

Abstract
Stimulation of the mouse mammary tumor virus with steroids results in the generation of a DNase I-hypersensitive region (HSR) spanning the hormone responsive element (HRE) in the long terminal repeat. Restriction enzymes were used to characterize the accessibility of various sites within the HSR of mouse mammary tumor virus long terminal repeat-reporter constructions in four different cell lines. The glucocorticoid-dependent HSR was found to span minimally 187 bases, a stretch of DNA longer than that associated with histones in the core particle. Although the 5′-most receptor binding site within the HRE is downstream of −190, hypersensitive sites were found further upstream to at least −295. The relationship in the accessibility between pairs of sites in the vicinity of the HSR was further examined in one cell line by a two-enzyme restriction access assay. In the uninduced state, the accessibilities at these sites were found to be independent of each other. In contrast, when stimulated with hormone, the accessibilities at these sites were observed to become linked. That is, once a distinct promoter was activated, all of the sites within the HSR of that molecule became accessible. The HSR formed along an invariant stretch of DNA sequence despite the multiplicity of nucleosome frames in the nucleosome B region, where the HRE is located. The results indicate that the macroscopic length of the HSR does not arise from core length-remodeling events in molecules containing Nuc-B in alternative positions.
Regulation of transcription of the mouse mammary tumor virus (MMTV) by steroid hormones is mediated through a hormone response element (HRE) located between positions −70 and −190 (16, 28, 33, 39). Four binding sites for steroid receptors have been mapped within the HRE (57, 61, 68, 82). Binding sites for other factors have also been detected within (30, 41, 73, 74) and immediately upstream of (17, 27, 44, 45, 49) this region, and they participate in the regulation of MMTV by steroids (17, 27, 44, 45, 74). Loading of transcription factors downstream of the HRE occurs upon activation of the promoter by the glucocorticoid or the progesterone receptors, including NF-1, OTF-1, and TFIID (5, 19, 43, 59, 76). Concomitant with this activation, hypersensitivity to DNase I, methidiumpropyl-EDTA · iron(II) [MPE-Fe(II)], and restriction enzymes is detected in the general location of the HRE (3, 9, 62, 67, 76, 88).
To understand the chromatin transition leading to hypersensitivity and thus the regulation of MMTV by steroids, it is necessary to understand the chromatin organization of the promoter in both the uninduced and induced states. The long terminal repeat (LTR) of MMTV is organized in an array of six nucleosomes termed Nuc-A through Nuc-F; the HRE and the transcription initiation site are in the Nuc-B and Nuc-A regions, respectively (67). Nucleosomes in these two regions of the LTR occupy multiple frames; that is, different copies of the LTR possess Nuc-A and Nuc-B in different positions (26). Although treatment with dexamethasone, a synthetic glucocorticoid, results in a hypersensitive Nuc-B region, the appearance of the nucleosomal ladders is the same as in uninduced templates, suggesting that the Nuc-B DNA sequence remains nucleosomal (67, 76). Consistent with this observation, the content of Nuc-B DNA in mononucleosomes (76), as well as the position and relative occupancy of nucleosome frames (26), also appears the same in control and steroid-treated cell populations. Furthermore, while a decrease in the content of the linker histone H1 is observed in the activated promoter, no comparable loss in the amount of the core histone H2B is detected (11). However, the observation of heterogeneity in the activation state of MMTV in steroid-treated populations (36, 78) introduces a measure of uncertainty on the nucleosomal status of the active promoter. The subpopulation of uninduced promoters can be expected to contribute to the background signal obtained in experimental assays of steroid-treated cells.
To advance our understanding of the steroid-dependent change in the chromatin structure of the Nuc-B region, we considered it necessary to further analyze the properties of the hypersensitive region (HSR). Here we report that the HSR stretches from at least −109 through −295, a DNA sequence longer than that associated with core histones in one nucleosome. Based on the premise that hypersensitivity in one molecule results from the transition or remodeling of the B nucleosome in that molecule, the above finding raised the possibility that different MMTV promoters containing Nuc-B in alternative positions would remodel different stretches of the promoter, i.e., different DNA sequences. Using a two-enzyme restriction access assay to examine the linkage between nonadjacent sites in the HSR, we asked whether the region of hypersensitivity was the result of transitions in distinct Nuc-B frames. The data show that in the uninduced state, digestion of one molecule by any one enzyme occurs mostly independently of the cutting at a nonadjacent site by another enzyme. In contrast, in the induced state, a molecule accessible at any site in the HSR is always accessible at a nonadjacent site in the HSR. This demonstrates that the length of the HSR is not the end result of core length transitions in alternative locations but, rather, that activation of any single promoter leads to the “opening” of the entire region between −109 and −295.
MATERIALS AND METHODS
Cell lines.
Cells were maintained as monolayers grown in Dulbecco modified Eagle medium containing 10% fetal bovine serum. At 24 h before use, the cells were switched to Dulbecco modified Eagle medium lacking phenol red and containing 10% charcoal-stripped serum. Induction was performed with 0.5 μM dexamethasone (Sigma).
Cell line 19g11.2 harbors 10 to 15 copies of the GR strain of MMTV in a rat hepatoma background and expresses elevated levels of the glucocorticoid receptor (78). The mouse cell lines harboring LTR constructs derived from the C3H strain of MMTV have been described previously (4, 26, 32, 56, 67). Cell lines 904.13 and 1471.1 carry BPV-based MMTV LTR episomes in a C127 mouse mammary cell background, and cell line 1361.5 carries a similar construct in NIH 3T3 cells. Line 3134 is a subclone of 904.13 containing about 200 copies of the plasmid in an integrated head-to-tail tandem array (32). The LTR drives the chloramphenicol acetyltransferase gene in the 1471.1 cell line and Ha-ras in 3134 and 1361.5 cells; the orientation of the LTR reporter relative to the BPV backbone differs in the last two lines. The 1361.5 and 1471.1 cells used in this study carry approximately 50 and 1,000 copies of the MMTV constructs, respectively (data not shown).
Preparation of nuclei.
Cells were harvested by being scraped into cold phosphate-buffered saline and centrifuged for 3 to 5 min at 1,000 × g. The cell pellet was resuspended and disrupted in a Dounce homogenizer with an A pestle in 0.3 M sucrose–2 mM magnesium acetate–3 mM CaCl2–1% Triton X-100–0.5 mM dithiothreitol (DTT)–0.5 mM phenylmethylsulfonyl fluoride–10 mM HEPES (pH 7.8). Homogenates were diluted 1:1 with 5 to 10 ml of PB (25% glycerol, 5 mM magnesium acetate, 0.1 mM EDTA, 5 mM DTT, and 10 mM HEPES [pH 7.8]), centrifuged at 1,000 × g for 15 min in a tabletop centrifuge through a 10- to 20-ml pad of PB, and resuspended in the same buffer containing 0.5 mM DTT, with 150 to 200 μl per 150-mm culture dish. The concentration of nuclei ranged from 5 to 10 mg of nucleic acid per ml as determined by measuring the optical density at 260 nm.
Restriction enzyme access assay.
Concentrated enzymes were purchased from New England Biolabs (Beverly, Mass.). We first compared the effect of various buffering conditions (31, 72, 85) on the digestion of nuclei by using various concentrations of SacI. In agreement with previous findings (72), we observed that plateau levels of cleavage were reached at high concentrations of enzyme; SacI digestion began to plateau at a concentration of 500 U/ml in both control and dexamethasone-treated samples regardless of the buffer used (data not shown). In addition, the fractional cleavage values observed in the buffer described by Workman and Langmore were higher overall, which has been attributed to the low concentration of magnesium (0.5 mM) present during the digestion (85). Four other enzymes tested in this buffer reached plateau levels of cleavage at concentrations similar to or lower than that of SacI (data not shown). We retained this low Mg2+ buffer since any differences in cleavage between the dexamethasone-treated and control samples might be more apparent. Further, to minimize the error between determinations, we chose an enzyme concentration of 1,000 U/ml for the single-enzyme restriction assays; restrictions were conducted with nuclei at a DNA concentration of 100 μg/ml (enzyme-to-DNA ratio, 10 U/μg of DNA) in 50 mM NaCl–0.5 mM MgCl2–1 mM β-mercaptoethanol–50 mM Tris-Cl (pH 8.0) (85) for 15 min at 37°C. Digestions were quickly chilled in an ice-water bath and stopped by the addition of 1 volume of 0.2 M NaCl–20 mM EDTA–2% sodium dodecyl sulfate–50 mM Tris-Cl (pH 8) and 2 volumes of H2O. The samples were digested with 100 to 200 μg of proteinase K per ml and extracted with phenol-chloroform, and the DNA was recovered by precipitation with isopropanol. For the purpose of quantitating the cleavage, the extracted DNA was cut to completion with a second restriction enzyme, phenol extracted, and ethanol precipitated prior to analysis of the digestion products by primer extension.
