

Abstract
Loss-of-function mutations of the X-chromosome gene UPF3B cause male neurodevelopmental disorders (NDDs) via largely unknown mechanisms. We investigated initially by interrogating a novel synonymous UPF3B variant in a male with absent speech. In silico and functional studies using cell lines derived from this individual show altered UPF3B RNA splicing. The resulting mRNA species encodes a frame-shifted protein with a premature termination codon (PTC) predicted to elicit degradation via nonsense-mediated mRNA decay (NMD). UPF3B mRNA was reduced in the cell line, and no UPF3B protein was produced, confirming a loss-of-function allele. UPF3B is itself involved in the NMD mechanism which degrades both PTC-bearing mutant transcripts and also many physiological transcripts. RNAseq analysis showed that ~1.6% of mRNAs exhibited altered expression. These mRNA changes overlapped and correlated with those we identified in additional cell lines obtained from individuals harbouring other UPF3B mutations, permitting us to interrogate pathogenic mechanisms of UPF3B-associated NDDs. We identified 102 genes consistently deregulated across all UPF3B mutant cell lines. Of the 51 upregulated genes, 75% contained an NMD-targeting feature, thus identifying high-confidence direct NMD targets. Intriguingly, 22 of the dysregulated genes encoded known NDD genes, suggesting UPF3B-dependent NMD regulates gene networks critical for cognition and behaviour. Indeed, we show that 78.5% of all NDD genes encode a transcript predicted to be targeted by NMD. These data describe the first synonymous UPF3B mutation in a patient with prominent speech and language disabilities and identify plausible mechanisms of pathology downstream of UPF3B mutations involving the deregulation of NDD-gene networks.
Introduction
Hemizygous loss-of-function mutations in UPF3B cause male intellectual disability of variable severity associated with a spectrum of non-penetrant behavioural disturbances including attention deficient hyperactive disorders (ADHD), childhood-onset schizophrenia (COS) and autism (OMIM: 300298) (1–6). UPF3B encodes a core factor in the nonsense-mediated mRNA decay (NMD) pathway (7,8). NMD degrades mutant transcripts bearing premature termination codons (PTCs). More recently, it was discovered that NMD also regulates up to 20% of the normal transcriptome and is essential in brain development and function (7,8). Here we experimentally resolve the pathogenicity of a synonymous variant identified via whole-exome sequencing in UPF3B in an individual with absent speech. The impact of this UPF3B variant on the individual’s transcriptome, compared to that we discovered in additional individuals with UPF3B mutations, led us to postulate new mechanisms of pathology underpinning UPF3B-associated neurodevelopmental disorders (NDDs).
The initial subject of this study was a Caucasian male born through an uneventful pregnancy following a non-consanguineous union. The individual’s older sister developed normally. The mother reportedly had minor motor delay as a child. The clinical features of the affected male were previously described (9). In brief, the major findings included (at age 32 months) motor and fine motor delay, absence of speech and delayed receptive language and, later (48 months), communication only via single words and gestures, accompanied with a formal diagnosis of autism. He was of normal height (75th percentile), normal head circumference (50th percentile), low weight (5–10th percentiles), low muscle tone and displayed joint hypermobility. Facial dysmorphisms included a prominent forehead and chin, deep-set eyes and a long thin face (9).
The subject’s genetic inquiry began with high-resolution (60 K) microarray-based comparative genomic hybridization, which identified a maternally inherited duplication at Xq26.3 spanning ~650Kb (9). Duplications overlapping this region have been identified in individuals with developmental delay phenotypes; however, in all cases where investigated, the duplication was assessed as benign or likely benign (9). Duplication of Xq26.3 is however associated with X-linked acrogigantism (X-LAG), but the individual did not display the elevated growth parameters and endocrine features that are its hallmark (9,10). Furthermore, the individuals’ Xq26.3 duplication is mapped outside the X-LAG minimal critical region and did not contain GPR101, strongly supported as the driver of the disease (9–12). As such, the Xq26.3 duplication was classified as likely benign. Subsequent whole-exome sequencing is performed as part of the Deciphering Developmental Disorders UK (DDD UK) study (13) which revealed a single variant with potential relevance to the individual’s symptoms, a maternally inherited novel synonymous variant in UPF3B.
In this study, we show that the synonymous UPF3B variant impacts a canonical splice donor site in UPF3B, resulting in an alternative splicing event that culminates in a non-functional allele and compromised NMD. This is the first synonymous change identified in UPF3B that causes a NDD, and herein supports UPF3B, and NMD in general, as an essential pathway involved in motor development, speech, language and communication. Our transcriptomic analysis of lymphoblastoid cell lines (LCLs) from this and additional UPF3B patients provide evidence of NMD-regulated NDD gene networks, providing new insights into pathological mechanisms responsible for neurological disease.
Results
A synonymous UPF3B variant that alters a canonical splice site
In addition to the individual’s likely benign Xq26.3 duplication encompassing 10 protein coding and 6 non-coding genes, whole-exome sequencing revealed a single synonymous variant of unknown significance in UPF3B: NC_000023.10 g.118975698C > T; NM_080632.3, c.624G > A; and NP_542199.3, p.(Gln208=) (Supplementary Material, Fig. S1). This variant is absent from the GnomAD and dbSNP databases. Examination of this variant revealed it altered the highly conserved −1 position of the 5′ canonical spice donor site of exon 6 (Supplementary Material, Fig. S2a). Canonical splice acceptor site (NYAG/G) and donor site (CAG/GUAAGU) sequences are strongly conserved and defined exon–intron and intron–exon boundaries, respectively (14). Variation from the canonical sequences can induce alternative splicing events (15). To investigate if the c.624G > A variant might disrupt splicing of UPF3B mRNA, we first assessed its impact using in silico variant and splicing predictive algorithms (Supplementary Material, Fig. S2b) (16–21). The CADD score of the c.624G > A variant was 20.4, classifying it amongst the top 1% of deleterious variants. All algorithms predicted a loss of the wild-type 5′ donor splice site. Algorithms reporting on ‘splice site strength’ (Human Splicing Finder, MaxEnt and Neural Network Splice Predictor) predicted the variant splice site to be less efficient than wild type (Supplementary Material, Fig. S2b).
