

Abstract
The United States Environmental Protection Agency has proposed a tiered testing strategy for chemical hazard evaluation based on new approach methods (NAMs). The first tier includes in vitro profiling assays applicable to many (human) cell types, such as high-throughput transcriptomics (HTTr) and high-throughput phenotypic profiling (HTPP). The goals of this study were to: (1) harmonize the seeding density of U-2 OS human osteosarcoma cells for use in both assays; (2) compare HTTr- versus HTPP-derived potency estimates for 11 mechanistically diverse chemicals; (3) identify candidate reference chemicals for monitoring assay performance in future screens; and (4) characterize the transcriptional and phenotypic changes in detail for all-trans retinoic acid (ATRA) as a model compound known for its adverse effects on osteoblast differentiation. The results of this evaluation showed that (1) HTPP conducted at low (400 cells/well) and high (3000 cells/well) seeding densities yielded comparable potency estimates and similar phenotypic profiles for the tested chemicals; (2) HTPP and HTTr resulted in comparable potency estimates for changes in cellular morphology and gene expression, respectively; (3) three test chemicals (etoposide, ATRA, dexamethasone) produced concentration-dependent effects on cellular morphology and gene expression that were consistent with known modes-of-action, demonstrating their suitability for use as reference chemicals for monitoring assay performance; and (4) ATRA produced phenotypic changes that were highly similar to other retinoic acid receptor activators (AM580, arotinoid acid) and some retinoid X receptor activators (bexarotene, methoprene acid). This phenotype was observed concurrently with autoregulation of the RARB gene. Both effects were prevented by pre-treating U-2 OS cells with pharmacological antagonists of their respective receptors. Thus, the observed phenotype could be considered characteristic of retinoic acid pathway activation in U-2 OS cells. These findings lay the groundwork for combinatorial screening of chemicals using HTTr and HTPP to generate complementary information for the first tier of a NAM-based chemical hazard evaluation strategy.
Keywords: High-throughput phenotypic profiling, Cell Painting, concentration-response, computational toxicology
Introduction
Thousands of chemicals in commerce and present in the environment are lacking toxicity information (Judson et al., 2009; NRC, 1984). As traditional animal experiments lack the necessary throughput for generating hazard information on thousands of chemicals, new approach methods (NAMs) based largely on in vitro assays in human cell lines or assays in simple model organisms, together with in silico modeling, have been proposed as alternatives to traditional in vivo testing (NRC, 2007). The United States Environmental Protection Agency (USEPA) recently proposed a tiered testing strategy for hazard evaluation using NAMs (Thomas et al., 2019). The first tier consists of high-throughput profiling (HTP) assays and in silico modeling to generate potency estimates for chemical bioactivity and predict mechanism-of-action (MoA). Chemicals of interest can then be investigated in greater detail using targeted in vitro high-throughput screening assays (tier 2) and, if needed, with more complex organotypic culture models (tier 3). Tier 1 ideally casts a broad net for capturing biological effects of chemicals across diverse cell types expressing varying complements of molecular targets and pathways. Two HTP assays currently being explored are high-throughput transcriptomics (HTTr) using whole transcriptome TempO-Seq, a form of targeted RNA-Seq (Harrill et al., 2021; Yeakley et al., 2017) and imaging-based high-throughput phenotypic profiling (HTPP) using the Cell Painting assay (Bray et al., 2016; Nyffeler et al., 2020). These two complementary approaches capture changes at different levels of cellular organization: while HTTr is focused on the molecular level (gene expression), HTPP measures changes at the cellular level (morphological changes in cells and their organelles).
Both HTP methods have been applied previously by USEPA and others to profile cellular responses to chemicals. With regards to HTPP, over 30,000 chemicals have been screened in U-2 OS cells at a single concentration (Bray et al., 2017; Gustafsdottir et al., 2013). More recently, USEPA tested 460 chemicals at multiple concentrations in U-2 OS cells (Nyffeler et al., 2020). Multiple studies have demonstrated that application of HTPP to other cell types is also possible (Gustafsdottir et al., 2013; Warchal, Dawson, & Carragher, 2016; Willis, Nyffeler, & Harrill, 2020). Similarly, ~20,000 chemicals have been transcriptionally profiled at a single concentration (Duan et al., 2014; Subramanian et al., 2017) in various cell types using the L1000 platform, which measures the expression of ~1000 genes. Further, USEPA tested 44 chemicals in concentration-response format using whole transcriptome TempO-Seq (Harrill et al., 2021). To date, only a few studies have combined the two HTP approaches to predict mechanisms (Lapins & Spjuth, 2019) or assay outcomes (Becker et al., 2020; Wawer et al., 2014). These studies were conducted at a single test concentration and overall concluded that HTPP and HTTr provide complementary information regarding perturbation of cellular biology by chemicals. However, comparison of potencies across assays was not possible due to lack of concentration-response data.
A goal of USEPA’s tier 1 screening strategy is to conduct concurrent HTTr and HTPP chemical bioactivity screens in a single test system (e.g., same cell type and culture conditions). Prior to this study, it was unknown whether HTTr and HTPP are comparable to one another with regards to potency estimation. Moreover, there is potential value in the ability to link phenotypic responses and changes in gene expression to specific molecular initiating events (MIE), such as nuclear receptor activation.
We previously applied HTTr to MCF7 cells (Harrill et al., 2021) and HTPP to U-2 OS cells (Nyffeler et al., 2020). In order to use both assays (HTTr, HTPP) concurrently for potency estimation and mechanistic prediction, there is a critical need to harmonize culture conditions across the assays for any given cell model. The present study was therefore undertaken to harmonize protocols for chemical screening in U-2 OS cells. Our previous U-2 OS HTPP screen was conducted at a low seeding density (i.e., 400 cells/well), while HTTr requires a higher cell density per well to generate enough RNA for TempO-Seq profiling in cell lysates. Therefore, the test system must be adapted to a cell seeding density that could accommodate both assay formats.
In addition, to monitor assay performance during screening campaigns, reference chemicals are tested on each assay plate (Nyffeler et al., 2020). Previously, these reference chemicals were tailored to HTPP, but in order to support both assays, reference chemicals suitable for monitoring performance would ideally exert robust phenotypic effects in a concentration-dependent manner, and induce robust changes in gene expression that are associated with a known and specific MoA. We thus tested a set of candidate reference chemicals in both assays and compared their qualitative and quantitative effects in both assays.
The U-2 OS cell line was selected because it is the cell line that was originally used to develop the Cell Painting assay and the one most commonly used for HTPP at US EPA (Bray et al., 2016; Nyffeler et al., 2020). U-2 OS cells are derived from an osteosarcoma, a bone cancer (Pautke et al., 2004). Osteoblasts and osteoclasts are involved in remodeling of bones. This process is modulated by all trans-retinoic acid (ATRA). ATRA and other retinoic acid (RA) derivatives are vertebrate hormones, that play a role in many other biological processes, particularly during the development of organs, limbs, and the nervous system (Ghyselinck & Duester, 2019; Janesick, Wu, & Blumberg, 2015). Thus, for the final part of this study, we explored the biology underlying changes in cell morphology and gene expression induced by a candidate phenotypic reference chemical, ATRA and its analogues.
Materials and Methods
Materials
Materials and reference chemicals used for the present studies were obtained from a number of different suppliers (Tables S1–S3). Chemical stock solutions (Tables S2 + S3) were prepared in dimethyl sulfoxide (DMSO). The first phase of this study involved testing a set of 11 candidate reference chemicals in both HTPP and HTTr (Table 1). The second phase of this study involved testing a collection of RA pathway modulators in a variety of follow-up experiments using HTPP and reverse transcription quantitative polymerase chain reaction (RT-qPCR) (Table 2).
Table 1:
List of candidate reference chemicals.
| Name | DTXSID | CASRN | Rationale | Mechanism / Target | Reference |
|---|---|---|---|---|---|
| Berberine chloride | DTXSID8024602 | 633-65-8 | HTPP reference chemical (Nyffeler et al., 2020) | redistribution of mitochondria | Gustafsdottir et al., 2013 |
| Ca-074-Me | DTXSID50881386 | 147859-80-1 | Cathepsin B inhibitor | Buttle et al., 1992 | |
| Etoposide | DTXSID5023035 | 33419-42-0 | Topoisomerase II inhibitor | Hande et al., 1998 | |
| Rapamycin | DTXSID5023582 | 53123-88-9 | mTOR inhibitor | Huang et al., 2003 | |
| Trichostatin A | DTXSID6037063 | 58880-19-6 | HTTr reference chemical | Histone deacetylase inhibitor | Yoshida et al., 1990 |
| Cycloheximide | DTXSID6024882 | 66-81-9 | Large transcriptomic effects in MCF-7 cells | protein synthesis inhibitor | Obrig et al., 1971 |
| all-trans-Retinoic acid | DTXSID7021239 | 302-79-4 | receptor expressed in U-2 OS cells | Retinoid | Marill et al., 2003 |
| Dexamethasone | DTXSID3020384 | 50-02-2 | Glucocorticoid | Brinks et al., 2018 | |
| Actinomycin D | DTXSID9020031 | 50-76-0 | Large magnitudes (RNA channel) | RNA polymerase inhibitor | Aktipis et al., 1981 |
| Cucurbitacin I | DTXSID501015546 | 2222-07-3 | Large magnitudes (AGP channel) | JAK2 /STAT3 inhibitor | Blaskovich et al., 2003 |
| Docetaxel | DTXSID0040464 | 114977-28-5 | Large magnitudes (DNA channel) | Microtubule stabilizer | Imran et al., 2020 |
| Staurosporine | DTXSID6041131 | 62996-74-1 | viability positive control | Protein kinase inhibitor | Tamaoki et al., 1986 |
| Dimethyl sulfoxide | DTXSID2021735 | 67-68-5 | solvent control | - | - |
Table 2:
List of retinoic acid pathway modulators.
| Name | DTXSID | CASRN | Mechanism / Target | Reference |
|---|---|---|---|---|
| all-trans-Retinoic acid | DTXSID7021239 | 302-79-4 | RAR agonist | Marill et al., 2003 |
| AM580 | DTXSID5040758 | 102121-60-8 | Kagechika et al., 1988 | |
| Arotinoid acid | DTXSID6040743 | 71441-28-6 | Loeliger et al., 1980 | |
| Bexarotene | DTXSID1040619 | 153559-49-0 | RXR agonist | Boehm et al., 1994 |
| Triphenyltin hydroxide | DTXSID1021409 | 76-87-9 | Kanayama et al., 2005, Nakanishi et al., 2005 | |
| Docosahexaenoic acid | DTXSID5040465 | 6217-54-5 | de Urquiza et al., 2000, Lengqvist et al., 2004 | |
| Methoprene acid | DTXSID10886034 | 53092-52-7 | Harmon et al., 1995 | |
| Liarozole dihydrochloride | DTXSID201026639 | 1883548-96-6 | RA metabolism inhibitor | van Wauwe et al., 1992+1994 |
| Citral * | DTXSID6024836 | 5392-40-5 | RA synthesis inhibitor | Connor et al., 1987, Tanaka et al., 1996 |
| CD 2665 * | DTXSID50168846 | 170355-78-9 | RAR antagonist (selectivity for b, g) | Szondy et al., 1997, Meister et al., 1998 |
| ER 50891 * | DTXSID501026640 | 187400-85-7 | RAR antagonist (selectivity for a) | Kikuchi et al., 2001, Ren et al., 2005 |
| UVI 3003 * | DTXSID501024375 | 847239-17-2 | RXR antagonist | Nahoum et al., 2007 |
| Dexamethasone | DTXSID3020384 | 50-02-2 | reference chemical | - |
| Etoposide | DTXSID5023035 | 33419-42-0 | - | |
| Saccharin | DTXSID5021251 | 81-07-2 | negative control | - |
| Sorbitol | DTXSID5023588 | 50-70-4 | - | |
| Staurosporine | DTXSID6041131 | 62996-74-1 | viability positive control | - |
| Dimethyl sulfoxide | DTXSID2021735 | 67-68-5 | solvent control | - |
In sequential treatment experiments, chemicals marked with an asterisk were used as ‘pre-treatments’, while all other chemicals were applied as ‘test chemicals’.