Two-enzyme restriction assays were performed with one enzyme held at 25 U/μg of DNA (2,500 U/ml; denoted Ea) and various concentrations of a second enzyme (denoted Eb), as specified in the figures. The DNA concentration, buffer, and incubation times were as above. Reaction mixes differing in Eb concentration were supplemented with the appropriate restriction enzyme storage buffer to match conditions exactly; the same precaution was observed with nucleus storage buffer when the concentration of nucleus stocks differed. After phenol-chloroform extraction, the protein-free DNA was digested to completion with a third restriction enzyme to allow quantitation of the fractional cleavage. The direction of primer extension was performed so that the site cut by Ea was distal to the primer. Otherwise, the relations shown below do not apply for low values of cleavage by Eb. The most informative data is collected at very low concentrations of Eb, where the slopes of the curves of the fractional cleavage, F(b), versus [Eb] are steep and thus susceptible to significant error in the F(b) values. This was of particular concern since the determinations conducted with nuclei from steroid-treated cells required a parallel analysis with control nuclei. Therefore, the data was analyzed (see below) only when it was obtained in assays in which the concentrations of the uninduced and induced nuclei were comparable during the restriction reaction, as evidenced by the intensity of the resulting primer extension signals.
Theoretical basis for restriction enzyme linkage analysis.
Treatment of a molecule of chromatin containing restriction sites a and b with enzymes Ea and Eb together results in one of four outcomes: a single cut at a, a single cut at b, cuts at both a and b, or no cuts. Let a be the collection of molecules accessible at site a, b be the collection of molecules accessible at site b, and A be the subgroup of a inaccessible at b, or
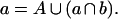 |
1 |
It follows from equation 1 that
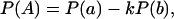 |
2 |
where P(a) and P(b) are the probabilities of cutting at the corresponding sites, P(A) is the probability of cutting molecules accessible at a and inaccessible at b, and k is the conditional probability of cutting a molecule containing an accessible site a given it contains an accessible site b. Similarly, for a population containing molecules in two mutually exclusive states,
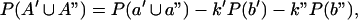 |
3 |
where the prime and double prime denote the two states.
The experimentally determined amount of cleavage is expressed as the fractional cleavage, F(x). Equations 2 and 3 are valid for all values of F(b) if molecules are randomly selected for restriction from b, which we assume to be the case. However, the analysis cannot be conducted at low values of F(a), because all the molecules in a must be accounted for, for k to equal the conditional probability. Since the fractional cleavage at any site reaches a plateau level at high concentrations of restriction enzyme (reference 72 and data not shown), we assume that F(a) = P(a) at high concentrations of Ea.
Analysis of the induced promoter by the two-enzyme restriction assay.
The F(A)STE for the expected outcomes was computed by using the determined value of F(a)STE, and the computed values of k′, k", F(b′)Eb, and F(b")Eb for each concentration of enzyme Eb according to equation 5. The subscript STE refers to the steroid-treated populations. Rectangular hyperbolas were fitted by nonlinear regression to plots of F(b) and F(b)STE versus [Eb], and the values of F(b)Eb and ΔF(b)Eb were obtained from the best curve and the difference between curves, respectively. F(b′)Eb, and F(b")Eb were then computed by using the best-fit values of F(b)Eb and ΔF(b)Eb according to equations 6 and 7. The value of k" was set to 0, F(a"), or 1 for mutually exclusive, independent, or linked relations in the induced state; F(a") was computed from F(a), ΔF(a), and wa according to an equation equivalent to equation 7. The value of k′ was taken as the product of wa and k, where k was obtained from the slope of the F(A) versus F(b) plot of the uninduced state. The r2 values of the regressions ranged from 0.96 to 0.99. Regressions were conducted with the program Prism (GraphPad Software, Inc.).
Oligonucleotide primers.
The oligonucleotides were designed based on the sequence of the LTR in the C3H strain of MMTV (GenBank accession no. J02274). Oligonucleotides were synthesized in an Applied Biosystems 392 DNA Synthesizer, dried, and ethanol precipitated. The oligonucleotides were gel purified, desalted in a Sep-Pak column (Waters), dried, ethanol precipitated, and stored in solution at −20°C. Their sequence, position, and orientation in the LTR sequence are summarized in Table 1.
TABLE 1.
Oligonucleotides used for the primer extension determinations of restriction enzyme access in the MMTV LTRa
| Oligonucleotide | Sequence | 5′ end | Strand | Site(s) probedb |
|---|---|---|---|---|
| 669 | ACAAGAGGTGAATGTTAGGACTGTTGC | +27 | − | −162, −109, −74 |
| 670 | TCAGAGCTCAGATCAGAACCTTTGATACC | −101 | − | −225, −295 |
| 730 | ATACCAAGGAGGGGACAGTG | −310 | + | −225, −162 |
| 750 | TGCGGGGGGACCCTCTGGAA | +98 | − | −74 |
| 752 | TTACTTAAGCCTTGGGAACC | −193 | − | −295 |
| 754 | CTTCTTGAGACAACATACAC | −389 | − | −560 |
| 756 | GTAAGAGGAAGTTGGCTGTG | −870 | − | −1084, −972 |
| 761 | AGGAGACAGGTGGTGGCAAC | −594 | + | −378, −295 |
| 764 | GTTGCCACCACCTGTCTCCT | −575 | − | −724 |
| 765 | CAGACCAACAGATGCCCCCT | −546 | + | −378, −295 |
| 766 | GAGTGTTCTATTTTCCTATG | −102 | + | +21, +50, +87 |
| 767 | CACAGCCAACTTCCTCTTAC | −889 | + | −638, −560 |
The design of the oligonucleotides was based on the sequence of the C3H LTR.
The positions indicate the first base (5′) of the sequence recognized by the various restriction enzymes used in this work; the enzymes are indicated in Fig. 2.
Primer extensions.
Oligonucleotide primers were phosphorylated with 100 μCi of [γ-32P]ATP per pmol of primer (5,000 to 6,000 Ci/mmol), using T4 kinase. The kinase was heat inactivated, and the labeled oligonucleotides were desalted by spin column chromatography through Sephadex G-25. Each primer extension reaction was done with 0.4 pmol of labeled oligonucleotide and 10 to 25 μg of template DNA in 50 mM KCl–3.5 mM MgCl2–0.1% Triton X-100–10 mM Tris-Cl (pH 8.3)–200 μM deoxynucleoside triphosphates–2.5 U of Taq polymerase in a reaction volume of 30 μl. The primer extension mixtures were thermally cycled to achieve a 30-fold linear amplification of the signal; temperatures 6°C above Tm were used for the annealing step. The Tms were estimated with the program OLIGO (National Biosciences). The annealing temperature was reduced 1.5°C per mismatch when the sequence of the C3H-based oligonucleotide primers differed from that of the GR strain of the LTR (GenBank accession no. V01175).
Reaction products were extracted and ethanol precipitated before being subjected to electrophoresis in 8% sequencing gels. The electrophoresed products were visualized with a PhosphorImager (Molecular Dynamics). Quantitation was performed with the ImageQuant program (Molecular Dynamics) by baseline-to-baseline area integration of lane-wide rectangular strips spanning the bands of interest in each lane. The fractional cleavage of the site assayed in chromatin was taken as the integrated intensity of the bank, normalized to the sum of the integrated intensities of the bands corresponding to the cuts in chromatin and naked DNA.
RESULTS
Time course of hypersensitivity.
Transcriptional activation of MMTV occurs within minutes of glucocorticoid administration (77). With the 3134 cell line, we first examined the kinetics of the hypersensitive response to determine an optimal time to characterize the HSR. This cell line contains an integrated LTR construction and is thus presumably more stable than the original episomal cell line from which it was derived (see Materials and Methods). A time course of SacI and HaeIII accessibility in the LTR-reporter construct in the 3134 cells is shown in Fig. 1A. SacI cuts in the Nuc-B region DNA at −109, and HaeIII cuts at −225. The two enzyme sites differed in their accessibility in the uninduced promoter; SacI cut about 10% of the templates, whereas HaeIII cut 45% (Fig. 1B). A change in the fractional cleavage, F(x), was detected at both the SacI and HaeIII sites within 8 min of dexamethasone treatment, the earliest time tested. The accessibility continued to increase to 1 h and thereafter decreased continuously through 23 h of treatment. However, it did not reach the level observed in the uninduced state at time zero. This is seen more clearly in Fig. 1C, where the change in fractional cleavage [ΔF(x)] is plotted against the time of induction. This time course of hypersensitivity is very similar to that obtained with DNase I in XC-derived cell lines (9). Significantly, the change in HaeIII accessibility paralleled very closely what was observed for SacI. In addition, the magnitude of the change in fractional cleavage was nearly the same for both enzymes, about 0.17 at 60 min of dexamethasone treatment. Subsequent experiments (below) were conducted with nuclei isolated from cells treated for 60 min with dexamethasone, the optimum time of induction observed for 3134 cells (Fig. 1) and 1471.1 cells (reference 4 and data not shown).
FIG. 1.

