

Abstract
Plasma lipids are modulated by gene variants and many environmental factors, including diet-associated weight gain. However, understanding how these factors jointly interact to influence molecular networks that regulate plasma lipid levels is limited. Here, we took advantage of the BXD recombinant inbred family of mice to query weight gain as an environmental stressor on plasma lipids. Coexpression networks were examined in both nonobese and obese livers, and a network was identified that specifically responded to the obesogenic diet. This obesity-associated module was significantly associated with plasma lipid levels and enriched with genes known to have functions related to inflammation and lipid homeostasis. We identified key drivers of the module, including Cidec, Cidea, Pparg, Cd36, and Apoa4. The Pparg emerged as a potential master regulator of the module as it can directly target 19 of the top 30 hub genes. Importantly, activation of this module is causally linked to lipid metabolism in humans, as illustrated by correlation analysis and inverse-variance weighed Mendelian randomization. Our findings provide novel insights into gene-by-environment interactions for plasma lipid metabolism that may ultimately contribute to new biomarkers, better diagnostics, and improved approaches to prevent or treat dyslipidemia in patients.
Keywords: BXD recombinant inbred mice, Plasma lipid, Modular specificity, Mendelian randomization, Bayesian network
1. Introduction
Plasma lipids are controlled by both genetic and environmental variables, particularly physical activity and diet. The main components of plasma lipids are triglycerides (TG), which are involved in energy storage, and total cholesterol (TC), which is mainly used to synthesize cell membranes and sterols. Epidemiological studies have shown that abnormal plasma lipid levels, such as elevated low-density lipoprotein cholesterol (LDL) or TG, are strong risk factors for metabolic syndrome [1], type two diabetes [2], atherosclerosis [3,4], coronary artery disease [5], and cardiovascular disease [6–8], the leading cause of death worldwide [9].
Genetic studies in humans have uncovered dozens of key regulators of plasma lipid concentrations, including LDLR, APOB, APOE, CETP, LPL, ABCA1, LCAT, and LPL [10]. Also, recent genome-wide association studies (GWAS) have linked hundreds of genetic variants and disease causing genes to plasma lipids levels [11–14]. For example, 826 variants across 118 novel and 268 previously identified loci were associated with lipid levels in 312,571 US veterans [14]. In addition to human studies, extensive studies on the genetic basis of plasma lipid levels have been conducted in mouse models. For instance, our previous studies [15,16] in the BXD mouse reference population have revealed several quantitative trait loci (QTLs) genes that are associated with plasma lipid levels. In addition, according to the Rat Genome Database (RGD, https://rgd.mcw.edu/) [17], about 100 TC, 1 LDL, 60 TG, and 140 high-density lipoprotein cholesterol (HDL)-related QTLs have been mapped on the mouse genome. However, previous studies, taken together, explained only a small fraction of the variation in plasma lipid levels due to potential unmeasured variables, termed “missing heritability,” such as gene-by-gene or gene-by-environment interactions [18–20]. Therefore, we aimed to quantify how individual genes interact and form molecular networks and how these interactions respond to changes in environment, such as the introduction of an obesogenic diet to address the need for clarity in genetic modifiers of plasma lipids.
Although reductions of dietary fat and cholesterol have positive effects on plasma lipids at the population level, much individual variability exists, a trend that could be explained by interactions between diet and gene polymorphisms [21]. However, analysis of these interactions in human populations is difficult as it is hard to control environmental factors and access relevant tissues for molecular analyses [22]. Murine models provide an alternative way to dissect gene-by-diet interactions by introducing controlled environmental perturbations into high-precision genetic reference populations, such as the BXD strains derived from a cross between C57BL/6J mice (B6) and DBA/2J mice (D2) with more than 20 consecutive generations [23]. Currently, more than 150 BXD stains are available and numerous phenome data sets have been published over the past decades, making it an invaluable resource for system genetics studies [24]. As each BXD strain has been stably inbred, each strain can be replicated in large numbers as desired, facilitating precise mapping of complex traits with low to moderate heritability [25] and the discovery of candidate genes and mechanisms related to many phenotypes, including plasma lipids.
The advantage of our approach is that compared to differential gene expression analysis which focuses on single genes, network-based analysis can directly link gene modules (sets of tightly correlated genes) to clinical traits. In addition, network analysis of gene expression under different conditions can be performed to identify differential coexpression networks or modules that are specific to particular environmental conditions [26]. To date, a number of algorithms have been developed for network based analysis of gene expression data sets [27], such as Weighted Gene Co-Expression Network Analysis (WGCNA) [28]. This approach has been successfully used to identify plasma lipid associated modules and hub genes in human populations with hyperlipidemia [29] and dyslipidemia [30] and in mouse F2 populations [31] and knockout models [15,32].
In this study, plasma lipid levels and liver transcriptomes were examined in 42 strains of the BXD mouse population, with roughly 10 individuals of each strain separated evenly into cohorts of control diet (CD) or high-fat diet (HFD). Diet-induced obesity (DIO) resulted in the differential expression of thousands of genes and altered the composition of the coexpressed gene modules that were identified using WGCNA. We analyzed network modules identified for both conditions to identify a DIO-induced gene module that was associated with plasma lipid concentrations which contained many genes that have functions related to lipid metabolism. Bayesian network based causative analysis was conducted to determine relationships among genetic variation, gene expression, and linked phenotypes. Finally, correlation analysis confirms coexpression of some of these genes in humans, and inverse-variance weighed Mendelian randomization with human Genotype-Tissue Expression (GTEx) normalized liver gene expressions and 249 body weight- and lipid-related traits suggests that the activation of these genes in humans might be linked to reduced free cholesterol and lower levels of TG in very large HDL.
2. Materials and methods
2.1. Mice and phenotyping
The BXD recombinant inbred (RI) mice were born and raised at the École Polytechnique Fédérale de Lausanne (EPFL) in Switzerland. Briefly, 10 males from 42 strains of the BXD family and both parental strains (C57BL/6J and DBA/2J) were grouped into two cohorts at 8 weeks of age, five animals per strain on a CD (6% kcal/fat, 20% protein, 74% carbohydrate, Harlan 2018) and 5 animals per strain on HFD (60% kcal/fat, 20% protein, 20% carbohydrate, Harlan 06,414). All mice were sacrificed at 29 weeks of age after fasting overnight. Under isoflurane anesthesia, blood was removed from the vena cava and animals were perfused with 4°C phosphate-buffered saline. Tissues were taken immediately and frozen in liquid nitrogen. The detailed procedure for plasma analysis and phenotype measurement can be found in our previous publications [33,34].
2.2. Histopathologic evaluation
Samples of the liver tissue were fixed in 4% paraformaldehyde, embedded in paraffin, cut, and stained with hematoxylin and eosin (H&E). Fat was identified morphologically as sharply demarcated clear cytoplasmic vacuoles. Abundant lipid produces large coalescing vacuoles that displace the nucleus to the cell margin (macrovesicular steatosis), while smaller amounts produce small spherical clear vacuoles scattered in the cytoplasm (microvesicular steatosis). Glycogen is differentiated morphologically as it produces ragged clear spaces that are not spherical and do not have sharp margins or displace nuclei.
2.3. RNA isolation and microarray
Total RNA was extracted from 100 mg pieces of liver tissue using Trizol reagent (Invitrogen) followed by a standard phase separation extraction using chloroform and precipitated by isopropanol. Individual RNA samples from all mice of the same strain and cohort (3–5 mice) were pooled equally (by microgram of RNA) into a single RNA sample. Pooled RNA samples were then purified with RNEasy (QIAGEN). Agilent 2,100 Bioanalyzer was used to evaluate RNA integrity and quality. Samples that passed quality control (RIN > 8.0) were run on Affymetrix Mouse Gene 1.0 ST at the University of Tennessee Health Science Center (UTHSC).
2.4. Microarray normalization and preprocessing
Raw microarray data from both cohorts were normalized together using the Robust Multichip Array (RMA) method [35]. The expression data were then renormalized using a modified Z score described in a previous publication [36]. Briefly, RMAs were first transformed into log2-values. Then the data of each single array was converted to Z-scores, multiplied by two, and a value of eight was added. Before microarray data was used in the analysis discussed below, the data was filtered to remove probes that lacked annotation or had low expression. Specifically, of the ~35,000 probe sets on Affymetrix Mouse Gene 1.0 ST, 18,453 probes (corresponding to 15,754 genes) remained after removing unannotated Affy probes and probes with mean expression level < 7. These probes were used in subsequent analysis.
2.5. Differential expression analysis
The Limma package from R Bioconductor [37] https://www.nature.com/articles/nature16,064-ref-CR35 was used for the normalized microarray data to analyze differentially expressed genes (DEGs) between HFD and CD cohorts. Genes below a Benjamini-Hochberg false discovery rate (FDR) [38] threshold of 5% were considered DEGs.
2.6. Gene set over-representation analysis
To investigate the biological functions of the gene sets of interest, such as the DEGs, WebGestalt (http://www.webgestalt.org/) [39], an open-source online analysis toolkit, was used for enrichment analysis for Gene Ontology (GO) biological processes and KEGG pathways. GO biological process terms were filtered to only include terms at hierarchical level nine to limit redundancy in the GO terms. Mouse genome was used as reference gene set and the minimum number of genes for a category was set to five. The FDR < 0.05 indicated significant overrepresentation in a category for the queried genes.
