

Abstract
We compare two cases of primary spinal atypical teratoid/rhabdoid tumor (AT/RT), which rarely occurs in adults marked by SMARCA4 inactivation, and SMARCB1 inactivation for pediatric cases. AT/RT represents a highly malignant neoplasm comprising poorly differentiated constituents and rhabdoid cells, with SMARCB1(INI1) or infrequently SMARCA4 (BRG1) inactivation. These tumors are predominantly found in children but are rare in adults. While AT/RT can arise anywhere in the central nervous system, spinal cord localization is comparatively scarce. Despite mutation or loss of SMARCB1 at the 22q11.2 locus serving as the genetic hallmark of AT/RTs, infrequent cases of SMARCA4 inactivation with intact SMARCB1 protein expression are significant. We present each case of primary spinal tumors in a child and an adult, showing loss of the SMARCB1 and SMARCA4 proteins, respectively. Both tumors met the AT/RT diagnostic criteria. The histopathology demonstrated the presence of rhabdoid cells in both cases. Diagnosing primary spinal AT/RT with SMARCB1 protein loss remains a challenge. Nevertheless, the presence of SMARCB1 positivity alone must be noted to be insufficient to exclude the possibility of AT/RT diagnosis. In cases in which the diagnosis of AT/RT is highly suspected clinically, additional testing is warranted, including SMARCA4 analysis.
Keywords: spinal tumor, AT/RT, spine, INI-1, BRG-1
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant tumor comprising poorly differentiated constituents, which often includes rhabdoid cells, with inactivation of SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily b, member 1; SMARCB1 (integrase interactor 1; INI1) or rarely SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4; SMARCA4 (Brahma-related gene 1; BRG1).1) These cases often occur in children under five years2) but rarely in adults.3) Although AT/RT typically has been reported in all central nervous system (CNS) regions,4) pediatric primary spinal AT/RTs cases are below 60, while adult primary spinal AT/RTs are even more scarce, with reported cases below 10.5) The extreme rarity of primary spinal AT/RT presents a diagnostic challenge, especially for adult patients.
The genetic hallmark of AT/RTs is the mutation or loss of the SMARCB1 locus at 22q11.2 in most cases,6,7) resulting in the complete abrogation of the SMARCB1 protein expression. Hence, the assessment through immunohistochemistry for SMARCB1(INI1) protein loss serves as a specific diagnostic clue.8) In rare cases, SMARCA4 mutations are reported, characterizing the loss of SMARCA4 expression while retaining SMARCB1 protein expression.9)
We report pediatric and adult spinal AT/RT cases. In the adult case, the diagnosis was challenging due to the atypical nature of the primary spinal AT/RT and the harbored SMARCA4 mutation.
Case Report
Case 1
A 5-year-old female presented with neck pain and difficulty in using her right hand. Clinical examination revealed right upper-limb weakness and cervical rotatory disorder. Magnetic resonance imaging (MRI) showed a dumbbell tumor of Eden type II, with heterogeneous enhancement, located within the intradural extramedullary space and paraspinal muscles between the C2 and C5 vertebral bodies. The tumor had compressed the cervical cord (Fig. 1). Furthermore, the tumor had encroached upon the extraforaminal paravertebral space, thus necessitating combining the anterior and posterior approaches to achieve gross total resection.
Fig. 1.

Magnetic resonance imaging of the spine showing a dumbbell tumor of Eden type II, with heterogeneous enhancement at the vertebral body level between C2 and C5. The tumor has spread from the intradural extramedullary to the intramuscular and compressed the cervical cord. (A) T1-weighted sagittal image, (B) T2-weighted sagittal image, (C) gadolinium-enhanced T1-weighted sagittal image, (D) gadolinium-enhanced T1-weighted C2/3 axial image, (E) gadolinium-enhanced T1-weighted C3/4 axial image.