Cell culture
U-2 OS human osteosarcoma cells (HTB-96®, Lot: 64048673, ATCC) were cultured as described in Nyffeler et al. (2020). Briefly, cells were cultured in media consisting of Dulbecco’s Modified Eagle Medium (DMEM) with 10% heat-inactivated fetal bovine serum and penicillin/streptomycin/glutamine, passaged using TrypLE Select and maintained at 37 °C in a humidified atmosphere of 5% CO2 in air.
A cryo stock of internal passage number 6 (P6) was generated as described in Nyffeler et al. (2020); experiments were performed using U-2 OS cells from P7 to P10. Cells were counted using a Countess II automated cell counter and seeded into CellCarrier-384 Ultra microplates at 400 cells/well or 3000 cells/well in 40 μl media per well. For experiments in 96-well plate format, cells were seeded into CellCarrier-96 Ultra microplates at 9000 cells/well in 120 μl media.
Chemical treatment in 384-well plates
For all experiments in 384-well plates, treatments were applied using a LabCyte Echo 550 acoustic dispenser (Beckman-Coulter, Indianapolis, IN). Plate layouts for treatments were uniquely randomized for each assay plate, i.e., technical replicates were distributed across the plate to avoid systematic edge-well effects. Dilution series for test chemicals were prepared prior to dosing in Echo qualified 384-well low dead volume (384LDV) plates using the Echo 550 acoustic dispenser. A Certus FLEX Microdispenser (Trajan Scientific Americas, Morrisville, NC) was used for backfilling with DMSO, as described previously (Nyffeler et al., 2020).
For experiments testing single chemical treatments per well, dilution series were prepared as 200x stock and 200 nl of chemical solution was dispensed (see Tables S2 + S3 for concentration ranges). For pharmacological blockade experiments, dilution series were prepared at 400x the desired test concentration and the pre-treatment was dispensed in a volume of 100 nl, followed by dispensing of 100 nl of the test chemical. The final concentration of DMSO in test wells was 0.5% for all experiments. Cells were treated with test chemicals 24 h after seeding.
Cell Viability (CV) Assay
For the experiment with the 11 candidate reference chemicals, parallel plates were prepared to measure cytotoxicity and cytostasis, as described previously (Nyffeler et al., 2020). Briefly, cells were live-labeled with propidium iodide (PI) and H-33342, then fixed with 3.5% paraformaldehyde for 10 min and rinsed with phosphate buffered saline (PBS). Plates were imaged on an Opera Phenix High Content Screening System (PerkinElmer, Waltham, MA) using a 10x objective and four fields per well. Images were analyzed to count the number of Hoechst-33342 labeled cells and the percentage of PI-positive cells. Well-level results were then modeled using concentration-response functions from the R package tpclfit2 (v0.1.0) (https://rdrr.io/github/USEPA/CompTox-ToxCast-tcplFit2/). Cytotoxicity data (i.e., % PI-positive cells) were fit to three functions: constant, Hill and Gain-Loss function, whereas cell count data were only modeled with the first two functions. The benchmark response (BMR) was set at 3 times the normalized median absolute deviation (nMAD, with nMAD = 1.4826*MAD) for cytotoxicity data, while for cell counts, the effective concentration 50% (EC50) was calculated. The lower of the two was defined as the cell viability benchmark concentration (BMC). Subsequently, the first test concentration above the BMC was defined as the lowest observed effect concentration (LOEC) and test concentrations above the LOEC were removed before concentration-response modelling of profiling data as described previously (Nyffeler et al., 2020). For the experiment with the 11 candidate reference chemicals, this included the highest tested concentration for actinomycin D and docetaxel and the two highest tested concentrations for trichostatin A.
For experiments with the RA pathway modulators, no cell viability plates were prepared. Instead, the relative cell count was obtained from the Cell Painting images and used to estimate the EC50.
High-throughput phenotypic profiling assay (HTPP)
Fluorescent labeling
Phenotypic profiling of cells was performed using the ‘Cell Painting’ method (Bray et al., 2016) as described in Nyffeler et al. (2020). First, MitoTracker Deep Red was applied to live cells 24 h after chemical treatment. After incubation for 30 min in the incubator, plates were fixed with 3.5% paraformaldehyde for 10 min and washed with PBS. Then, cells were permeabilized with 0.1% Triton X-100 for 30 min at room temperature, followed by rinsing with PBS. The remaining labeling reagents (Hoechst-33342, SYTO14, Concanavalin A-488, Alexa Fluor 568 Phalloidin and Wheat Germ Agglutinin Alexa Flour 555 Conjugate) were applied as a cocktail in PBS with 1% bovine serum albumin. After a 30 min incubation at room temperature, plates were rinsed four times with PBS and stored in PBS at 4 °C until the day of imaging.
Image Acquisition & Feature Extraction
Fluorescent images were acquired using an Opera Phenix High Content Screening System and Harmony® software (v4.9). Five images were acquired with a 20× water immersion objective in confocal mode (2 × 2 pixel binning) using four different excitation wavelengths: 405 nm (DNA), 488 nm (RNA, z-offset 1), 488 nm (ER, z-offset 2), 561 nm (actin, golgi, plasma membrane) (AGP), 640 nm (mitochondria) (Mito). For experiments at the lower seeding density, 9 fields per well were acquired, while for experiments at the higher seeding density 5 fields per well were acquired. Feature extraction was performed using Harmony® software as previously described (Nyffeler et al., 2020). Briefly, nuclei were segmented in the DNA channel. The nuclei were then used as “seeds”, i.e., starting points, for segmentation of the cells in the ER channel. Cells touching the image border were excluded from analysis. The protocol is provided in the supporting information. Of note, the chemical ER 50981 was auto-fluorescent in the DNA channel and interfered with accurate cell segmentation. Treatments with > 3 μM ER 50981 could not be properly segmented, and hence were excluded from further analysis.
Data analysis
For each cell, 1300 features were measured. Typically, approximately 250 cells/well and 1000 cells/well were analyzed for the low and high seeding densities, respectively. Cell-level data were aggregated to the well-level and normalized using R statistical software (v 3.6.2), as previously described (Nyffeler et al., 2020). Briefly, cells treated with the vehicle control were used to define a median and MAD for normalization. Well-level aggregates were calculated by computing the median from all cells. In a slight modification from previous reports (Nyffeler et al., 2021; Nyffeler et al., 2020; Willis et al., 2020), the well-level data were scaled further using the combined vehicle control wells of the corresponding plate group (typically 96 wells). A plate group consists of the four biological replicate plates that were administered the same treatments. These scaled values were used for heatmap visualizations with values within [−1, +1] (i.e., within 1 standard deviation of controls) being substituted with 0 to indicate that the feature was not substantially affected by the treatment. Profiles were compared by first averaging the biological replicates, then computing pairwise Pearson correlation.
In order to reduce the high-dimensional data to a single value, the ‘Global Mahalanobis distance’ approach was applied as described previously (Nyffeler et al., 2021). Briefly, Mahalanobis distance is similar to common Euclidean distance, but takes into account the correlation of features. To this end, scaled feature-level data were transformed using principal component analysis, then the covariance matrix was estimated. The covariance matrix was then used to calculate each well’s distance to the mean position of the corresponding (solvent) control wells. Thus, a large distance means that the well’s profile was very different from the control.
Concentration-response modeling
Mahalanobis distances were subjected to concentration-response modeling using the R package tpclfit2 (v0.1.0, https://github.com/USEPA/CompTox-ToxCast-tcplFit2) (Sheffield, Brown, Davidson, Friedman, & Judson, 2021). First, all concentrations above the cell viability LOEC were removed. Then the data were modeled with the following functions: constant, Hill, poly1, poly2, power and four exponential models (exp2-exp5). The baseline median and MAD was calculated using data from appropriate control wells. For the experiments with single treatments, these were data from the lowest tested concentration of test chemicals. For the experiments with sequential treatments, wells that received the same pre-treatment, but were not treated with the test chemical served as controls. For all experiments, the BMR was set at 1 nMAD and concentration-response fits exceeding this cutoff value were considered active. Chemicals with a BMC below the highest tested concentration were considered active; no BMCs were extrapolated above the tested range. BMCs below the tested range were set at half an order of magnitude below the lowest tested concentration. The BMC of the global Mahalanobis curve fit was defined as the phenotype altering concentration (PAC).
High-Throughput Transcriptomics (HTTr)
Sample Generation & Sequencing
For the experiment with the 11 candidate reference chemicals in U-2 OS cells seeded at high density (3000 cells/well), parallel plates were prepared for whole transcriptome profiling of cell lysates using TempO-Seq (Yeakley et al., 2017). Samples were prepared as previously described (Harrill et al., 2021), with some modifications: wells were washed with PBS, drained to 10 μL and lysed by addition of 10 μL of 2X Enhanced BioSpyder Lysis Buffer. Plates were temporarily sealed with an adhesive film, shaken for 30 s on the stage of MultiFlow FX microplate dispenser (BioTek, Winooski, VT) and then placed in a hybridization oven (37°C) for 10 min. The adhesive films were removed, plates were re-sealed with Nunc™ aluminum acrylate sealing tape and stored at −80°C. Prior to shipment, the tape was removed and reference samples (reference RNAs, bulk lysates and lysis buffer blank) (Harrill et al., 2021) were added in duplicate to each plate while keeping the plates frozen. The plates were then resealed with new Nunc™ aluminum acrylate sealing tape.
Plates were shipped to BioSpyder, Inc. frozen (on dry ice) where U-2 OS cell lysates were then analyzed using a custom-attenuated version of the TempO-Seq human whole transcriptome assay version 2 (hWTv2). Lysates were processed and the TempO-Seq assay was performed as previously described (Harrill et al., 2021). Ligation products generated from each sample using the TempO-Seq assay were “barcoded” with well coordinate-specific primer pairs containing universal adaptors for sequencing. Barcoded ligation products were then pooled into a combined sequencing library and distributed across multiple lanes of a HiSeq dual flow cell and analyzed on a HiSeq 2500 Ultra-High-Throughput Sequencing System (Illumina, San Diego, CA). The target depth for each test sample was 3 million mapped reads.
Data processing
TempO-Seq data were processed as previously described (Harrill et al., 2021). In brief, raw TempO-Seq data were aligned to probe sequences in the hWTv2 assay probe manifest using HISAT2 v.2.1.0 (Kim, Langmead, & Salzberg, 2015; Kim, Paggi, Park, Bennett, & Salzberg, 2019) with splice alignment disabled. The number of uniquely aligned reads for each probe were computed using SAMtools v1.9 (Li et al., 2009). The probe counts for each sample are provided via FigShare (https://doi.org/10.23645/epacomptox.16834519.v1). Raw and processed data are available through Gene Expression Omnibus (GEO) under accession GSE200845. Source code for all data processing steps is included in an open source package ‘httrpl’ (https://github.com/USEPA/httrpl_pilot).
