

Abstract
Pathologic complete response (pCR) to neoadjuvant chemoradiation for locally advanced esophageal adenocarcinoma (EAC) confers significantly improved survival. The ability to infer pCR may spare esophagectomy in some patients. Currently, there are no validated biomarkers of pCR. This study sought to evaluate whether a distinct signature of DNA copy number alterations (CNA) can be predictive of pCR in EAC. Pretreatment biopsies from 38 patients with locally advanced EAC (19 with pCR and 19 with pathologic partial/poor response) were assessed for CNA using OncoScan assay. A novel technique was employed where within every cytogenetic band, the quantity of bases gained by each sample was computed as the sum of gained genomic segment lengths weighted by the surplus copy number of each segment. A threefold cross-validation was used to assess association with pCR or pathologic partial/poor response. Forty patients with locally advanced EAC from The Cancer Genome Atlas (TCGA) constituted an independent validation cohort. Gains in the chromosomal loci 14q11 and 17p11 were preferentially associated with pCR. Average area under the receiver operating characteristic curve (AUC) for predicting pCR was 0.80 among the threefold cross-validation test sets. Using 0.3 megabases as the cutoff that optimizes trade-off between sensitivity (63%) and specificity (89%) in the discovery cohort, similar prediction performance for clinical and radiographic response was demonstrated in the validation cohort from TCGA (sensitivity 61%, specificity 82%). Copy number gains in the 14q11 and 17p11 loci may be useful for prediction of pCR, and, potentially, personalization of esophagectomy in EAC.
Keywords: copy number alterations, esophageal adenocarcinoma, neoadjuvant chemoradiation, pathologic complete response, The Cancer Genome Atlas
INTRODUCTION
Esophageal adenocarcinoma (EAC) is the most common type of esophageal cancer in the United States.1 The 5-year overall survival among patients with EAC is dismal at 19%.2,3 Approximately, half of all patients are eligible for treatment with curative intent at the time of EAC diagnosis. The standard of care for locally advanced EAC (stages II and III) is trimodality therapy, which involves neoadjuvant chemoradiation followed by esophagectomy.4,5
Assessment of pathologic complete response (pCR) plays an important role in the treatment of EAC. pCR is defined as the absence of tumor (i.e. ypT0N0, no primary or lymph node disease) in the esophagectomy specimen. Neoadjuvant chemoradiation leads to pCR in 25–30% of patients with EAC. In patients with locally advanced EAC treated with neoadjuvant chemoradiation, pCR nearly doubles 3-year overall survival, from 48 to 86% and 40 to 70% in two separate institutional experiences.6,7 Furthermore, establishment of pCR can spare patients both the morbidity and mortality associated with esophagectomy. Alternatively, the ability to infer ineffective neoadjuvant treatment may allow some patients to proceed directly to surgery (with the possibility of subsequent enrollment in a clinical trial) and thus avoid the adverse effects of chemoradiation and disease progression during unsuccessful neoadjuvant treatment.8,9 Thus, accurate prediction of pCR may be crucial for some patients with EAC.
To date, no pretreatment clinical or imaging features, singly or in combination, have been shown to accurately predict pCR in EAC. A nomogram-based approach that integrates post-treatment positron emission tomography data with pretreatment demographics, histology, and clinical stage was able to predict pCR with a modest 60% accuracy; however, this result has not been independently validated.10 Moreover, there is a growing consensus that a predictive model based solely on clinical/imaging variables is unlikely to be accurate.11 Other strategies to predict pCR include assessment of pretreatment biomarkers. Several reports have described gene-specific biomarkers that are associated with pCR.12–14 These studies focused on mutations or gene expression signatures, but to our knowledge none of these biomarkers have been independently validated.
The genomic landscape of EAC is dominated by prominent heterogeneity, and high levels of DNA copy-number alterations (CNA) appear to play an important role in EAC pathogenesis.15–18 Previous studies revealed relationships between CNA and survival in patients with EAC, although the results were inconsistent.19–22
To our knowledge, no studies have evaluated the relationship between CNA and pCR. Taking into account both the biology of EAC and positive association between pCR and overall survival,6 we were motivated to examine aggregate genomic alterations rather than changes in specific genes. We reasoned that the quantity or size of CNA taken in totality, assessed across large genomic intervals, may have value in predicting pCR. We hypothesize that specific pretreatment CNA signatures may increase the sensitivity of EAC to neoadjuvant treatment and serve as a predictive biomarker for pCR. The potential of CNA to predict pCR following neoadjuvant therapy in EAC could guide operative indication in patients with EAC. Such prediction of pCR would constitute a major step forward in improving management of locally advanced EAC.