The synonymous UPF3B variant alters UPF3B splicing
A lymphoblastoid cell line (LCL) was derived from the affected individual to experimentally assess UPF3B mRNA splicing. Human UPF3B gives rise to two protein-encoding isoforms: a long isoform (NM_080632.3) consisting of 11 exons (1452 bp) and an alternatively spliced shorter isoform (NM_023010.3) arising due to exon 8 skipping (in-frame, 1413 bp). Both isoforms are affected by the c.624G > A variant as it is positioned in exon 6 upstream of exon 8 (Fig. 1A). Variants at 5′ donor splice sites can lead to single exon skipping but can also result in the use of alternative ‘cryptic’ splice sites. To determine the consequence of UPF3B c.624G > A on splicing, cDNA was reverse transcribed from RNA of LCLs derived from control individuals and the affected individual and used as a template in a UPF3B-specific PCR. The primers amplified a product from exon 5 to exon 9, spanning the variant site (Fig. 1A). In control cell lines, PCR products representing both long and short isoforms were detected as expected, with the shorter isoform being most prominently expressed (Fig. 1B). The long and short isoform PCR products amplified from transcripts containing the UPF3B variant c.624G > A were both reduced in size and appeared reduced in intensity (Fig. 1B). Sanger sequencing of these PCR products revealed complete exclusion of exon 6 (44 bp) (Fig. 1C). These alternatively spliced transcripts lacking exon 6 are herein referred to collectively as UPF3BΔEx6 mRNA.
Figure 1 .
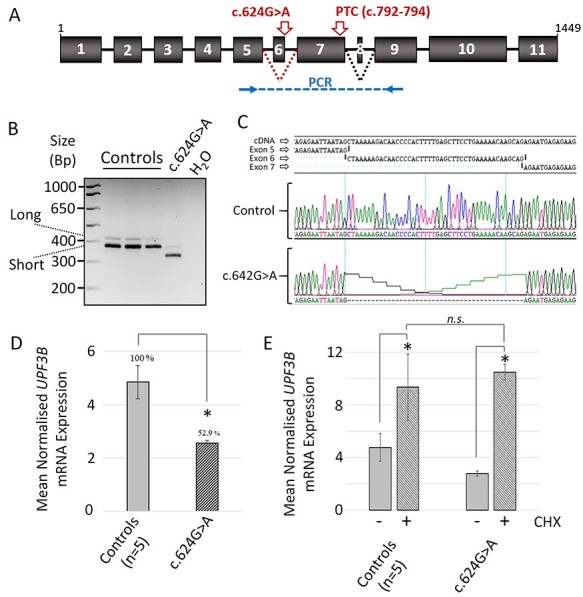
The c.624G > A UPF3B variant results in loss of exon 6 from UPF3B mRNA associated with reduced mRNA levels. (a) A schematic representation of UPF3B pre-mRNA structure. Black dotted lines represent alternative splicing of exon 8 which produces two major UPF3B isoforms (long and short). Position of c.624G > A variant shown in red. Red dotted line represents scenario of exon 6 skipping and the position of the premature termination codon (PTC) that would result. Position of PCR primers used in B is shown in blue. (b) Reverse transcribed cDNA from the RNA of LCLs derived from male control individuals (n = 3), and male individual with c.624G > A variant was subject to PCR using primers shown in A. Expected sizes of the UPF3B long and short isoforms are shown. (c) Sanger sequencing of PCR products in B. Data confirms the UPF3B c.624G > A variant mRNA species lack exon 6 (44 bp in length). (d) Quantitative PCR (qPCR) of UPF3B mRNA in LCLs. Data normalized to GAPDH mRNA expression. (e) Cycloheximide (CHX) treatment of LCLs restores UPF3B c.624G > A variant mRNA to control levels. LCLs were treated for 6 h with or without cycloheximide prior to isolation of RNA. qPCR analysis of UPF3B mRNA normalized to GAPDH. Note UPF3B mRNA from control LCLs is also upregulated also in response to cycloheximide treatment. qPCR data (D and E) represent the mean of triplicate experiments +/− standard deviation, *P < 0.05 by Student’s t-test; n.s.: not significant.
UPF3B ΔEx6 mRNA is expressed at a reduced level
The UPF3BΔEx6 mRNA encodes a frameshift and introduces a premature termination codon (PTC) within exon 7 (p.Ala194fs*57). This PTC is located >55 nt upstream of the terminal exon–exon junction and is thus predicted to be degraded by NMD (Fig. 1A) (7). But as UPF3B itself is a core factor of the NMD machinery, the NMD process may be inefficient (22,23). Quantitative real-time PCR was performed to compare the levels of UPF3B and UPF3BΔEx6 mRNA in control and c.624G > A LCLs, respectively (Fig. 1D). UPF3BΔEx6 mRNA was reduced by 47% compared to control UPF3B mRNA. To investigate if this downregulation depends on NMD, cells were incubated with cycloheximide, an inhibitor of translation and thus NMD (1,24). Cycloheximide treatment elevated the levels of UPF3BΔEx6 mRNA suggestive of NMD involvement (Fig. 1E). Cycloheximide treatment also modestly elevated the levels of wild-type UPF3B mRNA, a phenomena which has been previously reported (1,4), and likely reflects the NMD self-regulatory feedback mechanism in which the mRNAs encoding several NMD factors including UPF3B are themselves physiological NMD targets (25).
UPF3B protein is not detectably expressed from UPF3B ΔEx6 mRNA
Because ~53% of UPF3BΔEx6 mRNA persists in LCLs (Fig. 1D), these variant transcripts may be translated into a truncated protein (of predicted ~29.7 kDa size for both short and long isoforms; Fig. 2A). To investigate, protein was isolated from control and c.624G > A variant LCLs. As an additional control, protein was isolated from LCLs derived from an individual with a known loss-of-function UPF3B variant (c.867_868delAG; p.Gly290fs*2) (1). This individual has mild intellectual disability as previously described [Family 2, Individual III-5 in (1)]. Proteins were separated by polyacrylamide gel electrophoresis and analysed by western blot. To assess if any full-length or C-terminally truncated UPF3B proteins were expressed in the variant LCLs, an antibody to the N-terminus of UPF3B was used (Fig. 2B). Whilst full-length UPF3B protein was observed in control LCLs, no UPF3B peptides of any size were detected in LCLs harbouring either the c.624G > A or c.867_868delAG UPF3B variants. We further confirmed loss of full-length UPF3B using an antibody which binds a C-terminal epitope downstream of the PTCs (Fig. 2C). In the absence of UPF3B protein, the levels of the paralogous protein UPF3A are known to become elevated and proposed to support residual NMD activity (23,26). Consistent with this, UPF3A protein was upregulated in both the c.624G > A and c.867_868delAG variant LCLs compared to control LCLs (Fig. 2C). Of note also, the level of UPF3A protein, in particular it’s shorter isoform, was greater in the c.867_868delAG compared to the c.624G > A variant LCLs, which in support of previous studies inversely related with the severity of neurological features these two individuals (23). These data demonstrate that the UPF3BΔEx6 transcript fails to detectably express either full-length or truncated UPF3B protein species, and as such, the c.624G > A UPF3B synonymous variant is a loss-of-function allele.