Time course of accessibility in the HSR. (A) Nuclei were prepared from 3134 cells treated with dexamethasone for the times indicated. Aliquots containing a 10-μg equivalent of DNA were treated with either SacI (lanes 1 to 8) or HaeIII (lanes 9 to 16) at 1,000 U/ml and processed as described in Materials and Methods. Primer extension reactions were conducted with oligonucleotide 669 (lanes 1 to 8) or 670 (lanes 9 to 16), and the products were resolved in a sequencing gel. The times of dexamethasone treatment are shown above the lanes; the bands corresponding to the SacI and HaeIII cleavage sites are indicated (the doublet appearance is presumably due to the action of endogenous nucleases on the sticky ends). The upper band in each lane corresponds to the DpnII (lanes 1 to 8) or NlaIII (lanes 9 to 16) secondary cuts. (B) Cleavage at the SacI and HaeIII sites was quantitated and normalized to obtain the fractional cleavage, F(x). Duplicate data points from the first 5 h of a plot of F(x) as a function of time of dexamethasone treatment are plotted for the SacI and HaeIII sites, as indicated. (C) The change in fractional cleavage relative to time zero, ΔF(x), is plotted as a function of time. The means of the duplicates shown in panel B were used in this plot.
We had argued previously that the HaeIII site defined the upstream edge of the hormonally responsive nucleosome in the Nuc-B region because no change in access was judged to occur (3). However, analysis of enzyme access data in terms of the fold increase in accessibility is potentially misleading, since SacI could approach a 10-fold increase whereas maximum cleavage by HaeIII could be represented as only a 2-fold increase. As we show here, the change in accessibility at both restriction sites is practically identical.
The steroid-dependent HSR spans 187 bases.
A map of the LTR displaying the restriction enzyme sites that we examined is shown in Fig. 2A. To provide internal controls, a number of the restriction enzymes chosen cut multiple sites in the LTR. Furthermore, in addition to the 3134 cells, we examined uninduced and steroid-treated 1471.1, 1361.5, and 19g11.2 cells to ensure that the results were not particular to the 3134 cell line. These cells differ in the LTR-reporter, copy number, integration state, and genetic background (see Materials and Methods).
FIG. 2.

Analysis of the accessibility and hypersensitivity of the LTR. (A) Positions of the restriction sites on the LTR. Fo, FokI; Hp, HphI; Ap, ApoI; Ha, HaeIII; Av, AvaII; Nl, NlaIII; Al, AlwNI; Sa, SacI. The ApoI site shown at −74 is found in the C3H strain of the LTR; the GR strain has a HinfI site at this position. In the −370 region, there are two sites for NlaIII, at −378 and at −362; there is also one site for RsaI at −365. The low-resolution positions of nucleosomes A through F in the LTR are denoted by ellipses. (B) Primer extension analysis of cleavage at various sites of the LTR (GR strain) in untreated (−) or 60-min-dexamethasone-treated (+) 19g11.2 cells. The cuts by the various enzymes, indicated above each pair of lanes, are marked by an asterisk to the right of each pair of lanes; the nucleotide position of the recognition site is shown at the bottom. The top band in each lane represents the secondary enzyme cut used for quantitation. The primers used for the analysis shown were (from left to right) 756, 764, 767, 767, 761, 761, 669, 669, 669, 766, and 766. (C) The fractional cleavage, F(x), for each site in the uninduced cells is shown for the 3134, 1471.1, 1361.5, and 19g11.2 cell lines, as indicated at the bottom. The LTR of the GR strain (in the 19g11.2 cells) lacks sites for NlaIII at −252, HaeIII at −225, and HphI at +21; the low signal from HphI at −972 could not be quantitated in this cell line because of high background in the assays. (D) the ΔF(x) after a 60-min dexamethasone treatment is shown; sites and cell lines are as in panel C.
The results of an electrophoretic analysis of a typical restriction enzyme access assay are shown in Fig. 2B for the 19g11.2 cells. Each pair of lanes shows the enzymatic cleavage by the various enzymes in untreated and dexamethasone-treated cells; the primer extension product resulting from each restriction site assayed in chromatin is marked to the right of each lane. The fractional cleavage at the various sites in the uninduced 19g11.2 cells is shown in the chart in Fig. 2C, along with the results from the other cell lines examined. It is apparent that the accessibility varies widely along the LTR, ranging from 5 to 50% cleavage at various sites. However, with the clear exception of one site at +50, the accessibility at each site is similar from cell line to cell line.
The change in fractional cleavage, ΔF(x), between uninduced and induced cells is shown in Fig. 2D. It is evident that not all sites display hypersensitivity; only those in the vicinity of the promoter do so. In addition, the restriction sites that display a hormone-dependent ΔF(x) are the same in the four cell lines. In the Nuc-B region, these sites span the 187 nucleotides from the AlwNI site at −295 to the SacI site at −109 and includes FokI at −162, HaeIII at −225, and NlaIII at −252. Also examined in one experiment in the 3134 cell line was the AflII site at −201, which displayed an F(x) of 0.12 and a ΔF(x) of 0.15, and the Sau961 site at −225, which displayed an F(x) of 0.40 and a ΔF(x) of 0.15. This last result is in keeping with the F(x) determined for HaeIII at −225 and indicates that such a high fractional cleavage is a property of the site and not the enzyme. Finally, while the ΔF(x) clearly varies among the various cell lines, being highest in the 19g11.2 cells and lowest in the 1471.1 cells, it is very similar from site to site within any given cell line. It should be evident that in none of the steroid-treated samples does the cleavage reach 100%.
The nearest restriction site downstream of SacI at −109 is at −74 (ApoI in the C3H strain of the LTR and HinfI in the GR strain), while upstream of AlwNI at −295 the nearest measurable site is NlaIII at −362, whose accessibility does not change (data not shown). The length of the HSR as defined by restriction enzymes, 187 bp, may therefore be an underestimate.
The accessibility and the change in accessibility of the FokI site at +50 in Nuc-A displayed more variability. However, since this region does not display DNase I or MPE-Fe(II) hypersensitivity in the hormone-activated promoter, understanding the variation at this site was deemed beyond the scope of the present work.
Two-enzyme restriction assay for linkage between sites.
Because different molecules of MMTV contain Nuc-B at different positions (26), the prospect arose that in response to activation with glucocorticoids, distinct templates would remodel the chromatin associated with different DNA sequences. The detected hypersensitivity at any one restriction site would thus be the result of the cumulative hypersensitivity detected in molecules containing Nuc-B in different positions. Therefore, it might be the case that molecules containing the 3′-most of the hormone-responsive Nuc-B frames undergo a transition at the SacI site (at −109) but not at the AlwNI site (at −295), and so on, resulting in independent transitions at nonadjacent sites. Alternatively, the entire region from −109 to −295 may increase accessibility in any single promoter once it is hormonally activated, regardless of the position of the steroid-responsive Nuc-B.
We can differentiate between the above two scenarios in a two-enzyme digestion experiment. In this approach, a sample is treated simultaneously with two restriction enzymes, with the aim of quantitating the cleavage at both sites by using primer extension as a detection assay. The primer extension reaction will detect cleavage at the primer-distal site (site a) of a molecule only if the primer-proximal site (site b) of the same molecule is not cut. The various potential relations between accessible sites can thus be described by
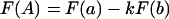 |
4 |
where F(a) is the fractional access at site a, F(b) is the fractional access at b and F(A) is the fractional access detected at a in the presence of potential interference by cutting site b (see equation 2). The constant k is the conditional probability of cleavage at a given an accessible site b; it has values of 0, F(a), or 1 for mutually exclusive, independent, and linked relationships, respectively. Equation 4 is valid only when F(a) approaches the value of the corresponding probability of cleavage P(a); we assume this to be the case when the concentration of enzyme Ea is high enough to observe a plateau level of F(a).
A point to be considered when analyzing induced MMTV is that not every promoter in the population is activated by steroid treatment. This was shown by Ko et al. (36), in an analysis of LTR-driven β-galactosidase activity by histochemical and fluorescence-activated cell sorter analysis of cells carrying a single-copy LTR–β-galactosidase construction. The incomplete conversion to a hypersensitive phenotype in the cell lines we examined (Fig. 1 and 2) (78) is consistent with molecules in the population being present in more than one state. We made the following assumptions to analyze this case. First, at the optimal time of induction with dexamethasone, the population of promoters exists in two mutually exclusive states: uninduced and fully induced. Second, any promoters that are not hypersensitive at the time of the assay are presumed to resemble the uninduced promoter. A relation analogous to equation 4 can be derived (see equation 3):
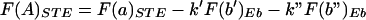 |
5 |
where F(b′)Eb and F(b")Eb indicate the fractional cleavage at site b of uninduced and induced templates, respectively, at a given concentration of enzyme Eb, and k′ and k" are the corresponding conditional probabilities; the subscript STE indicates the steroid-treated population. Assuming that a fraction, wb, of the molecules accessible at b in the uninduced state remains uninduced after treatment of the population with steroid, the fractional cleavage F(b′)Eb of the population of molecules remaining uninduced after hormone administration is given by
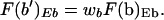 |
6 |
In the same manner, F(b")Eb reflects the sum of the contributions from the fraction (1 − wb) of the molecules that were accessible in the uninduced state and then recruited and cleaved in the induced state and the molecules that were inaccessible in the uninduced state and then recruited and cleaved in the induced state, according to
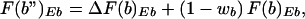 |
7 |
where the change in fractional access, ΔF(b)Eb, is a measure of the recruited molecules that were inaccessible in the uninduced state and cleaved at the given concentration of enzyme Eb. A parameter wa, similar to wb, has also been defined. It should be evident that analysis of induced MMTV requires a parallel analysis of the uninduced promoter. Of interest, we note that when wb = 1 − ΔF(b)/[1 − F(b)], the induction process selects at random from the accessible and inaccessible molecules at b (similarly for wa). As with equation 4, we assume the above relations to be valid at high concentrations of enzyme Ea.