MATERIAL AND METHODS
Patient population and tissue selection
All patients with EAC, including gastroesophageal junctional adenocarcinoma, treated with neoadjuvant chemoradiation and esophagogastrectomy from 1998 to 2017 were identified using the electronic databases of the Department of Pathology and Cancer Registry at Dartmouth-Hitchcock Medical Center. Inclusion criteria for the study were (i) availability of paired histologic material (slides and/or paraffin blocks) consisting of diagnostic pretreatment biopsies and esophagogastrectomy specimens; and (ii) availability of clinical and survival information. The primary study outcome was pCR, defined as complete absence of adenocarcinoma in the resection specimen including harvested lymph nodes. Pathologic partial/poor response (pPPR) was defined as residual adenocarcinoma with more than 10% of tumor cells remaining.23 This study was approved by the Institutional Review Board of Dartmouth-Hitchcock Medical Center (STUDY00030218).
Clinicopathologic data
Age, sex, tumor location, endoscopic tumor size, treatment regimens, follow-up time, pretreatment clinical tumor stage, pre-neoadjuvant therapy histologic tumor grade, and overall survival were ascertained. EAC was staged using the seventh edition of the American Joint Committee on Cancer Staging Manual. All de-identified clinical data used in this study are available in Supplementary Table S1.
Neoadjuvant chemoradiation regimen
Details of neoadjuvant chemotherapy and external beam radiotherapy are outlined in Table 1.
Table 1.
Clinicopathologic characteristics of study participants
| Mean ± SD* or count (%) | |||
|---|---|---|---|
| Pathologic complete response (N = 19) |
Pathologic poor response (N = 19) | P-value | |
| Age, years | 65 ± 11 | 68 ± 14 | 0.52 |
| Sex | 0.11 | ||
| Female | 4 (21) | 0 (0) | |
| Male | 15 (79) | 19 (100) | |
| Tumor Location | 0.52 | ||
| Distal esophagus | 10 (53) | 7 (37) | |
| Gastroesophageal junction | 9 (47) | 12 (63) | |
| Primary tumor size, cm | 5.1 ± 2.3 | 5.8 ± 2.6 | 0.45 |
| Pretreatment biopsy tumor grade | 1.00 | ||
| Low grade | 9 (47) | 9 (47) | |
| High grade | 10 (53) | 10 (53) | |
| Pretreatment clinical T stage | 0.45 | ||
| T2 | 6 (32) | 3 (16) | |
| T3 | 13 (68) | 16 (84) | |
| Pretreatment clinical N stage | 0.15 | ||
| N0 | 4 (21) | 5 (26) | |
| N1 | 13 (68) | 11 (58) | |
| N2 | 0 (0) | 3 (16) | |
| N3 | 2 (11) | 0 (0) | |
| Pretreatment AJCC 7 overall clinical stage | 0.74 | ||
| II | 8 (42) | 6 (32) | |
| III | 11 (47) | 13 (68) | |
| Neoadjuvant chemotherapy | 0.15 | ||
| Platinum agent/Taxane | 15 (79) | 11 (58) | |
| Fluoropyrimidine/Taxane | 2 (11) | 7 (37) | |
| Unknown | 2 (11) | 1 (5) | |
| Neoadjuvant radiotherapy dose, Gy | 50.0 ± 1.3 | 50.1 ± 1.3 | 0.96 |
| Esophagectomy surgical approach | 0.48 | ||
| Ivor-Lewis | 11 (58) | 14 (74) | |
| McKeon (three hole) | 5 (26) | 4 (21) | |
| Transhiatal | 3 (16) | 1 (5) | |
*Standard deviation.
DNA preparation and OncoScan assay
Tumor tissue was macro-dissected from unstained tissue slides (on average four slides per sample) identified in a corresponding hematoxylin and eosin-stained slide as containing an adequate concentration of tumor cells (at least 25%). Cases that did not have a clearly defined region of greater than 25% tumor cells, which could be easily macro-dissected were excluded. Genomic DNA was quantified using quantitative polymerase chain reaction. Eighty nanograms of genomic DNA were subjected to the OncoScan FFPE CNV assay (Affymetrix, Santa Clara, CA). The assay utilizes molecular inversion probe (MIP) technology, proven for identifying CNA including deletions, duplications, and unbalanced translocations, loss of heterozygosity, copy neutral loss of heterozygosity, and somatic mutations. Intensity of signal and allelic ratio of the genotypes at each single nucleotide polymorphism locus were used to determine copy number and loss of heterozygosity (allelic imbalance) data for further processing in Nexus Express for Oncoscan (BioDiscovery, El Segundo, CA) and Chromosome Analysis Suite software version 3 (Affymetrix).