2.7. Weighted gene coexpression network analysis (WGCNA)
We constructed gene coexpression networks for both cohorts (CD and HFD) separately using the WGCNA v1.63 package in R (v3.1.3) [40]. Briefly, a soft threshold power ( for HFD and for CD) was selected by pickSoftThreshold based on the criterion of approximate scale-free topology. Then, the signed adjacency matrix was calculated with the following equation:
where and are the ith and jth gene expression traits. Subsequently, a Topological Overlap Matrix (TOM) was constructed. Genes were aggregated into modules by hierarchical clustering based on TOM and further refined using the dynamic tree cut algorithm. We also evaluated the module-trait association by calculating the correlation coefficient between traits and module eigengenes (MEs), defined as the first principal component of a given module.
2.8. Criteria for identifying gene modules induced by HFD and associated with plasma lipids
Gene modules identified by WGCNA that were both induced by HFD and associated with plasma lipid levels were evaluated to select gene network modules. First, modules were identified in which at least 44.5% of the genes were identified as being differentially expressed (i.e., FDR < 0.05) because this cut-off value was twice the percentage of DEGs in the entire data set (22.3%). Second, modules were selected that were induced only after the introduction of HFD. We calculated the Jaccard coefficient, for each pair of modules from the two conditions with the following equation:
where and are the genes from the CD and HFD modules, respectively. Jaccard coefficient is a measure of similarity between two elements. The index ranges from 0–1. The closer to zero, the less similarity of the two data sets. For this, HFD modules with Jaccard coefficient < 0.15 when compared with each CD module were then selected as being induced by HFD. Third, modules were selected that were significantly enriched with genes associated with plasma lipid-related biological processes in GO. We performed GO enrichment analysis to identify biological processes that were significantly overrepresented (FDR < 0.05) among the genes in each module. Last, we examined associations between modules and plasma lipid phenotypes and identified modules in which the ME was significantly correlated with these phenotypes.
2.9. Identification of hub genes
Module hub genes are believed to be highly connected with other module genes and have the most significant biological impact for the associated traits. Module hub genes were identified and ranked using the following three parameters: (1) intramodular connectivity, which refers to the connectivity between genes in the same module; (2) gene significance, the correlation coefficient between the expression level of the gene and the phenotype; and (3) module membership, which was obtained by correlation analysis between the expression level of the gene and the MEs. Ideally, hub genes should have high values of IC and low P-values for both GS and MM. All three analyses were done with the WGCNA package.
2.10. Protein-protein interaction (PPI) network
We explored PPI networks for the top 30 hub genes in HFD module M9 in the STRING database (https://string-db.org/) [41] with a minimum interaction score of 0.4.
2.11. Human GWAS
The NHGRI GWAS Catalog (https://www.ebi.ac.uk/gwas/) [42] is a continuously updated public resource to facilitate researchers to quickly and efficiently access current GWAS results. In order to validate human GWAS with BXD RI model findings, we collected GWAS signals with P-values less than 1 × 10−6 for plasma lipid and fasting glucose levels.
2.12. Mammalian phenotype ontology (MPO)
Plasma lipid, body weight, and glucose-associated genes were retrieved from Mouse Genome Informatics (http://www.informatics.jax.org/) with the 32 MPO terms listed in Supplementary Data 1.
2.13. QTL mapping
QTL mapping was conducted with WebQTL in GeneNetwork (http://www.genenetwork.org/). The 7,320 informative SNP markers which segregated in BXD RI strains were used for interval mapping. Likelihood ratio statistics (LRS) were used to assess the association or linkage between differences in traits and differences in particular genotype markers. We report quantitative trait loci (QTLs) achieving significance (genome wide P-value < .05) based on 2,000 permutation tests. The 1.5-logarithm of the odds (LOD) confidence interval was used to filter the candidate genes. In addition, phenotype-associated QTL using the Haley and Knott method were further confirmed with GEMMA [43], a linear mixed model mapping algorithm that accounts for kinship among the BXD strains.
2.14. Causative analysis with Bayesian network modeling
In order to investigate pathways connecting QTL genotypes, gene expression, and phenotypes, we created Bayesian network models using the Bayesian Network Webserver (BNW; http://compbio.uthsc.edu/BNW/) [44,45]. Bayesian networks are graphical models that describe the conditional dependencies between data variables, where directed edges between network variables imply causal relationships. Bayesian network model structures were learned from the data using BNW with the following structure learning settings. Each variable had a maximum of four direct parents in any potential model, model averaging of the 1,000 highest scoring networks was performed, and directed edges with weights greater than 0.5 were included in the final network model.
2.15. Mapping mouse genes to human genes
To map the mouse genes to their corresponding human genes, we used UniProt’s “Similar proteins” field or protein BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi? PAGE=Proteins) if the former did not provide a human protein with at least 50% identity.
2.16. Genotype-tissue expression (GTEx) normalized liver gene expressions and eQTL analysis
Normalized gene expressions were calculated by the GTEx consortium [46] based on RNA-Seq count data in three sequential steps: (1) genes were selected that have ≥ 0.1 transcripts-permillion and ≥ 6 reads in ≥ 20% of the samples, (2) counts were normalized with edgeR’s TMM normalization, and (3) normalized expressions were inverse-normally transformed. These normalized gene expressions are publicly available from GTEx through https://storage.googleapis.com/gtex_analysis_v8/single_tissue_qtl_data/GTEx_Analysis_v8_eQTL_expression_matrices.tar.cis-eQTLs (adjusted P-value < 5%) were mapped by the GTEx consortium with FastQTL [47] based on the normalized gene expressions and the data are publicly available at the GTEx portal through https://storage.googleapis.com/gtex_analysis_v8/single_tissue_qtl_data/GTEx_Analysis_v8_eQTL.tar.
2.17. Mendelian randomization
We conducted Mendelian randomization analysis using the TwoSampleMR R package [48]. Wald ratio tests were used to calculate the associations of the individual genes with the outcomes. Associations between module activation and outcomes were assessed with the inverse-variance weighted method [49]. A Benjamini-Hochberg FDR < 5% was considered statistically significant.
3. Results
3.1. BXD RI mice on HFD have elevated blood lipid levels
To characterize the effect of diet on plasma lipids, we examined 42 BXD RI strains on two diets: 41 strains were fed CD and 40 strains were fed HFD, with 39 strains overlapping in both diets. These strains included 37 BXD strains and the two parental strains (C57BL/6J and DBA/2J) fed both diets, as well as two strains (BXD60 and BXD92a) fed only CD, and one strain (BXD50) fed only HFD. Diet was initiated at 8 weeks of age. Male littermates from breeding trios were used, with typically three trios per strain established simultaneously. Thirty-two strains yielded sufficient pups for both CD and HFD cohorts to be established concurrently (i.e., 7–10 males born within ±2 days), while 7 strains with poor breeding performance yielded CD and HFD cohorts that were separated by birthdate. Mice were sacrificed at 29 weeks of age after fasting overnight. Serum and liver were collected for phenotyping and mRNA expression profiling (Fig. 1A). Overall, BXD RI strains on HFD had a significant increase in body weight (Fig. 1B) and fat mass (Fig. 1C) that was accompanied by a decrease in lean mass (Fig. 1D). Concentrations of plasma lipids, including TC, HDL, and LDL (Fig. 1E–G), were significantly higher in HFD-fed mice compared to CD-fed controls. Diet did not have a significant impact on free fatty acid (FFA), TG, and basal plasma glucose levels (Fig. 1H–J).
Fig. 1.
