly selected fields (shown in bar graphs). *P < 0.005, compared with vehicle-treated controls (C). (B) Breast cancer cells were treated with 5 µM WFA and subjected to immunocytochemistry using an LC3B antibody. Scale bars: 20 µm. Representative immunofluorescence images are shown. Bar diagram shows number of LC3B puncta per cell. *P < 0.001, compared with vehicle-treated controls (C). (C) Breast cancer cells were treated with 5 µM WFA, and total cell lysates were immunoblotted for LC3B (14, 16 kDa) expression. ACTB (45 kDa) was used as a loading control. (D) Total protein lysates from MDA-MB-231-derived tumors from vehicle-treated and WFA-treated mice were examined for the expression of LC3B (14, 16 kDa). ACTB (45 kDa) was used as a loading control. (E) Immunoblot analysis of ATG5 (55 kDa), ATG7 (78 kDa), and BECN1 (60 kDa) in breast cancer cells treated with 5 µM WFA as indicated. ACTB (45 kDa) was used as a loading control. (F) Tumors from vehicle and WFA-treated mice were subjected to immunohistochemical (IHC) analysis using LC3B antibodies. Scale bars: 100 µm. Bar diagrams show quantification of IHC analysis. *P< 0.01, compared with control. (G) Breast cancer cells were treated with 5 µM WFA, and total RNA were examined for the expression of ATG5, ATG7 and BECN1. ACTB was used as a loading control. (H) Schematic diagram of the tfLC3 plasmid (upper panel). MDA-MB-231 and MCF7 cells were transfected with tfLC3 followed by treatment with 5 µM WFA or 200 nM rapamycin or Earle’s balanced salt solution. Earle’s balanced salt solution and rapamycin were used as positive controls for autophagic induction. Representative fluorescent images are shown. Scale bar: 20 μm.
WFA augments the fusion of autophagosomes and lysosomes
Late stage of autophagy, the fusion of autophagosomes with lysosomes, can be investigated by staining for GFP-LC3B (decorating autophagosomes) along with LysoTracker Red (an acidic pH marker for lysosomes). Confocal microscopy of WFA-treated cells showed increased overlap of GFP-LC3B and LysoTracker Red signals (increased yellow puncta) in comparison to control cells indicating an increased fusion of autophagosomes and lysosomes (Figure 2A). RAB7, a member of small GTPases, is one of the important molecules involved in the maturation of autolysosomes in both the endolysosomal and autophagic pathways (29,30). MCF7 and MDA-MB-231 cells were cotransfected with GFP-LC3B and RFP-Rab7 plasmids and autophagosomes (GFP positive/RFP negative; green puncta), autolysosomes (RFP positive/GFP negative; red puncta) and autolysosomes fusion (GFP positive/RFP positive; yellow puncta) were examined. Confocal analyses showed that withaferin treatment resulted in a significant overlap of LC3B and Rab7 signals compared with control cells indicating that WFA treatment led to fusion of autophagosomes and lysosomes (Figure 2B). Next, we evaluated autolysosomal pH using acridine orange (AO) staining as autolysosomes are characterized by acidic pH. Upon WFA treatment, nucleus and cytoplasm exhibited green fluorescence whereas the acidic compartments showed orange and red fluorescence. We observed that autolysosomes have the characteristics of acidic compartments in WFA-treated cells (Figure 2C and D). Together, WFA increases the fusion of autophagosomes and lysosomes to form autolysosomes.
Figure 2.

WFA induces the fusion of autophagosomes and lysosomes. (A) MCF7 and MDA-MB-231 cells were transfected with GFP-LC3B followed by treatment with 5 µM WFA or Earle’s balanced salt solution (2 h) and staining with LysoTracker Red. Cells were fixed and subjected to confocal microscopy. Representative fluorescent images are shown. Scale bar: 10 μm. Earle’s balanced salt solution was used as positive control for autophagic induction. (B) MCF7 and MDA-MB-231 cells were cotransfected with GFP-LC3B and RFP-RAB7 followed by treatment with 5 µM WFA or Earle’s balanced salt solution (2 h). Fixed cells were subjected to confocal microscopy. Representative fluorescent images are shown. Scale bars: 10 μM. (C) MCF7 and MDA-MB-231 cells were treated with 2.5 and 5 µM WFA followed by acridine orange staining. Representative images of MCF7 cells are shown. Scale bar: 10 μm. (D) Bar graph shows acridine orange punta per cell in MCF7 and MDA-MB-231 cells. *P< 0.01, compared with control.