Figure 2 .
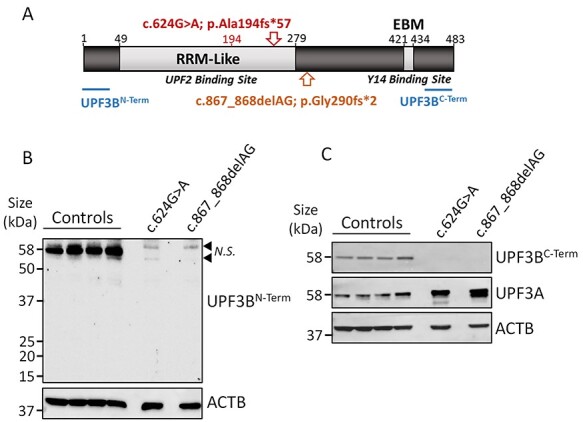
UPF3B protein is not expressed from UPF3BΔEx6 mRNA. (a) Schematic of UPF3B protein. Locations of the RRM-Like and EBM domains shown in light grey. The protein encoded by UPF3BΔEx6 mRNA as a result of the c.624G > A variant is shown in red, along with position of p.Ala194 and position of the premature termination codon (PTC; red arrow). Position of the PTC produced from mRNA housing the c.867_868delAG variant is shown in yellow. Epitopes of antibodies used in B and C are shown in blue. (b and c) Western-blot analysis of UPF3B protein isolated from LCLs derived from male control individuals (n = 4) and individuals harbouring the c.624G > A variant and the c.867_868delAG variant. ACTB used as a loading control. (b) UPF3B identified using an N-terminal antibody. Black arrow heads identify non-specific (N.S.) bands. (c) UPF3B identified using a C-terminal antibody. Blot was stripped and re-probed for UPF3A.
Identification of mRNAs misregulated in response to loss-of-function UPF3B
As aforementioned, UPF3B is a core NMD factor that promotes the decay of both mutant transcripts harbouring PTCs and endogenous mRNA species which harbour particular ‘NMD-targeting features’ (27). Such features include upstream reading frames, introns within 3’UTRs, PTC-like codons introduced via alternative splicing events and long (>1.5Kb) 3’UTRs (27). GADD45B, ATF4 and GAS5 mRNA have all been shown to be targeted by NMD in different contexts (1,22,25,28–30). We assessed the expression levels of these three mRNAs as an initial proxy for NMD activity in control and UPF3B c.624G > A variant LCLs (Fig. 3A). All three of these NMD target mRNAs were shown to be upregulated in the c.624G > A variant LCL compared to controls, with increases ranging from ~30% (GADD45B) to 160% (GAS5). These data are aligned with reduced NMD capacity in c.624G > A variant LCLs.
Figure 3 .
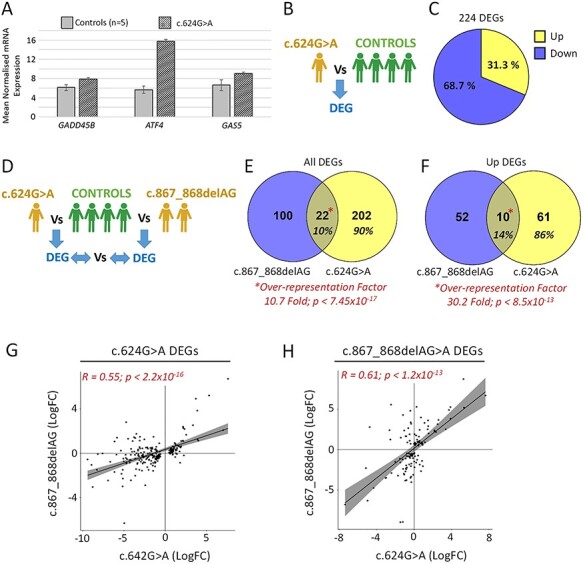
LCLs harbouring the c.624G > A variant show evidence of compromised NMD. (a) qPCR of NMD target genes GADD45B, ATF4 and GAS5 using RNA isolated from LCLs of control individuals (n = 5) and the individual with c.624G > A UPF3B variant. Data normalized to GAPDH expression. (b) Schematic of RNAseq comparison used in C; DEG: differentially expressed gene (fold change >2; P < 0.05). (c) Proportion of DEGs found up- and downregulated. (d) Schematic of RNAseq comparison used in E–F. (e and f) Overlap of DEGs and upregulated DEGs found in c.624G > A and c.867_868delAG variant LCLs. Transcriptome-wide over-representation factors provided; P-values represent an exact hypergeometric probability test, (g) Scatter plot and Pearson’s correlation of the expression of c.624A > G DEGs in c.867_868delAG LCLs. (h) Scatter plot and Pearson’s correlation of expression of c.867_868delAG DEGs in c.624A > G LCLs.
To investigate the potential defect in NMD further using a transcriptome-wide approach, RNAseq was conducted on RNA isolated from the c.624G > A variant LCLs and four male control LCLs. This identified differentially expressed genes (DEGs; fold change >2; P < 0.05; Fig. 3B, Supplementary Material, Fig. S3, Online Resource 1). Of the 224 DEGs (= ~1.6% of the LCL transcriptome), 31% were observed to be upregulated (Fig. 3C). Next we aimed to establish that this transcriptomic deregulation aligns with loss of UPF3B-dependent NMD. We compared the DEG profile of the c.624G > A variant LCL against DEG profiles obtained from LCLs from two brothers harbouring the known pathogenic UPF3B variant c.867_868delAG and four normal individuals (controls) (=122 DEGs; Fig. 3D, Supplementary Material, Fig. S3, Online Resource 1) (1). Although the c.867_868delAG and c.624G > A UPF3B variants are distinct, they both ultimately result in the complete loss of UPF3B protein [Fig. 2B and C and (1)]. We found 10% of DEGs (=22 genes) overlapped between the two UPF3B variant DEG sets, equating to a 10.7-fold over-representation (P = 7.45x10−17 exact hypergeometric probability test; Fig. 3E). In addition to UPF3B, this list included three other known NDD genes (ALDH7A1, DCHS1 and TBL1X; Online Resource 1). The percentage of overlapping DEGs was slightly higher (14%) when restricting analysis to upregulated DEGs, which equated to a 30.2-fold over-representation (P = 8.5 × 10−13 exact hypergeometric probability test; Fig. 3F). We next sought to correlate the expression of each of the DEG sets independently between the two UPF3B variant LCL transcriptomes. The expression of c.624A > G DEGs in c.867_868delAG LCLs was found to be significantly and positively correlated, and vice versa, the expression of c.867_868delAG DEGs in the c.624A > G LCL was also significantly and positively correlated (Fig. 3G–H). Thus, the DEGs between the different UPF3B variant transcriptomes were significantly overlapped, and in general, the direction and magnitude of transcriptional change of DEGs from both variant lines significantly correlated. Collectively, these data demonstrate reduced NMD function in p.624A > G LCLs.