Samples from treatments above the cell viability LOEC were excluded from transcriptomic analysis. In addition, quality control metrics for each sample were calculated as previously described (Harrill et al., 2021). The thresholds for sample exclusion based on the fraction of uniquely mapped reads and the number of uniquely mapped reads were set at ≥ 50% and < 10% (of the target read depth of 3 million mapped reads), respectively, as in Harrill et al. (2021). The thresholds for sample exclusion based on the number of probes with at least 5 mapped reads (NCov5), the number of probes capturing the top 80% of counts (NSig80) and the Gini coefficient (Graczyk, 2007; van Mierlo, Hyatt, & Ching, 2016) were set at approximately Tukey’s Outer Fence (Tukey, 1977), defined as 3x the inter-quartile range of the distribution of all test samples and vehicle controls. Thresholds for sample exclusion based on quality control metrics are illustrated in figure S1.
Differential expression analysis
Differential expression analysis was performed independently for each chemical as described previously (Harrill et al., 2021). For each chemical, read counts for all test samples across all concentrations tested and corresponding vehicle controls were tabulated. Probes with mean read count < 5 across the tabulated samples were removed. Counts for the remaining probes were modeled using DESeq2 v1.24 (Love, Huber, & Anders, 2014) by treating the experimental plate and treatment effect as independent factors (i.e., counts ~ plate + concentration). Size factors and dispersion were estimated using package defaults and model-fitting was performed with ‘betaPrior = FALSE’. Fold-change shrinkage was applied separately for each test concentration vs. vehicle control to compute moderated log2 fold-change (L2FC) values for each probe, adjusted to remove any average plate effects. For each chemical, the gene level L2FC values were determined by aggregating the probe level L2FC values for each gene and using the probe value with the highest fold change in either direction. All L2FC data for this study are included in the data release (https://doi.org/10.23645/epacomptox.16834519.v1).
Signature score calculation
L2FC data were used to generate transcriptomic signature scores that were then used as input for concentration-response modeling as previously described (Harrill et al., 2021), with modifications. The signature collection used in this study contained 10056 signatures collected from a variety of resources including the Molecular Signatures database (MSigDB) collections C2 and H (Liberzon et al., 2015; Liberzon et al., 2011), BioPlanet (R. Huang et al., 2019) and DisGeNET (Pinero et al., 2015). Directional signatures in the collection were derived from the Connectivity Map (CMAP) database (Subramanian et al., 2017) by identifying the 100 most highly downregulated and 100 most highly upregulated genes from each CMAP profile. Signatures in the collection were annotated with a target class inferred from the signature name and manually curated. The target class indicates, for example, the molecular target that would elicit the response (e.g., receptor), a process at the molecular level (e.g., transcription factor) or cellular level (e.g., apoptosis) or a disease state (e.g., cancer). Signatures were further aggregated into “super-targets” of biologically similar signatures to aid in mechanistic interpretation. The super-targets represent specific genes or a group of genes (e.g., RAR), biological entities (e.g., estrogens, antibiotics), or pathologies (e.g., adenocarcinoma). The full signature catalogue with annotations is included in the data release (https://doi.org/10.23645/epacomptox.16834519.v1).
The signature scoring analysis began by calculating the gene level L2FC matrix for all concentrations of all test chemicals. For a gene in the L2FC matrix to be retained for this analysis, at least 95% of conditions must contain data. For retained genes, any missing L2FC values were set to zero. For each chemical-concentration-signature combination, a signature score was computed using the Single-Sample Gene Set Enrichment Analysis (ssGSEA) method (Barbie et al., 2009). For directional signatures from CMAP, enrichment scores were calculated separately for up- and down-regulated signatures and a final score calculated as scoreup – scoredown. In this way, treatments that elicit both up- and downregulation of transcripts as part of a characteristic response were associated with larger signature scores.
Concentration-response modeling
For each combination of test chemical and signature, the concentration-response series of signature scores was modeled using tcplfit2 (v0.1.0) using the following functions: constant, Hill, poly1, poly2, power and four exponential models (exp2-exp5). The model with the lowest Akaike information criterion (Akaike, 1974) was selected as the preferred model. A chemical-signature was considered active if the signal exceeded a statistically defined noise threshold (‘cutoff’) by a given magnitude. The cutoff was estimated for each signature by calculating the 95% confidence interval around the signature scores for the two lowest concentrations across all chemicals. Active chemical-signatures were defined as those where the ‘top over cutoff’ was ≥ 1 and the hitcall was ≥ 0.9. Out of all active signatures that had a benchmark dose upper confidence limit (BMDU) over benchmark dose lower confidence limit (BMDL) (BMDU/BMDL) ratio < 40 for a given chemical, the BMC for the 5th most potent active signature (i.e., the one with the 5th lowest BMC value) was defined as the biological pathway altering concentration (BPAC) for that chemical. To provide insight into the biological activities of the test chemicals, active signatures were also aggregated to the super-target level. BMCs for all active signatures were grouped according to super-target annotation. Super-targets containing at least one active signature were then rank ordered by the median signature BMC and visualized as accumulation plots.
Pharmacological blockade with retinoic acid pathway modulators
For pharmacological blockade experiments, cells were treated 24 h after seeding with the first treatment (‘pre-treatment’), a single concentration of CD 2665 (10 μM), ER 50891 (1 μM), UVI 3003 (10 μM) or citral (100 μM). The concentration was selected to be high (i.e., above the inhibitory concentration 50 of the respective target) but non-cytotoxic. One hour later, the second treatment (‘test chemicals’) was applied, typically in concentration-response, for 24 h.
For the HTPP assay, cells were seeded in 384-well plates, as described above, and at the end of the exposure period, the cells were processed as described in the ‘High-throughput phenotypic profiling assay (HTPP)’ section. Global Mahalanobis distances were calculated as described above, by using the corresponding pre-treatment (with DMSO only as the ‘test chemical’) as the control. Thus, the distances of different pre-treatments cannot be compared quantitatively. Global Mahalanobis distances were then fit with tcplfit2 as described above.
Each biological replicate consisted of two plates, with two modulators plus DMSO per plate (citral and CD 2665 on one plate; ER 50891 and UVI 3003 on the other). Therefore, two curves are displayed for pre-treatment with DMSO in plots of these data. For one biological replicate, wells treated with ER 50981 were excluded from analysis, because the cells could not be properly segmented due to the ER 50981 chemical’s autofluorescence interfering with the Hoechst-33342 signal.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Cell culture for all RT-qPCR experiments was conducted in 96-well format. For testing the RT-qPCR primers, DMSO and ATRA dilutions were prepared as 5x concentrate in culture medium and added manually as a 30 μl volume, to obtain final concentrations of 0.5% DMSO and 500 nM ATRA in 0.5% DMSO. Four biological replicates (i.e., independent cultures) were conducted with one technical replicate (i.e., test well) each.
For all other RT-qPCR experiments, 20 μl medium was removed from each well before dosing. The pre-treatments and test chemicals were dispensed at a volume of 500 nl with the LabCyte Echo 550 acoustic dispenser into an empty plate, then back filled with 16.16 μl medium, of which 10 μl was added to the cells (corresponding to 300 nl of chemicals in 10 μl being applied). The final DMSO concentration after both treatments was 0.5%.
Twenty-four hours after applying the test chemicals, cells were harvested, RNA reverse transcribed, and qPCR conducted using the SYBR Green Fast Advanced Cells-to-CT Kit (Invitrogen) following manufacturer’s recommendations. Briefly, cells were washed once with ice-cold PBS, then lysed by addition of 50 μl lysis solution including DNase I. Samples were shaken for 30 s on a MultiFlo FX stage and incubated for 5 min at room temperature. Then, 5 μl stop solution was applied to each well, followed by another shake for 30 s and a 2 min incubation at room temperature. Samples were then stored at −80 °C until further use. Reverse transcription was carried out in 50 or 100 μl volumes, with 20 or 40 μl lysate as input.
The qPCR was carried out in 384-well MicroAmp Optical 384-well reaction plates. Reactions contained 1 μl cDNA, 5 μl PowerUp SYBR Green Master Mix and 250 nM of each forward and reverse primer in a total of 10 μl. Primers (Table S4) were designed to amplify a segment spanning two exons. A subset of primers has been used in previous studies (Nyffeler et al., 2018; Nyffeler et al., 2017). Reactions were carried out in duplicate, with averages used for analysis. Each biological replicate contained three cell culture wells that were treated with DMSO only. One of these replicates was used to normalize all data using the ΔΔCt method (Livak & Schmittgen, 2001), with ACTB as the reference gene.
Curve fitting was conducted with tcplfit2, as described above, with a BMR of 1 nMAD (derived from the remaining DMSO control wells that were not used for normalization) and an (arbitrary) cutoff of 0.5 cycles. For qPCR experiments with combined chemical treatments, the two DMSO control wells not used for normalization were further averaged to obtain one value per biological replicate. This value was then used in a two-sided paired t-test to determine statistically significant effects of both pre-treatments and test chemicals.
In parallel, a 384-well plate was cultured, treated with the sequential treatments and processed for HTPP. Data were analyzed as described in the ‘Pharmacological blockade with retinoic acid pathway modulators’ section. The technical replicates were then averaged to obtain one value per biological replicate to use in the two-sided paired t-test, analogous to the gene expression results.
Experimental Design
Typically, four technical replicates across four independent cultures (e.g., biological replicates) were performed for HTPP. For HTTr, four technical replicates were performed across two biological replicates. For test chemicals, 6–8 concentrations at half log10 spacing were tested (except for qPCR which was run at a single concentration). Detailed information about the number of biological and technical replicates, as well as the negative and reference chemicals run for reach experiment type can be found in Table S5.
Results
Phenotypic profiling of candidate reference chemicals at two seeding densities
In order to make the HTPP test system amenable for the Temp-O-Seq assay, the number of cells seeded per well had to be increased from that used in previous studies (Nyffeler et al., 2020). Preliminary experiments indicated that 3000 cells/well was a suitable density (data not shown), allowing cells to grow in a monolayer necessary for accurate cell segmentation with the Cell Painting assay, while also providing sufficiently concentrated lysates for TempO-Seq analysis. To confirm that the test system was suitable for detecting phenotypic changes at the higher seeding density, we tested a set of 11 candidate reference chemicals and a viability positive control (staurosporine) at both cell densities (400 cells/well and 3000 cells/well) (Table 1). Among the chemical set were four chemicals previously used as reference chemicals for HTPP (Nyffeler et al., 2020), two that previously yielded robust responses in HTTr (Harrill et al., 2021), as well as two agonists of receptors expressed in U-2 OS cells, and three chemicals that exerted large effects in previous HTPP screens (unpublished).
Chemicals were tested in concentration-response format at both seeding densities. The low-density screen was performed first, and some of the dose ranges were adapted for the high-density screen. Overall, we observed that the phenotypic profiles of all reference chemicals were qualitatively similar (Fig. 1A) and strongly correlated (Fig. 1B) across the two seeding densities. We further estimated BMCs for cytotoxicity, cytostasis and phenotypic effects and compared them across seeding densities (Fig. 1C). For most chemicals, PACs at the two seeding densities were within half an order of magnitude of each other. The largest difference was observed for dexamethasone (ca. 1.1 orders of magnitude). We therefore concluded that the higher seeding density yielded similar results to those observed at lower seeding density using the HTPP assay.
Figure 1: High-throughput phenotypic profiling of candidate reference chemicals at two seeding densities.