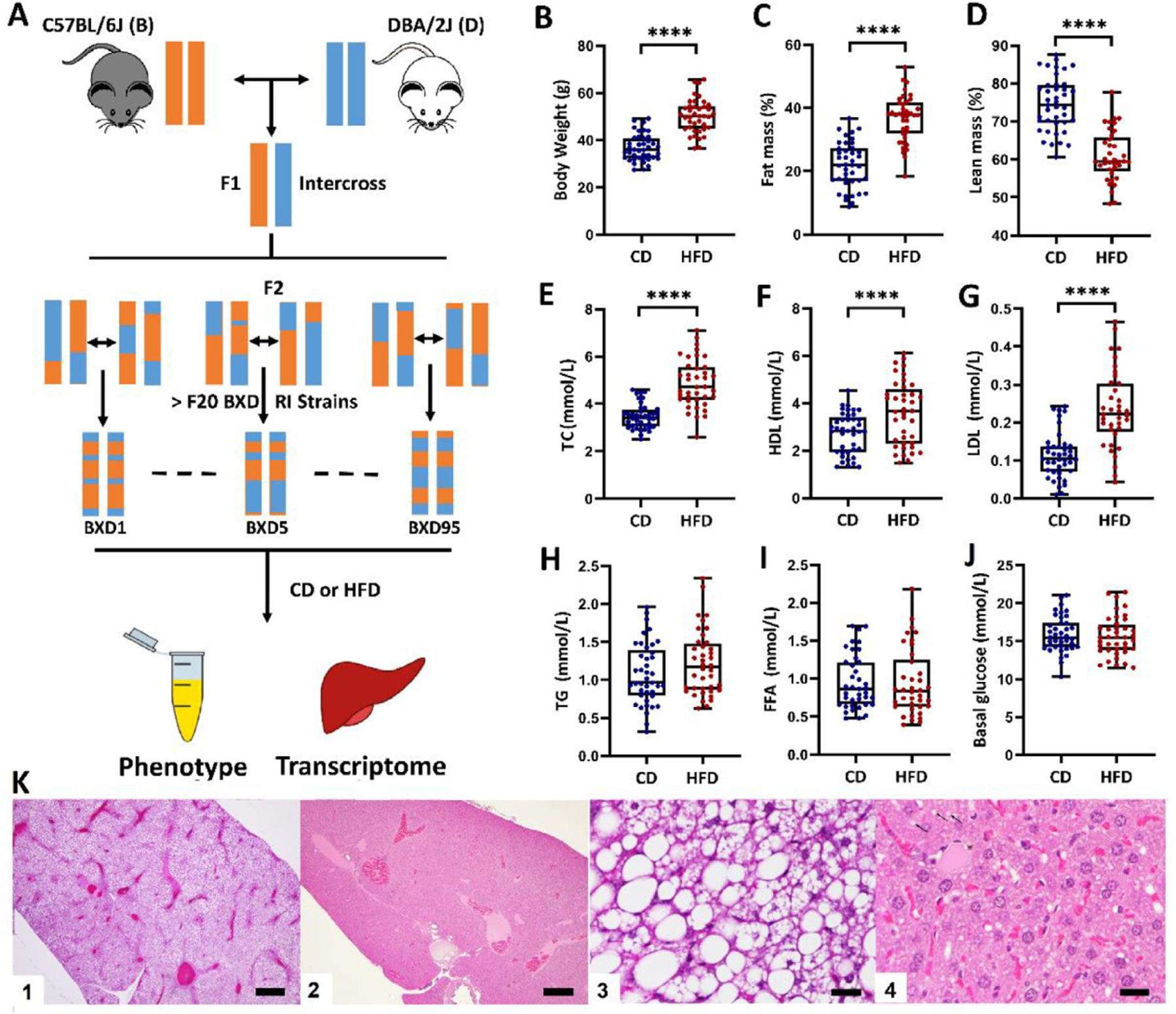
Effects of HFD on body weight, body composition, and plasma lipid. (A) Study design. BXD strains are crosses of C57BL/6J (B) and DBA/2J (D). BXDs and parental strains were divided into two diet groups. Blood serum phenotypes and liver transcriptomes of male mice were analyzed. (B–J) Boxplots showing differences between CD and HFD cohorts in obesity and plasma lipid-related phenotypes. ****P < 0.0001. (K) Histologic analysis shows heterogeneity in fatty liver across strains. Representative images for strains BXD214 (one and three) and BXD2 (two and four). Scale bars 500 microns (one and two) and 25 microns (three and four).
3.2. BXD RI mice on HFD develop hepatic steatosis of varying severity
In order to evaluate whether HFD promotes hepatic steatosis across the BXD strains, we performed hepatic H&E analysis. Results demonstrated that some strains, such as BXD214 (Fig. 1K), displayed diffuse marked macrovesicular steatosis (accumulation of large lipid droplets in hepatocytes), and some strains, such as BXD2 (Fig. 1K), remain resistant to steatosis.
3.3. Diet significantly altered lipid metabolism-related transcriptional profiles
To study the impact on the heterogeneity of gene expression induced by HFD across strains, liver transcriptomes of strains fed CD or HFD were analyzed. The expression levels of 3,513 genes (22.3%, Supplementary Data 2) were significantly altered in livers of mice fed HFD (FDR < 0.05) compared to controls. Expression of the DEGs was more likely to be upregulated in HFD than CD, as 2,109 genes of the DEGs were upregulated in HFD.
In general, the magnitude of differences in message expression between the two groups was relatively modest, as only seven genes had fold change (FC) expression differences greater than 2.0. The seven genes with FC > 2.0 and FDR < 0.01were Cyp2b9 (FC = 8.8, FDR = 1.76E-08), Lcn2 (FC = 3.3, FDR = 2.77E-06), Saa2 (FC= 2.3, FDR = 0.001), Serpina4-ps1 (FC = −2.3, FDR = 5.06E-05), 9030619P08Rik (FC = 2.2, FDR = 5.42E-09), Saa1 (FC = 2.1, FDR = 0.002), and Cd36 (FC = 2.0, FDR = 6.57E-09). In addition, around 420 of the DEGs had 1.2 ~ 2.0 FC with FDR < 0.05. This group included Apoa4, Cidea, Hamp, Gsta1, Gsta2, and mir-122.
In order to determine the overall functional relevance of both upregulated and downregulated DEGs, we performed gene set over-representation analysis, which identified 40 and 42 terms that were significantly enriched (FDR< 0.05) for GO biological processes at hierarchical tree level 9 and KEGG pathways, respectively. Notably, many of the enriched terms were related to metabolism and plasma lipids. For example, the most enriched GO biological process terms were Organic Acid Metabolic Process, Peptide Metabolic Process, and Lipid Metabolic Process. Similarly, the most highly enriched KEGG pathway was Metabolic Pathways (Table 1).
Table 1.
Top 10 enrichment terms of GO (biological process) and KEGG pathway for the DEGs induced by HFD in BXD RI mice.
| Term | Description | N. Genes | FDR |
|---|---|---|---|
|
| |||
| GO (Biological Process) | |||
| GO:0002376 | immune system process | 444 | < 2.06E-14 |
| GO:0002682 | regulation of immune system process | 239 | < 2.06E-14 |
| GO:0006082 | organic acid metabolic process | 254 | < 2.06E-14 |
| GO:0006518 | peptide metabolic process | 174 | < 2.06E-14 |
| GO:0006629 | lipid metabolic process | 321 | < 2.06E-14 |
| GO:0006631 | fatty acid metabolic process | 113 | < 2.06E-14 |
| GO:0006915 | apoptotic process | 362 | < 2.06E-14 |
| GO:0006952 | defense response | 268 | < 2.06E-14 |
| GO:0006955 | immune response | 255 | < 2.06E-14 |
| GO:0008219 | cell death | 384 | < 2.06E-14 |
| KEGG pathway | |||
| mmu01100 | Metabolic pathways | 345 | 2.03E-11 |
| mmu03010 | Ribosome | 60 | 6.65E-09 |
| mmu04142 | Lysosome | 49 | 4.93E-06 |
| mmu05204 | Chemical carcinogenesis | 39 | 1.36E-05 |
| mmu00260 | Glycine, serine and threonine metabolism | 20 | 4.58E-04 |
| mmu04210 | Apoptosis | 47 | 6.27E-04 |
| mmu00140 | Steroid hormone biosynthesis | 33 | 8.87E-04 |
| mmu00830 | Retinol metabolism | 33 | 1.34E-03 |
| mmu00980 | Metabolism of xenobiotics by cytochrome P450 | 26 | 1.79E-03 |
| mmu05150 | Staphylococcus aureus infection | 22 | 2.04E-03 |
3.4. Diet influences gene modules identified by WGCNA
It has been widely recognized that coexpressed genes are commonly involved in similar biological pathways or processes [50]. Therefore, we constructed gene coexpression networks using WGCNA with a soft-thresholding power ( for HFD and for CD) determined by scale-free topology (Fig. 2A–C and Fig. S1). A total of 15,754 genes were parsed into 18 and 35 distinct coexpression modules for the HFD and CD cohorts, respectively (Supplementary Data 2). The modules identified by WGCNA have a wide range of sizes, with some modules containing thousands of genes and other containing only dozens (Supplementary Data 2). To identify HFD modules that are impacted by diet, we determined the percentage of DEGs from above within each module. The percentages of DEGs in the individual HFD modules ranged from 8.2%–70.1% (Table 2 and Supplementary Data 2). Compared to the percentage of DEGs among all genes of 22.3%, there were several HFD modules that were enriched with DEGs, including 5 modules (M7, M8, M9, M13, and M16) that had percentages of DEGs that were more than twice the overall DEG percentage.
Fig. 2.