The accessibilities of sites within the HSR are linked.
Determining the relationship between the AlwNI site (−295) and the SacI site (−109) was of particular interest since these two sites are in the vicinity of the 5′ and 3′ boundaries of the HSR. The gel in Fig. 3A shows the result of an AlwNI-SacI two-enzyme experiment. Nuclei from uninduced (lanes 1 to 7) or induced (lanes 8 to 14) cells were treated with a constant amount of SacI and increasing amounts of AlwNI. The cleavage at both sites was then examined in a primer extension reaction with an oligonucleotide priming from position −546 in the LTR. Visual inspection of the gel shows that increasing the amount of AlwNI cleavage reduced the intensity of the signal at the SacI site in both the uninduced (lanes 1 to 7) and induced (lanes 8 to 14) states, indicating that both populations contain molecules with both sites simultaneously accessible.
FIG. 3.

AlwNI-SacI two-enzyme digestion of 3134 cell nuclei. (A) aliquots of nuclei (5 μg of DNA) from uninduced or induced 3134 cells were digested in a volume of 50 μl with SacI at 2,500 U/ml and AlwNI at 0 (lanes 1 and 8), 10 (lanes 2 and 9), 25 (lanes 3 and 10), 50 (lanes 4 and 11), 100 (lanes 5 and 12), 250 (lanes 6 and 13), and 500 (lanes 7 and 14) U/ml. After digestion and extraction of the DNA, the MspII site at +103 was cut and quantitation was performed with oligonucleotide 765 for primer extension. The electrophoretic analysis of the extension products shows, from the bottom of the gel, the bands corresponding to cuts at the AlwNI, SacI, and MspII sites (indicated to the right); lanes 1 to 7, uninduced cells; lanes 8 to 14, dexamethasone-induced cells. The image is overexposed to highlight the SacI and AlwNI bands. This does not affect the phosphorimager quantitation but gives rise to the appearance that SacI cuts more than we report in panel C (e.g., over 50% by inspection of lane 8, in contrast to a quantitated value of 37%). (B) The F(SACI) in uninduced cells is plotted against the F(AlwNI) (squares); the broken line indicates the least-squares line through the data. The F(SACI) expected for independent (I), mutually exclusive (ME), and fully linked (L) outcomes was computed from equation (4), with the appropriate values of k, as described in the text. (C) The F(SACI) in induced cells is plotted against the F(AlwNI) (squares). The computed family of solutions differing in the value of the partition parameters wSacI and wAlwNI for the independent (I), mutually exclusive (ME), and fully linked (L) outcomes of F(SACI) versus F(AlwNI) are shaded. The boundary of the set of linked outcomes closest to the data has wSacI and wAlwNI values of 1; the line shown was computed with values of 0.8.
The plot in Fig. 3B displays the fractional cleavage detected at the SacI site, F(SACI), in the uninduced promoter as a function of the fractional cleavage at the AlwNI site (Fig. 3A, lanes 1 to 7). There is clearly a linear decrease in F(SACI) as a function of F(AlwNI); the broken line indicates the best fit obtained by linear regression. For comparison, the lines describing the expected F(SACI) for mutually exclusive, independent, and linked relationships between the SacI and AlwNI sites are also shown. The independent scenario best describes the relationship between AlwNI and SacI cutting in the uninduced promoter, although it is evident that there is a certain degree of linkage.
The analysis of the SacI-AlwNI pair in the steroid-stimulated promoter population (Fig. 3A, lanes 8 to 14) is shown in Fig. 3C. The shaded regions in the graph represent the set of solutions for the mutually exclusive, independent, and linked outcomes. Specific solutions for each outcome fall within the shaded regions and differ in the values of the parameters wSacI and wAlwNI; the boundaries of the shaded regions were estimated by using alternatively values of 0 and 1 for both wSacI and wAlwNI. Notice there is no overlap between the three families of solutions. The detected F(SACI)STE measured as a function of the fractional cleavage F(AlwNI)STE, fell within the estimated set of linked outcomes, near the solution specified by wSacI = wAlwNI = 1. These values of wSacI and wAlwNI correspond to the case where none of the molecules accessible at either site in the uninduced state is recruited to the induced state. However, we cannot conclude that this is the situation, since there is insufficient accuracy to rule out other cases, for instance wSacI = wAlwNI ≈ 0.8 (illustrated as the solid line in the linked family of outcomes), where the induction process selects randomly from the accessible and inaccessible populations. Of course, we cannot determine the value of the parameter w except in the situation where it equals 1 for both sites. In any case, this result indicates that the hypersensitivities at the AlwNI and SacI sites do not occur independently but, rather, are fully linked.
To determine whether the linkage we observed between the SacI and AlwNI sites was specific to this enzyme pair, we performed two-enzyme restriction assays with the FokI-SacI (positions −162 and −109 [Fig. 4]) and FokI-HaeIII (positions −162 and −225 [data not shown]) enzyme pairs. For the uninduced promoter (Fig. 4A), the accessibility between the FokI and SacI sites matched the independent scenario more closely than with the AlwNI-SacI enzyme pair. In the induced state, in contrast, the curve described by plotting F(SACI) versus F(FokI) values fell within the linked family of solutions (Fig. 4B), and, as before, there was no overlap between the curves describing the various outcomes. Thus, as with AlwNI-SacI, the accessibilities of the SacI and FokI sites in the induced promoter are linked; the transitions are not independent of each other. Similar results were observed for the FokI-HaeIII enzyme pair (data not shown).
FIG. 4.

FokI-SacI two-enzyme digestion of 3134 cell nuclei. (A) Aliquots of nuclei (10 μg of DNA) from uninduced or induced 3134 cells were digested in a volume of 100 μl with SacI at 2,500 U/ml and FokI at 0, 5, 10, 20, 30, 40, 50, 100, and 250 U/ml; the ApoI site at −74 was subsequently cut for the quantitation with oligonucleotide 730. The F(SACI) in uninduced cells is plotted against F(FokI) (squares). The computed F(SACI) outcomes expected from independent (I), mutually exclusive (ME), and fully linked (L) relations with AlwNI are indicated. The broken line indicates the best line through the data. (B) The F(SACI) in induced cells is plotted against the F(FokI) (squares). The family of solutions computed for each outcome of F(SACI) versus F(FokI) is shaded; the line through the data was computed with wSacI and wFokI values of 0.85; other details are as in Fig. 3.
Linkage is limited to pairs of sites within the HSR.
A reasonable expectation is that for a site outside the HSR, linkage to a hypersensitive site should not develop upon activation of the promoter by steroid. Between such a pair of sites, the same relationship should be observed in both the uninduced and induced states. To test this, we examined the RsaI site at −365 in combination with the AlwNI site at −295 (Fig. 5); the RsaI site lies outside the HSR of the induced promoter, while AlwNI lies within. As with the other enzyme pairs examined, cleavage of the RsaI site in the uninduced promoter occurred mostly independently of cleavage at the AlwNI site (Fig. 5A); the F(RSAI)-versus-F(AlwNI) data was scattered around the line defining the independent outcome. However, unlike the other enzyme pairs examined, the accessibilities at the RsaI and AlwNI sites did not become linked upon stimulation with dexamethasone. The decrease in F(RSAI) as a function of F(AlwNI) fit the independent family of solutions (Fig. 5B). As predicted, the relationship between this pair of sites remained as in the uninduced state.
FIG. 5.

Linkage analysis of sites outside the HSR. (A) Aliquots of nuclei from uninduced or induced 3134 cells were digested with 2,500 U of RsaI per ml and increasing amounts of AlwNI, as in Fig. 3A; a HinfI site at −758 was subsequently cut for the quantitation with oligonucleotide 670. The F(RSAI) in uninduced cells is plotted against F(AlwNI) (squares). The computed F(RSAI) outcomes expected from independent (I), mutually exclusive (ME), and fully linked (L) relations with AlwNI are indicated. The best line through the data coincided with the independent outcome. (B) The F(RSAI) in induced cells is plotted against the F(AlwNI) (squares). The family of solutions computed for each outcome of F(RSAI) versus F(AlwNI) is shown shaded; the line shown was computed with wRsaI and wAlwNI of 0.25; details are as in Fig. 3.
The decrease in the detected cleavage of primer-distal sites, F(A), is not due to dynamic events.