Pathologic evaluation
Tumor histologic grade in the biopsies was classified as low-grade or high-grade. Tumor regression in the resection specimens was classified using the Becker system as complete absence of tumor; <10% of residual tumor; 10–50% of residual tumor; and >50% of residual tumor.23,24 The amount and histologic grade of tumor in diagnostic biopsies, as well as tumor regression in the resection specimens, were reviewed by one board-certified gastrointestinal subspecialty-trained pathologist with 12 years of experience (M.L.).
Evaluation of regions of differing genomic gains between samples with pCR and pPPR
Adhering to the manufacturer’s recommended quality control filters for OncoScan FFPE CNV assay, only samples with OS-MAPD <0.30 were included. We accepted the location, length, and classification (i.e. gain versus loss) of CNA events automatically called by the Nexus Express software using a P-value threshold of less than 1 × 10−5 and coverage of at least 10 probes.25 Within every cytoband of the Genome Reference Consortium Human Build 37 coordinate system, the total quantity of bases gained by each sample was computed as the sum of gained genomic segment lengths weighted by the surplus copy number of each segment (Fig. 1).26 Samples were then randomly partitioned, with two-thirds allocated to a training set and one-third allocated to a test set for threefold cross-validation. During each round of cross-validation, total genomic gains in up to two distinct cytobands were compared between samples with pCR and pPPR of the training set using two-sided t-test and false discovery rate (FDR) as multiple hypothesis testing correction.27 All permutations of one to two cytobands were evaluated. Cytobands found to exhibit the most significant differences between samples with pCR and pPPR were then assessed on the corresponding test set samples using area under the receiver operating characteristic curve (AUC). A combination of no more than two different cytobands was evaluated at a time to avoid potential overfitting.
Fig. 1.
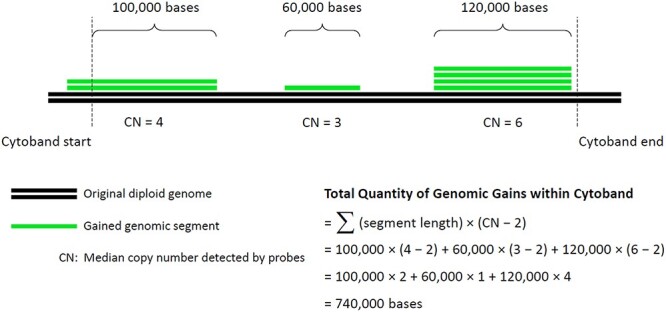
Schematic diagram. Total quantity of bases gained within each cytoband was computed as the sum of gained genomic segment lengths weighted by the surplus copy number of every segment.
Candidate gene and pathway analysis
For every CNA event of every sample, we identified whether it overlapped with a gene, mapped involved genes to all gene sets of the Molecular Signatures Database (MSigSB, http://software.broadinstitute.org/gsea/msigdb/collections.jsp), and tested whether certain gene sets were more enriched with CNA-affected genes among complete responders versus incomplete responders. At the individual gene level, threefold cross-validation was performed to identify candidate genes associated with pCR/pPPR status, and their encompassing MSigDB gene sets were evaluated using Gene Set Enrichment Analysis with FDR control.26 Beyond well-established biologic pathways, we were broadly interested in interrogating gene sets of shared molecular function and co-localization as well, so all 17,000+ gene sets within MSigDB were tested.
External validation in The Cancer Genome Atlas
Clinical and level 3 CNA data in The Cancer Genome Atlas (TCGA) for 185 patients with esophageal carcinoma17 were downloaded from Firebrowse hosted by the Broad Institute.28 The 40 patients with stage II or III EAC and documented responses to neoadjuvant chemoradiation were included for analysis. We recorded response to chemoradiation using the provided categories ‘complete response’, ‘partial response’, or ‘progressive disease’. TCGA data dictionary did not explicitly mention pathologic assessment of response. However, progressive disease was further characterized by ‘clinical progressive disease’ or ‘radiographic progressive disease’. So we conservatively assumed that treatment response in TCGA was documented clinically and radiographically. Patients with missing response statuses were excluded. Considerable quality control measures had already been performed on TCGA level 3 data, so the only additional filtering of CNA event calls was for coverage of at least 10 probes. We defined a CNA event as a copy number gain if its probes possess mean log2 (copy number/2) >0.5, a commonly implemented threshold.29 Total genomic gains in the regions of interest for the 40 EAC samples were then computed as above for the discovery dataset.
Bioinformatics and statistical analyses
All genomic analyses were performed in R (http://www.R-project.org/) using the packages ROCR, survival, and ggplot2. The distributions of baseline characteristics were compared between groups using a two sample t-test for continuous variables and Chi-squared test for categorical variables. Continuous variables are presented as the mean ± standard deviation.