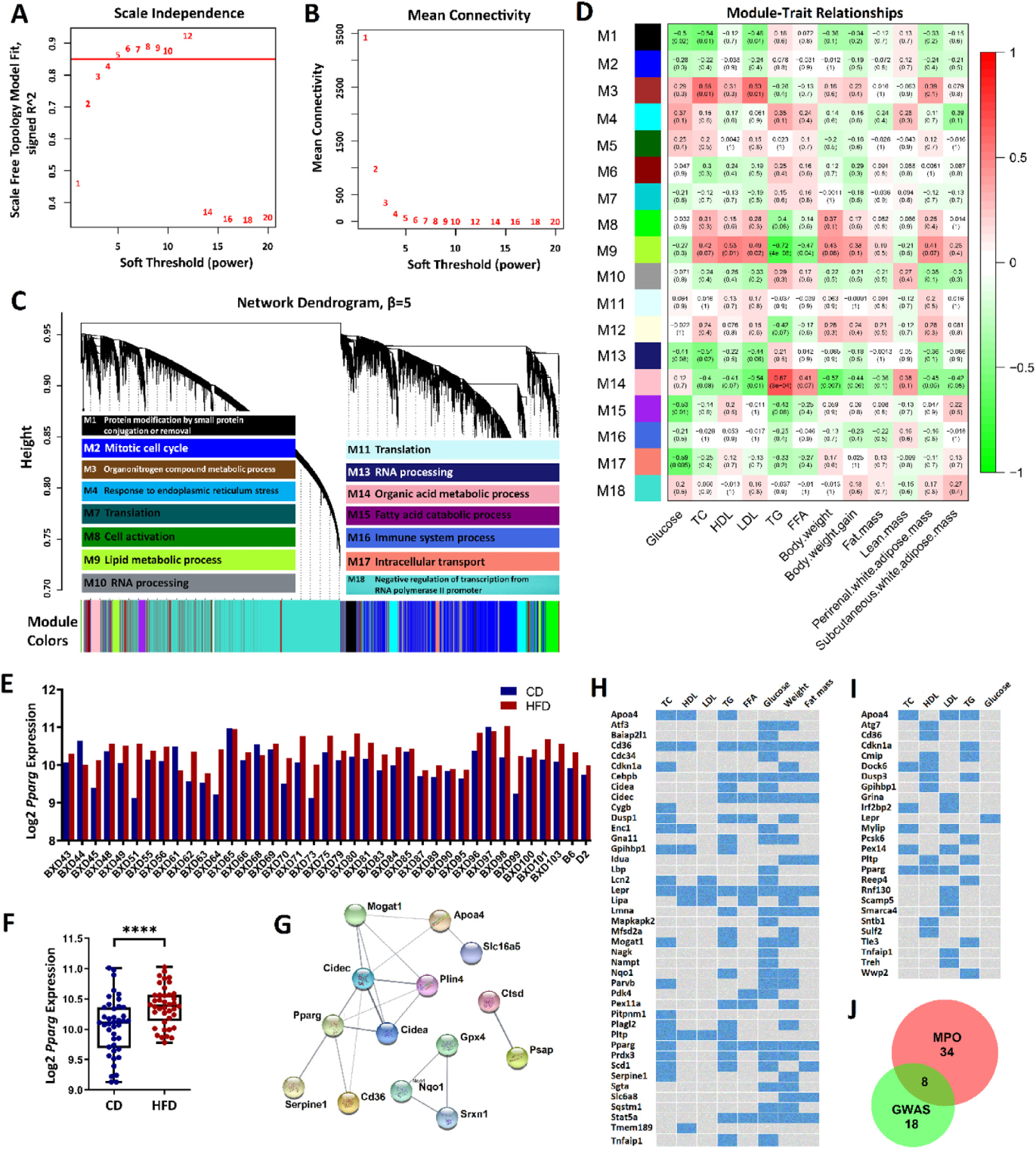
HFD module M9 is responsive to diet and associated with plasma lipids. (A) Soft thresholding index R2 as a function of soft-thresholding power indicated scale-free topology. (B) Mean connectivity (degree) as a function of . (C) 18 coexpression modules identified from HFD liver transcriptome data by dendrogram branch cutting and their most significantly enriched GO term among discrete biological processes. (D) Associations (Pearson correlation r, with FDR in parentheses) between HFD module eigengenes and plasma lipid phenotypes. (E) Pparg expression across BXD strains. (F) Mean difference in Pparg expression between diets. ****FDR < 0.0001. (G) Protein-protein interaction subnetworks from the top 30 hub genes in module M9. 14 genes have predicted interaction scores > 0.4 (medium confidence). (H-I) Module M9 gene-trait heatmap retrieved from the MPO (H) and human NHGRI GWAS Catalog (I). Blue cells represent genes with functional roles critical or associated with the corresponding trait(s). (J) Venn diagram of gene overlap between MPO and GWAS.
Table 2.
Size and condition specificity of HFD modules.
| HFD module | Number of genes in module | % of module genes that are DEGs | Maximum Jaccard coeffa | Most similar CD moduleb | Number of genes in most similar CD module |
|---|---|---|---|---|---|
|
| |||||
| M1 | 356 | 35.39 | 0.06 | M4 | 261 |
| M2 | 3893 | 22.99 | 0.40 | M2 | 3955 |
| M3 | 1402 | 29.32 | 0.14 | M12 | 589 |
| M4 | 761 | 32.85 | 0.12 | M13 | 142 |
| M5 | 28 | 28.57 | 0.04 | M18 | 271 |
| M6 | 28 | 42.86 | 0.04 | M31 | 83 |
| M7 | 12 | 58.33 | 0.02 | M24 | 197 |
| M8 | 828 | 54.83 | 0.27 | M1 | 366 |
| M9 | 340 | 54.12 | 0.13 | M23 | 109 |
| M10 | 1203 | 23.19 | 0.11 | M2 | 3955 |
| M11 | 49 | 8.16 | 0.05 | M28 | 310 |
| M12 | 52 | 26.92 | 0.04 | M20 | 110 |
| M13 | 86 | 53.49 | 0.09 | M9 | 88 |
| M14 | 354 | 26.84 | 0.18 | M6 | 129 |
| M15 | 245 | 31.84 | 0.27 | M26 | 263 |
| M16 | 87 | 70.11 | 0.38 | M32 | 97 |
| M17 | 119 | 34.45 | 0.07 | M8 | 299 |
| M18 | 6719 | 9.23 | 0.74 | M33 | 6339 |
Maximum value of the Jaccard coefficient between given HFD module and any CD module.
Identity of the CD module with the maximum Jaccard coefficient with the HFD module.
To examine the condition-specificity of the modules in the CD and HFD cohorts, we calculated the Jaccard coefficient between each of the modules for the two conditions. In general, the Jaccard coefficients between the modules in the two conditions were relatively low, indicating that WGCNA identified distinct modules in the HFD and CD data (Table 2 and Supplementary Data 3 and 4). Specifically, the maximum Jaccard coefficient between a HFD module and any of the CD modules was ≤ 0.15 for 12 of the HFD modules, thus we considered these 12 modules as HFD-specific. However, there were some pairs of HFD and CD modules with high Jaccard coefficients, indicating that the module was shared between the two diets. Module pairs with a high amount of overlap for the two diets included the two largest modules for each diet (HFD module M18 and CD module M33, HFD module M2 and CD module M2), but there were also some small modules with relatively large Jaccard coefficients. For example, HFD module M16 and CD module M32, each of which contained ~100 genes, had a Jaccard coefficient of 0.38.
Combining the modular specificity as defined by the Jaccard coefficient with the percentages of DEGs in each module can further identify modules that are impacted or created by a HFD. Specifically, 5 HFD modules (M1, M6, M7, M9, and M17) had a high percentage (> 44.5%) of genes that were DEGs and low (≤ 0.15) Jaccard coefficients when compared with all CD modules. Therefore, the genes within these modules are likely sensitive to changes in diet. In contrast, HFD module M18, for example, had a low percentage of DEGs and high Jaccard coefficient with a CD module, indicating that the expression and interactions of the genes in this module were highly responsive to the different environments of CD and HFD.
3.5. Several modules identified by WGCNA are associated with plasma lipid levels in HFD condition
In order to investigate biological functions related to the HFD modules, we conducted GO enrichment analysis, which identified 15 modules that were significantly enriched (FDR < 0.05) in different biological processes (Fig. 2C and Supplementary Data 5). Notably, two modules, M9 and M15, were enriched with genes involved in the lipid metabolic process and fatty acid catabolic process, respectively (Fig. 2C). The genes of modules M5, M6, and M12 were not significantly enriched with genes for any functional annotations in our analysis. In addition, we correlated the MEs for each module to the external phenotypes. Seven modules showed significant correlation (FDR < 0.05) with at least one of the investigated traits. Module M9 was associated with several of the phenotypes of interest, with MEs that were significantly correlated with HDL, LDL, TG, and FFA (Fig. 2D).
3.6. M9 is a HFD-induced specific module that is significantly associated with plasma lipid levels
Based on analysis of both the condition specificity of CD and HFD modules and associations between module genes and plasma lipids, HFD module M9 was identified as a module that was induced and specific to HFD. Module M9 was the only HFD module that met the following criteria: it had a Jaccard coefficient ≤ 0.15 when compared with all CD modules; more than 44.5% of the genes in the module were identified as DEGs; it was significantly associated with most plasma lipid phenotypes; and it was enriched with genes involved in a relevant biological process after GO enrichment analysis. Therefore, we sought to further investigate key genes and interactions in module M9.
3.7. M9 module hub genes are associated with plasma lipid levels
Among the 340 genes in M9 module, we computed and ranked the genes with intramodular connectivity, gene significance, and module membership (see materials and methods). The top 30 hub genes are shown in Table 3. Many of these, including Cidec, Cd36, Mogat1, Slc6a8, Apoa4, Serpine1, Cidea, Pparg, and Nqo1, are known to regulate genes for plasma lipid metabolism (http://www.informatics.jax.org/). Additionally, all the 30 genes showed strong correlations (P < .05, Supplementary Data 6) with hepatic steatosis parameters Alanine transaminase and Aspartate transaminase. Moreover, Pparg, a ligand-dependent nuclear hormone transcription factor, could be a master regulator since it regulates the transcription of 19 out of the 30 hub genes (Cidec, Cd36, Serpine1, Pcx, Cidea, Adcy6, Prune, Slc16a5, Plin4, Acot2, Slc25a10, Nqo1, Ctsd, Srxn1, Gpx4, Slc6a8, Ly6d, Slc16a7, and Morc4) directly according to the PPAR gene database (http://www.ppargene.org/) [51]. The expression level of Pparg in liver shows large variability across the BXD strains in response to HFD (Fig. 2E). For instance, PParg mRNA levels in BXD90 show resistance to HFD (9.8 in CD and 9.9 in HFD), but vulnerable in BXD51 (9.1 in CD and 10.6 in HFD). Overall, Pparg showed a 1.3-fold increase in HFD mice compared with CD mice (Fig. 2F).