Impairment of autophagic process fails to protect cells against WFA induced growth inhibition and apoptosis induction
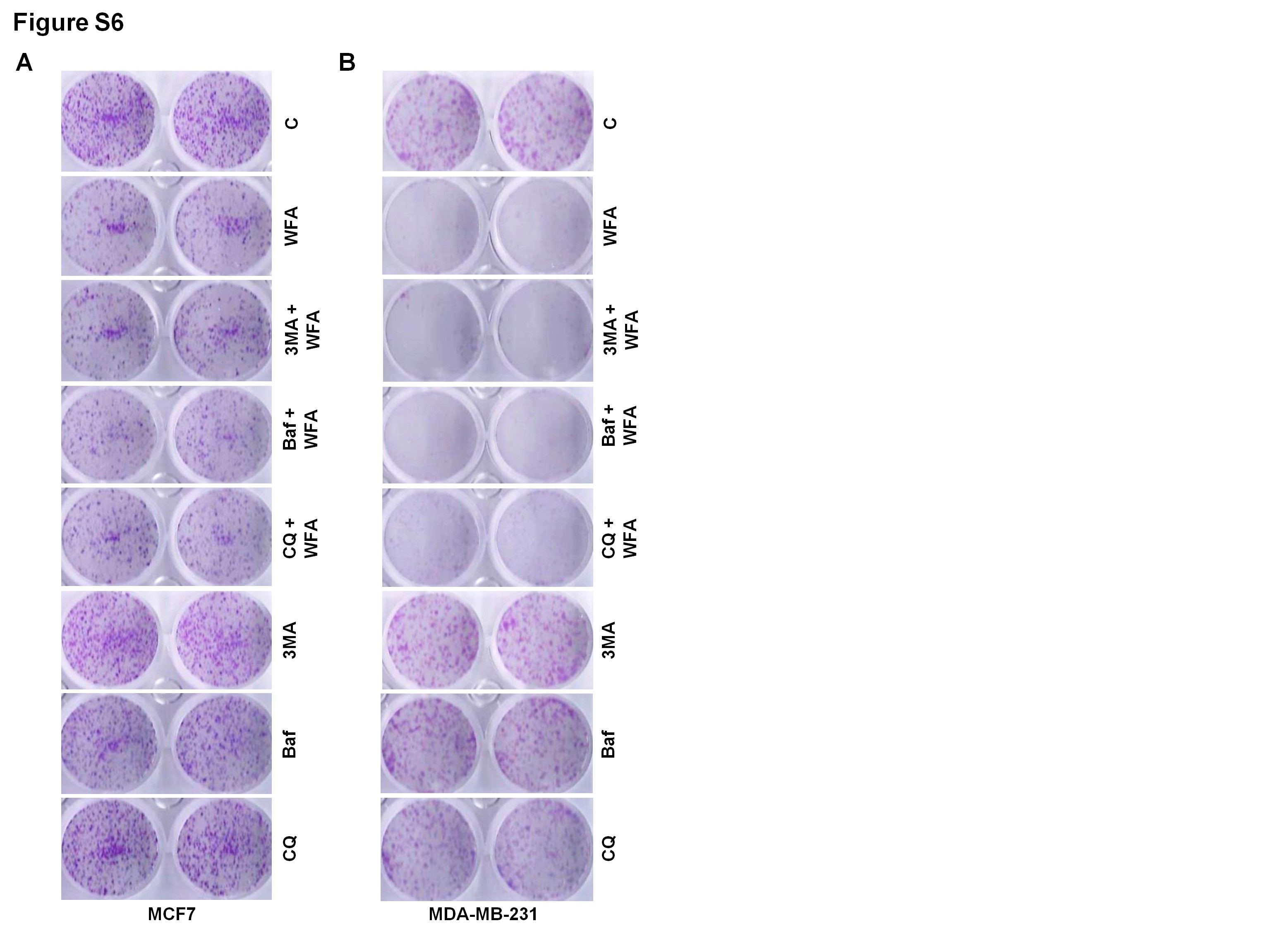
Having observed WFA-mediated increased autophagosome turnover and autolysosome formation, we evaluated the role of autophagy in WFA-induced apoptotic induction and growth inhibition. We blocked autophagy in WFA-treated cells using 3-MA, a phosphatidylinositol 3-kinase (PtdIns3K/Vps34) inhibitor that impedes autophagosome formation (31) or Baf, a specific vacuolar type H+-ATPase inhibitor that prevents autophagosome–lysosome fusion (32) or CQ, a weak base that elevates lysosomal pH. Interestingly, WFA-mediated inhibition of growth of breast cancer cells remained unaltered upon combined treatment with 3-MA, CQ or Baf (Figure 3A, Supplementary Figure S6, available at Carcinogenesis Online). Also, the magnitude of apoptotic cell death observed upon WFA treatment was unaffected when breast cancer cells were co-treated with autophagy inhibitors 3-MA, CQ or Baf (Figure 3B). Immunoblot analysis of MCF7 and MDA-MB-231 cells treated with combination of WFA and 3-MA showed equivalent levels of cleavage of PARP1 in comparison to WFA treatment alone (Figure 3C). Concomitant actions of many ATG proteins are required in autophagy (18). As a genetic intervention, we knocked out (KO) BECN1 and ATG7 in MCF7 cells using CRISPR/Cas9 technology (Figure 3D). Treatment with WFA-induced PARP1 cleavage in vector control cells as well as in BECN1-KO and ATG7-KO cells (Figure 3E). Moreover, WFA-mediated inhibition of clonogenic growth of breast cancer cells was not impacted upon BECN1 and ATG7 knockout as BECN1-KO and ATG7-KO MCF7 cells showed similar growth inhibition upon WFA treatment as vector-control MCF7 cells (Figure 3F). BECN1-KO and ATG7-KO MCF7 cells also showed significant decrease in cell viability upon WFA treatment analogous to vector control MCF7 cells (Figure 3G). Collectively, these data provide evidence that WFA induced cell death is not an ‘autophagic cell death’.