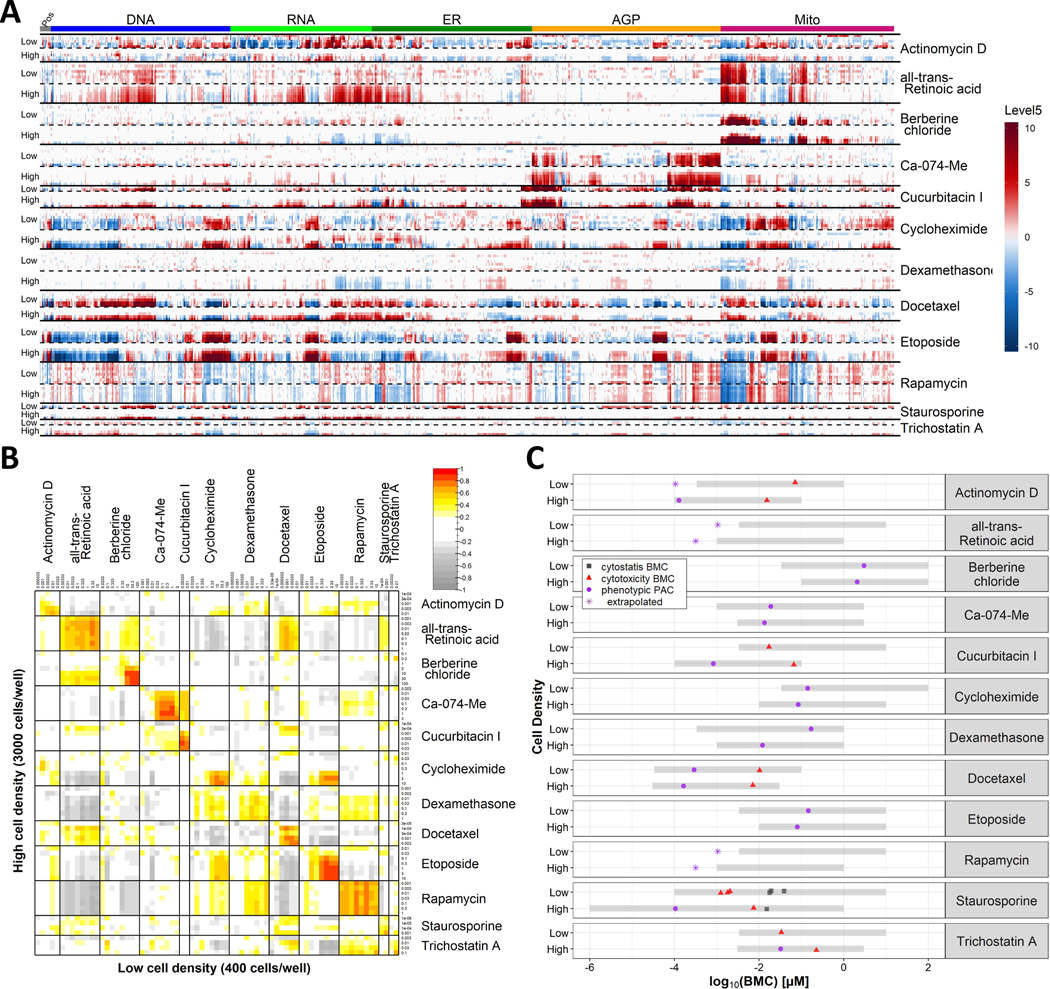
Test chemicals were screened in concentration-response using the HTPP assay in U-2 OS cells seeded at low (400 cells/well) or high (3000 cells/well) densities. Data were collected across four independent cell cultures. The exposure duration was 24 h. (A) The columns represent the 1300 features measured per cell, arranged within the fluorescent channel, as indicated by the color key on top. Phenotypic profiles for each chemical are visualized in rows, with increasing test concentrations arrayed from top to bottom in each horizontal section of the heatmap. Only non-cytotoxic, non-cytostatic concentrations are shown. Results from low and high seeding densities for each chemical are shown in consecutive horizontal sections of the heatmap. (B) Correlation matrix of the similarity of phenotypic profiles, as measured with Pearson correlation. A Pearson correlation of > 0.75 (red) is considered a strong correlation. (C) Potency estimates for each chemical, expressed as benchmark concentration (BMC). The cytotoxicity BMC (red triangles) is defined as an increase in % propidium-iodide positive cells. The cytostasis BMC (gray squares) is the EC50 of the normalized cell count. The phenotype altering concentration (PAC) (purple circles) is the concentration at which the phenotype was different from control. The gray shading indicates the tested concentration range for each chemical, which can be different between the two seeding densities. If the potency is below the tested range, it is set at ½ an order of magnitude below the lowest tested concentration and marked accordingly. For three chemicals (cucurbitacin I, staurosporine, trichostatin A) a PAC could not be estimated for the lower seeding density because too few non-cytotoxic concentrations were available for modeling. Abbreviations: AGP: actin, golgi, plasma membrane; ER: endoplasmic reticulum; Mito: mitochondria; Pos: position (features not associated with a particular channel).
Comparison of transcriptional and phenotypic effects
The 11 candidate reference chemicals were then tested under the same high seeding density conditions (i.e., 3000 cells/well, 384-well format) using the Temp-O-Seq assay. Initially, HTTr data were analyzed with two approaches: BMDExpress (as described in Harrill et al. (2021)) and modeling of signature scores with tcplfit2. For 10/11 chemicals, the HTTr signature analysis resulted in more potent estimates of bioactivity (i.e., BPACs); for four chemicals the difference was one order of magnitude or more (Fig. S2). Therefore, the signature analysis was selected for further comparisons. For all but one chemical, the PAC and BPAC were within half an order of magnitude of each other (Fig. 2A). For dexamethasone, HTTr was more sensitive by a factor of 5.
Figure 2: Comparison of high-throughput transcriptomics and high-throughput phenotypic profiling.

Test chemicals were screened in concentration-response using the HTTr assay in U-2 OS cells seeded at high (3000 cells/well) density. The exposure duration was 24 h. (A) Comparison of the chemical-wise BPAC from HTTr (green squares), PAC from HTPP (purple circles), and cell viability BMC (red crosses). The numbers to the right indicate the difference between the HTPP and HTTr potency on the log scale. The gray shaded area indicates the tested concentration range of each chemical. Chemicals with BPACs below the tested concentration range were set to ½ an order of magnitude below the lowest tested concentration. (B) Accumulation plots of active signatures from HTTr, summarized by super target. Each graph displays the 30 most sensitive super target classes. The point and stems represent the median and the 10th and 90th percentile, respectively. The numbers in parentheses are the number of affected signatures and the number of total signatures in the signature catalog associated with the super target, respectively. The size of the points corresponds to the number of active signature and the shade of the point and stem (from light to dark) represents the percentage of signatures that were active in each super target class. The gray shaded area indicates the tested concentration range for each chemical. (C) Concentration-response curves from HTPP modeling global Mahalanobis distances. The gray shaded area indicates the noise level (median ± nMAD). (D) Efficacy measures (expressed as ‘top over cutoff’) for concentration-response modelled signature score data of all active signatures. The number of active signatures (n) is indicated on the plot.
In order to visualize mechanistic information underlying the transcriptional responses, active signatures were grouped into super targets (Fig. 2B, S3). This visualization revealed that for ATRA, many of the most potently affected pathways were related to the retinoic acid receptor (RAR) (e.g., fourth and sixth super target). For dexamethasone, the glucocorticoid receptor (GR) super target was only ranked as 9th most potent, and only two out of 56 signatures of this super target were active. However, the overall most potent signature belonged to the GR super target and had a BMC below the tested range. For etoposide, many super targets were affected. Several of those were related to chromatin remodeling and DNA replication. For some of the other chemicals, the interpretation of gene expression effects was less clear, or their expected effect on gene expression was not known. Therefore, these three chemicals (ATRA, dexamethasone, etoposide) were chosen as reference chemicals for subsequent experiments. It is also notable that these three chemicals vary in their effect sizes both for HTPP and HTTr. The largest concentration-responsive increases in global Mahalanobis distance were observed with etoposide, followed by ATRA and dexamethasone (Fig. 2C). Similarly, the largest number of active HTTr signatures was observed etoposide, followed by ATRA and dexamethasone (Fig. 2D).
Phenotypic profiling of retinoic acid pathway modulators
All-trans-retinoid acid was selected for more detailed analysis for multiple reasons: (1) it had a distinct phenotypic profile in HTPP, (2) it activated the retinoic acid (RA) pathway as one of the most sensitive biological effects in HTTr, (3) both effects happened at sub-nanomolar concentrations, and (4) it is known to modulate bone homeostasis (Henning, Conaway, & Lerner, 2015; Herlin et al., 2021). Therefore, it served as an interesting case study to investigate whether the observed morphological phenotype was directly linked to the receptor activation. To this end, we tested other RAR agonists in the HTPP assay (Table 2). As RAR and retinoid X receptor (RXR) heterodimerize to activate gene expression (Marill, Idres, Capron, Nguyen, & Chabot, 2003), we also evaluated whether RXR agonists produce a similar phenotype as RAR agonists. In addition to RAR and RXR agonists, we also tested RAR and RXR inhibitors, as well as a RA metabolism inhibitor and an RA synthesis inhibitor, to investigate how perturbing RA signaling and homeostasis would affect cellular phenotypes. All chemicals were tested at eight to twelve concentrations.
The two additional RAR agonists (AM580 and arotinoid acid) produced the same phenotype as ATRA (Fig. 3A+B). This phenotype includes robust changes in mitochondrial morphology (Fig. 3C) and more subtle changes in the DNA and RNA fluorescent channels. Exploration of cell-level data revealed that there is a population-level shift in mitochondrial compactness, with more cells having higher compactness upon ATRA treatment (Fig. 3D). Moreover, two of the RXR agonists (bexarotene, methoprene acid) displayed the same phenotype as the RAR agonists, albeit at higher test concentrations (Fig. 3A+3B). Triphenyltin hydroxide, another RXR agonist, had very weak effects but was also similar to the aforementioned RAR and RXR agonists. The RXR agonist docosahexaenoic acid had a different phenotypic profile, which was only observable at the highest non-cytotoxic concentration (Fig. S4). The RA metabolism inhibitor (liarozole dihydrochloride) and RA synthesis inhibitor (citral) had rather weak effects (at non-cytotoxic concentrations) that did not resemble the retinoid phenotype. Interestingly, the RAR antagonist CD 2665 and the RXR antagonist UVI 3003 also induced profiles similar to those of retinoids. Lastly, the RAR antagonist ER 50891 resulted in a very distinct phenotype. ER 50891 is autofluorescent in the blue (DNA) channel, which led to interference with nuclei segmentation at higher concentrations. This resulted in an apparent increase in cell counts (see Fig. S4). At concentrations ≥ 10 μM nuclei segmentation was considerably affected; hence these conditions were excluded from analysis.
Figure 3: High-throughput phenotypic profiling of retinoic acid pathway modulators.