RESULTS
General characterization of patients
We identified 169 patents with clinical stage II–III EAC, including gastroesophageal junctional adenocarcinoma, treated with neoadjuvant chemoradiation and esophagectomy/esophagogastrectomy from 1998 to 2017. pCR was observed in resection specimens of 42 (25%) patients. Diagnostic pretreatment biopsies were unavailable or did not have enough tumor tissue for molecular analysis in 18 of the 42 patients with pCR. The remaining 24 patients constituted the group with pCR. The pPPR group was chosen to have the least amount of tumor regression and consisted of 30 patients. Two patients with pPPR had 90% residual tumor, 3 patients had 50–90% residual tumor, and the remaining 25 patients had 10–50% residual tumor. DNA was then extracted from macro-dissected tumor tissue of pretreatment biopsies and processed using the OncoScan assay. Five samples with pCR and 11 samples with pPPR did not pass OncoScan’s quality control filter (OS-MAPD < 0.30) and were excluded. The final set of patients with pCR and pPPR consisted of 19 patients each.
Clinicopathologic and survival characterization of patients with pCR and pPPR
Patients in the pCR and pPPR groups were statistically similar with regard to all relevant clinical variables, including age, sex, clinical T or N category, clinical stage, tumor location, tumor size, type of neoadjuvant chemotherapy, neoadjuvant radiation therapy dose, surgical technique, and tumor grade in pretreatment biopsies (Table 1). The patients with pCR were found to have superior overall survival compared to those with pPPR (hazard ratio = 0.382, P = 0.029), with median overall survival of 52 months versus 20 months (Fig. 2).
Fig. 2.
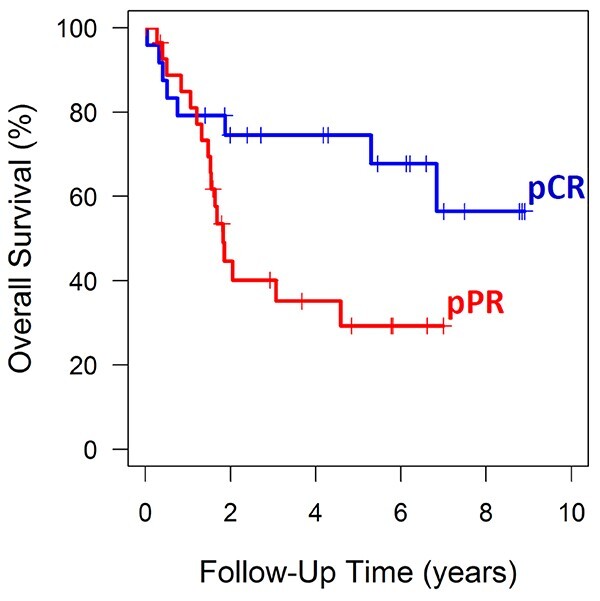
Overall survival stratified by pathologic response status. Kaplan–Meier curves were plotted to show the difference in survival between patients with pathologic complete response (pCR, blue curve) and pathologic partial/poor response (pPPR, red curve).
Inference of pCR or pPPR
In threefold cross-validation, genomic gains within cytobands 14q11 and 17p11 (Supplementary Table S2) displayed the most significant difference between pCR and pPPR samples across all three training sets (P < 0.05, FDR < 0.05). The average AUC for correctly predicting pCR in the test set samples was 0.80 (Fig. 3). Repeating this process with genomic losses in cytobands yielded poor discrimination between pCR and pPPR statuses, and the top cytobands were inconsistent across cross-validation training sets. Similarly, this theme was observed with total CNA events and genes involving CNA events as well (Supplementary Table S3).
Fig. 3.

Prediction accuracy among the test sets of threefold cross validation. Areas under the receiver operating characteristic curve (AUCs) from all three test sets of threefold cross-validation using total genomic gains in the cytobands 14q11 and 17p11.
Within our dataset as a whole (Fig. 4), setting a cutoff anywhere between 1 and 300 kilobases yields a reasonable balance between sensitivity (63%) and specificity (89%). Incorrectly predicting partial/poor response as complete response should be viewed as a more serious error than incorrectly predicting complete response as partial/poor response. In the latter scenario, standard of care esophagectomy would follow. In the former scenario, a patient mistakenly inferred to have achieved a complete response after chemoradiation may progress to unsalvageable disease in the setting of suboptimal surveillance. Furthermore, this case–control study was designed to genomically characterize tumors with the polar extremes of treatment response after chemoradiation. Pathologic discernment between complete response and poor response may be less clear among the general population of patients with stage II or III EAC who receive neoadjuvant chemoradiation. In parallel, their distributions of total genomic gains within the 14q11 and 17p11 loci may not be as distinct. For these reasons, we preferred to set a decision cutoff that slightly favors capturing pPPR: if genomic gains in chromosomes 14q11 and 17p11 exceed 0.3 megabases, infer pCR; otherwise infer pPPR.