Figure 3.

Inhibition of autophagy does not impact WFA-mediated inhibition of cell survival and apoptosis induction in breast cancer cells. (A) MCF7 and MDA-MB-231 cells were treated with 5 µM WFA alone or in combination with 200 nM Baf, 25 μM CQ and 4 mM 3-MA as indicated and subjected to MTT assay. *P< 0.001, compared with control. (B) Breast cancer cells were treated with 5 µM WFA alone or in combination with 25 μM CQ and 4 mM 3-MA as indicated and subjected to DNA fragmentation assay. *P< 0.01, compared with control. (C) MCF7 and MDA-MB-231 cells were treated with 5 µM WFA and 4 mM 3-MA and total cell lysates were immunoblotted for cleaved PARP1 (cPARP1, 89 kDa), PARP1 (116 kDa) and ACTB (45 kDa) as indicated. (D) CRISPR/Cas9 was used to knockout BECN1 and ATG7 in MCF7 cells and total cell lysates were immunoblotted for BECN1 (60 kDa) and ATG7 (78 kDa). ACTB (45 kDa) was used as loading control. (E) Vector-control, BECN1-KO and ATG7-KO MCF7 cells were treated with 5 µM WFA for indicated time intervals and total cell lysates were immunoblotted for cleaved-PARP1 (89 kDa) and total-PARP1 (116 kDa) expression levels. ACTB (45 kDa) was used as loading control. (F) Clonogenicity of control, BECN1-KO and ATG7-KO MCF7 cells treated with 5 µM WFA as indicated. (G) Cell viability of control, BECN1-KO and ATG7-KO MCF7 cells treated with 5 µM WFA was examined using MTT assay. Representative pictures of cells are shown.
WFA treatment inhibits lysosomal activity
A key step in autophagic process is the degradation of cargo in the autolysosomes. We questioned whether WFA treatment affected lysosomal activity in breast cancer cells, therefore, lysosomal proteolytic activity was assayed using DQ-BSA (a derivative of BSA whose green fluorescence is quenched except when it is cleaved by proteolytic enzymes) assay. Earle’s balanced salt solution starvation and rapamycin treatment exhibited stimulated lysosomal activity as evident by dequenching of DQ-BSA in lysosomes. WFA treated cells showed decreased DQ-BSA emission (green puncta) in lysosomes (red puncta) as indicated by colocalization with LysoTracker Red (merge showing increased red puncta) (Figure 4A and B). Reduction in SQSTM1/p62 level is linked with increased autophagic activity as SQSTM1/p62 interacts with ubiquitin protein and LC3B-II and gets degraded in the autolysosomes, therefore, we investigated the levels of SQSTM1/p62 in breast cancer cells treated with WFA. WFA-treated breast cancer cells and MDA-MB-231 xenografts showed increased SQSTM1/p62 levels (Figure 4C and D). Immunohistochemical analysis of MDA-MB-231 xenograft tumors from WFA-treated mice showed increased expression of SQSTM1/p62 in comparison to tumors from vehicle-treated mice (Figure 4E). Cathepsin (CTS) proteases are implicated as important lysosomal hydrolases that degrade the sequestered components. Using lysosomes isolated from WFA-treated cells, we found that WFA impaired the maturation of Cathepsin D (CTSD) (Figure 4F). Inhibition of CTSD maturation also indicated reduced CTSB and CTSL activity as they are essential for the cleavage of CTSD. WFA treated cells show reduced levels of CTSD (Figure 4G). Together, these results present that while WFA does not inhibit the fusion of autophagosomes and lysosomes, it impairs autophagic flux by inhibiting lysosomal activity in breast cancer cells.
Figure 4.