Chemicals were screened in concentration-response using the HTPP assay in U-2 OS cells seeded at high (3000 cells/well) density. The exposure duration was 24 h. Data were collected across four independent cell cultures. (A) The columns represent the 1300 features, arranged within the fluorescent channel, as indicated by the color key on top. Phenotypic profiles for each chemical are visualized in rows, with increasing test concentrations arrayed from top to bottom in each horizontal section of the heatmap. Saccharin and sorbitol served as negative controls, while dexamethasone and etoposide served as reference chemicals (assay controls). (B) Correlation matrix of the similarity of phenotypic profiles, as measured with Pearson correlation. (C) Representative images of cells treated with ATRA (1 μM in 0.5% DMSO) or solvent alone (0.5% DMSO) for 24 h. ATRA produced subtle, but reproducible, changes in mitochondrial morphology. The radial distribution of mitochondria in the perinuclear region tends to be less symmetrical and more compact in ATRA treated cells. Compare the patterns in cells marked with arrowheads. Top row: Pseudo color composite image displaying the nucleus and mitochondria fluorescent channels only. Bottom row: Transformed image of mitochondria fluorescent labeling using background intensity subtraction. (D) Cell-level results of cells treated with ATRA (1 μM) or solvent alone (0.5% DMSO) for 24 h. The feature ‘Mito_Cells_Morph_STAR_Threshold_Compactness_40%_SP-Filter’ corresponds to a measure of compactness measured in the transformed image using the sliding parabola filter. Abbreviations: AGP: actin, golgi, plasma membrane; ER: endoplasmic reticulum; Mito: mitochondria; Pos: position (features not associated with a particular channel).
While potency estimation was not the primary focus of this experiment, we performed concentration-response analysis for all chemicals (Fig. S4). Of note, the three RAR agonists were effective at sub-nanomolar concentrations, while not affecting cell numbers up to 1 μM. In contrast to this, chemicals like docosahexaenoic acid and citral had a very small window of phenotypic activity before cytostasis was observed. As several of the RAR and RXR agonists resulted in the same phenotypes, we concluded that this phenotype is likely to be specific for RA pathway activation and less likely to be an ‘off-target’ effect.
Pharmacological blockade of retinoic acid pathway: Phenotypic effects
To determine whether the observed phenotype is a result of activation of the RA signaling pathway, varying concentrations of the RAR and RXR agonists, as well as the RA metabolism inhibitor liarozole were applied to cells pre-treated with a single concentration of RAR antagonists, RXR antagonists or synthesis inhibitors. In accordance with the previous experiments, several RAR and RXR agonists produced the characteristic phenotype, in a concentration-response manner (Fig. S5). However, pre-treatments with certain inhibitors prevented this concentration-dependent phenotype from manifesting (Fig. 4A, Fig. S5). For example, for ATRA, the concentration-dependent phenotype is apparent in samples pre-treated with DMSO only or UVI 3003 and overlaid with the phenotype of citral. But upon treatment with CD 2665 or ER 50891, no concentration-dependent effects of ATRA were detectable, only the phenotypic effects of the inhibitor itself (see Fig. 4A, black arrows).
Figure 4: Combined effects of retinoic acid pathway modulators on phenotypic profiles.

U-2 OS cells were pre-treated with a single concentration of the RAR antagonists CD 2665 (10 μM), ER 50891 (1 μM), the RXR antagonist UVI 3003 (10 μM) or the RA synthesis inhibitor citral (100 μM) for 1 h, prior to treatment with the test chemicals listed in Table 2 (e.g. RAR/RXR agonists, RA metabolism inhibitor, reference chemicals, negative control) in concentration-response for an additional 24 h. Results are displayed as the average of four biological replicates, with the exception of treatments with ER 50891, which was interfering with cell segmentation in one biological replicate. (A) Phenotypic profiles for reference chemicals, and ATRA. The columns represent the 1300 features, arranged within the fluorescent channel, as indicated by the color key on top. Profiles are arranged in rows for individual treatments, with increasing chemical concentration from top to bottom within each horizontal section of the heatmap. The black arrows highlight two groups of features that are affected by ATRA treatment, but not affected when cells were pretreated with RAR antagonists. (B) Concentration-response curves for test chemicals pre-treated with the different modulators. Global Mahalanobis distance of each well was calculated relative to the mean of wells (n=24 per biological replicate) of the corresponding pre-treatment (in absence of the test chemical). In this graph, the Mahalanobis distances of the pre-treatment wells is subtracted, so that all curves start at 0. The stars indicate the phenotype altering concentration (PAC, i.e. the concentration at which the signal exceeded 1 * nMAD of the noise). (C) Overview of the PACs for the curves displayed in (B). The gray boxes indicate the tested concentration range. The error bar indicates the lower and upper bound (95% confidence interval) of the potency estimates. The pre-treatments were spread across two plates per biological replicate, hence there are two values for pre-treatment with DMSO. Sequential treatments that did not result in a PAC are displayed as open circles ½ an order of magnitude above the highest tested concentration. Abbreviations: AGP: actin, golgi, plasma membrane; ER: endoplasmic reticulum; Mito: mitochondria; PAC: phenotype altering concentration; Pos: position (features not associated with a particular channel).
To quantify these observations, concentration-response modeling was performed following transformation of the phenotypic effects into Mahalanobis distances (Nyffeler et al., 2021). Briefly, Mahalanobis distances are similar to Euclidean distances, but take into account the correlation amongst features. In the present experiment, distances were calculated relative to wells treated with the pre-treatment but not with the second treatment. Hence, if an inhibitor suppresses the retinoid phenotype, no concentration-response is expected. For the three RAR agonists (ATRA, AM580, arotinoid acid), citral and UVI 3003 pre-treatments were ineffective (i.e., did not prevent the appearance of the retinoid phenotype with increasing agonist concentration), while pre-treatment with the two RAR antagonists (CD 2665, ER 50891) right-shifted the concentration-response by two orders of magnitude or more (Fig. 4B+C). Analogously, UVI 3003 was able to shift the concentration-response of the RXR agonists bexarotene and methoprene acid. For methoprene acid, RAR antagonist ER 50891 led also to a slight right-shift but did not affect efficacy to the same extent as RXR antagonist UVI 3003. Responses to docosahexaenoic acid, triphenyltin and liarozole were not blocked by any inhibitor.
As apparent in figure 4A, only certain features were responsive to retinoid treatment. In order to identify feature categories that were affected by retinoid treatment, we applied the category-level Mahalanobis approach as described previously (Nyffeler et al., 2021) and compared the efficacy of the concentration-responses (Fig. S6). The two categories with the largest effects were related to the MitoTracker channel and concerned mitochondrial compactness and radial distribution of the intensity signal (Fig. S7). For both categories, the effect was suppressed by pre-treating cells with either RAR antagonist, and was similar to the results obtained with the ‘global Mahalanobis’ approach.
To summarize, we saw that effects of RAR agonists (ATRA, AM580, arotinoid acid) can be blocked by pre-treatment with RAR antagonists (CD 2665, ER 50891), and effects of RXR agonists can be blocked by pre-treatment with the RXR antagonist (UVI 3003), as expected. Of interest, we observed that RAR agonists and RXR agonists (bexarotene, methoprene acid) produced the same phenotype, although they apparently act on different receptor types.
Pharmacological blockade of retinoic acid pathway: Transcriptional effects
Phenotypic profiling with sequential antagonist/agonist treatments demonstrated that the observed phenotype is related to RA signaling. However, it was not clear whether the phenotype is a consequence of the receptor activation or preceded receptor activation. We therefore monitored receptor activation by measuring expression of genes of the RA pathway, as some of these genes are known to exert auto-regulation: i.e., changes in mRNA expression of a receptor in response to modulation of that receptor pathway at the protein level (de The, Vivanco-Ruiz, Tiollais, Stunnenberg, & Dejean, 1990; Haq, Pfahl, & Chytil, 1991; Kato et al., 1992; Martin, Ziegler, & Napoli, 1990; Nyffeler et al., 2018; Ocaya et al., 2011; Ray, Bain, Yao, & Gottlieb, 1997; Rossant, Zirngibl, Cado, Shago, & Giguere, 1991; Rowe, Richman, & Brickell, 1991).
In a first step, a broad set of genes was evaluated using RT-qPCR to identify genes whose expression is affected by ATRA treatment. The set included the three RAR subtypes, three RXR subtypes, vitamin D receptor, three CYP26 isoforms (as CYP26 metabolizes RA) and three aldehyde dehydrogenases (ALDH, as these enzymes synthesize RA) (Marill et al., 2003). We also evaluated two ‘off-target’ genes (CTNNB1, PTK2) and two housekeeping genes (ACTB, GADPH) for normalization. Overall, the measured expression level correlated well with public data from the human protein atlas (http://www.proteinatlas.org) (Uhlen et al., 2017). Of those genes, all ALDH and two CYP26 forms (CYP26A1, CYP26C1) had very low expression, and their expression was not increased upon ATRA treatment (Fig. S8A). Only two genes were found to be induced upon treatment with 500 nM ATRA: RARB and CYP26B1. This is consistent with the literature (de The et al., 1990; Haq et al., 1991; Kato et al., 1992; Martin et al., 1990; Nyffeler et al., 2018; Ocaya et al., 2011; Ray et al., 1997; Rossant et al., 1991; Rowe et al., 1991). Subsequently, primers for RARA, RARB, RARG, RXRA and RXRB as well as CYP26B1 were chosen for follow-up experiments characterizing the concentration-dependent upregulation of gene expression in response to ATRA. We observed a concentration-dependent increase in RARB and CYP26B1 expression following ATRA treatment, paralleling the phenotypic changes measured by global Mahalanobis distance modeling of HTPP data (Fig. 5A, Fig. S9). The BMC for phenotypic changes and for CYP26B1 upregulation was ~ 1 nM, while RARB was already upregulated at concentrations below 100 pM.
Figure 5: Gene expression changes upon treatment with retinoic acid pathway modulators.

U-2 OS cells were treated with the RA pathway modulators as explained in Figure 4. Experiments for HTPP and gene expression were conducted in parallel in 384-well and 96-well plates, respectively. Data were collected across four independent experiments, with six technical replicates for HTPP. (A) Concentration-response of cells treated with ATRA for 24 h. The left panel (in purple) represents HTPP results displayed as global Mahalanobis distance relative to vehicle control wells. The shaded area indicates the noise band (1 * nMAD of n=144 vehicle control wells). The dashed vertical line indicates the PAC. The two right panels (in blue) represent qPCR results for two genes, RARB and CYP26B1, expressed as difference in cycles (ΔΔCt) relative to the housekeeping gene ACTB and DMSO treatment. Positive numbers indicate upregulation relative to DMSO treatment. The shaded area indicates the noise band (1 * nMAD, derived from n=8 vehicle control wells that were not used for normalization). The dashed vertical lines indicate the BMC. The dashed horizontal line is at 0.5, indicating the threshold for a marked biological effect. (B) Combined effects of RA pathway modulators on phenotypic profiles (HTPP) and gene expression (RARB, CYP26B1). U-2 OS cells were pre-treated with the RAR antagonists CD 2665 (10 μM), ER 50891 (1 μM), or the RXR antagonist UVI 3003 (10 μM) for 1 h prior to treatment with the RAR agonist ATRA (100 nM) or RXR agonist bexarotene (100 nM) for 24 h. HTPP results are expressed as global Mahalanobis distances relative to the corresponding pre-treatment. The qPCR results are expressed as ΔΔCt values, as in (A). The filling of the bars corresponds to the test chemicals, while the multicolored outlines correspond to the pre-treatment. The bars represent mean +/− standard deviation of the four biological replicates. In these graphs, technical replicates within a biological replicate (e.g., plate) are averaged and represented as a single point. Statistical significance was calculated using paired, two-tailed t-tests. p-values for addition of the pre-treatment (vs. pre-treatment with DMSO alone) is indicated above the bars. p-values for addition of the test chemical (vs. DMSO alone) are indicated below the bars. *: p<0.05; **: p<0.01; ***: p<0.001.