Fig. 4.
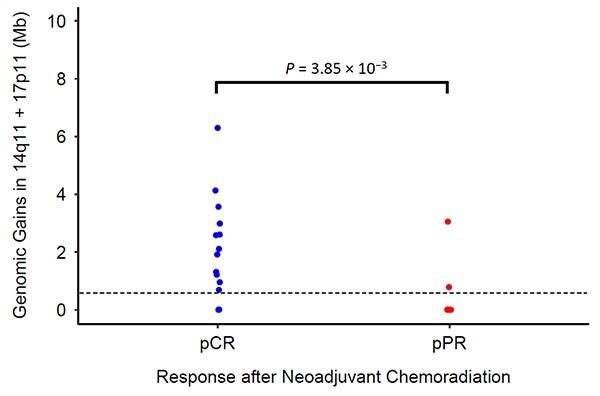
Total genomic gains (in megabases). pCR, pathologic complete response, blue; pPPR, pathologic partial/poor response, red.
Validation using The Cancer Genome Atlas data
We sought to validate our findings in an independent cohort of patients with locally advanced EAC treated by neoadjuvant chemoradiation and esophagectomy. In TCGA, there are 40 patients with stage II–III EAC and documented responses to neoadjuvant chemoradiation (Table 2). Of the 23 patients who each displayed a complete response by clinical and radiographic assessment, 14 patients had pretreatment tumor genomic gains in chromosomes 14q11 and 17p11 that surpassed 0.3 megabases (sensitivity 61%). Of the 17 patients with incomplete responses, 14 patients had genomic gains less than 0.3 megabases (specificity 82%) (Supplementary Table S4). Overall accuracy was 70% and AUC across all cutoffs of genomic gains was 0.72.
Table 2.
Characteristics of patients with locally advanced EAC from The Cancer Genome Atlas
| Mean ± SD* or count (%) | |||
|---|---|---|---|
| Complete response (N = 23) |
Not complete response (N = 17) |
P-value | |
| Age, years | 56 ± 8 | 60 ± 9 | 0.22 |
| Sex | 0.35 | ||
| Female | 3 (13) | 0 (0) | |
| Male | 20 (87) | 17 (100) | |
| Primary tumor location | 1.00 | ||
| Distal esophagus | 23 (100) | 17 (100) | |
| Gastroesophageal junction | 0 (0) | 0 (0) | |
| Tumor grade | 0.10 | ||
| 1 | 5 (22) | 0 (0) | |
| 2 | 10 (43) | 13 (76) | |
| 3 | 7 (30) | 3 (18) | |
| Unknown | 1 (4) | 1 (6) | |
| Pretreatment AJCC 7 overall clinical stage | 0.72 | ||
| II | 15 (65) | 12 (71) | |
| III | 8 (35) | 5 (29) | |
| Neoadjuvant chemotherapy | 0.63 | ||
| Platinum agent/Fluoropyrimidine | 12 (52) | 7 (41) | |
| Platinum agent/Taxane | 2 (9) | 3 (18) | |
| Carboplatin/Gemcitabine | 1 (4) | 0 (0) | |
| Unknown | 8 (35) | 7 (41) | |
| Neoadjuvant radiotherapy dose, Gy | 48.7 ± 2.6 | 49.3 ± 1.5 | 0.75 |
*Standard deviation.
DISCUSSION
We performed genome-wide assessment of pretreatment CNAs in locally advanced EAC and analyzed their correlation with pCR/pPPR status following neoadjuvant chemoradiation, seeking to develop a method that could prospectively identify patients who may or may not need esophagectomy. Multiple methods of analyzing CNA data were attempted. We first compared basic CNA types (i.e. quantities of CNA gains, CNA losses, and total CNA events). We also attempted to identify whether CNA events affecting specific genes or gene sets may be predictive of pCR/pPPR status. However, even after enforcing P < 0.05 and FDR < 0.05 in the training sets during threefold cross-validation, the top identified CNA events as well as CNA-involved genes and gene sets emerged as poor predictors of neoadjuvant therapy response in the test sets; the importance of validation accuracy supersedes that of association significance observed in the training sets. CNA events in local genomic regions (i.e. cytobands) fulfilled this priority. We show that genomic gains in the loci 14q11 and 17p11 exceeding 0.3 megabases were able to predict pathologic complete response with sensitivity of 63%, specificity of 89%, and average AUC of 0.80 in the cross-validation test sets.