WFA inhibits protein degradation in lysosomes. (A, B) MCF7 and MDA-MB-231 cells were incubated with 10 μg/ml DQ-BSA for 2 h followed by washing with medium and treatment with 5 µM WFA or 200 nM rapamycin or starving in Earle’s balanced salt solution. The cells were fixed and stained with LysoTracker Red followed by confocal microscopy. Earle’s balanced salt solution and rapamycin were used as positive controls for autophagic induction. Representative fluorescent images are shown. Scale bars: 10 μM. (C) Breast cancer cells were treated with 5 µM WFA for indicated time intervals and total lysates were immunoblotted for SQSTM1 (62 kDa) expression levels. ACTB (45 kDa) was used as loading control. (D) Total protein lysates from MDA-MB-231-derived xenograft tumors from vehicle-treated and WFA-treated mice were examined for the expression of SQSTM1 (62 kDa). ACTB (45 kDa) was used as a loading control. (E) Tumors from vehicle and WFA-treated mice were subjected to immunohistochemical (IHC) analysis using SQSTM1 antibodies. Scale bars: 100 µm. Bar diagrams show quantification of IHC analysis. *P < 0.01, compared with control. (F) MDA-MB-231 and SUM159 breast cancer cells were treated with 5 µM WFA followed by lysosome extraction. Total lysates and lysosomes were immunoblotted for cathepsin D (CTSD). LAMP2 (130 kDa) was used as control. (G) Immunocytochemical analysis of cathepsin D in breast cancer cells treated with WFA.
WFA induces growth inhibition and apoptotic induction by energy depletion in breast cancer cells
It is important to note that blockade in lysosomal proteolytic activity reduces the recycling of cellular fuels, that can result in insufficient energy leading to growth inhibition and cell death (33). To examine whether WFA affects cellular energy levels in breast cancer cells, we measured the intracellular production of ATP levels in breast cancer cells treated with WFA and found that ATP levels were 70–80% reduced within 24 h post-treatment (Figure 5A, Supplementary Figure S3E, available at Carcinogenesis Online). AMPK, sensor of cellular energy status, gets activated in response to the modulation of AMP/ATP ratio to maintain cellular energy homeostasis. Indeed, Thr172 phosphorylation of the PRKAA1, catalytic subunit of AMPK, was increased in WFA-treated MCF7 and MDA-MB-231 breast cancer cells (Figure 5B) and MDA-MB-231 xenograft tumors from WFA-treated mice (Supplementary Figure S7A, available at Carcinogenesis Online). Inhibition of AMPK activation with compound C reduced anti-cancer efficacy of WFA whereas AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) ameliorated the effects of WFA (Supplementary Figure S7B–E, available at Carcinogenesis Online). 2-DG is a glucose analog that decreases cellular ATP production resulting in AMPK activation leading to inhibition of cancer cell growth (29). Indeed, combined treatment of WFA and 2-DG resulted in a significant decrease in cellular ATP levels (Figure 5C) and increased apoptosis induction and growth inhibition of breast cancer cells in comparison to either treatment alone (Figure 5D, F and G). WFA and 2-DG treatment also showed higher cleavage of PARP1 in breast cancer cells (Figure 5E). Importantly, WFA and 2-DG induced synergistic cytotoxic effect in HCC1569, HCC1806, Hs578t, MCF7, MDA-MB-231 and MDA-MB-468 cells. Combination index (CI) analyses was performed using CompuSyn software to analyze the combined effect of WFA and 2-DG in breast cancer cells. CI < 1 shows synergism, CI = 1 shows additivity and CI > 1 shows antagonism and notably, in multiple combinations of WFA and 2-DG, CI values were less than 1 (Figure 6A and B).
Figure 5.

WFA induces energy impairment with AMPK activation. (A) Breast cancer cells were treated with 5 µM WFA for the indicated time intervals and intracellular ATP production was measured. Relative ATP levels are expressed with respect to the control. *P< 0.005, compared with control. (B) Breast cancer cells were treated with 5 µM WFA for indicated time intervals and total lysates were immunoblotted for phospho-PRKAA1 (p-PRKAA1) and total PRKAA1 (62 kDa) expression levels. ACTB (45 kDa) was used as loading control. (C) SUM149 and SUM159 cells were treated with 5 µM WFA alone or in combination with 2-DG as indicated. Intracellular ATP production was measured. Relative ATP levels are expressed with respect to the control. *P< 0.005, compared with control; **P< 0.01, compared with WFA alone. (D) SUM149 and SUM159 cells were treated with 5 µM WFA alone or in combination with 2-DG as indicated followed by Hoechst 33342 staining apoptosis detection. Mean number of apoptotic cells are presented in bar graphs. *P< 0.01, compared with control; **P< 0.01, compared with WFA alone. (E) Breast cancer cells were treated with 5 µM WFA alone or in combination with 2-DG as indicated. Total lysates were immunoblotted for the expression of cPARP1 (89 kDa) and PARP1 (116 kDa). ACTB (45 kDa) was used as loading control. (F, G) SUM149 and SUM159 cells were treated with 5 µM WFA alone or in combination with 2-DG as indicated and subjected to Trypan Blue exclusion assay. Bar graph shows percentage of alive cells. *P< 0.01, compared with control; **P< 0.005, compared with WFA alone.
Figure 6.