Finally, the effect of ATRA (100 nM) and bexarotene (100 nM) on gene expression was examined in presence and absence of RAR/RXR inhibitors. Results from the HTPP assay run in parallel agreed with the previous results: CD 2665 and ER 50891 suppressed the cellular phenotype invoked with ATRA but not bexarotene. In contrast, UVI 3003 suppressed the phenotype invoked with bexarotene, but not ATRA (Fig. 5B, left). ATRA-induced upregulation of RARB did not occur with ER 50891 pre-treatment, but did occur with CD 2665 or UVI 3003 pre-treatment (Fig. 5B, right, Fig. S10). CD 2665 slightly inhibited RARB upregulation in response to ATRA, but as CD 2665 treatment itself increased RARB expression, these results are difficult to interpret. Upregulation of RARB by bexarotene was only suppressed by UVI 3003. Of note, the magnitude of upregulation by bexarotene is smaller than the one induced by ATRA (1 qPCR cycle vs 4 qPCR cycles). Interestingly, bexarotene and CD 2665 had a synergistic effect on RARB expression. CYP26B1 expression was not affected by bexarotene. The ATRA-induced CYP26B1 expression was suppressed by treatment with the two RAR antagonists CD 2665 and ER 50891. Unlike for RARB, CYP26B1 expression was not induced by CD 2665 treatment alone. Overall, these gene expression changes are consistent with the phenotypic changes; both can be suppressed by applying RAR and RXR specific antagonists for RAR and RXR agonists, respectively.
Discussion
In this study, we aimed to combine two HTP assays, phenotypic profiling and transcriptomics, for use in U-2 OS cells. We have demonstrated that (1) HTPP yielded very similar results if conducted at different cell densities, both in terms of potency and profiles of phenotypic effects; (2) for most of the chemicals tested, PACs for perturbation of cell morphology and BPACs for perturbation of gene expression were within half an order of magnitude; (3) retinoids induced a characteristic phenotypic profile, that could be prevented by pre-treating cells with appropriate RAR or RXR antagonists and (4) these phenotypic changes coincided with changes in gene expression known to occur upon perturbation of RA pathway signaling, including autoregulation of RARB.
In order to harmonize the conditions for the two HTP assays, the cell seeding density had to be increased in relation to those used in previous HTPP studies (Nyffeler et al., 2020; Willis et al., 2020). For HTTr, a higher seeding density is beneficial for generating cell lysates that are compatible with the TempO-Seq assay in terms of RNA content per unit volume. In contrast, for HTPP, a higher seeding density can decrease the ability to accurately segment and profile individual cells. In the present study, we have increased the seeding density of U-2 OS cells nearly ten-fold as compared to previous studies (Nyffeler et al., 2020; Willis et al., 2020), without considerably affecting the resulting phenotypic profiles or PACs (Fig. 1). The differences in PACs across the two seedings densities were mostly < ½ order of magnitude, and thus comparable to the intra-screen variability of PACs from repeatedly tested reference chemicals (Nyffeler et al., 2020). While consistency of phenotypes across seeding densities is often debated, this is – to our knowledge – the first study to show their equivalence. In addition, using methods established previously (Harrill et al. 2021), lysates generated using the higher seeding density of U-2 OS cells (3000 cells input per well) yielded TempO-Seq whole transcriptome data of high quality with only a few sporadic exceptions (Fig. S1).
Upon harmonization of the culture conditions, testing of the same chemical set in both assays in parallel enabled us to identify chemicals that yield both robust phenotypic effects and changes in gene expression (Fig. 2B). ATRA, dexamethasone, and etoposide were identified as promising reference chemicals for future HTTr/HTPP studies in U-2 OS cells. These chemicals were selected for multiple reasons: First, the PACs for phenotypic effects and BPACs for gene expression changes were well below the threshold for cytotoxicity for each of these chemicals in U-2 OS cells exposed for 24 hours. In fact, cytotoxicity was not observed within the tested concentration range for any of these three chemicals. Second, the phenotypic and transcriptional effects are either strong (etoposide), moderate (ATRA) or weak (dexamethasone) with regard to the number of phenotypic features or gene expression signatures affected as well as response magnitudes associated with each. While a strong reference chemical might be the more obvious choice for monitoring assay performance, combinatorial use of multiple reference chemicals ranging from weak to strong and with varying potencies for perturbing cellular biology might allow for more comprehensive monitoring of HTP assay performance compared to a single chemical. Third, all three of these chemicals have specific MoA, with dexamethasone and ATRA targeting specific nuclear receptors known to be expressed in U-2 OS cells. These molecular targets (RAR for ATRA; GR for dexamethasone; DREAM cell cycle regulator complex, E2F and topoisomerase inhibitor for etoposide) were represented amongst the most sensitive super target classes for each of these chemicals in the present study.
To date, only a few studies have combined HTTr and HTPP (Becker et al., 2020; Lapins & Spjuth, 2019; Wawer et al., 2014). However, these studies did not focus on concentration responsiveness. In the present study, we compared the potency estimates from HTPP and HTTr for a small set of chemicals and found that, in general, the PAC for phenotypic effects and the BPAC for gene expression changes were within half an order of magnitude (Fig. 2A). While results from testing of this small chemical set indicate that potencies from HTTr and HTPP are highly comparable, it is unclear if this trend is extensible to larger, more structurally diverse sets of chemicals. This will be a topic for investigation in future chemical screening studies.
When considering each HTP assay separately, interpretation of biological effects from HTTr data is often more straightforward than interpretation of biological effects from HTPP. This is because the HTTr assay measures discrete biological molecules that, in a majority of cases, are annotated with regards to cellular function and membership in established signaling pathways. In contrast, HTPP data are based upon measurement of cellular features computationally derived from patterns of fluorescent labels applied to cells. These features are not discrete biological molecules, but rather emergent properties that are not annotated to any particular functional protein or signaling pathway. Effects associated with changes in the morphology of a labeled organelle may be due to a variety of upstream molecular initiating events; thus, mechanistic interpretation of phenotypic profiles often involves consideration of other data types as well as comparison of effect profiles to those produced by well-annotated reference chemicals evaluated in the same test system.
Phenotypic and transcriptional responses to ATRA were studied in more detail because of ATRA’s relevance in the U-2 OS cell model: U-2 OS cells are osteosarcoma cells, and ATRA plays a role in bone remodeling (Henning et al., 2015; Herlin et al., 2021). In order to gain confidence that the phenotypic response to ATRA is linked to modulation of RA signaling, follow-up studies were conducted using a variety of approaches. First, we tested several different RAR agonists, as well as some RXR agonists in the HTPP assay. A majority of those chemicals resulted in the same characteristic phenotype as ATRA, which supports that this phenotype is related to activation of the RAR/RXR nuclear receptor complex. In contrast, treatment with the RAR antagonists CD 2665 and ER 50891 resulted in phenotypes distinct from one another. This could be indicative of very specific on-target effects on different RAR receptor isoforms, or unrelated off-target effects. It is unclear whether the RAR receptor is active in untreated U-2 OS cells and thus whether it could be inhibited by RAR antagonists in the absence of exogenous ligand. In a similar manner, the RXR agonists docosahexaenoic acid and triphenyltin hydroxide did not result in the characteristic phenotype produced by other RAR and RXR agonists. The phenotypes produced by these chemicals could be due to the interaction of activated RXR with nuclear receptors other than RAR (Gilardi & Desvergne, 2014). However, effects of those chemicals were also not blocked by the RXR antagonist. Both chemicals have biological activity at other molecular targets within the same concentration range required for RXR activation (triphenyltin hydroxide: https://comptox-prod.epa.gov/dashboard/dsstoxdb/results?search=triphenyltin%20hydroxide#invitrodb, docosahexaenoic acid: https://www.guidetopharmacology.org/GRAC/LigandActivityRangeVisForward?ligandId=1051), which could explain the appearance of a non-retinoid phenotype.
Second, we conducted pharmacological blockade experiments via sequential treatment of U-2 OS cells with RAR or RXR antagonists followed by RAR or RXR agonists. While RAR and RXR agonists resulted in the same phenotype, only application of the respective antagonist was able to prevent the occurrence of the phenotype. These results indicate that the phenotypic effects are likely downstream effects of the receptor activation. Interestingly, effects of the RXR agonist methoprene acid were mitigated by RXR antagonist UVI 3003 and partly by the RAR antagonist ER 50891. These results could be explained by either methoprene acid not being a “pure” RXR agonist as previously reported (Harmon, Boehm, Heyman, & Mangelsdorf, 1995), or by the fact that RXR agonists cannot activate the heterodimer alone requiring an active RAR to elicit phenotypic effects (le Maire, Teyssier, Balaguer, Bourguet, & Germain, 2019). Of note, RA synthesis and metabolism inhibitors did not result in profound phenotypes, which indicates that U-2 OS cells might not have the capacity to synthesize ATRA.
Third, we applied RT-qPCR as an orthogonal assay. Upon binding of the ligand, the RAR/RXR heterodimer induces transcription of target genes. The pathway is known to autoregulate, and thus several RA pathway genes are transcriptionally regulated upon receptor activation (de The et al., 1990; Haq et al., 1991; Kato et al., 1992; Martin et al., 1990; Nyffeler et al., 2018; Ocaya et al., 2011; Ray et al., 1997; Rossant et al., 1991; Rowe et al., 1991). The most well-known autoregulated target gene is RARB. We identified two RA pathway genes that were upregulated upon ATRA treatment and served as biomarkers of effects: RARB and CYP26B1. ATRA-induced transcriptional upregulation of RARB and CYP26B1 was concentration-dependent and paralleled the phenotypic changes. As the BMC for RARB upregulation (~40 pM) was lower than the PAC (~400 pM), one could hypothesize that some changes in gene expression precede the phenotypic effects. Upregulation of RARB and CYP26B1 by ATRA was prevented by treatment with RAR antagonists, consistent with ATRA’s known MoA (Marill et al., 2003). At the same nominal concentration, RXR agonist bexarotene had less pronounced phenotypic and transcriptional effects, possibly because bexarotene is less potent than ATRA or because bexarotene targets RXR and not RAR. Of interest, RAR antagonist CD 2665 alone upregulated RARB, and led to a synergistic effect together with bexarotene. This indicates that CD 2665 seems to activate the RA pathway, possibly in a counter-regulatory manner, and consistent with the observed phenotypic effects in HTPP.
To summarize, the combination of RT-qPCR and HTPP showed that the transcriptional changes and phenotypic effects co-occur and appear to be driven by modulation of RAR/RXR nuclear receptor signaling. It is unknown whether the phenotypic changes occur up- or downstream of the transcriptional activation as these effects were measured at only a single point in time. ATRA can exert biological effects in different ways: (1) through a classical nuclear receptor mechanism involving binding of the activated RAR/RXR heterodimer to genomic retinoic acid response elements (RARE); (2) through an alternative genomic mechanism with lower affinity by binding to peroxisome proliferator-activated receptors (PPAR)/RXR or retinoid-related orphan receptors (ROR); (3) or through a non-genomic mechanism such as phosphorylation of cAMP response element binding protein (CREB) or inhibition of translation by RARα (Conaway, Henning, & Lerner, 2013). Our results do not allow us to definitively conclude through which pathway ATRA exerts its effect on cell morphology. However, the pharmacological blockade experiments provide evidence that activation of the RAR/RXR receptors is necessary for the phenotype to occur as preventing ligands from activating these receptors prevents emergence of the phenotype. As both RAR and RXR agonists resulted in the same phenotype, this suggests that the observed phenotype is likely related to the classical genomic pathway where RAR and RXR form heterodimers.