UPF3B-dependent NMD regulates NDD gene networks
The finding that all three UPF3B variant cell lines lack detectable UPF3B protein and exhibit an overlapped and correlated pattern of mRNA deregulation provided an opportunity to identify a high-confidence UPF3B-dependent gene network. To this end, we grouped all three UPF3B variant transcriptomes together to provide more robust identification of DEGs related to loss of UPF3B (Fig 4A, Online Resource 1). We identified 102 DEGs in common to these 3 LCLs lacking UPF3B (~0.7% of the LCL transcriptome; Fig. 4B). Half of the DEGs were upregulated, and half were downregulated. Upregulated mRNAs are candidates to be direct NMD targets. In support that many of these upregulated mRNAs may be direct targets, >75% of them had known NMD-inducing feature, compared with only ~35% of the downregulated mRNAs (Fig. 4C). In total there were 40 upregulated genes, and 83 transcripts associated with those genes, which have an NMD-targeting feature consisting of either a long 3′UTRs (>1.2 kb), an upstream open reading frame (uORFs) or an exon–exon junction downstream of physiological termination codons (dEJs; Online Resource 1). These genes, and their NMD-transcripts, are thus high-confidence UPF3B-NMD targets. Half of these genes expressed multiple transcript species with NMD features, and >30% of the NMD-targeted transcripts had multiple NMD-targeting features (Fig. 4D). We used these high-confidence UPF3B-NMD-targeted transcripts to explore how UPF3B might select its mRNA targets. The predominant NMD-targeting features were a long 3′UTR (found in 65.1% of transcripts) or uORFs (54.3%). The presence of a dEJs (15.6%) was the least prevalent, and 31.4% of transcripts had multiple combinations of features (Fig. 4D). These data identify high-confidence UPF3B-NMD targets and suggest that long 3′UTRs and uORF are the most frequent NMD-targeting features that direct degradation via UPF3B-NMD in LCLs.
Figure 4 .
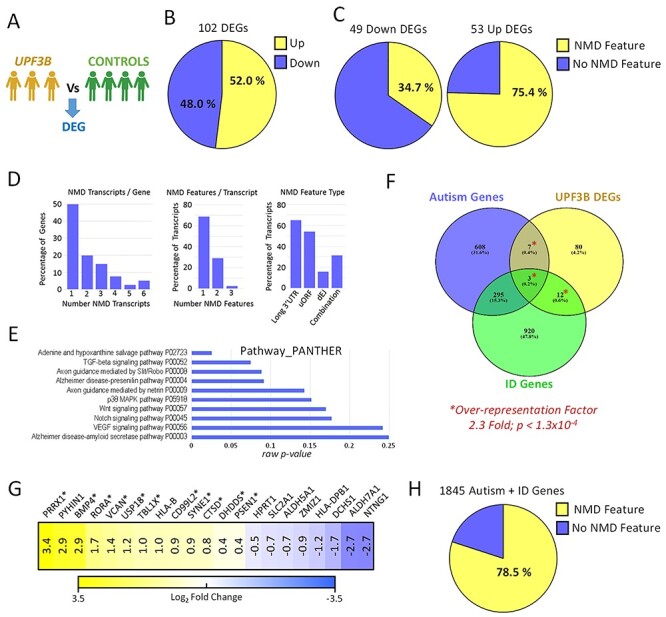
UPF3B-dependent NMD regulates networks of neurodevelopmental disorder genes. RNA was isolated from LCLs derived from four male control individuals and three individuals harbouring loss-of-function UPF3B variants including c.624G > A (singleton) and c.867_868delAG (brothers). (a) Schematic of RNAseq comparison applied. DEG: differentially expressed gene (fold change >2; P < 0.05). (b) Proportion of DEGs found up- and downregulated. (c) Percentage of up- and downregulated DEGs with transcripts harbouring NMD-targeting mRNA features. (d) Analysis of the 40 upregulated genes which express transcripts harbouring NMD-targeting features (NMD transcripts). (e) Gene pathway analysis of the 102 DEGs. Raw P-values are graphed. Note adjusted P-values for all data displayed are >0.05 (not significant). (f) Overlap of DEGs with known autism and intellectual disability (ID) genes (collectively termed neurodevelopmental disorders genes; NDD genes). Transcriptome wide over representation factor is provided. P-value represents an exact hypergeometric probability test. (g) Expression of NDD-related DEGs in UPF3B mutant LCLs. Asterisk designates upregulated NDD genes with NMD-targeting features. (h) Percentage of autism and ID related NDD genes with transcripts that harbour NMD-targeting features.
We performed gene ontology and gene pathway analysis using various different algorithms (DAVID, ENRICHR, GOSEQ, CLUSTER PROFILE) (31–34) on the 102 genes significantly differentially expressed between the 3 UPF3B variant LCLs and the control LCLs. Almost universally, these algorithms failed to identify statistically enriched terms or pathways (adjusted P-values were >0.05; Fig. 4E, and data not shown), probably because of the small number of DEGs (102) that were analysed. The one exception was an enrichment of genes involved in the ‘immune response’ biological pathway (Online Resource 1). We then manually inspected the DEGs and found that 27 of them were associated with OMIM terms, 12 of which involving neurological manifestation (Online Resource 1). We then tested whether NDD genes known to be expressed in LCLs were significantly enriched (see Materials and Methods and Online Resource 1). A total of 22/102 DEGs were known intellectual disability and/or autism genes, which equated to a 2.2-fold genome wide over-representation (P = 3.3 x ×10−4 via Exact Hypergeometric Probability test; Fig. 4F). These data reveal that UPFB-dependent NMD targets a large network of NDD genes. Of these 22 NDD genes, 13 were upregulated of which 11 contained NMD-targeting features, i.e. were high-confidence direct UPF3B-NMD targets (Fig. 4G). Finally we sought to extrapolate this observation and conducted a search of NMD features in transcripts derived from all known ID and autism genes. We discovered that ~78.5% of these 1845 NDD genes contained transcripts with annotated NMD-targeting features (Fig. 4G).