Combined treatment with WFA and 2-DG synergistically inhibits breast cancer cells. WFA induces energy impairment with reducing LDHA and higher expression of AMPK and low expression of LDHA correlates with increased overall survival. (A, B) HCC1569, HCC1806, HS578t, MDA-MB-231 and MDA-MB-468 breast cancer cells were treated with various concentrations of WFA (1.0, 2.5, 5.0. 7.5, 10.0 µM) in combination with 4.0 mM of 2-DG. MCF7 cells were treated with various concentrations of WFA in combination with 2.0 mM 2-DG. Cells were subjected to XTT assay and combination index values were calculated using CompuSyn software. CI < 1 represents synergism. Table shows combination index of different concentrations of WFA and 2-DG. (C) MCF7 cells were treated with 5 µM WFA or 10 mM MP as indicated and intracellular ATP production was measured. Relative ATP levels are expressed with respect to the control. *P< 0.01, compared with control; **P< 0.005, compared with WFA alone. (D) MCF7 cells were treated with 5 µM WFA or 10 mM MP as indicated and subjected to Trypan Blue exclusion assay. Bar graph shows fold change of alive cells. *P< 0.01, compared with control; **P< 0.001, compared with WFA alone. (E) MCF7 cells were treated with 5 µM WFA or 10 mM MP as indicated followed by Hoechst 33342 staining apoptosis detection. Mean number of apoptotic cells are presented as fold change in bar graphs. *P < 0.01, compared with control; **P< 0.01, compared with WFA alone. (F) Total lysates were immunoblotted for the expression of cPARP1 (89 kDa) and PARP1 (116 kDa). ACTB (45 kDa) was used as loading control. (G) Schematic representation of TCA cycle and aerobic glycolysis in proliferating cancer cells. Role of LDHA is noted. (H) Breast cancer cells were treated with 5 µM WFA for various time intervals as indicated. Total RNA was examined for the expression of LDHA using RT-PCR. ACTB was used as loading control. (I) Breast cancer cells were treated with 5 µM WFA for various time intervals as indicated. Total lysates were immunoblotted for the expression of LDHA (37 kDa) and phospho-LDHA (p-LDHA). ACTB (45 kDa) was used as loading control. (J) In breast cancer patients, higher expression of AMPK and lower expression of LDHA correlate to better prognosis (n = 4,374, HR = 0.75, P = 1.3e-05 and HR = 1.59, P = 1.6e-16, respectively). When investigating ER negative HER2 negative patients, the correlation to better survival was more prominent in LDHA (HR = 1.64, P = 4.7e-05) but was smaller for AMPK (HR = 0.81, P = 0.1). The plots display relapse free-survival, ER and HER2 status were determined using gene expression.
Decrease in cellular ATP levels might reflect an insufficiency of TCA carbon substrates; therefore, we incubated breast cancer cells with methyl pyruvate (MP) as a source of substrate for the TCA cycle. Excess MP not only restored ATP levels in WFA-treated cells (Figure 6C) but also protected breast cancer cells against WFA-induced growth inhibition and apoptotic induction (Figure 6D, E). As expected, MP inhibited WFA-induced cleavage of PARP1 in breast cancer cells (Figure 6F). Normal proliferating cells produce ATP through oxidative phosphorylation in the mitochondria while proliferating cancer cells largely depend on aerobic glycolysis to meet their high energy requirements in the presence of adequate oxygen. Glucose gets converted to pyruvate during glycolysis and cancer cells preferentially transform pyruvate to lactate in the cell cytoplasm instead of utilizing TCA cycle in mitochondria (Figure 6G). LDHA is an important enzyme that drives the increased flow of metabolites through this aberrant metabolic pathway by catalyzing the conversion of pyruvate to lactate (30). LDHA is considered an ideal target to mitigate the cellular energy supply in cancer cells (34). We found that WFA treatment effectively reduces the expression of LDHA in breast cancer cells (Figure 6H and I). Tyrosine phosphorylation of LDHA promotes cellular metabolism and plays an important role in tumor growth promotion (35), therefore, we investigated the impact of WFA treatment on LDHA phosphorylation levels. Interestingly, WFA treatment effectively reduced Y10 phosphorylation of LDHA in MDA-MB-231 and MCF7 breast cancer cells (Figure 6I). We found that WFA treatment increases AMPK and decreases LDHA in breast cancer cells and intriguingly, high-AMPK and low-LDHA expression associate with better relapse-free survival in breast cancer patients (Figure 6J). The entire combined dataset contained 4374 patients, of which 696 were estrogen receptor (ER) negative and HER2 negative (hence on termed as TNBC cohort). The mean follow-up of all patients was 71.5 months. In all patients, higher expression of AMPK correlated to better survival (n = 4374, P = 1.3e-05, HR = 0.75) and when restricted to TNBC patients, the correlation trend remained (n = 696, P = 0.1, HR = 0.81). At the same time, an opposite association was observed for LDHA as higher expression resulted in worse survival outcomes (in all patients P = 1.6e-16, HR = 1.59). The effect of LDHA was more pronounced in TNBC patients (P = 4.9e-05, HR = 1.64) (Figure 6J).