The two-enzyme analysis for linkage assumes a static relationship between the accessibilities of nonadjacent sites, at least during the period of the assay. The above analysis could not be performed if cutting at one site affected the accessibility and thus the cleavage of the second site. Possible events affecting secondary cleavage are release of superhelical strain (35, 38), DNA unwrapping from the nucleosome (64, 65), and nucleosomal mobility (48, 60) in the absence of ATP (58, 79). In experiments with all the pairwise combinations of enzymes we report here, we determined that the accessibility at primer-distal sites was not affected by cutting the primer-proximal sites. This is shown for the AlwNI-SacI enzyme pair in Fig. 6. After treatment of nuclei with AlwNI and various concentrations of SacI, the DNA was extracted, restricted with RsaI, and split prior to the primer extension analysis, as schematized in Fig. 6A. Half of the sample was analyzed with oligonucleotide 752, which directly detects cleavage at the AlwNI site, and the other half was analyzed with oligonucleotide 669, which primer extends through the SacI site before scoring the AlwNI site (Fig. 6A). The plot in Fig. 6C shows that the cutting detected at the AlwNI site, F(ALWNI), using oligonucleotide 669, decreases as the concentration of SacI is increased in both control and dexamethasone-treated samples. However, Fig. 6B shows that the actual cutting at AlwNI, F(AlwNI), is unaffected, as determined directly with oligonucleotide 752 in the primer extension.
FIG. 6.

The decrease in F(A) is attributed to secondary cleavage interfering with detection. (A) Scheme used to assess the effect of cutting the SacI site on the accessibility of the AlwNI site. Induced and uninduced 3134 nuclei were digested with AlwNI at 1,000 U/ml and SacI at the concentrations shown in panels B and C. The extracted DNA was restricted with RsaI and extracted, and aliquots were taken for analysis by primer extension with either oligonucleotide 752 or 669. (B) F(AlwNI) plotted as a function of the SacI concentration with oligonucleotide 752 for analysis; triangles (Dex), induced nuclei; squares (Cont), uninduced nuclei. (C) F(ALWNI) plotted as a function of SacI concentration with oligonucleotide 669 for analysis; other details are as in panel B.
DISCUSSION
Accessibility profile of the uninduced LTR.
The pattern of restriction enzyme access of the LTR is essentially the same in four cell lines examined. These cell lines differ in genetic background, copy number, and reporter gene sequence (see Materials and Methods). Nevertheless, the pattern of accessibility is very similar and therefore the factors restricting access to the LTR in the four cell lines must be nearly identical. This is not surprising since positioning of nucleosomes is a known constraint on the access of enzymes and the low-resolution positioning of nucleosomes on the LTR is the same in the 3134, 1361.5, and 1471.1 cell lines (reference 67 and data not shown). The characterization of the MMTV chromatin in the 19g11.2 cells is not as complete as in the other lines, but the present evidence suggests an equivalent structure.
We did not find any restriction sites in the Nuc-A or Nuc-B region that were either entirely protected or fully accessible. Such partial protection against cleavage might be expected given that, although nucleosomal DNA is resistant to restriction (1, 2, 46, 50, 84), nucleosomes in this region of the LTR are not uniquely positioned (26). In general, the accessibility at most of the sites examined in the MMTV promoter was inversely correlated with our rough estimate of the nucleosome frame occupancy, e.g., HaeIII at −225, SacI at −109, and AflII at −201. However, the accessibility at other sites did not, e.g., FokI at −162. Because of the lack of adequate restriction enzyme isoschizomers, we could not attribute such unusual accessibilities either to the enzymes or to the sites themselves. This leaves open the possibility that factors in addition to the translational position of nucleosomes affect the accessibility of specific sites within the uninduced MMTV promoter (72; but see also reference 29).
Interestingly, the two-enzyme restriction experiments demonstrated that the accessibilities of nonadjacent sites in uninduced MMTV are mostly independent of each other. At present, we can only speculate that this might be an indication of the variability in the length of the spacer DNA between nucleosomes in the promoter population (53, 66, 71). This is in addition to the variability in nucleosome positions that exists in this region of the MMTV LTR (26). The deviation we observed between the actual and expected independent outcome of the SacI-AlwNI pair, separated by approximately one nucleosomal repeat length, might be interpreted within this context as a tendency of neighboring nucleosomes to maintain a certain spacing (37).
Accessibility in the steroid-dependent HSR.
In the four cell lines examined, the stretch of hypersensitive chromatin is at least 187 nucleotides in length, bounded by an AlwNI site at −295 and a SacI site at −109. The present results extend previous estimates of the length of the HSR to include sites upstream of the HRE and the HaeIII site at −225 (3, 6, 52). That the HSR may be even longer than this estimate is suggested by a previous analysis of the DNase I cuts on the hypersensitive promoter in 904.13 cells, which indicated an increased accessibility of the sequences downstream of −109 (11). Our inability to detect hypersensitivity in the NF-1 site at −74 by ApoI in the C3H strain of MMTV or by HinfI in the GR strain might reflect a hindrance to restriction posed by bound NF-1. Restriction access has been used as a tool to examine factor site occupancy in transcriptionally active simian virus 40 complexes (22, 23, 29). The results reported here, as well as those reported by others (3, 6, 52), are in contrast to a recent report that concluded that the HSR was very short and was localized in the immediate vicinity of the SacI site at −109 (76). This conclusion was partly derived from examining multiple sites within the HRE cut simultaneously by a single restriction enzyme. However, unless linkage between sites is considered, this would have failed to uncover hypersensitivity at distal sites. Nevertheless, these investigators also reported observing hypersensitivity at the HaeIII site at −225 (76), in agreement with our results.
At the macroscopic level in a population of templates, and given the presence of multiple Nuc-B frames, the HSR could result from a chromatin transition in distinct nucleosome frames in different members of the MMTV population. There are at least two consequences to be predicted from this scenario. First, the central region of the HSR, i.e., the stretch of DNA common to the nucleosome frames that undergo the transition, should exhibit a higher change in fractional access [ΔF(x)] in the population of templates. However, the ΔF(x) at a number of sites throughout the hypersensitive region is clearly not larger in the center relative to the edge of the HSR. Second, the change in accessibility associated with a transition at a specific site should occur independently of the change in accessibility at another site. Using the two-enzyme restriction assay described in this work, we found the converse; the transition at one site is linked to the transition at nonadjacent sites. Based on these two observations, we conclude that the location and the length of the steroid-dependent HSR are invariant in the MMTV population: they do not differ between molecules despite the organization of the HRE in a nucleosome (Nuc-B) that differs in position among the various molecules of the population. The entire region between −109 and −295 becomes hypersensitive once the MMTV promoter is activated by the glucocorticoid receptor.
Is the HSR histone free?
DNase I-hypersensitive regions have generally been considered to reflect a nonnucleosomal architecture in chromatin (see reference 24 for a review). We doubt whether the MMTV HSR is free of histones, but uncertainties still remain. We previously analyzed the H1 and H2B content of the induced relative to the uninduced MMTV promoter by using a cross-linking–immunoprecipitation approach (11). The amount of H1 cross-linked to an HaeIII-generated tetranucleosome fragment containing the promoter decreased by about 45%, while the H2B content decreased by only 11%. However, a quantitative depletion of H2B from Nuc-B could account for the observed decrease if only 15 to 20% of the promoters in the population respond to stimulation by dexamethasone. The present observation that the HaeIII site at −225 is hypersensitive further complicates the interpretation of those findings, since it introduces the issue of selection of templates recovered for analysis. Truss et al. also addressed this issue by using filter hybridization to examine the content of Nuc-B DNA in preparations of mononucleosomal DNA from a high-copy-number cell line (76). No significant change in the hybridization signal was observed in induced relative to control cells. However, hybridization controls for recovery were not used. In addition, the proportion of active promoters was estimated by using a DNase I hypersensitivity assay, which is prone to error since the normalization standard is also cleaved. Ligation-mediated PCR was utilized to answer the same question in a single-copy cell line, thus eliminating some of the issues related to heterogeneity (76). Examination of the 3′ edge of the Nuc-B region in a footprinting reaction did not uncover differences between uninduced and induced templates (76). However, the micrococcal nuclease digestion might have been too extensive, so the resulting double-stranded micrococcal nuclease cuts scored in the assay could have reflected instead the 5′ ends of Nuc-A nucleosomes frames (25, 26).
The results of the two-enzyme experiments are inconsistent with the loss of histones from multiple Nuc-B frames in the hypersensitive state. Assuming that histone loss results in hypersensitivity, this scenario should have resulted in statistical independence in the accessibility of nonadjacent sites, which we clearly did not observe. On the other hand, if the number of adjacent Nuc-B frames losing histones is limited to two or three and we postulate a length of >185 bp per overlapping hypersensitive region, the assay may not have uncovered transition independence. However, introducing this restriction should have rendered histone loss detectable in an analysis of the nucleosome frames in this region of the LTR, and this was not observed (26). It is unclear whether such frame analysis would have uncovered histone loss if the number of adjacent frames is raised beyond four. Although the present findings are consistent with the notion that Nuc-B remains nucleosomal, these experiments only indirectly assess the status of Nuc-B. A conclusive answer to this question requires approaches that, as above (11, 76), directly examine nucleosomal material, histones, and/or DNA but with a higher degree of precision.