Previous studies concluded that HTPP and HTTr provide complimentary information. Wawer et al. (2014) reported that while a large portion of tested chemicals were active in HTPP and HTTr, there was not complete overlap in the chemical sets. Similarly, other studies found that HTPP and HTTr assays are complementary in predicting MoA of chemicals (Lapins & Spjuth, 2019) and assay outcomes (Becker et al., 2020). Although the present study investigated only a small number of chemicals, we observed that the two assays are susceptible to different artifacts: ER 50891 appeared to be phenotypically active (due to autofluorescence) but did not affect gene expression of ATRA target genes. In contrast, CD 2665 was more amenable to HTPP, while modulating expression of ATRA target genes. Thus, combining assays can help understand and overcome these limitations.
The present study with 11 chemicals suggests that HTPP and HTTr have comparable sensitivity for detecting perturbations in cellular biology. Consideration of HTTr and HTPP data in combination allows on one hand for establishment of linkages between morphological phenotypes and putative initiating events revealed via gene expression profiling, and on the other hand for associating gene expression changes to apical cellular effects observed at a higher level of biological complexity. Moreover, HTPP is more costeffective than HTTr and more readily applicable to cell models from other species. Recently, Becker et al. (2020) demonstrated that HTPP augments the ability of quantitative structure activity-based models to predict assay outcomes more than HTTr. The results of the present study demonstrate the advantages of combining in silico methods, HTPP and HTTr, in the first tier of the USEPA’s tiered testing strategy for hazard evaluation (Thomas et al., 2019). Future studies will use this modified U-2 OS cell test system to evaluate chemicals of interest to the USEPA.
Supplementary Material
Highlights.
HTPP conducted at two different cell seeding densities lead to comparable results
Phenotypic profiling and transcriptomics resulted in comparable potency estimates
Three test chemicals were selected as suitable phenotypic reference chemicals
Multiple retinoids produced the same phenotypic profile and upregulated RARB
Pre-treatment with RAR antagonists blocked phenotypic and gene expression effects
Acknowledgements
The authors would also like to thank Terri Fairley, Daniel Hallinger, and Sandra Roberts for operations support activities during conduct of this research. Finally, the authors would like to thank Drs. Thomas Knudsen, Daniel Villeneuve, Jocylin Pierro, Kimberly Slentz-Kesler and Sid Hunter for their insightful comments during review of this manuscript.
Funding Information
The USEPA through its Office of Research and Development provided funding for this research. J.N. was supported by an appointment to the Research Participation Program of the USEPA, Office of Research and Development, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the USEPA.
Abbreviations
- 384LDV
Echo qualified 384-well low dead volume plate
- AGP
actin, golgi, plasma membrane
- ALDH
aldehyde dehydrogenases
- ATRA
all trans-retinoic acid
- BMC
benchmark concentration
- BMD
benchmark dose
- BMDL
benchmark dose lower confidence limit
- BMDU
benchmark dose upper confidence limit
- BMR
benchmark response
- BPAC
biological pathway altering concentration
- CMAP
connectivity map
- DMSO
dimethyl sulfoxide
- DMEM
Dulbecco’s Modified Eagle Medium
- EC50
effective concentration 50%
- GEO
Gene Expression Omnibus
- GR
glucocorticoid receptor
- HTP
high-throughput profiling
- HTPP
high-throughput phenotypic profiling
- HTTr
high-throughput transcriptomics
- hWTv2
TempO-Seq human whole transcriptome assay version 2
- L2FC
log2 fold-change
- LOEC
lowest observed effect concentration
- MAD
median absolute deviation
- MIE
molecular initiating event
- Mito
mitochondria
- MoA
mechanism-of-action
- MSigDB
Molecular Signatures database
- NAMs
new approach methods
- nMAD
normalized median absolute deviation
- PAC
phenotype altering concentration
- PBS
phosphate-buffered saline
- PI
propidium iodide
- P6
passage number 6
- RA
retinoic acid
- RAR
retinoic acid receptor
- RT-qPCR
reverse transcription quantitative polymerase chain reaction
- RXR
retinoid X receptor
- ssGSEA
single-sample gene set enrichment analysis
- USEPA
United States Environmental Protection Agency
Footnotes
Conflict of Interest
The authors declare no conflict of interest. This manuscript has been reviewed by the Center for Computational Toxicology and Exposure, Office of Research and Development, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Bibliography
- Akaike H. (1974). New Look at Statistical-Model Identification. Ieee Transactions on Automatic Control, Ac19(6), 716–723. doi:Doi 10.1109/Tac.1974.1100705 [DOI] [Google Scholar]
- Aktipis S, & Panayotatos N. (1981). A kinetic study on the mechanism of inhibition of RNA synthesis catalyzed by DNA-dependent RNA polymerase. Differences in inhibition by ethidium bromide, 3,8-diamino-6-ethylphenanthridinium bromide and actinomycin d. Biochim Biophys Acta, 655(3), 278–290. doi: 10.1016/0005-2787(81)90038-1 [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, . . . Hahn WC (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462(7269), 108–112. doi: 10.1038/nature08460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Yang K, Caicedo JC, Wagner BK, Dancik V, Clemons P, . . . Carpenter AE (2020). Predicting compound activity from phenotypic profiles and chemical structures. bioRxiv, 2020.2012.2015.422887. doi: 10.1101/2020.12.15.422887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, & Sebti SM (2003). Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res, 63(6), 1270–1279. [PubMed] [Google Scholar]
- Boehm MF, Zhang L, Badea BA, White SK, Mais DE, Berger E, . . . Heyman RA (1994). Synthesis and Structure-Activity-Relationships of Novel Retinoid-X Receptor-Selective Retinoids. Journal of Medicinal Chemistry, 37(18), 2930–2941. doi:DOI 10.1021/jm00044a014 [DOI] [PubMed] [Google Scholar]
- Bray MA, Gustafsdottir SM, Ljosa V, Singh S, Sokolnicki KL, Bittker JA, . . . Carpenter AE (2017). A dataset of images and morphological profiles of 30,000 small-molecule treatments using the Cell Painting assay. Gigascience. doi: 10.1093/gigascience/giw014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, . . . Carpenter AE (2016). Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc, 11(9), 1757–1774. doi: 10.1038/nprot.2016.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinks J, van Dijk EHC, Habeeb M, Nikolaou A, Tsonaka R, Peters HAB, . . . Boon CJF (2018). The Effect of Corticosteroids on Human Choroidal Endothelial Cells: A Model to Study Central Serous Chorioretinopathy. Invest Ophthalmol Vis Sci, 59(13), 5682–5692. doi: 10.1167/iovs.18-25054 [DOI] [PubMed] [Google Scholar]
- Buttle DJ, Murata M, Knight CG, & Barrett AJ (1992). CA074 methyl ester: a proinhibitor for intracellular cathepsin B. Arch Biochem Biophys, 299(2), 377–380. doi: 10.1016/0003-9861(92)90290-d [DOI] [PubMed] [Google Scholar]
- Conaway HH, Henning P, & Lerner UH (2013). Vitamin a metabolism, action, and role in skeletal homeostasis. Endocr Rev, 34(6), 766–797. doi: 10.1210/er.2012-1071 [DOI] [PubMed] [Google Scholar]
- Connor MJ, & Smit MH (1987). Terminal-group oxidation of retinol by mouse epidermis. Inhibition in vitro and in vivo. Biochem J, 244(2), 489–492. doi: 10.1042/bj2440489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, & Dejean A. (1990). Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature, 343(6254), 177–180. doi: 10.1038/343177a0 [DOI] [PubMed] [Google Scholar]
- de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, & Perlmann T. (2000). Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science, 290(5499), 2140–2144. doi: 10.1126/science.290.5499.2140 [DOI] [PubMed] [Google Scholar]
- Duan Q, Flynn C, Niepel M, Hafner M, Muhlich JL, Fernandez NF, . . . Ma’ayan A. (2014). LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res, 42(Web Server issue), W449–460. doi: 10.1093/nar/gku476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghyselinck NB, & Duester G. (2019). Retinoic acid signaling pathways. Development, 146(13). doi: 10.1242/dev.167502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilardi F, & Desvergne B. (2014). RXRs: collegial partners. Subcell Biochem, 70, 75–102. doi: 10.1007/978-94-017-9050-5_5 [DOI] [PubMed] [Google Scholar]
- Graczyk PP (2007). Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J Med Chem, 50(23), 5773–5779. doi: 10.1021/jm070562u [DOI] [PubMed] [Google Scholar]
- Gustafsdottir SM, Ljosa V, Sokolnicki KL, Anthony Wilson J, Walpita D, Kemp MM, . . . Shamji AF (2013). Multiplex cytological profiling assay to measure diverse cellular states. PLoS One, 8(12), e80999. doi: 10.1371/journal.pone.0080999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hande KR (1998). Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer, 34(10), 1514–1521. doi: 10.1016/s0959-8049(98)00228-7 [DOI] [PubMed] [Google Scholar]
- Haq R, Pfahl M, & Chytil F. (1991). Retinoic acid affects the expression of nuclear retinoic acid receptors in tissues of retinol-deficient rats. Proc Natl Acad Sci U S A, 88(18), 8272–8276. doi: 10.1073/pnas.88.18.8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon MA, Boehm MF, Heyman RA, & Mangelsdorf DJ (1995). Activation of mammalian retinoid X receptors by the insect growth regulator methoprene. Proc Natl Acad Sci U S A, 92(13), 6157–6160. doi: 10.1073/pnas.92.13.6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill JA, Everett LJ, Haggard DE, Sheffield T, Bundy JL, Willis CM, . . . Judson RS (2021). High-Throughput Transcriptomics Platform for Screening Environmental Chemicals. Toxicol Sci, 181(1), 68–89. doi: 10.1093/toxsci/kfab009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning P, Conaway HH, & Lerner UH (2015). Retinoid receptors in bone and their role in bone remodeling. Front Endocrinol (Lausanne), 6, 31. doi: 10.3389/fendo.2015.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlin M, Sánchez-Pérez I, Esteban J, Korkalainen M, Barber X, Finnilä MAJ, . . . Håkansson H. (2021). Bone toxicity induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and the retinoid system: A causality analysis anchored in osteoblast gene expression and mouse data. Reproductive Toxicology, 105, 25–43. doi: 10.1016/j.reprotox.2021.07.013 [DOI] [PubMed] [Google Scholar]
- Huang R, Grishagin I, Wang Y, Zhao T, Greene J, Obenauer JC, . . . Austin CP (2019). The NCATS BioPlanet - An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front Pharmacol, 10, 445. doi: 10.3389/fphar.2019.00445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Bjornsti MA, & Houghton PJ (2003). Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther, 2(3), 222–232. doi: 10.4161/cbt.2.3.360 [DOI] [PubMed] [Google Scholar]
- Imran M, Saleem S, Chaudhuri A, Ali J, & Baboota S. (2020). Docetaxel: An update on its molecular mechanisms, therapeutic trajectory and nanotechnology in the treatment of breast, lung and prostate cancer. Journal of Drug Delivery Science and Technology, 60. doi:ARTN 101959 10.1016/j.jddst.2020.101959 [DOI] [Google Scholar]