Discussion
Here we report, and functionally validate, the pathogenicity of a novel synonymous variant in UPF3B in a male paediatric case with a NDD. The mechanism of action involves disruption of the 5′ splice donor site of exon 6, leading to an exon 6-skipped transcript encoding a truncated UPF3B. We could not detect this truncated UPF3B species, but even if sufficiently stable to be expressed at low level, it would lack functional UPF2- and Y14-binding sites and thus be non-functional.
Loss-of-function UPF3B mutations are an established cause of a range of X-linked NDDs, including developmental delay, intellectual disability, ADHD, COS, communication disorders, autism and obsession (1–6). Presentation of these features is highly variable, even within families. Missense variants in UPF3B can also be pathogenic and further expanding clinical variability (1,3). Here, the affected individual’s phenotype overlaps with many of the clinical features initially reported in individuals with complete loss-of-function UPF3B mutations. These phenotypes include intellectual disability, autism, slender build and poor muscle tone, a long and thin face with prominent forehead and chin and deep-set eyes (1,9). However, the most striking features of the individual in our study are the motor defects and almost complete absence of speech and language skills, overall making the clinical presentation in this individual severe. Whilst delay in motor development has been reported in another case, ongoing motor disability is a novel finding. Likewise, delays in speech and language have been reported in some individual cases (2,5), but the ongoing absence of speech and language in this individual is a much more severe outcome. Of note, the complete absence of speech was also recently described in another family with loss-of-function UPF3B mutation (6). These data thus expand the UPF3B-associated phenotypical spectrum to include motor and severe speech language and communication disabilities. Interestingly, heterozygous loss-of-function mutations in UPF2 encoding another core component of the NMD machinery have also been recently implicated in individuals with speech and language disorders (35).
Synonymous variants are historically challenging to interpret. Whilst it is widely acknowledged that synonymous SNVs can sometimes contribute to disease phenotypes by altering splicing patterns, miRNA binding, pre-mRNA structure and translation dynamics, they ultimately require experimental validation (36–44). Synonymous variants impacting splice sites, such as the one we study here, are often classified as variants of unknown significance in a diagnostic setting without functional investigations (14,15). Here, for the first time, we functionally validate a UPF3B splice variant. Another UPF3B variant (c.846 + 1G > A) affecting the splice donor site of exon 7 has been reported but awaits functional interrogation (44). This male presented with mild ID, no language capability, macrocephaly, tall stature, hypotonia, facial dysmorphisms and dilated Virchow–Robin spaces.
In multiple animal species, loss of the core NMD factors, including UPF1 and UPF2, causes embryonic lethality (45). In contrast, UPF3B is not required for survival of either mice or humans (1,46), probably because UPF3B is specifically required for a branch of the NMD pathway, not the entire pathway (22). In the absence of UPF3B, the protein product of its gene paralog, UPF3A, is dramatically stabilized, leading to its upregulation (26). Indeed, we found that to be the case for the c.624G > A LCL studied here. This has also been observed in other cell types, including those of the developing embryo and brain (23,26,28,46). In the absence of UPF3B, stabilized UPF3A is speculated to partially, but not fully, rescue NMD (23). Furthermore, the level of UPF3A stabilization has been shown to inversely correlate with disease severity across UPF3B several patients (23). Our data provides additional support of this relationship.
Multiple lines of evidence suggest that c.624G > A variant LCL has compromised NMD: (i) ~50% decrease UPF3B mutant mRNA itself (carrying a PTC) which could be rescued with a NMD inhibitor; (ii) upregulation of mRNA of the well-established NMD target mRNAs GADD45B, ATF4 and GAS5 and (iii) deregulation of ~1.6% of the transcriptome, which (iv) significantly overlapped and correlated gene expression changes with other loss-of-function UPF3B variant LCLs. Heterozygous loss-of-function mutations in UPF2 in individuals with NDDs have also been shown to have a similar molecular impact (23,35). CNVs in several NMD and exon junction complex genes, namely, UPF2, UPF3A, RBM8A, SMG6, eIF4AIII and RNPS1, are statistically enriched in individuals with NDDs also (47). Thus compromised NMD is an established cause of NDD in humans (8). In further support, multiple neural cell- and mouse-based models of brain development show a requirement of full NMD activity for normal function and learning, memory and speech in particular (8,28,35,46,48). Whilst these models have begun to elucidate some of the underlying molecular and cellular mechanisms, much remains unknown, particularly in the human patient context.
We leveraged our UPF3B loss-of-function LCLs to investigate the downstream pathogenic mechanisms by which UPF3B-NMD acts. Although LCLs are of haematopoietic origin, remarkably, their transcriptomes are similar with brain transcriptomes (23). We also found 1312/1845 NDD genes were expressed in LCLs (Online Resource 1). In the LCLs with the c.624G > A variant, FOXP1, a gene involved in speech and communication, was found deregulated (49,50). Deregulation of FOXP1 was also recently reported in LCLs of UPF2 patients, which also exhibit prominent speech and language deficits (35). By comparison, FOXP1 was not found deregulated in c.867_868delAG LCLs, whom do not present with severe speech and language features. Thus, we speculate that deregulation specifically of NMD-regulated NDD gene networks can contribute to the patient phenotype. Interestingly, a total 21 NDD genes in addition to UPF3B were significantly deregulated in LCLs from the severely affected individual with the c.624G > A variant, whilst only 6 NDD genes were found deregulated in the more mildly affected individuals with c.867_868delAG variants. By comparison, gene ontology-based approaches failed to identify any significant terms or pathways that could otherwise explain meaningful differences between the c.624G > A and c.867_868DelAG DEGs (Online Resource 1).
By integrating the RNAseq data from the independent LCL lines with loss-of-function UPF3B alleles, we identified RNAs deregulated in all three lines (representing ~0.7% of the transcriptome). The majority of the upregulated transcripts harbour NMD-inducing features, which suggests they could be direct NMD targets. Gene ontology analysis of all DEGs identified the immune response pathway as the only significantly enriched term (Online Resource 1). Intriguingly, an activated brain immune response was recently identified as the mechanism driving neurological dysfunction in Upf2 brain-specific knockout mice. Treating the mice with anti-inflammatory drugs ameliorated the phenotype (35). Other genes that were deregulated encode TGFβ signalling components and axon guidance proteins, which have been shown to be functionally linked with the role of UPF3B and/or NMD in brain development in previous studies (8,28,48,51,52). The most significant finding from our analysis was that genes involved in NDDs were over-represented amongst the UPF3B-regulated mRNAs and that a large number of these NDD mRNAs have NMD-inducing features, suggesting they may be direct NMD targets. Studies involving human brain tissue, for example, derived from patient-induced pluripotent stem cells, will be required to interrogate the phenomena of NMD-regulated NDD gene networks in detail with view of understanding the disease mechanisms.