In addition to ensuring internal consistency, we externally validated our findings using TCGA. Among patients with locally advanced EAC in TCGA, genomic gains in the loci 14q11 and 17p11 predicted complete clinical and radiographic response following neoadjuvant chemoradiation with sensitivity of 61% and specificity of 82%, using the same stratification cutoff of 0.3 megabases, which was fixed a priori. Certain differences between our discovery dataset and the available data in TCGA may explain the small difference in prediction performance. Our discovery patient cohort features the curated extremes of either pCR or pPPR, whereas TCGA consists of a general population with locally advanced EAC. Many patients in TCGA would be expected to exhibit treatment responses ‘in between’ pCR and pPPR. It is possible that differences in neoadjuvant chemotherapy may have also contributed to the difference in prediction performance. Platinum/taxane regimens predominate in our institutional dataset, whereas it appears that platinum/fluoropyrimidine regimens predominate in TCGA. However, this may not necessarily be true given that chemotherapy information is missing for over one-third of the patients in TCGA. Moreover, regardless of the differences in neoadjuvant chemotherapy in the context of fairly similar neoadjuvant radiotherapy doses across several studies, the rate of pCR has been stable at 25–29% over the last two decades.7,30,31 Platinum/taxane and platinum/fluoropyrimidine regimens are both considered Category 1 recommendations for neoadjuvant systemic treatment of esophageal cancer by the National Comprehensive Cancer Network.32
We did explore other gene-specific relationships that have been previously reported in EAC. Patients with lower immunohistochemical indices of ALDH1A1 and Gli1 were shown to be more likely to have pCR. Higher levels of CCL28 and DKK3 mRNA as measured by polymerase chain reaction were also associated with pCR.12–14 These expression-based findings appear to be independent of our CNA findings, as none of the genes localize to either 14q11 or 17p11.
We also analyzed the association of pretreatment clinicopathologic features with treatment response to potentially build a multivariate model to predict pCR in EAC. However, none of the evaluated variables were significantly associated with treatment response status. Therefore, our predictive model remains simple: if genomic gains in chromosomes 14q11 and 17p11 exceed 0.3 megabases, infer pCR; otherwise infer pPPR.
It is biologically interesting that the quantity and location of genomic gains in various loci appear to be more influential than the identities of CNA-affected genes for inferring EAC treatment response following neoadjuvant chemoradiation. It is possible that accumulated gains at specific parts of the architecturally complex three-dimensional genome may be triggering destabilizing cellular mechanisms in a manner not yet understood. On the other hand, there are many ways for a set of CNA-affected genes to achieve a given quantity of gains (e.g. 300,000 base-pairs); no pattern of gene identities is necessary, as long as a critical sum of gained base-pairs is reached. Similarly, total mutation burden in melanomas, non-small cell lung cancer, and urothelial carcinomas have been shown to confer favorable response to immune checkpoint inhibitors33–35; however, a common or consistent mutated set of genes/neoantigens has yet to be identified.36
Chromosomal instability, leading to prominent genomic heterogeneity that is characteristic of EAC, may both promote and hinder tumor survival and growth. For example, estrogen receptor-negative breast cancers with extreme chromosomal instability (highest quartile) were found to have better prognosis compared to their counterparts in the bottom three quartiles.37 The paradoxical association between extreme chromosomal instability and improved prognosis has also been observed in gastric, ovarian, and non-small cell lung cancers.37 Ionizing radiation can increase chromosomal instability by introducing mitotic chromosome segregation errors, leading to aneuploidy and decreased cell viability.38 These data suggest that cancer cells may tolerate only a certain level of chromosomal instability beyond which there is a deleterious effect on cell viability, and this effect may be augmented by antineoplastic treatments.
Several studies have explored the relationship between CNA and survival in EAC with encouraging but variable results.19,21,39 A significant negative correlation between the number of CNA and overall survival was shown using targeted screening of chromosomal and gene loci.19 Likewise, the highly aberrant genomes of EAC cells that have metastasized to bone or lymph nodes are hallmarks of poorer overall survival.22 In contrast, genome-wide screening using SNP arrays showed that EAC tumors in the lowest or highest quartiles of segmental copy number gains and losses had significantly better overall survival compared to those in the middle quartiles. This suggests that sensitivity to neoadjuvant therapy in EAC may depend on the levels of CNA.20 Findings from our study align more closely with this latter hypothesis, and advance the field by implicating specific genomic regions that may be most important.
Our study has several limitations. It is retrospective and the possibility of selection bias cannot be excluded. In addition, multiple DNA samples did not pass OncoScan quality control. This deficiency was likely due to degradation of DNA in older stored tissues; this problem should not be encountered in freshly fixed biopsy specimens. Also, many patients with EAC from TCGA had incompletely annotated data, which could have led to selection bias as well. Furthermore, TCGA did not document pathologic treatment response, as in our discovery cohort, only clinical and radiographic treatment response. Finally, the discovery and validation cohorts had small sample sizes and additional larger scale prospective studies are warranted.