Subtotal removal of the spinal tumor was scheduled to decompress the spinal cord. The surgical procedure was conducted with the patient in the prone position, and a skin incision was made from the C1 to C5 spinous processes. Grayish cyst-like tumor tissue was discerned in the right paraspinal muscle, extending into the right C4/5 intervertebral foramen. The tumor was also detected in the right side of the spinal canal. Though dissected from the spinal cord and surrounding tissue, the tumor seemed to invade the right C5 dorsal and anterior roots. Furthermore, it had extended to the contralateral extramedullary space via the ventral aspect of the spinal cord, which was difficult to remove. Therefore, the right C5 dorsal root was amputated, while the right C5 anterior root was preserved with the residual tumor. Subtotal removal of the foraminal and the paravertebral tumor was subsequently accomplished via a posterior approach (Fig. 2A and B). The foraminal tumor was removed extensively from posterior foraminotomy with medial 1/3 of lateral mass resection. The paravertebral tumor connecting to the foraminal lesion was also removed, including the surrounding tumor-invasive muscle without any surgical margin, and was amputated just on the surface of the lateral edge of the lateral mass.
Fig. 2.
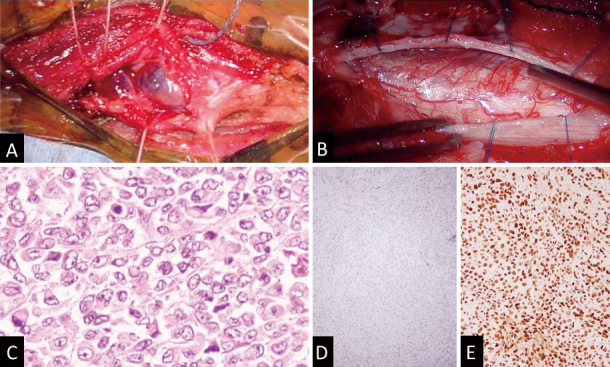
Surgical findings and pathological findings. (A) Surgical findings showing a grayish cyst-like tumor tissue in the muscle layer. (B) An incision in the dura revealing a thinned spinal cord and tumor mass. (C) Hematoxylin and eosin staining showing a malignant neoplasm composed of rhabdoid cells. (D) Although the INI1 protein is negative. (E) the BRG1 protein is positive on immunohistochemistry.
Pathological investigation of the excised tumoral tissues revealed a malignant neoplasm comprising rhabdoid cells, endothelial proliferation, and necrosis (Fig. 2C). Immunohistochemical examination confirmed positive vimentin and pan-CKAE1/AE3 expression. Tumorous cells were partly positive for synaptophysin, smooth-muscle actin, and CD56. SMARCA1 (BRG1) was also positive. However, SMARCB1(INI-1) was negative (Fig. 2D and E). Over 40% of tumor cells demonstrated positivity for Ki-67. These observations corroborated the diagnostic criteria for AT/RT.
Following the surgical intervention, the patient received chemotherapy (MTX iT (intrathecal) + VDC/ICE; methotrexate, vincristine, doxorubicin, cyclophosphamide, ifosfamide, cisplatin, and etoposide) and proton therapy (54 Gy/30 fractions). Although asymptomatic, neoplastic lesions recurred within the intradural extramedullary space near Th10 eight months post-surgery. Since the tumor invaded the dorsal root of the spinal cord, a subtotal resection was performed. Notwithstanding the chemotherapy cycle (MTX iT), the tumor recurred in the corpus callosum and the cerebellopontine angle 10 months following the initial surgery.
The tumor was refractory to treatment and had multiple disseminated lesions In the spinal cord. The patient is presently receiving palliative care.
Case 2
A 49-year-old man presented with right-sided chest pain. The patient had an unremarkable medical history, and no neurological deficits were identified. MRI revealed an Eden type III dumbbell tumor with heterogeneous enhancement at the T9-10 vertebral body level. The tumor exhibited contact with the pleura from the right side of the T9-10 spinal canal through the intervertebral foramen (Fig. 3).
Fig. 3.