That compromised NMD causes NDDs is clear from multiple human genetic and model system studies (1,8,35,47). Ongoing investigations of the NMD mechanism, and its downstream mRNA targets, and impact on brain function are required to resolve the key processes of pathology and allow the development of future precision therapy options. Furthermore, as NMD is as a major modifier of the 12% of all genetic diseases which feature PTC-type mutations, such knowledge may benefit our understanding of the role of NMD in genetic disease more generally (53–55).
Materials and Methods
Ethical compliance
This study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. This study was approved by the Women’s and Children’s Health Network Human Research Ethics Committee, South Australia, Australia (HREC786–07-2020), and UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). This study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed and written parental consent was obtained as previously described (9).
Whole-exome sequencing
Whole-exome sequencing was performed as part of the DDD UK as previously described (13).
Cell culture
The Epstein–Barr virus-immortalized lymphoblastoid cell lines (LCLs) were established and cultured as previously described (56).
RNA extraction, PCR and Sanger sequencing
Total RNA was obtained from cell pellets using an automated extraction protocol on the Maxwell® RSC Instrument and Maxwell® RSC simply RNA Cell kit as per manufactures instructions (Promega, Madison, WI, USA). cDNA was synthesized from 1.5 μg of RNA using random hexamer primers 5x First-Strand Buffer and Superscript III reverse transcriptase (Invitrogen, Thermo Fisher, Waltham, MA, USA).
PCR was performed using Taq DNA polymerase as per manufactures instruction (Roche, Basel, Switzerland) using cDNA template. Primers against UPF3B were Forward, TGACATCTACTCCAGAGACAC, and Reverse GGCTCTTTCATCACTGAGATTC. Cycling conditions were as follows: 94°C for 5 min, 30 cycles of 94°C for 30 s, 58°C for 30s and 72 for 10 s with a final extension at 72°C for 10 min. PCR products were subjected to sequencing reactions using Bright Dye Terminator (MCLAB, San Francisco, CA, USA) and analysed using automated capillary sequencing. Sequences were aligned using SeqMan module of the Lasergene DNA and protein analysis software package (DNAStar, Inc., Madison, WI, USA).
For quantitative PCR (qPCR), the Fast SYBR green (Applied Biosystems, Thermo Fisher) reagents were used. Reactions were run on the StepOne Plus qPCR machine (Applied Biosystems, Thermo fisher). GAPDH was used for gene normalization. Relative mRNA expression was calculated using the ΔΔCt method where Ct represents cycle threshold. Primer sequences were previously described (1).
Immunoblotting
Cells were lysed with RIPA buffer (65.3 mM Tris, 150 mM NaCl, 1%(v/v) Nonidet P-40) supplemented with 40 μL/ml of 25x protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO) and 5 μL/ml of 200 mM Na2VO4, 200 mM NaF and 200 mM phenylmethylsulfonyl (PMSF) as previously described (57). Protein concentration was determined by Bradford Assay and cell lysate was then processed for immunoblotting and visualized with ECL substrate (Bio-Rad, Hercules, CA, USA) as per manufacturer’s protocol. The following antibodies were used: primary antibodies—Sheep-UPF3B 901 [1/1000; in house, (1)], Rabbit-UPF3B (1/250; Sigma-Aldrich), Rabbit-UPF3A (1/1000; Sigma-Aldrich) and Mouse-βactin (1/3000, Sigma-Aldrich). The secondary antibodies were as follows: HRP-Conjugated donkey anti-sheep IgG (1/2000, Chemicon, Temecula, CA, USA), HRP-conjugated Goat anti-mouse (1/2000, DAKO, Agilent, Santa Clara, CA, USA) and HRP conjugated goat anti-rabbit (1/2000, DAKO).
RNA sequencing
LCLs were cultured in parallel and mRNA isolated at the same time. Poly-A+ mRNA was isolated and subjected to paired-end RNA sequencing (RNA Seq) on the Illumina NextSeq platform. RNA sequencing data are available from the corresponding author (RNA Seq counts data is in Online Resource 1). The quality of the reads was assessed using the FastQC package and segments of low-quality reads trimmed using Trim Galore. Greater than 48 million reads were obtained per sample. Normalization and differential gene expression analysis were performed using the DESeq2 pipeline (58). P-values were adjusted to control for the false discovery rate (FDR) using the Benjamini–Hochberg method. A differentially expressed gene (DEG) was defined by a fold change >2 and an adjusted P-value <0.05. DEGs were analysed using various gene ontology packages (31–34). List of autism genes was obtained from the Simons Foundation Autism Research Initiative (SFARI) Human Gene Module (https://www.sfari.org/resource/sfari-gene/; accessed 31/1/2020). List of intellectual disability genes was obtained from Nijmegen Genome Diagnostics (https://www.genomediagnosticsnijmegen.nl/index.php/en/; accessed 31/1/2020). The lists of these genes and the reduction of the list to those expressed in LCLs are provided in Online Resource 1. Statistical significance of over-representation factors (number over expected number of overlapping genes) was calculated using a hypergeometric probability test (https://nemates.org/MA/progs/overlap_stats.html), where the whole-gene population was defined as the 13 284 genes found expressed in LCLs, and NDD gene lists used limited to the 1312 NDD genes expressed in LCLs (Online Resource 1).
Supplementary Material
Acknowledgement
The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC).
Conflict of Interest statement. The authors have no conflicts of interest to declare.
Contributor Information
Deepti Domingo, University of Adelaide and Robinson Research Institute, Adelaide, SA 5005, Australia.
Urwah Nawaz, University of Adelaide and Robinson Research Institute, Adelaide, SA 5005, Australia.
Mark Corbett, University of Adelaide and Robinson Research Institute, Adelaide, SA 5005, Australia.
Josh L Espinoza, J Craig Venter Institute, La Jolla, CA 92093, USA.
Katrina Tatton-Brown, St George’s University of London, London SW17, UK; Southwest Thames Regional Genetics Centre, St George’s Healthcare NHS Trust, London SW17, UK.
David Coman, School of Medicine, University of Queensland, Brisbane, QLD 4072, Australia.
Miles F Wilkinson, Department of Reproductive Medicine, School of Medicine, University of California, San Diego, La Jolla, CA 92093, USA; Institute of Genomic Medicine, University of California, San Diego, La Jolla, CA 92093, USA.