The strengths of our study include the discovery of a novel approach to pCR prediction, grounded in the biology of EAC that is characterized by importance of CNA. Other strengths of our study are rigorous statistical analysis of the data and validation of the results in an independent cohort of patients that included independent detection of CNA.
In conclusion, the data suggest that genomic gains in chromosomes 14q11 and 17p11 may be useful for prediction of pCR or pPPR in patients with locally advanced EAC. To our knowledge, it is the first such biomarker validated in an independent cohort of patients. Clinical interrogation of this CNA signature may open the possibility in the future to help tailor the indication for esophagectomy in locally advanced EAC. We believe that our study is an important next step in the pursuit of personalized approaches to management of EAC, opening an avenue for preclinical large-scale prospective validation studies.
Funding
This work was supported by Dartmouth-Hitchcock Norris Cotton Cancer Center Prouty Pilot Developmental Funds to Joel Lefferts, Bassem Zaki, and Mikhail Lisovsky and by funds from the Department of Pathology and Laboratory Medicine.
Conflict of Interest
None.
Data availability
Research data used in this study is publicly available at https://bit.ly/3D4NarN.
Supplementary Material
Contributor Information
David C Qian, Department of Radiation Oncology, Winship Cancer Institute of Emory University, Atlanta, GA, USA.
Joel A Lefferts, Department of Pathology and Laboratory Medicine, Dartmouth Hitchcock Medical Center, Lebanon, NH, USA.
Bassem I Zaki, Department of Medicine, Dartmouth-Hitchcock Medical Center, Lebanon, NH, USA.
Elizabeth B Brickley, Department of Infectious Disease Epidemiology, London School of Hygiene & Tropical Medicine, London, UK.
Christopher R Jackson, Department of Pathology and Laboratory Medicine, Dartmouth Hitchcock Medical Center, Lebanon, NH, USA.
Juliana Andrici, Department of Pathology and Laboratory Medicine, Dartmouth Hitchcock Medical Center, Lebanon, NH, USA.
Aravindhan Sriharan, Department of Pathology and Laboratory Medicine, Dartmouth Hitchcock Medical Center, Lebanon, NH, USA.
Mikhail Lisovsky, Department of Pathology and Laboratory Medicine, Dartmouth Hitchcock Medical Center, Lebanon, NH, USA.
References
- [1]. Pohl H, Sirovich B, Welch H G. Esophageal adenocarcinoma incidence: are we reaching the peak? Cancer Epidemiol Biomarkers Prev 2010; 19(6): 1468–70. 10.1158/1055-9965.EPI-10-0012. [DOI] [PubMed] [Google Scholar]
- [2]. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- [3]. Hongo M, Nagasaki Y, Shoji T. Epidemiology of esophageal cancer: orient to occident. Effects of chronology, geography and ethnicity. J Gastroenterol Hepatol 2009; 24: 729–35. [DOI] [PubMed] [Google Scholar]
- [4]. Walsh T N, Noonan N, Hollywood D et al. A comparison of multimodal therapy and surgery for esophageal adenocarcinoma. N Engl J Med 1996; 335: 462–7. [DOI] [PubMed] [Google Scholar]
- [5]. Gebski V, Burmeister B, Smithers B M, Foo K, Zalcberg J, Simes J. Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. Lancet Oncol 2007; 8: 226–34. [DOI] [PubMed] [Google Scholar]
- [6]. Alnaji R M, Du W, Gabriel E et al. Pathologic complete response is an independent predictor of improved survival following neoadjuvant chemoradiation for esophageal adenocarcinoma. J Gastrointest Surg 2016; 20: 1541–6. [DOI] [PubMed] [Google Scholar]
- [7]. Schwameis K, Zehetner J, Hagen J A et al. Esophageal adenocarcinoma stage III: survival based on pathological response to neoadjuvant treatment. Surg Oncol 2017; 26: 522–6. [DOI] [PubMed] [Google Scholar]
- [8]. Bronson N W, Diggs B S, Bakis G et al. Molecular marker expression is highly heterogeneous in esophageal adenocarcinoma and does not predict a response to neoadjuvant therapy. J Gastrointest Surg 2015; 19: 2105–10. [DOI] [PubMed] [Google Scholar]
- [9]. Gaur P, Hunt C R, Pandita T K. Emerging therapeutic targets in esophageal adenocarcinoma. Oncotarget 2016; 7: 48644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Ajani J A, Correa A M, Hofstetter W L et al. Clinical parameters model for predicting pathologic complete response following preoperative chemoradiation in patients with esophageal cancer. Ann Oncol 2012; 23: 2638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]. Wu A J, Goodman K A. Clinical tools to predict outcomes in patients with esophageal cancer treated with definitive chemoradiation: are we there yet? J Gastrointest Oncol 2015; 6: 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. Ajani J A, Wang X, Song S et al. ALDH-1 expression levels predict response or resistance to preoperative chemoradiation in resectable esophageal cancer patients. Mol Oncol 2014; 8: 142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Wadhwa R, Wang X, Baladandayuthapani V et al. Nuclear expression of Gli-1 is predictive of pathologic complete response to chemoradiation in trimodality treated oesophageal cancer patients. Br J Cancer 2017; 117: 