Collectively, our data uncovers a sequence of events where WFA blocks autophagic flux by inhibiting proteolytic lysosomal activity and leads to insufficient substrates for TCA cycle, inhibits LDHA, leading to energy insufficiency and consequently to growth inhibition and apoptotic induction.
Discussion
Finding new therapeutic strategies that can inhibit carcinogenesis in an effective, non-toxic and non-endocrine manner is a major goal of breast cancer research. Autophagy is important for the survival of cancer cells as it helps maintain the cellular energy supply and homeostasis in rapidly proliferating cancer cells and helps evade various environmental and cellular stresses (36). In this study, we show that WFA is a potent inhibitor of lysosomal proteolytic activity, blocks autophagic flux and exerts antitumor activity in multiple subtypes of breast cancer including luminal A, luminal B, basal, claudin-low, and HER2 subtypes. We established a connection between autophagy inhibition, energy depletion and cell death in WFA-treated breast cancer cells. Upon observing increased autophagosome formation, LC3B conversion and LC3B puncta in WFA-treated breast cancer cells, we further investigated the effects of WFA on autophagic flux using several different approaches as limiting the assessment to the number of autophagosomes and LC3B levels is insufficient to gauge the entire autophagic process and its biological impact. Autophagy can be limited due to the delayed trafficking of autophagosomes to lysosomes, the fusion of autophagosomes and lysosomes, or defects in lysosomal proteolytic activity (27). Confocal analyses of tandem GFP-RFP-tagged LC3B fluorescence, LysoTracker Red assay and the colocalization of GFP-LC3B and RFP-RAB7 showed that WFA increases the fusion of autophagosomes and lysosomes. Autophagosome and lysosome fusion is regulated by the internal pH in acidic organelles (37) and indeed, AO staining shows that lysosomes display the characteristics of acidic compartments in WFA-treated cells. By definition, autophagy is self-eating process that recycles damaged cellular organelles, long-lived cytoplasmic protein and glycogen in a lysosome-dependent process (17,18). These cellular components are delivered to the autolysosome followed by their breakdown by lysosomal hydrolases—cathepsin proteases. Our data from DQ Green BSA assay showed that WFA abrogates lysosomal function and inhibits the maturation of cathepsins. With careful analyses of multiple steps of autophagic process, we uncovered that although WFA-treated breast cancer cells show the upregulation of initial steps of autophagy, WFA blocks autophagic flux by inhibiting lysosomal proteolytic activity.
Interestingly, our data excludes an ‘autophagic cell death’ in WFA-treated cells as inhibiting autophagy with 3-MA, CQ, Baf or ATG7 and BECN1 knockdown fails to rescue WFA-treated cells, and instead shows the involvement of energetic impairment in WFA-mediated growth inhibition and apoptosis induction. Indeed, MP treatment could protect WFA-treated breast cancer cells. Going in line with the notion that blocking autophagic flux would impact the availability of substrates for both biosynthesis and energy generation (36), our results demonstrate that WFA reduces cellular ATP production altering the AMP:ATP ratio leading to a significant increase in PRKAA1 phosphorylation in breast cancer cells. Inhibition of PRKAA1 with compound C reduces WFA-mediated growth inhibition and apoptotic induction while PRKAA1 activator AICAR potentiates WFA’s effect in breast cancer cells indicating the functional importance of PRKAA1 in WFA action. 2-DG, an analog of glucose that is effectively taken up by cells but inhibits hexokinase, leads to inhibition of glycolysis and intracellular ATP depletion (38). Targeting tumor energy pathways with 2-DG treatment has been shown to induce PRKAA1 activation leading to growth inhibition in multiple preclinical models (39,40). Our data show that combined treatment with 2-DG and WFA further reduces ATP levels and exhibits more effective reduction of cell growth and induction of apoptotic cells. Clinical studies utilizing 2-DG to target tumor metabolism have shown safety and efficacy of 2-DG in prostate cancer (41), advanced solid tumors (42) and cerebral gliomas (43). In light of our observations, combination of 2-DG and WFA might be useful in targeting metabolic fragility of breast cancer cells.