Jozef Gecz, University of Adelaide and Robinson Research Institute, Adelaide, SA 5005, Australia; South Australian Health and Medical Research Institute, Adelaide, SA 5000, Australia.
Lachlan A Jolly, University of Adelaide and Robinson Research Institute, Adelaide, SA 5005, Australia.
Funding
National Health and Medical Research Council (NHMRC) of Australia including Project Grant (1008077) to J.G. and L.J. and Program Grant (628952) and Research Fellowship (1041920) to J.G.; Australian Research Council (ARC) Fellowship (DE160100620 to L.J.); Health Innovation Challenge Fund (grant number HICF-1009-003); a parallel funding partnership between Wellcome and the Department of Health and the Wellcome Sanger Institute (grant number WT098051).
References
- 1. Tarpey, P.S., Raymond, F.L., Nguyen, L.S., Rodriguez, J., Hackett, A., Vandeleur, L., Smith, R., Shoubridge, C., Edkins, S., Stevens, C.et al. (2007) Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet., 39, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lynch, S.A., Nguyen, L.S., Ng, L.Y., Waldron, M., McDonald, D. and Gecz, J. (2012) Broadening the phenotype associated with mutations in UPF3B: two further cases with renal dysplasia and variable developmental delay. Eur. J. Med. Genet., 55, 476–479. [DOI] [PubMed] [Google Scholar]
- 3. Laumonnier, F., Shoubridge, C., Antar, C., Nguyen, L.S., Van Esch, H., Kleefstra, T., Briault, S., Fryns, J.P., Hamel, B., Chelly, J.et al. (2010) Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry, 15, 767–776. [DOI] [PubMed] [Google Scholar]
- 4. Xu, X., Zhang, L., Tong, P., Xun, G., Su, W., Xiong, Z., Zhu, T., Zheng, Y., Luo, S., Pan, Y.et al. (2013) Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin. Genet., 83, 560–564. [DOI] [PubMed] [Google Scholar]
- 5. Addington, A.M., Gauthier, J., Piton, A., Hamdan, F.F., Raymond, A., Gogtay, N., Miller, R., Tossell, J., Bakalar, J., Inoff-Germain, G.et al. (2011) A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol. Psychiatry, 16, 238–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tejada, M.I., Villate, O., Ibarluzea, N., de laHoz, A.B., Martinez-Bouzas, C., Beristain, E., Martinez, F., Friez, M.J., Sobrino, B. and Barros, F. (2019) Molecular and clinical characterization of a novel nonsense variant in exon 1 of the UPF3B gene found in a large Spanish Basque family (MRX82). Front. Genet., 10, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kurosaki, T., Popp, M.W. and Maquat, L.E. (2019) Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol., 20, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jaffrey, S.R. and Wilkinson, M.F. (2018) Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat. Rev. Neurosci., 19, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trivellin, G., Sharwood, E., Hijazi, H., Carvalho, C.M.B., Yuan, B., Tatton-Brown, K., Coman, D., Lupski, J.R., Cotterill, A.M., Lodish, M.B.et al. (2018) Xq26.3 duplication in a boy with motor delay and low muscle tone refines the X-linked acrogigantism genetic locus. J. Endocr. Soc., 2, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trivellin, G., Daly, A.F., Faucz, F.R., Yuan, B., Rostomyan, L., Larco, D.O., Schernthaner-Reiter, M.H., Szarek, E., Leal, L.F., Caberg, J.H.et al. (2014) Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med., 371, 2363–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trivellin, G., Hernandez-Ramirez, L.C., Swan, J. and Stratakis, C.A. (2018) An orphan G-protein-coupled receptor causes human gigantism and/or acromegaly: molecular biology and clinical correlations. Best Pract. Res. Clin. Endocrinol. Metab, 32, 125–140. [DOI] [PubMed] [Google Scholar]
- 12. Iacovazzo, D., Caswell, R., Bunce, B., Jose, S., Yuan, B., Hernandez-Ramirez, L.C., Kapur, S., Caimari, F., Evanson, J., Ferrau, F.et al. (2016) Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol. Commun., 4, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deciphering Developmental Disorders, S (2015) Large-scale discovery of novel genetic causes of developmental disorders. Nature, 519, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anna, A. and Monika, G. (2018) Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet., 59, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wai, H., Douglas, A.G.L. and Baralle, D. (2019) RNA splicing analysis in genomic medicine. Int. J. Biochem. Cell Biol., 108, 61–71. [DOI] [PubMed] [Google Scholar]
- 16. Ohno, K., Takeda, J.I. and Masuda, A. (2018) Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip. Rev. RNA, 9, e1451. [DOI] [PubMed] [Google Scholar]
- 17. Reese, M.G., Eeckman, F.H., Kulp, D. and Haussler, D. (1997) Improved splice site detection in genie. J. Comput. Biol., 4, 311–323. [DOI] [PubMed] [Google Scholar]
- 18. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwarz, J.M., Cooper, D.N., Schuelke, M. and Seelow, D. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- 20. Mort, M., Sterne-Weiler, T., Li, B., Ball, E.V., Cooper, D.N., Radivojac, P., Sanford, J.R. and Mooney, S.D. (2014) MutPred splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol., 15, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desmet, F.O., Hamroun, D., Lalande, M., Collod-Beroud, G., Claustres, M. and Beroud, C. (2009) Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res., 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan, W.K., Huang, L., Gudikote, J.P., Chang, Y.F., Imam, J.S., MacLean, J.A., 2nd and Wilkinson, M.F. (2007) An alternative branch of the nonsense-mediated decay pathway. EMBO J., 26, 1820–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nguyen, L.S., Jolly, L., Shoubridge, C., Chan, W.K., Huang, L., Laumonnier, F., Raynaud, M., Hackett, A., Field, M., Rodriguez, J.et al. (2012) Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol. Psychiatry., 17, 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carter, M.S., Doskow, J., Morris, P., Li, S., Nhim, R.P., Sandstedt, S. and Wilkinson, M.F. (1995) A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J. Biol. Chem., 270, 28995–29003. [DOI] [PubMed] [Google Scholar]
- 25. Huang, L., Lou, C.H., Chan, W., Shum, E.Y., Shao, A., Stone, E., Karam, R., Song, H.W. and Wilkinson, M.F. (2011) RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol. Cell, 43, 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan, W.K., Bhalla, A.D., Le Hir, H., Nguyen, L.S., Huang, L., Gecz, J. and Wilkinson, M.F. (2009) A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat. Struct. Mol. Biol., 16, 747–753. [DOI] [PubMed] [Google Scholar]