Table 3.
Top 30 hub genes in M9 module.
| Symbol | Intramodular connectivity | Module membership significance | Gene significance* | Rank |
|---|---|---|---|---|
|
| ||||
| Ly6d | 1.00 | 0.00 | 9 | 1 |
| Cidec | 0.88 | 5.61E-12 | 8 | 2 |
| Ctsd | 0.87 | 4.50E-12 | 8 | 3 |
| Prune | 0.85 | 2.37E-11 | 6 | 4 |
| Srxn1 | 0.85 | 1.85E-11 | 8 | 5 |
| Slc16a5 | 0.83 | 1.01E-10 | 10 | 6 |
| Gpx4 | 0.82 | 2.17E-10 | 11 | 7 |
| Srd5a3 | 0.82 | 6.82E-11 | 7 | 8 |
| Cd36 | 0.82 | 2.80E-10 | 9 | 9 |
| Plin4 | 0.81 | 2.15E-10 | 4 | 10 |
| Acot2 | 0.80 | 8.95E-10 | 7 | 11 |
| Mogat1 | 0.80 | 1.30E-09 | 6 | 12 |
| Slc41a3 | 0.79 | 2.45E-09 | 4 | 13 |
| Wfdc2 | 0.78 | 3.96E-09 | 6 | 14 |
| Slc6a8 | 0.77 | 2.77E-09 | 8 | 15 |
| Slc16a7 | 0.77 | 5.19E-09 | 10 | 16 |
| Apoa4 | 0.76 | 6.54E-09 | 8 | 17 |
| Morc4 | 0.76 | 5.22E-09 | 4 | 18 |
| Serpine1 | 0.75 | 5.86E-09 | 7 | 19 |
| Pcx | 0.75 | 1.31E-08 | 4 | 20 |
| Abhd4 | 0.74 | 1.12E-08 | 3 | 21 |
| Cidea | 0.74 | 3.77E-08 | 8 | 22 |
| Pparg | 0.73 | 3.13E-08 | 5 | 23 |
| Slc25a10 | 0.72 | 3.97E-08 | 7 | 24 |
| Adcy6 | 0.72 | 3.67E-08 | 5 | 25 |
| Golt1a | 0.72 | 2.13E-08 | 4 | 26 |
| Nqo1 | 0.72 | 3.86E-08 | 7 | 27 |
| Myo1d | 0.72 | 4.72E-08 | 2 | 28 |
| Tuba8 | 0.72 | 3.49E-08 | 8 | 29 |
| Psap | 0.71 | 8.07E-08 | 5 | 30 |
Bold indicates known involvement in plasma lipid regulation.
Number of traits (TC, HDL, LDL, TG, FFA, body weight, body weight gain, and perirenal white adipose mass) significantly associated with the gene.
In addition, by searching PPI networks from the STRING database, we identified three subnetworks for the top 30 hub genes (Fig. 2G). The largest one included nine genes in which Pparg directly interacts with Cidec, Cidea, Plin4, Serpine1 and Cd36, and with Apoa4 and Mogat1 through Cidec. The second subnetwork included three genes: Gpx4, Nqo1, and Srxn1, which are all involved in antioxidant defense. These genes harbor antioxidant response elements that are targeted by nuclear factor erythroid two–related factor two (Nrf2) [52–55]. The last subnetwork contained only two genes: Ctsd and Psap.
3.8. M9 module genes implicated in previous GWAS and mouse models
Among the 340 M9 genes, 42 were found to be associated with either TC, HDL, LDL, TG, FFA, glucose, body weight, and fat mass (Fig. 2H) in the MPO database. Furthermore, 26 genes were identified as functional genes in TC, HDL, LDL, TG, or glucose GWAS (Fig. 2I). Eight genes (Tnfaip1, Apoa4, Pparg, Pltp, Cdkn1a, Gpihbp1, Cd36, and Lepr) met both of these criteria (Fig. 2J).
3.9. Bayesian network-based causative analysis for M9 module genes
In order to investigate how M9 module genes cause phenotype variation, we created Bayesian network models that included a QTL genotype, expression of M9 module genes, and phenotypes. First, we performed QTL mapping to identify a genomic locus (Chr11: rs26890724) that controls body weight (GN:17562) and perirenal white adipose mass (GN: 17790) (Fig. 3A–B). Five M9 module genes (LOC728392, Myo1c, Flot2, Rab34, and Tnfaip1) were selected that were located within this Chr 11 QTL interval. A Bayesian network model was created from these 8 variables (including the Chr 11 QTL genotype, 5 gene expression traits, and two phenotypes) (Fig. 3C). The structure of the network was learned from the data, and, thus, the causal relationships (only included the genes that on the paths from the QTL to the phenotype) shown in the network were implied by the data. Expression of Flot2 did not influence the phenotypes or any other network variable. For clarity, we removed Flot2 expression from the network; the removal of this node did not alter any edges between the other network variables.
Fig. 3.
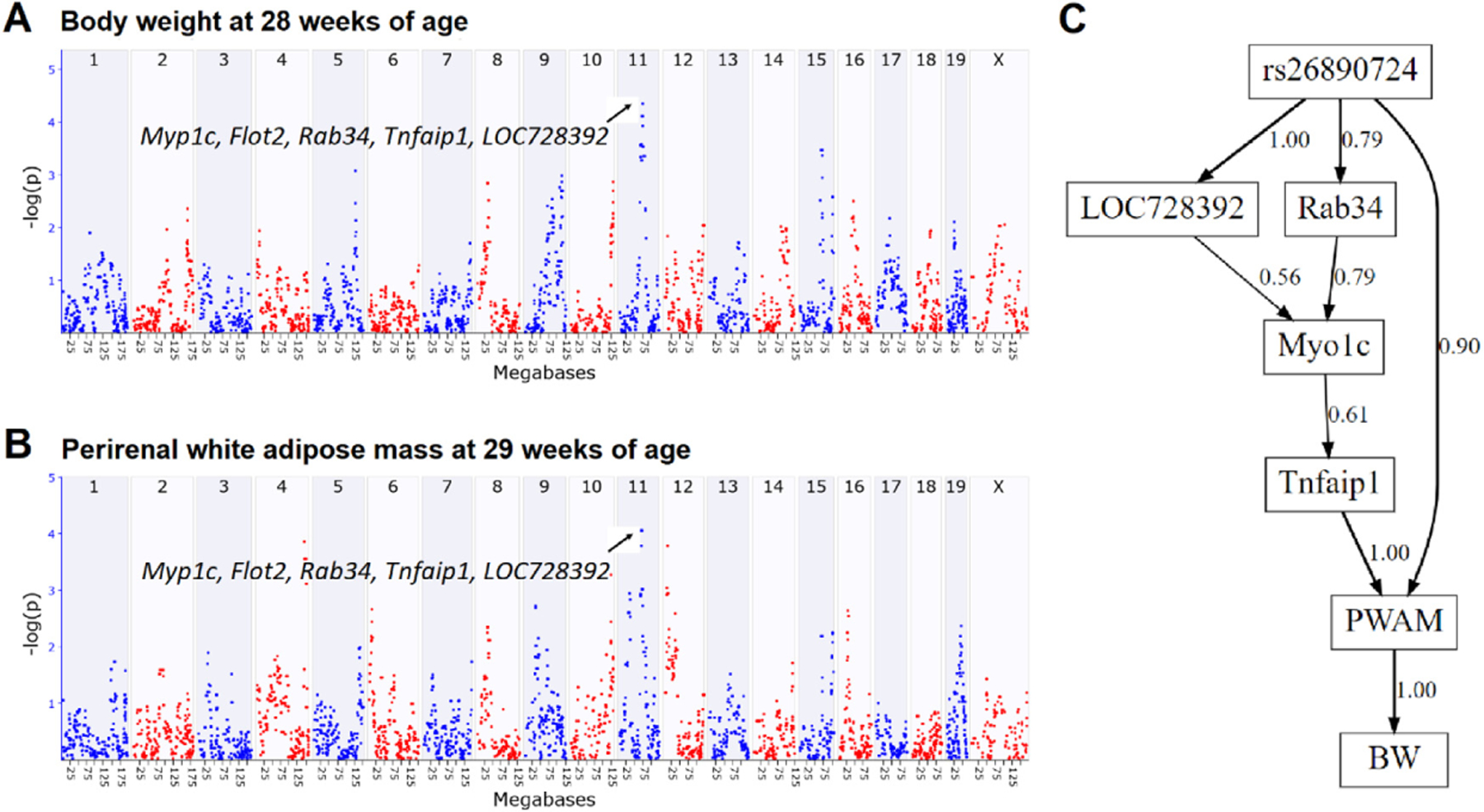
Causal analysis of M9 module genes (A-B) QTL mapping for body weight and perirenal white adipose mass on chromosome 11 (C) Bayesian network structure connecting QTL genotype (rs26890724), gene expression, and body weight (BW) and perirenal white adipose mass (PWAM) phenotypes. Numbers next to each network edges indicate edge confidence from model averaging of the 1,000 highest scoring networks.