Cancer cells preferentially utilize aerobic glycolysis to metabolize glucose in contrast to normal proliferating cells that produce ATP via oxidative phosphorylation in the mitochondria (30). Conversion of pyruvate and NADH to lactate and NAD+ during glycolysis is catalyzed by LDHA, a key enzyme that promotes the metabolic switch to aerobic glycolysis (44). Cancer cells exhibit higher expression levels of LDHA along with elevated tyrosine phosphorylation of LDHA (45,46). Phosphorylation of LDHA is more abundant in breast tumors in comparison to normal adjacent tissues and correlates with increased invasive and metastatic potential. Intriguingly, we found that WFA inhibits LDHA expression as well as phosphorylation at Y10 in breast cancer cells (47). These findings have important implications as LDHA is fast emerging as an anti-cancer target bridging the gap between metabolic perturbations and metastatic progression of cancer cells. Recognition of LDHA as an anticancer target has fueled the interest in developing LDHA inhibitors but very few LDHA inhibitors with in vivo efficacy have been reported so far. Earliest reported LDH inhibitor is oxamate that inhibits LDH activity by blocking the pyruvate binding site but it is a weak inhibitor. Diallyl trisulfide (DATS), an organo-sulfur compound from garlic, was reported to decrease LDHA in triple negative breast cancer cells. A small molecule inhibitor of LDHA, GNE-140, has shown effective inhibition of LDHA in pancreatic tumor cells. Some new inhibitors have also been developed but only few have shown in vivo efficacy (47). In this regard, recognition of WFA as an LDHA inhibitor in breast cancer cells is important as WFA has shown in vivo efficacy in multiple breast cancer models (12).
Conclusions
In conclusion, our study presents that WFA impairs proteolytic lysosomal activity causing blockade of autophagic flux resulting in energy depletion leading to growth inhibition and apoptotic induction. Our results also show that WFA is an AMPK activator that works synergistically with 2-DG to inhibit breast cancer growth and is also an inhibitor of LDHA. These attributes of WFA support its development as a potential cancer chemopreventive and chemotherapeutic agent for breast cancer.
Supplementary material
Supplementary data are available at Carcinogenesis online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by National Institute of Health National Cancer Institute [R01CA204555], Breast Cancer Research Foundation [90047965] and The Fetting Fund (to D.S.).
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- 2-DG
2-deoxy-d-glucose
- 3-MA
3-methyladenine
- ACTB
actin
- AMPK
AMP-activated protein kinase
- ATG5
autophagy related 5
- ATG7
autophagy related 7
- ATP
adenosine triphosphate
- BECN1
beclin 1
- CQ
chloroquine
- GFP
green fluorescent protein
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 beta
- LDHA
lactate dehydrogenase A
- MP
methyl pyruvate
- mRFP
monomeric red fluorescent protein
- PARP1
poly(ADP-ribose) polymerase1
- PRKAA1
protein kinase
- AMP
activated, alpha 1 catalytic subunit
- RFP
red fluorescent protein
- SQSTM1/p62
sequestosome 1
- WFA
withaferin A
Contributor Information
Nethaji Muniraj, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Sumit Siddharth, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Arumugam Nagalingam, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Alyssa Walker, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Juhyung Woo, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Balázs Győrffy, MTA TTK Momentum Cancer Biomarker Research Group, Budapest, Hungary; 2nd Department of Pediatrics, Semmelweis University, Budapest, Hungary.
Ed Gabrielson, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Neeraj K Saxena, Early Detection Research Group, National Cancer Institute, Rockville, MD, USA.
Dipali Sharma, Department of Oncology and the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
References
- 1. Mirjalili M.H. et al. (2009) Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules, 14, 2373–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vanden Berghe W. et al. (2012) Molecular insight in the multifunctional activities of Withaferin A. Biochem. Pharmacol., 84, 1282–1291. [DOI] [PubMed] [Google Scholar]
- 3. Mishra L.C. et al. (2000) Scientific basis for the therapeutic use of Withania somnifera (ashwagandha): a review. Altern. Med. Rev., 5, 334–346. [PubMed] [Google Scholar]
- 4. Misra L. et al. (2008) Withanolides from Withania somnifera roots. Phytochemistry, 69, 1000–1004. [DOI] [PubMed] [Google Scholar]
- 5. Chaurasiya N.D. et al. (2008) Analysis of withanolides in root and leaf of Withania somnifera by HPLC with photodiode array and evaporative light scattering detection. Phytochem. Anal., 19, 148–154. [DOI] [PubMed] [Google Scholar]
- 6. Stan S.D. et al. (2008) Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res., 68, 7661–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Srinivasan S. et al. (2007) Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer Res., 67, 246–253. [DOI] [PubMed] [Google Scholar]
- 8. Oh J.H. et al. (2009) Withaferin A inhibits tumor necrosis factor-alpha-induced expression of cell adhesion molecules by inactivation of Akt and NF-kappaB in human pulmonary epithelial cells. Int. Immunopharmacol., 9, 614–619. [DOI] [PubMed] [Google Scholar]
- 9. Palliyaguru D.L. et al. (2016) Withaferin A induces Nrf2-dependent protection against liver injury: role of Keap1-independent mechanisms. Free Radic. Biol. Med., 101, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vedi M. et al. (2016) Assessment of hepatoprotective and nephroprotective potential of withaferin A on bromobenzene-induced injury in Swiss albino mice: possible involvement of mitochondrial dysfunction and inflammation. Cell Biol. Toxicol., 32, 373–390. [DOI] [PubMed] [Google Scholar]