Alternative mechanisms that could explain the enhanced accessibility of the HSR without a concomitant displacement of histones have been investigated recently. Partially purified SWI/SNF complexes facilitate the access of transcription factors and alter the access of nucleases to nucleosomal DNA, apparently without displacing histones from the complex or eliminating all of the rotational constraints characteristic of a nucleosomal organization (21, 34, 55). Evidence has been presented suggesting that SWI/SNF facilitates glucocorticoid receptor action (15, 51, 87). Histone acetylation is another mechanism by which an active MMTV promoter could retain a nucleosomal organization and still exhibit increased access (42, 81). Although high concentrations of histone deacetylase inhibitors eliminate the response of MMTV to steroids (12), low concentrations activate transcription (8). Steroid receptor coactivators and integrators exhibit histone acetyltransferase activity (7, 18, 54, 69, 86), as first shown for the transcriptional adapter GCN5 (13, 40). The targeted acetylation afforded by these proteins in complexes with the glucocorticoid receptor could play a role in the formation of the HSR. Finally, Steger and Workman observed that binding of Sp1 and NF-κB to a fragment of the human immunodeficiency virus promoter reconstituted into a nucleosomal array resulted in DNase I hypersensitivity without histone loss (70). Since the glucocorticoid receptor binds to mononucleosomal Nuc-B reconstituted in vitro (3, 61, 63), it would be interesting to assess whether a similar effect occurs upon binding to an array.
Factors affecting the localization of the HSR.
The chromatin transition leading to formation of the MMTV HSR is not simply a nucleosome core remodeling event. It involves additional events adjacent to any Nuc-B that might be remodeled. As a framework for further investigation, we considered the formation of an HSR from the perspective of two disparate activation models not exclusive of each other (Fig. 7). In one class of models, the location of the HSR is fixed by the position of the steroid-responsive Nuc-B nucleosome(s), but the chromatin disruption that leads to formation of the HSR is global (nucleosome-repeat size; Fig. 7A). A very small number of adjacent Nuc-B frames, or perhaps even only one frame, responds to the steroid receptor. The position of this frame dictates the position of the HSR once the nucleosome core is remodeled. Further steps in the activation pathway, such as the loss of histone H1 or a localized decondensation of the chromatin fiber, results in a specific HSR length. Bresnick et al. previously reported H1 loss from chromatin fragments containing the activated MMTV promoter (11); interactions between the core histones and linker DNA have been documented (8a).
FIG. 7.

Two alternative classes of models for the generation of the steroid-dependent HSR of MMTV. (A) Disturbance in the chromatin structure is restricted to a Nuc-B nucleosomal repeat. Binding of the glucocorticoid receptor (GR) to Nuc-B (left) triggers a series of changes such as decreased interactions between histone H1 and Nuc-B and/or altered contacts between the core histones and the adjacent linker-DNA. Such changes, in combination with differences between the higher-order chromatin structures of the steroid receptor-bound and unbound states, result in an increased accessibility of the entire Nuc-B-containing nucleosomal repeat (right, delimited by arrows). The newly accessible linker DNA could be either 5′, 3′, or both 5′ and 3′ of the glucocorticoid-responsive Nuc-B frame according to the mechanistic details. A very small set of adjacent Nuc-B frames, perhaps just one, responds to the steroid receptor. (B) Highly localized disturbance in the vicinity of a chromatin-bound transcription factor. cis elements are organized in various molecules in different chromatin configurations, in Nuc-B, Nuc-C, and the B/C linker, due to the existence of multiple nucleosome frames (top). GR binds to its cognate sites and further recruits additional trans activators. The chromatin in the immediate vicinity of various factors is disrupted. The end result of multiple localized disruptions is increased accessibility throughout the Nuc-C/Nuc-B chromatin stretch containing the bound trans activators, perhaps delimited by the bound GR (bottom).
In a second class of models, the location of the HSR is dictated by the cis-acting elements of the promoter and the chromatin disruption is local (in the vicinity of bound transcription factors [Fig. 7B]). Various groups have reported detection of regulatory elements upstream of the minimal HRE (17, 27, 44, 45, 49), including binding sites for the glucocorticoid receptor (57), and an effect of this region on the hormonal response of MMTV has been documented (17, 27, 33, 44, 45, 49). Cooperative loading of transcription factors to an entire stretch of chromatin could partly explain the linkage between sites in the HSR. Support for the notion that cis elements could delimit an HSR comes from recent work by Boyes and Felsenfeld on the chicken βA/ɛ globin enhancer (10) and by Lu et al. on the Drosophila hsp26 promoter (47), where the length or position of the hypersensitive region was affected by the number or location of factor binding sites.
Determination of the collection of transcription factors and histones present in the active state will be crucial to our understanding of the activation of MMTV by steroids. Moreover, it is also clear that understanding the generation and the architecture of the HSR will require an examination of the effect of cis elements on the accessibility of the induced promoter. Whether steroid responsiveness is restricted to a subset of Nuc-B frames remains to be resolved. The method we describe here to analyze the linkage between the sites in an HSR should be useful in other studies, since both multiplicity of nucleosome frames (14, 20, 75, 80) and heterogeneity in the functional state of a promoter (reference 83 and references therein) have been documented in systems other than MMTV.
ACKNOWLEDGMENTS
Cell line 19g11.2 was kindly provided by K. Yamamoto. We thank Ron Wolford for technical assistance and Barbour Warren, Scot Roberts, Connie Myers, and Julia Michelotti for critically reviewing the manuscript.
REFERENCES
- 1.Almer A, Horz W. Nuclease hypersensitive regions with adjacent positioned nucleosomes mark the gene boundaries of the PHO5/PHO3 locus in yeast. EMBO J. 1986;5:2681–2687. doi: 10.1002/j.1460-2075.1986.tb04551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almer A, Rudolph H, Hinnen A, Horz W. Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J. 1986;5:2689–2696. doi: 10.1002/j.1460-2075.1986.tb04552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Archer T K, Cordingley M G, Wolford R G, Hager G L. Transcription factor access is mediated by accurately positioned nucleosomes on the mouse mammary tumor virus promoter. Mol Cell Biol. 1991;11:688–698. doi: 10.1128/mcb.11.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Archer T K, Lee H-L, Cordingley M G, Mymryk J S, Fragoso G, Berard D S, Hager G L. Differential steroid hormone induction of transcription from the mouse mammary tumor virus promoter. Mol Endocrinol. 1994;8:568–576. doi: 10.1210/mend.8.5.8058066. [DOI] [PubMed] [Google Scholar]
- 5.Archer T K, Lefebvre P, Wolford R G, Hager G L. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science. 1992;255:1573–1576. doi: 10.1126/science.1347958. [DOI] [PubMed] [Google Scholar]
- 6.Archer T K, Zaniewski E, Moyer M L, Nordeen S K. The differential capacity of glucocorticoids and progestins to alter chromatin structure and induce gene expression in human breast cancer cells. Mol Endocrinol. 1994;8:1154–1162. doi: 10.1210/mend.8.9.7838148. [DOI] [PubMed] [Google Scholar]
- 7.Bannister A J, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 8.Bartsch J, Truss M, Bode J, Beato M. Moderate increase in histone acetylation activates the mouse mammary tumor virus promoter and remodels its nucleosome structure. Proc Natl Acad Sci USA. 1996;93:10741–10746. doi: 10.1073/pnas.93.20.10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Bavykin S G, Usachenko S I, Zalensky A O, Mirzabekov A D. Structure of nucleosomes and organization of internucleosomal DNA in chromatin. J Mol Biol. 1990;212:495–511. doi: 10.1016/0022-2836(90)90328-J. [DOI] [PubMed] [Google Scholar]
- 9.Boronat S, Richard-Foy H, Pina B. Specific deactivation of the mouse mammary tumor virus long terminal repeat promoter upon continuous hormone treatment. J Biol Chem. 1997;272:21803–21810. doi: 10.1074/jbc.272.35.21803. [DOI] [PubMed] [Google Scholar]
- 10.Boyes J, Felsenfeld G. Tissue-specific factors additively increase the probability of the all-or-none formation of a hypersensitive site. EMBO J. 1996;15:2496–2507. [PMC free article] [PubMed] [Google Scholar]