The Bayesian network model linking the Chr 11 QTL with body weight and perirenal white adipose mass has a couple of interesting features. First, the only direct parent of body weight in the network is perirenal white adipose mass, implying that the QTL and genes in the network do not directly influence body weight. Instead, these variables indirectly impact body weight through their effects on perirenal white adipose mass. Second, one pathway links QTL genotype and phenotype through four of the five M9 genes located at the Chr 11 QTL. In this pathway, TNF Alpha Induced Protein one (Tnfaip1) is the direct parent of perirenal white adipose mass, indicating that Tnfaip1 is the gene in the network that most strongly influences the phenotypes. This result is in line with our investigation of the correlations between the M9 module genes in the network and the phenotypes, as the correlation coefficient between Tnfaip1 and perirenal white adipose mass is greater than the correlation coefficient between any of the other genes and phenotypes in the network. Finally, a network edge directly links the QTL genotype with perirenal white adipose mass, indicating that the influence of the genotype on the phenotypes was not entirely explained by the expression of the genes in M9 model that were located near the QTL and included in the network model. Indeed, genomic regulation of body weight is extremely complex, with many genes interacting with each other [18–20]. Thus, other factors, such as the expression of other genes like Pld2 that is also encoded within our QTL region and linked to body mass, as well as proteomic or other molecular level traits, may need to be considered to fully explain the link between the Chr 11 QTL and perirenal white adipose mass.
3.10. Expression and association with lipid metabolism of the top module M9 genes in humans
To assess the validity of module M9 in humans, we mapped the top 30 mouse genes in module M9 (Table 3 and Supplementary Data 7) to their corresponding human genes. Of these 30 human genes, 11 had at least one significant cis-eQTL in liver in the human Genotype-Tissue Expression (GTEx) database (FDR < 5%, Supplementary Data 7). We then calculated the eigengene of these 11 human genes with WGCNA. All genes, except ACOT2, correlate significantly with the eigengene, and out of 66 pairwise comparisons (11 genes and the eigengene), 51 had a significant Pearson correlation (FDR < 5%, Fig. 4A). This indicates that the eigengene captures the module’s expression well and that these genes tend to be coexpressed in humans. CTSD is the only gene that significantly correlates with all other 10 genes and the eigengene. GPX4 and MOGAT2 are the only two genes with a significant negative correlation with both the eigengene and CTSD. This overall strong degree of correlation in human liver gene expression suggests that these 11 genes might form part of a gene coexpression module in humans as well.
Fig. 4.
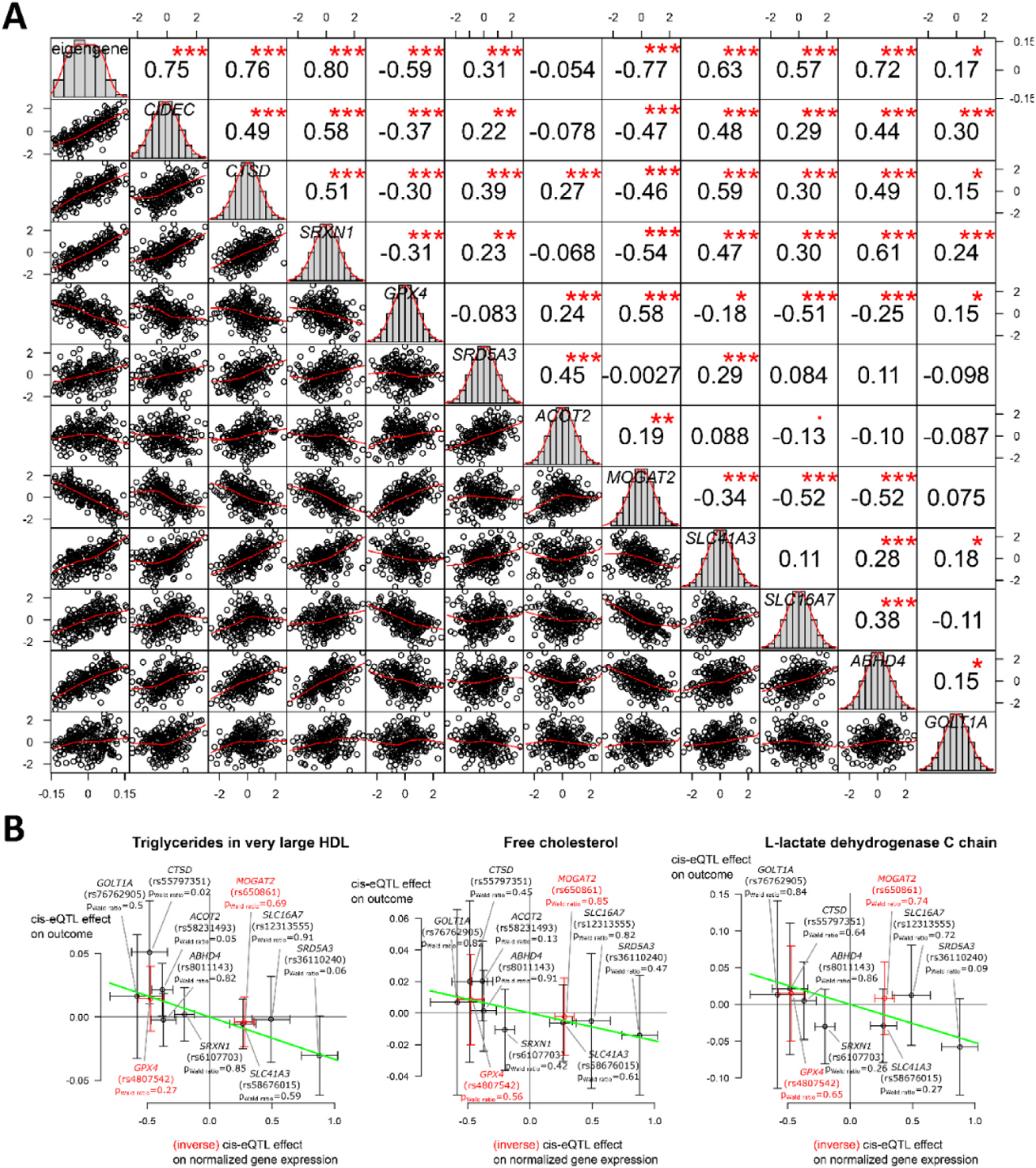
M9 hub genes are linked to lipid metabolism in humans. (A) Correlations between expression of human homologs of 11 M9 hub genes with each other and their eigengene. Diagonal panels show histograms and kernel density estimates of normalized expression in human liver in GTEx data. Panels in the lower triangle show bivariate scatter plots with a LOWESS smoother with locally-weighted polynomial regression as implemented in R’s lowess function. Panels in the upper triangle show Pearson correlation and significance after Benjamini-Hochberg correction (FDRs are denoted by: “no symbol”: > 0.1; “.”:< 0.1; “*”:< 0.05; “**”:< 0.01; “***”:< 0.001). (B) Inverse-variance weighted Mendelian randomization suggests upregulation of M9 genes in humans reduces TG in very large HDL, free cholesterol, and L-lactate dehydrogenase C chain. The horizontal axis shows the effect of the most significant eQTL in liver for each gene on its expression. The vertical axis shows the effects of these eQTLs on the outcomes. P-values for single-SNP Wald ratio tests are given; whiskers are asymptotic 95% confidence intervals. Green slopes denote estimated causal effects: −0.0334, −0.176, and −0.0518 (FDR: 0.04 each).
To assess potentially causative effects of the genes in module M9 on lipid metabolism and weight gain in humans, we used inverse-variance weighed Mendelian randomization to link their liver gene expressions to a set of 249 body weight- and lipid-related traits (Supplementary Data 8) through their most significant GTEx liver eQTLs. Our Mendelian randomization analysis relies on four assumptions: (1) that the 11 genes from part of a gene module in humans, (2) that each of the SNPs affect the expression of this gene module, (3) that the SNPs influence the outcomes only through activation of the module, and (4) that the SNPs are not associated with measured or unmeasured confounders. For each gene g, the most significant cis-eQTL in liver was selected. Fig. 4B shows the point estimates for each gene whereby is shown on the x-axis and on the y-axis.
is a summary statistic for the most significant cis-eQTL of gene g, obtained from GTEx as explained higher. It denotes the average difference in normalized GTEx liver gene expression between the minor and the major allele of the corresponding SNP. It is positive if the expression of gene g is higher in the minor allele and negative if the expression of gene g is lower in the major allele, except for GPX4 and MOGAT2, where we inverted the sign because these genes correlate negatively with both the eigengene and CTSD, the only gene with a significant correlation with all other genes (Fig. 4A). ACOT2 does not significantly correlate with the eigengene, but has a significant positive correlation with CTSD, hence, we opted not to invert its sign.
is a summary statistic obtained from various sources through the “available_outcomes” function from the TwoSampleMR R package [48]. denotes the average difference in outcome between the minor and the major allele of the corresponding SNP. It is positive if the expression of gene g is higher in the minor allele and negative if the expression of gene g is lower in the major allele for all genes, including GPX4 and MOGAT2.
The error bars in the horizontal and vertical directions denote the asymptotic 95% confidence intervals of and , respectively (assuming normality). These were calculated based on their estimated standard errors that were provided as summary statistics together with the point estimates.