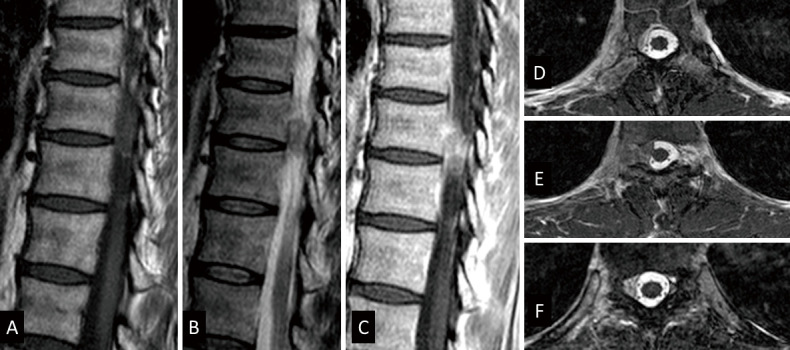
Magnetic resonance imaging of the spine showing a dumbbell tumor of Eden type III, with heterogeneous enhancement at T9 and T10 vertebral body levels. The tumor was in contact with the pleura from the right side of the T9/10 spinal canal through the intervertebral foramen. (A) T1-weighted sagittal image, (B) T2-weighted sagittal image, (C) gadolinium-enhanced T1-weighted sagittal image, (D) gadolinium-enhanced T1-weighted T9 axial image, (E) gadolinium-enhanced T1-weighted T9/10 axial image, (F) gadolinium-enhanced T1-weighted T10 axial image.
The surgical procedure was conducted with the patient in the prone position. A 7-cm skin incision was performed, and a laminectomy of T8-10 was conducted. A distinct, grayish, elastic, and soft neoplastic lesion was observed in the extradural and intradural space adjacent to the T9-10 intervertebral foramen. The intradural lesion adhered to the T9 dorsal root, slightly swollen with abnormal color, while the T9 anterior root appeared unaltered. Consequently, the T9 root was amputated, and the tumor was entirely excised (Fig. 4A and B).
Fig. 4.
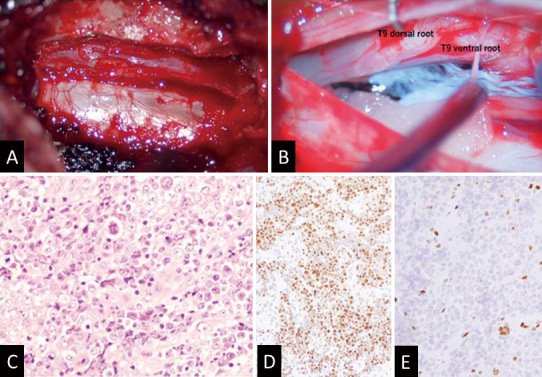
Surgical findings and pathological findings. (A) A grayish, elastic, soft, well-defined neoplastic lesion was confirmed epidurally in the T9-10 intervertebral foramen. (B) The dura is incised, a yellow tumor piece adheres to the dorsal root of T9, and the ventral root is slightly swollen with abnormal color. (C) Hematoxylin and eosin staining showing a malignant neoplasm partly composed of rhabdoid cells with necrosis. (D) Although the INI1 protein is positive, (E) the BRG1 protein is negative on immunohistochemistry.
Histological examination revealed a malignant neoplasm partially comprising rhabdoid cells with necrotic areas (Fig. 4C). Immunohistochemical examination demonstrated positive pan-CKAE1/AE3 expression, with over 80% of the tumor cells exhibiting Ki-67 positivity. Due to the rhabdoid or epithelioid features of tumoral cells, cancer metastasis was initially suspected, including lung cancer. However, the cytokeratin immunostaining pattern, CK20 (+)/CK7 (−), did not align with the pulmonary origin. Given the spinal cord localization, epithelioid malignant peripheral nerve sheath tumor (MPNST) was also considered. Nevertheless, the S-100 protein expression was not distinctive, even though focal and faint immune reactions were seen for vimentin, synaptophysin, and CD56. Subsequently, SMARCB1 (INI1) and SMARCA4(BRG1) expressions were examined. While SMARCB1 expression was retained, SMARCA4 was undetectable (Fig. 4D and E). These findings substantiated the diagnostic criteria for AT/RT. Despite the patient being asymptomatic, conventional radiation therapy (66 Gy/30 fractions) was initiated 37 days after the surgery.
Unfortunately, disseminated lesions were identified in the cerebellopontine angle, hypothalamus, lateral ventricle, and cisterna magna 7 months post-surgery. Although chemotherapy (modified ICE: ifosfamide/carboplatin/etoposide therapy) was initiated, it was discontinued due to adverse effects. The patient expired approximately 11 months post-surgery.