- 11.Bresnick E H, Bustin M, Marsaud V, Richard-Foy H, Hager G L. The transcriptionally-active MMTV promoter is depleted of histone H1. Nucleic Acids Res. 1992;20:273–278. doi: 10.1093/nar/20.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bresnick E H, John S, Berard D S, Lefebvre P, Hager G L. Glucocorticoid receptor-dependent disruption of a specific nucleosome on the mouse mammary tumor virus promoter is prevented by sodium butyrate. Proc Natl Acad Sci USA. 1990;87:3977–3981. doi: 10.1073/pnas.87.10.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brownell J E, Zhou J, Ranalli T, Kobayashi R, Edmondson D G, Roth S Y, Allis C D. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 14.Buttinelli M, Di Mauro E, Negri R. Multiple nucleosome positioning with unique rotational setting for the Saccharomyces cerevisiae 5S rRNA gene in vitro and in vivo. Proc Natl Acad Sci USA. 1993;90:9315–9319. doi: 10.1073/pnas.90.20.9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns B R, Levinson R S, Yamamoto K R, Kornberg R D. Essential role of Swp73p in the function of yeast Swi/Snf complex. Genes Dev. 1996;10:2131–2144. doi: 10.1101/gad.10.17.2131. [DOI] [PubMed] [Google Scholar]
- 16.Cato A C, Miksicek R, Schütz G, Arnemann J, Beato M. The hormone regulatory element of mouse mammary tumour virus mediates progesterone induction. EMBO J. 1986;5:2237–2240. doi: 10.1002/j.1460-2075.1986.tb04490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavin C, Buetti E. Tissue-specific and ubiquitous factors binding next to the glucocorticoid receptor modulate transcription from the mouse mammary tumor virus promoter. J Virol. 1995;69:3759–3770. doi: 10.1128/jvi.69.6.3759-3770.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H, Lin R J, Schiltz R L, Chakravarti D, Nash A, Nagy L, Privalsky M L, Nakatani Y, Evans R M. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 19.Cordingley M G, Riegel A T, Hager G L. Steroid-dependent interaction of transcription factors with the inducible promoter of mouse mammary tumor virus in vivo. Cell. 1987;48:261–270. doi: 10.1016/0092-8674(87)90429-6. [DOI] [PubMed] [Google Scholar]
- 20.Costanzo G, Di Mauro E, Negri R, Pereira G, Hollenberg C. Multiple overlapping positions of nucleosomes with single in vivo rotational setting in the Hansenula polymorpha RNA polymerase II MOX promoter. J Biol Chem. 1995;270:11091–11097. doi: 10.1074/jbc.270.19.11091. [DOI] [PubMed] [Google Scholar]
- 21.Cote J, Quinn J, Workman J L, Peterson C L. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science. 1994;265:53–60. doi: 10.1126/science.8016655. [DOI] [PubMed] [Google Scholar]
- 22.Eadara J K, Hadlock K G, Lutter L C. Chromatin structure and factor site occupancies in an in vivo-assembled transcription elongation complex. Nucleic Acids Res. 1996;24:3887–3895. doi: 10.1093/nar/24.20.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eadara J K, Lutter L C. Determination of occupancies of the SPH and GT-IIC transcription factor binding motifs in SV40: evidence for two forms of transcription elongation complex. Virology. 1996;223:120–131. doi: 10.1006/viro.1996.0461. [DOI] [PubMed] [Google Scholar]
- 24.Felsenfeld G. Chromatin as an essential part of the transcriptional mechanism. Nature. 1992;355:219–224. doi: 10.1038/355219a0. [DOI] [PubMed] [Google Scholar]
- 25.Fragoso G, Hager G L. Analysis of in vivo nucleosome positions by determination of nucleosome-linker boundaries in crosslinked chromatin. Methods Companion Methods Enzymol. 1997;11:246–252. doi: 10.1006/meth.1996.0411. [DOI] [PubMed] [Google Scholar]
- 26.Fragoso G, John S, Roberts M S, Hager G L. Nucleosome positioning on the MMTV LTR results from the frequency-biased occupancy of multiple frames. Genes Dev. 1995;9:1933–1947. doi: 10.1101/gad.9.15.1933. [DOI] [PubMed] [Google Scholar]
- 27.Gouilleux F, Sola B, Couette B, Richard-Foy H. Cooperation between structural elements in hormono-regulated transcription from the mouse mammary tumor virus promoter. Nucleic Acids Res. 1991;19:1563–1569. doi: 10.1093/nar/19.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gowland P L, Buetti E. Mutations in the hormone regulatory element of mouse mammary tumor virus differentially affect the response to progestins, androgens and glucocorticoids. Mol Cell Biol. 1989;9:3999–4008. doi: 10.1128/mcb.9.9.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadlock K G, Lutter L C. T-antigen is not bound to the replication origin of the simian virus 40 late transcription complex. J Mol Biol. 1990;215:53–65. doi: 10.1016/S0022-2836(05)80094-4. [DOI] [PubMed] [Google Scholar]
- 30.Hartig E, Nierlich B, Mink S, Nebl G, Cato A C. Regulation of expression of mouse mammary tumor virus through sequences located in the hormone response element: involvement of cell-cell contact and a negative regulatory factor. J Virol. 1993;67:813–821. doi: 10.1128/jvi.67.2.813-821.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewish D R, Burgoyne L A. Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem Biophys Res Commun. 1973;52:504–510. doi: 10.1016/0006-291x(73)90740-7. [DOI] [PubMed] [Google Scholar]
- 32.Htun H, Walker D, Hager G L. Optical imaging approach to study protein-DNA interaction and nuclear organization in cultured living cells. In: Sarma R H, Sarma M H, editors. Structure, motion, interaction, and expression of biological macromolecules. Schenectady, N.Y: Adenine Press; 1998. pp. 157–166. [Google Scholar]
- 33.Hynes N, van Ooyen A J, Kennedy N, Herrlich P, Ponta H, Groner B. Subfragments of the large terminal repeat cause glucocorticoid-responsive expression of mouse mammary tumor virus and of an adjacent gene. Proc Natl Acad Sci USA. 1983;80:3637–3641. doi: 10.1073/pnas.80.12.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imbalzano A N, Kwon H, Green M R, Kingston R E. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature. 1994;370:481–485. doi: 10.1038/370481a0. [DOI] [PubMed] [Google Scholar]
- 35.Jupe E R, Sinden R R, Cartwright I L. Stably maintained microdomain of localized unrestrained supercoiling at a Drosophila heat shock gene locus. EMBO J. 1993;12:1067–1075. doi: 10.1002/j.1460-2075.1993.tb05748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko M S, Nakauchi H, Takahashi N. The dose dependence of glucocorticoid-inducible gene expression results from changes in the number of transcriptionally active templates. EMBO J. 1990;9:2835–2842. doi: 10.1002/j.1460-2075.1990.tb07472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kornberg R D, Stryer L. Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism. Nucleic Acids Res. 1988;16:6677–6690. doi: 10.1093/nar/16.14.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramer P R, Sinden R R. Measurement of unrestrained negative supercoiling and topological domain size in living human cells. Biochemistry. 1997;36:3151–3158. doi: 10.1021/bi962396q. [DOI] [PubMed] [Google Scholar]
- 39.Kuhnel B, Buetti E, Diggelmann H. Functional analysis of the glucocorticoid regulatory elements present in the mouse mammary tumor virus long terminal repeat: a synthetic distal binding site can replace the proximal binding domain. J Mol Biol. 1986;190:367–378. doi: 10.1016/0022-2836(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 40.Kuo M H, Brownell J E, Sobel R E, Ranalli T A, Cook R G, Edmondson D G, Roth S Y, Allis C D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- 41.Langer S J, Ostrowski M C. Negative regulation of transcription in vitro by a glucocorticoid response element is mediated by a trans-acting factor. Mol Cell Biol. 1988;8:3872–3881. doi: 10.1128/mcb.8.9.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee D Y, Hayes J J, Pruss D, Wolffe A P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 43.Lee H-L, Archer T K. Nucleosome-mediated disruption of transcription factor: chromatin initiation complexes at the mouse mammary tumor virus long terminal repeat in vivo. Mol Cell Biol. 1994;14:32–41. doi: 10.1128/mcb.14.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee K I, Reddy E P, Reddy C D. Cellular factors binding to a novel cis-acting element mediate steroid hormone responsiveness of mouse mammary tumor virus promoter. J Biol Chem. 1995;270:24502–24508. doi: 10.1074/jbc.270.41.24502. [DOI] [PubMed] [Google Scholar]
- 45.Le Ricousse S, Gouilleux F, Fortin D, Joulin V, Richard-Foy H. Glucocorticoid and progestin receptors are differently involved in the cooperation with a structural element of the mouse mammary tumor virus promoter. Proc Natl Acad Sci USA. 1996;93:5072–5077. doi: 10.1073/pnas.93.10.5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linxweiler W, Horz W. Reconstitution of mononucleosomes: characterization of distinct particles that differ in the position of the histone core. Nucleic Acids Res. 1984;12:9395–9413. doi: 10.1093/nar/12.24.9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Q, Wallrath L L, Elgin S C. The role of a positioned nucleosome at the Drosophila melanogaster hsp26 promoter. EMBO J. 1995;14:4738–4746. doi: 10.1002/j.1460-2075.1995.tb00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meersseman G, Pennings S, Bradbury E M. Mobile nucleosomes—a general behavior. EMBO J. 1992;11:2951–2959. doi: 10.1002/j.1460-2075.1992.tb05365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meulia T, Diggelmann H. Tissue-specific factors and glucocorticoid receptors present in nuclear extracts bind next to each other in the promoter region of mouse mammary tumor virus. J Mol Biol. 1990;216:859–872. doi: 10.1016/S0022-2836(99)80006-0. [DOI] [PubMed] [Google Scholar]