CIDEC was excluded from the analysis because its only liver cis-eQTL, rs4582033, was palindromic with intermediate allele frequencies, leaving 10 genes for the analysis. We assumed that increased liver expression for each of these genes is a proxy for increased module activation, except for GPX4 and MOGAT2, whose decreased expressions are assumed to be linked to module activation because of their significant negative correlations with the eigengene and CTSD. Four traits were significant at 5% FDR: “TG in very large HDL,” “Free cholesterol,” “L-lactate dehydrogenase C chain” (negative associations with increased gene expressions in liver (Fig. 4B), as well as “Urinary albumin-to-creatinine ratio” (positive association with gene expressions). The last trait was however only based on eQTLs for ABHD4 and MOGAT2 and this result is therefore less representative for the whole gene set.
4. Discussion
In order to investigate gene-by-diet interactions in BXD strains, we first analyzed the impact of an obesogenic diet on plasma lipid-related phenotypes. HFD exposure significantly increased body weight, adiposity, and plasma lipid concentrations (e.g., cholesterol, HDL, LDL, and TG). In addition, long-term HFD feeding induced fatty liver in a heterogeneous manner, including marked macrovesicular steatosis in some strains, such as BXD214. This phenotypic variability could be largely modulated by genetic factors, as the BXD parental strain B6 is highly susceptible to diet-induced nonalcoholic fatty liver disease (NAFLD) [56,57]. Thus, BXD strains could serve as a reference population to mimic NAFLD susceptibility in natural human populations.
Phenotypic changes in fatty liver presented herein were accompanied by significant changes in the liver transcriptome, resulting in the differential expression of thousands of genes. Those genes were significantly involved in regulating lipid levels through metabolic pathways as highlighted in Table 1. The expression of Cyp2b9, a member of the cytochromes P450 2b family, was most affected by HFD compared to control [58]. Cytochrome P450s are essential for oxidizing steroids, fatty acids, and xenobiotics in liver. RNAi-mediated Cyp2b knockdowns have enlarged livers and increased abdominal fat deposition [59]. Furthermore, Cyp2b-knockdown mice treated intraperitoneally with 100 μL corn oil had elevated plasma TC, TG, HDL, HDL, and very low-density lipoprotein [60]. Additionally, the expression of several other cytochrome P450s genes, such as Cyp1a2, Cyp26a1, Cypa22, and Cyp2b10, changed significantly after HFD in our study, suggesting the cytochrome P450 family plays a significant role in maintenance of lipid homeostasis. The expression of transcripts such as Lcn2, Saa1, and Saa2 were also elevated more than two-fold in DIO mice compared to lean mice. Lcn2, which encodes Lipocalin two, is widely expressed in various tissues, including lung, liver, thymus, kidney, small intestine, adipocytes, and macrophages [61]. Mice carrying null Lcn2 alleles show potentiated DIO, dyslipidemia, fatty liver disease, and insulin resistance [62]. The expression of Lcn2 is considered a potential biomarker for metabolic diseases [63,64]. Saa1 and Saa2 encode the apolipoproteins serum amyloid A1 and A2, respectively. Altered expression of these genes has been associated with inflammatory diseases including atherosclerosis [65]. Saa1- and Saa2-deficient mice have increased levels of circulating TC, but not HDL [66]. We also found several miRNAs whose expression was impacted by diet, including miR-122. miR-122 is the first known lipid metabolism-related miRNA [67] and has been associated with hyperlipidemia [68] and fatty liver [69]. miR-122 inhibition in normal mice resulted in reduced TC levels, increased hepatic fatty-acid oxidation, and a decrease in hepatic fatty-acid and cholesterol synthesis rates [70]. miR-122 inhibition in a DIO mouse model resulted in decreased TC levels and a significant improvement in liver steatosis [70].
However, examining differential expression of individual genes does not fully leverage the power of the BXD family to investigate the genetic basis of disease and phenotype differences, as this approach does not consider how conditions, such as the DIO studied here, disrupt genetic networks [71]. In our study, we used 42 BXD RI strains in two diet groups, where each isogenic strain has a unique genetic background inherited from the parental B6 and D2 strains. This design provides the sample size and genetic variability required for the construction of robust genetic networks using WGCNA, a powerful tool for the identification of gene coexpression networks [40]. After using WGCNA to identify coexpression modules for both CD and HFD groups, we used a variety of tools, including modular specificity analysis, GO, PPI networks, and phenotype correlations, to investigate how these coexpression modules respond to changes in diet and to identify the hub genes that drive these responses.
The coexpression modules identified by WGCNA after HFD indicated strong relationships between gene expression, the quantitative phenotypes investigated here, and biological processes. Of the 18 coexpression modules identified by WGCNA in the HFD expression data, module M9 was identified as an HFD-induced module that may contain some of the keys to elucidating the relationship between changes in liver gene expression and plasma lipid phenotypes after DIO. In the PPI subnetworks constructed from the top 30 M9 module hub genes, Pparg, Cidec, and Cidea have the most connections with other genes. As a sensor of lipid status in a cell, PPARγ regulates the expression of a large number of genes in many physiological and pathological processes, including adipogenesis, inflammation [72], atherosclerosis [73], insulin resistance [74], glucose metabolism [75], lipid metabolism [76,77], and lifespan [78]. Our results showed that 19 out of the top 30 hub genes in M9 are directly regulated by Pparg, suggesting that Pparg could be a master regulator of plasma lipid metabolism in response to diet. In fact, genetic variants in Pparg interact with diet to regulate plasma lipid levels [21]. In addition, transcriptional activation of Pparg in the liver induces the lipogenic program to store fatty acids in lipid droplets. Knockout of the hepatic Pparg gene ameliorates hepatic steatosis induced by diet or genetic manipulations [79]. Hepatic PPARγ expression is robustly induced in NAFLD patients and experimental models [80]. Liver PPARγ regulates triglyceride homeostasis, contributing to hepatic steatosis, but protects other tissues from triglyceride accumulation and insulin resistance [81]. Currently, there are no FDA-approved medications for the treatment of NAFLD, hence further understanding the regulatory factors for PPARγ expression and activity will help develop preventative and therapeutic agents [79,80]. Both Cidea and Cidec belong to the cell death-inducing DNA fragmentation factor-α-like effector (CIDE) family. CIDE are lipid droplet-related proteins which are highly expressed in liver and brown and white adipose tissues. CIDE proteins’ main function is to participate in lipid storage, lipid droplet formation, and lipolysis, which impact the development of obesity, diabetes, and liver steatosis [82–86]. Although several studies have shown that the expression of CIDEC is positively related to PPARγ [87,88], the underlying interaction remains unclear.
In addition to the Pparg subnetwork, we identified an antioxidant subnetwork which contains three genes: Gpx4, Nqo1, and Srxn1. These genes harbor antioxidant response elements (ARE) that are targeted by NRF2 (nuclear factor, erythroid derived two, like two) [52–55]. As a major metabolic tissue, the liver can continuously produce reactive oxygen species which could cause cell damage and lead to various liver diseases such as liver inflammation, alcoholic and nonalcoholic steatohepatitis, fibrosis, and cirrhosis. The progression from inflammation to diseased tissue is highly impacted by oxidative stress [89]. The transcription factor Nrf2 is a master regulator of the oxidative stress pathway that binds to the ARE, an upstream promoter region located in genes with anti-oxidant functions [90,91]. NRF2 inhibits lipid accumulation in mouse liver after an HFD [92] and modulates metabolic diseases such as obesity, type two diabetes mellitus, and atherosclerosis [93]. For example, a potent NRF2-regulated antioxidant defense enzyme, glutathione peroxidase four (GPX4) is essential for reducing hydroperoxides in membrane lipids and lipoproteins [94]. Decreased expression of Gpx4 in mice resulted in increased hepatocyte apoptosis and mitochondrial damage, accompanied with body weight loss and death within 2 weeks [94]. Mice with deficiency of the NRF2-dependent Nqo1, a member of the NAD(P)H dehydrogenase family, have decreased plasma glucose, and increased TG levels [95]. Last, the NRF2-dependent sulfiredoxin (Srxn1) is associated with oxidative stress but is less well studied.
To more fully exploit the potential of the BXD RI family, we created a Bayesian network model that describes how M9 module genes may link genotype and phenotype (Fig. 3). Such models are based on the idea that the genetic variation within a diverse population, such as BXD mice, naturally provides perturbations that can be used to infer causal relationships between quantitative traits such as gene expression and phenotypes [96]. These relationships can be learned and represented using Bayesian networks, probabilistic graphical models in which a group variables are linked by directed edges that indicate dependencies between the variables [97]. We trained a Bayesian network here after identifying several M9 module genes that were located near a QTL (rs26890724 on Chr 11) that regulated two plasma lipid-related phenotypes (body weight and perirenal white adipose mass). Thus, the Bayesian network model integrated three levels of data (genotypes, gene expression, and higher order phenotypes) and indicated several interesting relationships about the pathway linking genotype and phenotype. First, the genotype at this locus directly influenced the expression of Rab34 and LOC728392; the other M9 module genes in the network depended on the expression of these two genes. Second, Tnfaip1 was the M9 module gene near the QTL that had the most direct impact on the phenotypes. Third, body weight depended on perirenal white adipose mass and was not otherwise directly influenced by the QTL genotype or other M9 module genes in the network. Finally, the Bayesian network model suggested that the influence of the QTL genotype on perirenal white adipose mass was not entirely explained by the expression of the five M9 module genes located near the QTL; thus, factors that were not included in this network model are likely involved in the molecular pathway linking this genotype and phenotype such as visceral white adipose depots or brown fat. By mapping the top genes of module M9 to their human counterparts, we were able to demonstrate that the expressions of these genes also tend to correlate in human livers, which suggests that module M9 might be conserved in humans. With inverse-variance weighted Mendelian randomization, we showed that the activation of such a module in the liver is causally linked to decreased TG in very large HDL, decreased free cholesterol, and decreased L-lactate dehydrogenase C chain in blood. This indicates that activating our module might reduce cholesterol in humans and thus forms an interesting target against cardiovascular disease.