- 27. Isken, O. and Maquat, L.E. (2008) The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat. Rev. Genet., 9, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jolly, L.A., Homan, C.C., Jacob, R., Barry, S. and Gecz, J. (2013) The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum. Mol. Genet., 22, 4673–4687. [DOI] [PubMed] [Google Scholar]
- 29. Mendell, J.T., Sharifi, N.A., Meyers, J.L., Martinez-Murillo, F. and Dietz, H.C. (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet., 36, 1073–1078. [DOI] [PubMed] [Google Scholar]
- 30. Weischenfeldt, J., Damgaard, I., Bryder, D., Theilgaard-Monch, K., Thoren, L.A., Nielsen, F.C., Jacobsen, S.E., Nerlov, C. and Porse, B.T. (2008) NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev., 22, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang da, W., Sherman, B.T. and Lempicki, R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 32. Kuleshov, M.V., Jones, M.R., Rouillard, A.D., Fernandez, N.F., Duan, Q., Wang, Z., Koplev, S., Jenkins, S.L., Jagodnik, K.M., Lachmann, A.et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young, M.D., Wakefield, M.J., Smyth, G.K. and Oshlack, A. (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol., 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu, G., Wang, L.G., Han, Y. and He, Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson, J.L., Stoica, L., Liu, Y., Zhu, P.J., Bhattacharya, A., Buffington, S.A., Huq, R., Eissa, N.T., Larsson, O., Porse, B.T.et al. (2019) Inhibition of Upf2-dependent nonsense-mediated decay leads to behavioral and neurophysiological abnormalities by activating the immune response. Neuron, 104, 665–679 e668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cartegni, L., Chew, S.L. and Krainer, A.R. (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet., 3, 285–298. [DOI] [PubMed] [Google Scholar]
- 37. Duan, J., Wainwright, M.S., Comeron, J.M., Saitou, N., Sanders, A.R., Gelernter, J. and Gejman, P.V. (2003) Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum. Mol. Genet., 12, 205–216. [DOI] [PubMed] [Google Scholar]
- 38. Chamary, J.V., Parmley, J.L. and Hurst, L.D. (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet., 7, 98–108. [DOI] [PubMed] [Google Scholar]
- 39. Kimchi-Sarfaty, C., Oh, J.M., Kim, I.W., Sauna, Z.E., Calcagno, A.M., Ambudkar, S.V. and Gottesman, M.M. (2007) A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science, 315, 525–528. [DOI] [PubMed] [Google Scholar]
- 40. Tsai, C.J., Sauna, Z.E., Kimchi-Sarfaty, C., Ambudkar, S.V., Gottesman, M.M. and Nussinov, R. (2008) Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol., 383, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartoszewski, R.A., Jablonsky, M., Bartoszewska, S., Stevenson, L., Dai, Q., Kappes, J., Collawn, J.F. and Bebok, Z. (2010) A synonymous single nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J. Biol. Chem., 285, 28741–28748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brest, P., Lapaquette, P., Souidi, M., Lebrigand, K., Cesaro, A., Vouret-Craviari, V., Mari, B., Barbry, P., Mosnier, J.F., Hebuterne, X.et al. (2011) A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat. Genet., 43, 242–245. [DOI] [PubMed] [Google Scholar]
- 43. Wang, Y., Qiu, C. and Cui, Q. (2015) A large-scale analysis of the relationship of synonymous SNPs changing MicroRNA regulation with functionality and disease. Int. J. Mol. Sci., 16, 23545–23555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cherot, E., Keren, B., Dubourg, C., Carre, W., Fradin, M., Lavillaureix, A., Afenjar, A., Burglen, L., Whalen, S., Charles, P.et al. (2018) Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experience of 2 clinical units and 216 patients. Clin. Genet., 93, 567–576. [DOI] [PubMed] [Google Scholar]
- 45. Hwang, J. and Maquat, L.E. (2011) Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr. Opin. Genet. Dev., 21, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang, L., Shum, E.Y., Jones, S.H., Lou, C.H., Dumdie, J., Kim, H., Roberts, A.J., Jolly, L.A., Espinoza, J.L., Skarbrevik, D.M.et al. (2018) A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry., 23, 1773–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nguyen, L.S., Kim, H.G., Rosenfeld, J.A., Shen, Y., Gusella, J.F., Lacassie, Y., Layman, L.C., Shaffer, L.G. and Gecz, J. (2013) Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet., 22, 1816–1825. [DOI] [PubMed] [Google Scholar]
- 48. Lou, C.H., Shao, A., Shum, E.Y., Espinoza, J.L., Huang, L., Karam, R. and Wilkinson, M.F. (2014) Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep., 6, 748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hamdan, F.F., Daoud, H., Rochefort, D., Piton, A., Gauthier, J., Langlois, M., Foomani, G., Dobrzeniecka, S., Krebs, M.O., Joober, R.et al. (2010) De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am. J. Hum. Genet., 87, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takahashi, H., Takahashi, K. and Liu, F.C. (2009) FOXP genes, neural development, speech and language disorders. Adv. Exp. Med. Biol., 665, 117–129. [DOI] [PubMed] [Google Scholar]
- 51. Lou, C.H., Dumdie, J., Goetz, A., Shum, E.Y., Brafman, D., Liao, X., Mora-Castilla, S., Ramaiah, M., Cook-Andersen, H., Laurent, L.et al. (2016) Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Reports, 6, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Colak, D., Ji, S.J., Porse, B.T. and Jaffrey, S.R. (2013) Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell, 153, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mort, M., Ivanov, D., Cooper, D.N. and Chuzhanova, N.A. (2008) A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat., 29, 1037–1047. [DOI] [PubMed] [Google Scholar]
- 54. Nguyen, L.S., Wilkinson, M.F. and Gecz, J. (2014) Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci. Biobehav. Rev., 46(Pt 2), 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Miller, J.N. and Pearce, D.A. (2014) Nonsense-mediated decay in genetic disease: friend or foe? Mutat. Res. Rev. Mutat. Res., 762, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jolly, L.A., Sun, Y., Carroll, R., Homan, C.C. and Gecz, J. (2018) Robust imaging and gene delivery to study human lymphoblastoid cell lines. J. Hum. Genet., 63, 945–955. [DOI] [PubMed] [Google Scholar]
- 57. Jolly, L.A., Nguyen, L.S., Domingo, D., Sun, Y., Barry, S., Hancarova, M., Plevova, P., Vlckova, M., Havlovicova, M., Kalscheuer, V.M.et al. (2015) HCFC1 loss-of-function mutations disrupt neuronal and neural progenitor cells of the developing brain. Hum. Mol. Genet., 24, 3335–3347. [DOI] [PubMed] [Google Scholar]
- 58. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.