- 50.Morse R H. Nucleosomes inhibit both transcriptional initiation and elongation by RNA polymerase III in vitro. EMBO J. 1989;8:2343–2351. doi: 10.1002/j.1460-2075.1989.tb08362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muchardt C, Yaniv M. A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J. 1993;12:4279–4290. doi: 10.1002/j.1460-2075.1993.tb06112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mymryk J S, Berard D, Hager G L, Archer T K. Mouse mammary tumor virus chromatin in human breast cancer cells is constitutively hypersensitive and exhibits steroid hormone independent loading of transcription factors in vivo. Mol Cell Biol. 1995;15:26–34. doi: 10.1128/mcb.15.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noll M, Kornberg R D. Action of micrococcal nuclease on chromatin and the location of histone H1. J Mol Biol. 1977;109:393–404. doi: 10.1016/s0022-2836(77)80019-3. [DOI] [PubMed] [Google Scholar]
- 54.Ogryzko V V, Schiltz R L, Russanova V, Howard B H, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 55.Ostlund Farrants A K, Blomquist P, Kwon H, Wrange O. Glucocorticoid receptor-glucocorticoid response element binding stimulates nucleosome disruption by the SWI/SNF complex. Mol Cell Biol. 1997;17:895–905. doi: 10.1128/mcb.17.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ostrowski M C, Richard-Foy H, Wolford R G, Berard D S, Hager G L. Glucocorticoid regulation of transcription at an amplified, episomal promoter. Mol Cell Biol. 1983;3:2045–2057. doi: 10.1128/mcb.3.11.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Payvar F, DeFranco D, Firestone G L, Edgar B, Wrange O, Okret S, Gustafsson J A, Yamamoto K R. Sequence-specific binding of glucocorticoid receptor to MTV DNA at sites within and upstream of the transcribed region. Cell. 1983;35:381–392. doi: 10.1016/0092-8674(83)90171-x. [DOI] [PubMed] [Google Scholar]
- 58.Pazin M J, Bhargava P, Geiduschek E P, Kadonaga J T. Nucleosome mobility and the maintenance of nucleosome positioning. Science. 1997;276:809–812. doi: 10.1126/science.276.5313.809. [DOI] [PubMed] [Google Scholar]
- 59.Pennie W D, Hager G L, Smith C L. Nucleoprotein structure influences the response of the mouse mammary tumor virus promoter to activation of the cyclic AMP signalling pathway. Mol Cell Biol. 1995;15:2125–2134. doi: 10.1128/mcb.15.4.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pennings S, Meersseman G, Bradbury E M. Mobility of positioned nucleosomes on 5 S rDNA. J Mol Biol. 1991;220:101–110. doi: 10.1016/0022-2836(91)90384-i. [DOI] [PubMed] [Google Scholar]
- 61.Perlmann T, Wrange O. Specific glucocorticoid receptor binding to DNA reconstituted in a nucleosome. EMBO J. 1988;7:3073–3079. doi: 10.1002/j.1460-2075.1988.tb03172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peterson D O. Alterations in chromatin structure associated with glucocorticoid-induced expression of endogenous mouse mammary tumor virus genes. Mol Cell Biol. 1985;5:1104–1110. doi: 10.1128/mcb.5.5.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pina B, Brüggemeier U, Beato M. Nucleosome positioning modulates accessibility of regulatory proteins to the mouse mammary tumor virus promoter. Cell. 1990;60:719–731. doi: 10.1016/0092-8674(90)90087-u. [DOI] [PubMed] [Google Scholar]
- 64.Polach K J, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 65.Polach K J, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 66.Prunell A, Kornberg R D. Variable center to center distance of nucleosomes in chromatin. J Mol Biol. 1982;154:515–523. doi: 10.1016/s0022-2836(82)80010-7. [DOI] [PubMed] [Google Scholar]
- 67.Richard-Foy H, Hager G L. Sequence specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J. 1987;6:2321–2328. doi: 10.1002/j.1460-2075.1987.tb02507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scheidereit C, Geisse S, Westphal H M, Beato M. The glucocorticoid receptor binds to defined nucleotide sequences near the promoter of mouse mammary tumour virus. Nature. 1983;304:749–752. doi: 10.1038/304749a0. [DOI] [PubMed] [Google Scholar]
- 69.Spencer T E, Jenster G, Burcin M M, Allis C D, Zhou J, Mizzen C A, McKenna N J, Onate S A, Tsai S Y, Tsai M J, O’Malley B W. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 70.Steger D J, Workman J L. Stable co-occupancy of transcription factors and histones at the HIV-1 enhancer. EMBO J. 1997;16:2463–2472. doi: 10.1093/emboj/16.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Strauss F, Prunell A. Nucleosome spacing in rat liver chromatin. A study with exonuclease III. Nucleic Acids Res. 1982;10:2275–2293. doi: 10.1093/nar/10.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tack L C, Wassarman P M, DePamphilis M L. Chromatin assembly. Relationship of chromatin structure to DNA sequence during simian virus 40 replication. J Biol Chem. 1981;256:8821–8828. [PubMed] [Google Scholar]
- 73.Tanaka H, Dong Y, Li Q, Okret S, Gustafsson J A. Identification and characterization of a cis-acting element that interferes with glucocorticoid-inducible activation of the mouse mammary tumor virus promoter. Proc Natl Acad Sci USA. 1991;88:5393–5397. doi: 10.1073/pnas.88.12.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanaka H, Dong Y, McGuire J, Okret S, Poellinger L, Makino I, Gustafsson J A. The glucocorticoid receptor and a putative repressor protein coordinately modulate glucocorticoid responsiveness of the mouse mammary tumor virus promoter in the rat hepatoma cell line M1.19. J Biol Chem. 1993;268:1854–1859. [PubMed] [Google Scholar]
- 75.Tanaka S, Livingstone-Zatchej M, Thoma F. Chromatin structure of the yeast URA3 gene at high resolution provides insight into structure and positioning of nucleosomes in the chromosomal context. J Mol Biol. 1996;257:919–934. doi: 10.1006/jmbi.1996.0212. [DOI] [PubMed] [Google Scholar]
- 76.Truss M, Bartsch J, Schelbert A, Hache R J, Beato M. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995;14:1737–1751. doi: 10.1002/j.1460-2075.1995.tb07163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ucker D S, Yamamoto K R. Early events in the stimulation of mammary tumor virus RNA synthesis by glucocorticoids. Novel assays of transcription rates. J Biol Chem. 1984;259:7416–7420. [PubMed] [Google Scholar]
- 78.Vanderbilt J N, Miesfeld R, Maler B A, Yamamoto K R. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol. 1987;1:68–74. doi: 10.1210/mend-1-1-68. [DOI] [PubMed] [Google Scholar]
- 79.Varga-Weisz P D, Blank T A, Becker P B. Energy-dependent chromatin accessibility and nucleosome mobility in a cell-free system. EMBO J. 1995;14:2209–2216. doi: 10.1002/j.1460-2075.1995.tb07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Venditti P, Costanzo G, Negri R, Camilloni G. ABFI contributes to the chromatin organization of Saccharomyces cerevisiae ARS1 B-domain. Biochim Biophys Acta. 1994;1219:677–689. doi: 10.1016/0167-4781(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 81.Vettese-Dadey M, Grant P A, Hebbes T R, Crane-Robinson C, Allis C D, Workman J L. Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- 82.von der Ahe D, Janich S, Scheidereit C, Renkawitz R, Schütz G, Beato M. Glucocorticoid and progesterone receptors bind to the same sites in two hormonally regulated promoters. Nature. 1985;313:706–709. doi: 10.1038/313706a0. [DOI] [PubMed] [Google Scholar]
- 83.Walters M C, Fiering S, Eidemiller J, Magis W, Groudine M, Martin D I K. Enhancers increase the probability but not the level of gene expression. Proc Natl Acad Sci USA. 1995;92:7125–7129. doi: 10.1073/pnas.92.15.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wasylyk B, Chambon P. Studies on the mechanism of transcription of nucleosomal complexes. Eur J Biochem. 1980;103:219–226. doi: 10.1111/j.1432-1033.1980.tb04306.x. [DOI] [PubMed] [Google Scholar]
- 85.Workman J L, Langmore J P. Efficient solubilization and partial purification of sea urchin histone genes as chromatin. Biochemistry. 1985;24:4731–4738. doi: 10.1021/bi00339a004. [DOI] [PubMed] [Google Scholar]
- 86.Yang X J, Ogryzko V V, Nishikawa J, Howard B H, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 87.Yoshinaga S K, Peterson C L, Herskowitz I, Yamamoto K R. Roles of SWI1, SWI2, and SWI3 proteins for transcriptional enhancement by steroid receptors. Science. 1992;258:1598–1604. doi: 10.1126/science.1360703. [DOI] [PubMed] [Google Scholar]
- 88.Zaret K S, Yamamoto K R. Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell. 1984;38:29–38. doi: 10.1016/0092-8674(84)90523-3. [DOI] [PubMed] [Google Scholar]