Discussion
Incidence of AT/RT in the adult spinal cord
AT/RT represents a rare and highly malignant neoplasm, with most cases occurring in children below 5 years old.2) Consequently, its prevalence in adults is exceptionally low.3) AT/RT accounts for 1.6% of all pediatric CNS tumors, and 4.4% of those occur in children aged 0-5 years. While AT/RT has been typically reported in various CNS regions, encompassing the supratentorial region, its occurrence in the cerebellopontine angle cistern, meninges, cranial nerves,4) and spinal canal is rare.5) To date, merely seven adult cases and 53 pediatric cases have been documented.10)
Histologically, AT/RT comprises rhabdoid tumor cells, with additional but variable components of neuroectodermal, epithelial, and mesenchymal origin. Some tumors may consist only of rhabdoid cells, whereas others show a combination of rhabdoid cells with other cell types.2,11) In our first case, the proliferation of rhabdoid cells was notably observed, presenting a relatively typical pathological image. In contrast, in the second case, even though the tumoral cells exhibited rhabdoid or epithelioid features, the unusual tumoral localization and the patient's age rendered the differential diagnosis of AT/RT challenging.
Different molecular pathological characteristics of AT/RT
Immunohistochemical investigation is crucial for AT/RT diagnosis. AT/RT shows variable expression of GFAP, S-100, and cytokeratin to exclude candidates in the differential diagnosis, such as epithelioid MPNST and rhabdoid meningioma.12) Recently developed INI1 antibody has been introduced as a useful marker to show the lack of expression of INI1 protein in tumor cells of AT/RT.12,13) INI1, the gene product of SMARCB1, is typically expressed in all normal tissues and most neoplasms. However, deletion or mutation of the SMARCB1 locus on 22q11.2 leads to the loss of this protein in AT/RTs.14) Nearly all AT/RT cases exhibit loss of INI1 protein, and approximately 70% of all cases detect SMARCB1 loss, whereas loss of SMARCA4 expression is seen in a small number of cases.15) Therefore, SMARCB1 positivity alone cannot rule out the diagnosis of AT/RT. In cases in which the diagnosis of AT/RT is strongly suspected clinically, additional testing is warranted, including SMARCA4 analysis.
SMARCA4 expression for an ATPase subunit of the SWI/SNF chromatin-remodeling complex associated with inherited germline mutations is responsible for rhabdoid tumors.16) As previous reports of AT/RT with SMARCA4 mutations suggest that SMARCA4-mutated tumors are biologically aggressive,15,16) as case 2 showed, where the tumor rapidly disseminated. Even if age or tumor site are atypical, additional testing, including SMARCA4 or SMARCA4 (BRG1) protein analysis, is warranted in cases where the tumoral cells show rhabdoid or epithelioid features.
Therapeutic strategy for primary spinal AT/RT
Given the lack of standardized treatment for spinal AT/RT, especially in adults, treatment protocols are often extrapolated intracranial pediatric AT/RT management. Surgical resection constitutes the primary therapeutic approach, complemented by chemotherapy and radiotherapy.5) Despite aggressive treatment, the prognosis of spinal AT/RT is considerably worse than that of intracranial AT/RT, with a median survival time of under six months.5,17) Although these approaches have yielded a modest improvement in overall survival, therapy-induced toxicity remains a critical problem, as case 2 illustrated. Recently, clinical trials of the enhancer of Zeste 2 (EZH2) inhibitor, Tazemostat, are being promoted. While the role of EZH2 is being evaluated, previous studies have suggested that EZH2 disruption alters cell-cycle progression and may be an important new therapeutic target, particularly in combination with radiation in AT/RT with INI-1 loss.18)
The primary causes of death resulting from AT/RTs include recurrence and dissemination.5) The rate of leptomeningeal dissemination in spinal AT/RT ranges from 20% to 40%.3) Interestingly, in our cases involved tumors originating from nerve roots recurred in a similar cranial region, specifically the cerebellopontine angle. Although some case reports have detailed AT/RT originating from the cranial nerves,4,19,20) the dissemination pattern from the spinal cord to the cerebellopontine angle had not been previously reported.