648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. McLaren P J, Barnes A P, Terrell W Z et al. Specific gene expression profiles are associated with a pathologic complete response to neoadjuvant therapy in esophageal adenocarcinoma. Am J Surg 2017; 213:915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Ross-Innes C S, Becq J, Warren A et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat Genet 2015; 47: 1038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Secrier M, Li X, de Silva N et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat Genet 2016; 48: 1131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Cancer Genome Atlas Research N, Analysis Working Group: Asan U, Agency B C C et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017; 541: 169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Nones K, Waddell N, Wayte N et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun 2014; 5: 5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Pasello G, Agata S, Bonaldi L et al. DNA copy number alterations correlate with survival of esophageal adenocarcinoma patients. Mod Pathol 2009; 22: 58–65. [DOI] [PubMed] [Google Scholar]
- [20]. Davison J M, Yee M, Krill-Burger J M et al. The degree of segmental aneuploidy measured by total copy number abnormalities predicts survival and recurrence in superficial gastroesophageal adenocarcinoma. PLoS One 2014; 9: e79079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Frankel A, Armour N, Nancarrow D et al. Genome-wide analysis of esophageal adenocarcinoma yields specific copy number aberrations that correlate with prognosis. Genes Chromosomes Cancer 2014; 53: 324–38. [DOI] [PubMed] [Google Scholar]
- [22]. Schumacher S, Bartenhagen C, Hoffmann M et al. Disseminated tumour cells with highly aberrant genomes are linked to poor prognosis in operable oesophageal adenocarcinoma. Br J Cancer 2017; 117: 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Becker K, Mueller J D, Schulmacher C et al. Histomorphology and grading of regression in gastric carcinoma treated with neoadjuvant chemotherapy. Cancer 2003; 98: 1521–30. [DOI] [PubMed] [Google Scholar]
- [24]. Schmidt T, Sicic L, Blank S et al. Prognostic value of histopathological regression in 850 neoadjuvantly treated oesophagogastric adenocarcinomas. Br J Cancer 2014; 110: 1712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nexus Express, 2013. https://assets.thermofisher.com/TFS-Assets/LSG/manuals/oncoscan_nexus_express_software_manual.pdf.
- [26]. Burrell R A, McClelland S E, Endesfelder D et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013; 494: 492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 1995; 57: 289–300. [Google Scholar]
- [28]. Deng M, Brägelmann J, Kryukov I, Saraiva-Agostinho N, Perner S. FirebrowseR: an R client to the broad Institute's firehose pipeline. Database (Oxford) 2017; 2017. 10.1093/database/baw160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Ivakhno S, Tavaré S. CNAnova: a new approach for finding recurrent copy number abnormalities in cancer SNP microarray data. Bioinformatics 2010; 26: 1395–402. [DOI] [PubMed] [Google Scholar]
- [30]. Chirieac L R, Swisher S G, Ajani J A et al. Posttherapy pathologic stage predicts survival in patients with esophageal carcinoma receiving preoperative chemoradiation. Cancer 2005; 103: 1347–55. [DOI] [PubMed] [Google Scholar]
- [31]. de Jongh M, Eyck B M, van der Werf L R et al. Pattern of recurrence in patients with a pathologically complete response after neoadjuvant chemoradiotherapy and surgery for oesophageal cancer. BJS Open 2021; 5: zrab022. 10.1093/bjsopen/zrab022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Ajani J A, D’Amico T A, Bentrem D J et al. Esophageal and Esophagogastric junction cancers, version 2.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2019; 17: 855–83. [DOI] [PubMed] [Google Scholar]
- [33]. Snyder A, Makarov V, Merghoub T et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371: 2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Rizvi N A, Hellmann M D, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348: 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Rosenberg J E, Hoffman-Censits J, Powles T et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016; 387: 1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Garcia-Garijo A, Fajardo C A, Gros A. Determinants for Neoantigen Identification. Front Immunol 2019; 10: 1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Birkbak N J, Eklund A C, Li Q et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 2011; 71: 3447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Bakhoum S F, Kabeche L, Wood M D et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat Commun 2015; 6: 5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Goh X Y, Rees J R, Paterson A L et al. Integrative analysis of array-comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut 2011; 60: 1317–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Research data used in this study is publicly available at https://bit.ly/3D4NarN.