- 11. Chengappa K.N.R. et al. (2018) Adjunctive use of a standardized extract of Withania somnifera (Ashwagandha) to treat symptom exacerbation in schizophrenia: a randomized, double-blind, placebo-controlled study. J. Clin. Psychiatry, 79, 1–9. [DOI] [PubMed] [Google Scholar]
- 12. Nagalingam A. et al. (2014) Mechanistic elucidation of the antitumor properties of withaferin A in breast cancer. Cancer Res., 74, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hahm E.R. et al. (2013) Metabolic alterations in mammary cancer prevention by withaferin A in a clinically relevant mouse model. J. Natl. Cancer Inst., 105, 1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hahm E.R. et al. (2011) Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One, 6, e23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee J. et al. (2010) Withaferin A inhibits activation of signal transducer and activator of transcription 3 in human breast cancer cells. Carcinogenesis, 31, 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J. et al. (2012) Withaferin A causes activation of Notch2 and Notch4 in human breast cancer cells. Breast Cancer Res. Treat., 136, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagelkerke A. et al. (2015) Therapeutic targeting of autophagy in cancer. Part II: pharmacological modulation of treatment-induced autophagy. Semin. Cancer Biol., 31, 99–105. [DOI] [PubMed] [Google Scholar]
- 18. Nagelkerke A. et al. (2015) Therapeutic targeting of autophagy in cancer. Part I: molecular pathways controlling autophagy. Semin. Cancer Biol., 31, 89–98. [DOI] [PubMed] [Google Scholar]
- 19. Knight B.B. et al. (2011) Survivin upregulation, dependent on leptin-EGFR-Notch1 axis, is essential for leptin-induced migration of breast carcinoma cells. Endocr. Relat. Cancer, 18, 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagalingam A. et al. (2012) Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis, 33, 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saxena N.K. et al. (2008) Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res., 68, 9712–9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ohri S. et al. (2002) Modulation of c-myc and c-fos gene expression in regenerating rat liver by 2-mercaptopropionylglycine. Cell Biol. Int., 26, 187–192. [DOI] [PubMed] [Google Scholar]
- 23. Avtanski D.B. et al. (2014) Honokiol inhibits epithelial-mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E-cadherin axis. Mol. Oncol., 8, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taliaferro-Smith L. et al. (2009) LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene, 28, 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prat A. et al. (2013) Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res. Treat., 142, 237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Green D.R. et al. (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell, 157, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mizushima N. et al. (2010) Methods in mammalian autophagy research. Cell, 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimura S. et al. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy, 3, 452–460. [DOI] [PubMed] [Google Scholar]
- 29. Wokoun U. et al. (2017) Co-treatment of breast cancer cells with pharmacologic doses of 2-deoxy-D-glucose and metformin: starving tumors. Oncol. Rep., 37, 2418–2424. [DOI] [PubMed] [Google Scholar]
- 30. Bui T. et al. (2006) Cancer’s sweet tooth. Cancer Cell, 9, 419–420. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y.T. et al. (2010) Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem., 285, 10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rubinsztein D.C. et al. (2009) In search of an “autophagomometer”. Autophagy, 5, 585–589. [DOI] [PubMed] [Google Scholar]
- 33. Yang Z.J. et al. (2011) The role of autophagy in cancer: therapeutic implications. Mol. Cancer Ther., 10, 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fantin V.R. et al. (2006) Attenuation of LDH A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell, 9, 425–434. [DOI] [PubMed] [Google Scholar]
- 35. Fan J. et al. (2011) Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell. Biol., 31, 4938–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Janku F. et al. (2011) Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol., 8, 528–539. [DOI] [PubMed] [Google Scholar]
- 37. Kawai A. et al. (2007) Autophagosome–lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy, 3, 154–157. [DOI] [PubMed] [Google Scholar]
- 38. Aft R.L. et al. (2002) Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br. J. Cancer, 87, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim S.M. et al. (2013) Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol. Cancer Ther., 12, 2145–2156. [DOI] [PubMed] [Google Scholar]
- 40. Cheong J.H. et al. (2011) Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther., 10, 2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stein M. et al. (2010) Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate, 70, 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raez L.E. et al. (2013) A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol., 71, 523–530. [DOI] [PubMed] [Google Scholar]
- 43. Mohanti B.K. et al. (1996) Improving cancer radiotherapy with 2-deoxy-D-glucose: phase I/II clinical trials on human cerebral gliomas. Int. J. Radiat. Oncol. Biol. Phys., 35, 103–111. [DOI] [PubMed] [Google Scholar]
- 44. Kroemer G. et al. (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell, 13, 472–482. [DOI] [PubMed] [Google Scholar]
- 45. Xian Z.Y. et al. (2015) Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumour Biol., 36, 8093–8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J. et al. (2016) Suppression of lactate dehydrogenase A compromises tumor progression by downregulation of the Warburg effect in glioblastoma. Neuroreport, 27, 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miao P. et al. (2013) Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. IUBMB Life, 65, 904–910. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.