Conclusions
We presented rare pediatric and adult spinal tumor cases, which meet the AT/RT diagnostic criteria. Primary spinal AT/RT was atypical. Particularly, our adult primary spinal AT/RT with loss of SMARCA4 expression is a rare case. There is no standard treatment for spinal AT/RT, especially in adults. Further clinicopathological and genetic studies are warranted to unravel AT/RT.
Consent for Publication
All the patients have consented to submitting the case report for consideration by the journal.
Conflicts of Interest Disclosure
The authors declare no conflicts of interest to declare.
References
- 1).Louis DN, Perry A, Reifenberger G, et al. : The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131(6): 803-820, 2016 [DOI] [PubMed] [Google Scholar]
- 2).Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85(1): 56-65, 1996 [DOI] [PubMed] [Google Scholar]
- 3).Sinha P, Ahmad M, Varghese A, et al. : Atypical teratoid rhabdoid tumour of the spine: report of a case and literature review. Eur Spine J 24(Suppl 4): S472-S484, 2015 [DOI] [PubMed] [Google Scholar]
- 4).Oh CC, Orr BA, Bernardi B, et al. : Atypical teratoid/rhabdoid tumor (ATRT) arising from the 3rd cranial nerve in infants: a clinical-radiological entity? J Neurooncol 124(2): 175-183, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Xu X, Li J, Zheng Y, Li F: Primary atypical teratoid/rhabdoid tumor of spinal canal in a child: Case report and the literature review. Interdisciplinary Neurosurgery 18, 2019 [Google Scholar]
- 6).Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer research 59(1): 74-79, 1999 [PubMed] [Google Scholar]
- 7).Versteege I, Sevenet N, Lange J, et al. : Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394(6689): 203-206, 1998 [DOI] [PubMed] [Google Scholar]
- 8).Johann PD, Erkek S, Zapatka M, et al. : Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29(3): 379-393, 2016 [DOI] [PubMed] [Google Scholar]
- 9).Hasselblatt M, Nagel I, Oyen F, et al. : SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128(3): 453-456, 2014 [DOI] [PubMed] [Google Scholar]
- 10).Li D, Heiferman DM, Syed HR, et al. : Pediatric primary spinal atypical teratoid rhabdoid tumor: a case series and review of the literature. J Neurosurg Pediatr 1-17, 2019 [DOI] [PubMed] [Google Scholar]
- 11).Athale UH, Duckworth J, Odame I, Barr R: Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 31(9): 651-663, 2009 [DOI] [PubMed] [Google Scholar]
- 12).Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M: Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol 88(3): 321-330, 2008 [DOI] [PubMed] [Google Scholar]
- 13).Medjkane S, Novikov E, Versteege I, Delattre O: The tumor suppressor hSNF5/INI1 modulates cell growth and actin cytoskeleton organization. Cancer Res 64(10): 3406-3413, 2004 [DOI] [PubMed] [Google Scholar]
- 14).Biswas A, Kashyap L, Kakkar A, Sarkar C, Julka PK: Atypical teratoid/rhabdoid tumors: challenges and search for solutions. Cancer Manag Res 8: 115-125, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Hasselblatt M, Gesk S, Oyen F, et al. : Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35(6): 933-935, 2011 [DOI] [PubMed] [Google Scholar]
- 16).Bookhout C, Bouldin TW, Ellison DW: Atypical teratoid/rhabdoid tumor with retained INI1 (SMARCB1) expression and loss of BRG1 (SMARCA4). Neuropathology 38(3): 305-308, 2018 [DOI] [PubMed] [Google Scholar]
- 17).Buscariollo DL, Park HS, Roberts KB, Yu JB: Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118(17): 4212-4219, 2012 [DOI] [PubMed] [Google Scholar]
- 18).Alimova I, Birks DK, Harris PS, et al. : Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol 15(2): 149-160, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Wykoff CC, Lam BL, Brathwaite CD, et al. : Atypical teratoid/rhabdoid tumor arising from the third cranial nerve. J Neuroophthalmol 28(3): 207-211, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Verma A, Morriss C: Atypical teratoid/rhabdoid tumor of the optic nerve. Pediatr Radiol 38(10): 1117-1121, 2008 [DOI] [PubMed] [Google Scholar]