- Janesick A, Wu SC, & Blumberg B. (2015). Retinoic acid signaling and neuronal differentiation. Cell Mol Life Sci, 72(8), 1559–1576. doi: 10.1007/s00018-014-1815-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson R, Richard A, Dix DJ, Houck K, Martin M, Kavlock R, . . . Smith E. (2009). The toxicity data landscape for environmental chemicals. Environ Health Perspect, 117(5), 685–695. doi: 10.1289/ehp.0800168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagechika H, Kawachi E, Hashimoto Y, Himi T, & Shudo K. (1988). Retinobenzoic acids. 1. Structure-activity relationships of aromatic amides with retinoidal activity. J Med Chem, 31(11), 2182–2192. doi: 10.1021/jm00119a021 [DOI] [PubMed] [Google Scholar]
- Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, & Nishikawa J. (2005). Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Molecular Pharmacology, 67(3), 766–774. doi: 10.1124/mol.104.008409 [DOI] [PubMed] [Google Scholar]
- Kato S, Mano H, Kumazawa T, Yoshizawa Y, Kojima R, & Masushige S. (1992). Effect of retinoid status on alpha, beta and gamma retinoic acid receptor mRNA levels in various rat tissues. Biochem J, 286 ( Pt 3), 755–760. doi: 10.1042/bj2860755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Tagami K, Hibi S, Yoshimura H, Tokuhara N, Tai K, . . . Nagai M. (2001). Syntheses and evaluation of quinoline derivatives as novel retinoic acid receptor a antagonists. Bioorganic & Medicinal Chemistry Letters, 11(9), 1215–1218. doi:Doi 10.1016/S0960-894x(01)00177-9 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, & Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods, 12(4), 357–360. doi: 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, & Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol, 37(8), 907–915. doi: 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapins M, & Spjuth O. (2019). Evaluation of Gene Expression and Phenotypic Profiling Data as Quantitative Descriptors for Predicting Drug Targets and Mechanisms of Action. bioRxiv, 580654. doi: 10.1101/580654 [DOI] [Google Scholar]
- le Maire A, Teyssier C, Balaguer P, Bourguet W, & Germain P. (2019). Regulation of RXR-RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells, 8(11). doi: 10.3390/cells8111392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengqvist J, Mata De Urquiza A, Bergman AC, Willson TM, Sjovall J, Perlmann T, & Griffiths WJ (2004). Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor alpha ligand-binding domain. Mol Cell Proteomics, 3(7), 692–703. doi: 10.1074/mcp.M400003-MCP200 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, . . . Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, & Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 1(6), 417–425. doi: 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, & Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27(12), 1739–1740. doi: 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods, 25(4), 402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Loeliger P, Bollag W, & Mayer H. (1980). Arotinoids, a New Class of Highly-Active Retinoids. European Journal of Medicinal Chemistry, 15(1), 9–15. [Google Scholar]
- Love MI, Huber W, & Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15(12), 550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marill J, Idres N, Capron CC, Nguyen E, & Chabot GG (2003). Retinoic acid metabolism and mechanism of action: a review. Curr Drug Metab, 4(1), 1–10. doi: 10.2174/1389200033336900 [DOI] [PubMed] [Google Scholar]
- Martin CA, Ziegler LM, & Napoli JL (1990). Retinoic acid, dibutyryl-cAMP, and differentiation affect the expression of retinoic acid receptors in F9 cells. Proc Natl Acad Sci U S A, 87(12), 4804–4808. doi: 10.1073/pnas.87.12.4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister B, Fink FM, Hittmair A, Marth C, & Widschwendter M. (1998). Antiproliferative activity and apoptosis induced by retinoic acid receptor-gamma selectively binding retinoids in neuroblastoma. Anticancer Research, 18(3a), 1777–1786. [PubMed] [Google Scholar]
- Nahoum V, Perez E, Germain P, Rodriguez-Barrios F, Manzo F, Kammerer S, . . . Bourguet W. (2007). Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc Natl Acad Sci U S A, 104(44), 17323–17328. doi: 10.1073/pnas.0705356104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi T, Nishikawa J, Hiromori Y, Yokoyama H, Koyanagi M, Takasuga S, . . . Tanaka K. (2005). Trialkyltin compounds bind retinoid X receptor to alter human placental endocrine functions. Mol Endocrinol, 19(10), 2502–2516. doi: 10.1210/me.2004-0397 [DOI] [PubMed] [Google Scholar]
- NRC. (1984). Toxicity Testing: Strategies to Determine Needs and Priorities. Washington (DC). [PubMed] [Google Scholar]
- NRC. (2007). Toxicity testing in the 21st century: a vision and a strategy. Reprod Toxicol, 25(1), 136–138. doi: 10.1016/j.reprotox.2007.10.013 [DOI] [PubMed] [Google Scholar]
- Nyffeler J, Chovancova P, Dolde X, Holzer AK, Purvanov V, Kindinger I, . . . Leist M. (2018). A structure-activity relationship linking non-planar PCBs to functional deficits of neural crest cells: new roles for connexins. Arch Toxicol, 92(3), 1225–1247. doi: 10.1007/s00204-017-2125-4 [DOI] [PubMed] [Google Scholar]
- Nyffeler J, Haggard DE, Willis C, Setzer RW, Judson R, Paul-Friedman K, . . . Harrill JA (2021). Comparison of Approaches for Determining Bioactivity Hits from High-Dimensional Profiling Data. SLAS Discov, 26(2), 292–308. doi: 10.1177/2472555220950245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyffeler J, Karreman C, Leisner H, Kim YJ, Lee G, Waldmann T, & Leist M. (2017). Design of a high-throughput human neural crest cell migration assay to indicate potential developmental toxicants. ALTEX, 34(1), 75–94. doi: 10.14573/altex.1605031 [DOI] [PubMed] [Google Scholar]
- Nyffeler J, Willis C, Lougee R, Richard A, Paul-Friedman K, & Harrill JA (2020). Bioactivity screening of environmental chemicals using imaging-based high-throughput phenotypic profiling. Toxicol Appl Pharmacol, 389, 114876. doi: 10.1016/j.taap.2019.114876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, & Hardesty B. (1971). The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem, 246(1), 174–181. [PubMed] [Google Scholar]
- Ocaya PA, Elmabsout AA, Olofsson PS, Torma H, Gidlof AC, & Sirsjo A. (2011). CYP26B1 plays a major role in the regulation of all-trans-retinoic acid metabolism and signaling in human aortic smooth muscle cells. J Vasc Res, 48(1), 23–30. doi: 10.1159/000317397 [DOI] [PubMed] [Google Scholar]
- Pautke C, Schieker M, Tischer T, Kolk A, Neth P, Mutschler W, & Milz S. (2004). Characterization of osteosarcoma cell lines MG-63, Saos-2 and U-2 OS in comparison to human osteoblasts. Anticancer Research, 24(6), 3743–3748. [PubMed] [Google Scholar]
- Pinero J, Queralt-Rosinach N, Bravo A, Deu-Pons J, Bauer-Mehren A, Baron M, . . . Furlong LI (2015). DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford), 2015, bav028. doi: 10.1093/database/bav028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray WJ, Bain G, Yao M, & Gottlieb DI (1997). CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J Biol Chem, 272(30), 18702–18708. doi: 10.1074/jbc.272.30.18702 [DOI] [PubMed] [Google Scholar]
- Ren MQ, Pozzi S, Bistulfi G, Somenzi G, Rossetti S, & Sacchi N. (2005). Impaired retinoic acid (RA) signal leads to RAR beta 2 epigenetic silencing and RA resistance. Molecular and Cellular Biology, 25(23), 10591–10603. doi: 10.1128/Mcb.25.23.10591-10603.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J, Zirngibl R, Cado D, Shago M, & Giguere V. (1991). Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev, 5(8), 1333–1344. doi: 10.1101/gad.5.8.1333 [DOI] [PubMed] [Google Scholar]
- Rowe A, Richman JM, & Brickell PM (1991). Retinoic acid treatment alters the distribution of retinoic acid receptor-beta transcripts in the embryonic chick face. Development, 111(4), 1007–1016. [DOI] [PubMed] [Google Scholar]
- Sheffield T, Brown J, Davidson S, Friedman KP, & Judson R. (2021). tcplfit2: an R-language general purpose concentration-response modeling package. Bioinformatics. doi: 10.1093/bioinformatics/btab779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, . . . Golub TR (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell, 171(6), 1437–1452 e1417. doi: 10.1016/j.cell.2017.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szondy Z, Reichert U, Bernardon JM, Michel S, Toth R, Ancian P, . . . Fesus L. (1997). Induction of apoptosis by retinoids and retinoic acid receptor gamma-selective compounds in mouse thymocytes through a novel apoptosis pathway. Molecular Pharmacology, 51(6), 972–982. [DOI] [PubMed] [Google Scholar]
- Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, & Tomita F. (1986). Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem Biophys Res Commun, 135(2), 397–402. doi: 10.1016/0006-291x(86)90008-2 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Tamura K, & Ide H. (1996). Citral, an inhibitor of retinoic acid synthesis, modifies chick limb development. Dev Biol, 175(2), 239–247. doi: 10.1006/dbio.1996.0111 [DOI] [PubMed] [Google Scholar]
- Thomas RS, Bahadori T, Buckley TJ, Cowden J, Deisenroth C, Dionisio KL, . . . Williams AJ (2019). The Next Generation Blueprint of Computational Toxicology at the U.S. Environmental Protection Agency. Toxicol Sci, 169(2), 317–332. doi: 10.1093/toxsci/kfz058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukey JW (1977). Exploratory data analysis. Reading, Mass. [u.a.]: Addison-Wesley. [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, . . . Ponten F. (2017). A pathology atlas of the human cancer transcriptome. Science, 357(6352). doi: 10.1126/science.aan2507 [DOI] [PubMed] [Google Scholar]
- van Mierlo T, Hyatt D, & Ching AT (2016). Employing the Gini coefficient to measure participation inequality in treatment-focused Digital Health Social Networks. Netw Model Anal Health Inform Bioinform, 5(1), 32. doi: 10.1007/s13721-016-0140-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wauwe J, Coene MC, Cools W, Goossens J, Lauwers W, Le Jeune L, . . . Van Nyen G. (1994). Liarozole fumarate inhibits the metabolism of 4-keto-all-trans-retinoic acid. Biochem Pharmacol, 47(4), 737–741. doi: 10.1016/0006-2952(94)90137-6 [DOI] [PubMed] [Google Scholar]
- Van Wauwe J, Van Nyen G, Coene MC, Stoppie P, Cools W, Goossens J, . . . Janssen PA (1992). Liarozole, an inhibitor of retinoic acid metabolism, exerts retinoid-mimetic effects in vivo. J Pharmacol Exp Ther, 261(2), 773–779. [PubMed] [Google Scholar]
- Warchal SJ, Dawson JC, & Carragher NO (2016). Development of the Theta Comparative Cell Scoring Method to Quantify Diverse Phenotypic Responses Between Distinct Cell Types. Assay Drug Dev Technol, 14(7), 395–406. doi: 10.1089/adt.2016.730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawer MJ, Li K, Gustafsdottir SM, Ljosa V, Bodycombe NE, Marton MA, . . . Clemons PA (2014). Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc Natl Acad Sci U S A, 111(30), 10911–10916. doi: 10.1073/pnas.1410933111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis C, Nyffeler J, & Harrill JA (2020). Phenotypic Profiling of Reference Chemicals Across Biologically Diverse Cell Types Using the Cell Painting Assay. SLAS Discovery, (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeakley JM, Shepard PJ, Goyena DE, VanSteenhouse HC, McComb JD, & Seligmann BE (2017). A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling. PLoS One, 12(5), e0178302. doi: 10.1371/journal.pone.0178302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, & Beppu T. (1990). Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem, 265(28), 17174–17179. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.