In summary, by applying systems genetics analysis, we provide evidence that plasma lipid metabolism is significantly regulated by both HFD, which is well known, but herein we have identified key functional gene networks that provide novel insight into lipid regulation that was previously unappreciated. One of the key findings of the study is that, using the power of the BXD RI family, we uncovered a plasma lipid related gene coexpression module that was induced by DIO. Although further functional analysis is needed to understand the biological mechanisms behind the module, our findings could be beneficial for clinical diagnosis and prevention. The study highlights the benefit of integrating differential expression, gene coexpression networks, analysis of module hub genes, and Bayesian networks in investigations of the impact of environment on gene expression and treatment. Such an integrated analysis could be applied to the investigation of other phenotypes and diseases to examine the changes in gene expression and gene networks that underlie links between variations in genotypes and phenotypes.
Supplementary Material
Funding
This work was supported by grants from the École Polytechnique Fédérale de Lausanne (EPFL), the European Research Council (AdG-787702), the Swiss National Science Foundation (SNSF 31003A-179435), and the NIH R01AG043930 to JA; NIH R01DK120567 to LL and LDQ; CITG fund from UTHSC to RWW; American Heart Association 19TPA34910232 and R01CA253329, to LM; R01CA262112 to LM, RWW, and LL; the Swiss Government Excellence Scholarship (FCS ESKAS-Nr. 2019.0009) to LJEG; the Binzhou Medical University Research Start-up Fund (50012305190) to FX. We also thank Dr. Robert Read for his histologic work of livers.
Abbreviations:
- CD
Control diet
- DIO
Diet induced obesity
- DEGs
Differentially expressed genes
- FDR
False discovery rate
- FFA
Free fatty acid
- GWAS
Genome-wide association studies
- GTEx
Genotype-Tissue Expression
- GO
Gene Ontology
- HFD
High-fat diet
- HDL
High-density lipoprotein cholesterol
- LDL
Low-density lipoprotein cholesterol
- MPO
Mammalian phenotype ontology
- QTL
Quantitative trait locus
- TG
Triglycerides
- TC
Total cholesterol
- WGCNA
Weighted Gene Coexpression Network Analysis
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Ethical approval
All animal experiments in this study were approved by the Swiss cantonal veterinary authorities of Vaud under licenses 2257.0 and 2257.1 and the UTHSC animal care and use committee.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jnutbio.2023.109398.
Data availability
Both phenotype and liver transcriptome data of the BXD mice used in this study were uploaded to our GeneNetwork (http://www.genenetwork.org) and systems-genetics (https://www.systems-genetics.org/) websites [33,34].
The phenotype data can be accessed with the following GN accession number for HFD: 17,562, 17,572, 17,576, 17,602, 17,604, 17,788, 17,790, 17,800, 17,802, 17,804, 17,806, 17,808, 17,810, and 17,812; and for CD: 17,561, 17,571, 17,575, 17,601, 17,787, 17,789, 17,799, 17,801, 17,803, 17,807, 17,809, and 17,811.
Raw microarray data is available on GEO (https://www.ncbi.nlm.nih.gov/geo/)) under the identifier GSE60149. The normalized data is available on GeneNetwork under the “BXD” group and “Liver mRNA” type with the identifier “EPFL/LISP BXD CD Liver Affy Mouse Gene 1.0 ST (August 18) RMA” for the control diet cohort, and “EPFL/LISP BXD HFD Liver Affy Mouse Gene 1.0 ST (Aug18) RMA” for the high-fat diet cohort.
References
- [1].Aguilera CM, Gil-Campos M, Canete R, Gil A. Alterations in plasma and tissue lipids associated with obesity and metabolic syndrome. Clinical Science 2008;114(3):183–93. [DOI] [PubMed] [Google Scholar]
- [2].Haase CL, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. HDL cholesterol and risk of type 2 diabetes: a Mendelian randomization study. Diabetes 2015;64(9):3328–33. [DOI] [PubMed] [Google Scholar]
- [3].Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Current Cardiology Reports 2011;13(6):544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: process, indicators, risk factors and new hopes. International Journal of Preventive Medicine 2014;5(8):927. [PMC free article] [PubMed] [Google Scholar]
- [5].Kannel WB, Castelli WP, Gordon T, McNamara PM. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. Ann Intern Med 1971;74(1):1–12. [DOI] [PubMed] [Google Scholar]
- [6].Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. The Lancet 2014;384(9943):626–35. [DOI] [PubMed] [Google Scholar]
- [7].Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease: a scientific statement from the. American Heart Association. Circulation 2011;123(20):2292–333. [DOI] [PubMed] [Google Scholar]
- [8].Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nature genetics 2013;45(11):1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Benjamin EJ, Muntner P, Bittencourt MS. Heart disease and stroke statistics-2019 update: a report from the. American Heart Association. Circulation 2019;139(10):e56–e528. [DOI] [PubMed] [Google Scholar]
- [10].Dron JS, Hegele RA. Genetics of lipid and lipoprotein disorders and traits. Current Genetic Medicine Reports 2016;4(3):130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466(7307):707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nature Genetics 2013;45(11):1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, et al. Exome-wide association study of plasma lipids in>300,000 individuals. Nature Genetics 2017;49(12):1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, et al. Genetics of blood lipids among~ 300,000 multi-ethnic participants of the Million Veteran Program. Nature Genetics 2018;50(11):1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jha P, McDevitt MT, Gupta R, Quiros PM, Williams EG, Gariani K, et al. Systems analyses reveal physiological roles and genetic regulators of liver lipid species. Cell Systems 2018;6(6) 722–733. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jha P, McDevitt MT, Halilbasic E, Williams EG, Quiros PM, Gariani K, et al. Genetic regulation of plasma lipid species and their association with metabolic phenotypes. Cell Systems 2018;6(6) 709–721. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shimoyama M, De Pons J, Hayman GT, Laulederkind SJ, Liu W, Nigam R, et al. The rat genome database 2015: genomic, phenotypic and environmental variations and disease. Nucleic Acids Research 2014;43(D1):D743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proceedings of the National Academy of Sciences 2012;109(4):1193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ober C, Vercelli D. Gene–environment interactions in human disease: nuisance or opportunity? Trends in Genetics 2011;27(3):107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Twins Kaprio J. and the mystery of missing heritability: the contribution of gene–environment interactions. Journal of Internal Medicine 2012;272(5):440–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rudkowska1 I, Vohl M-C. Interaction between diets, polymorphisms and plasma lipid levels. Clinical Lipidology 2010;5(3):421–38. [Google Scholar]
- [22].Lusis AJ, Seldin MM, Allayee H, Bennett BJ, Civelek M, Davis RC, et al. The hybrid mouse diversity panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. Journal of Lipid Research 2016;57(6):925–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, et al. A platform for experimental precision medicine: The extended BXD mouse family. Cell Systems 2021;12(3):235–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Williams RWWEG. Resources systems genetics. Methods Mol Bio 2017;1488:3–29. [DOI] [PubMed] [Google Scholar]
- [25].Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 2004;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].De la Fuente A From ‘differential expression’to ‘differential networking’–identification of dysfunctional regulatory networks in diseases. Trends in Genetics 2010;26(7):326–33. [DOI] [PubMed] [Google Scholar]
- [27].Tieri P, Farina L, Petti M, Astolfi L, Paci P, Castiglione F et al. , Network inference and reconstruction in Bioinformatics. 2019: p. 805–813. [Google Scholar]
- [28].Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical Applications in Genetics and Molecular Biology 2005;4(1). [DOI] [PubMed] [Google Scholar]
- [29].Miao L, Yin RX, Pan SL, Yang S, Yang D-Z, Lin W-X. Weighted gene co-expression network analysis identifies specific modules and hub genes related to hyperlipidemia. Cellular Physiology and Biochemistry 2018;48(3):1151–63. [DOI] [PubMed] [Google Scholar]
- [30].Miao L, Yin RX, Pan SL, Yang S, Yang D-Z, Lin W-X. Circulating miR-3659 may be a potential biomarker of dyslipidemia in patients with obesity. Journal of Translational Medicine 2019;17(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Langfelder P, Castellani LW, Zhou Z, Paul E, Davis R, Schadt EE, et al. A systems genetic analysis of high density lipoprotein metabolism and network preservation across mouse models. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 2012;1821(3):435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Deshpande V, Sharma A, Mukhopadhyay R, Thota LNR, Ghatge M, Vangala RK, et al. Understanding the progression of atherosclerosis through gene profiling and co-expression network analysis in Apobtm2SgyLdlrtm1Her double knockout mice. Genomics 2016;107(6):239–47. [DOI] [PubMed] [Google Scholar]
- [33].Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014;158(6):1415–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, et al. Systems proteomics of liver mitochondria function. Science 2016;352(6291):aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003;19(2):185–93. [DOI] [PubMed] [Google Scholar]
- [36].Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nature Genetics 2005;37(3):233. [DOI] [PubMed] [Google Scholar]
- [37].Smyth GK. Limma: linear models for microarray data. In: Bioinformatics and computational biology solutions using R and Bioconductor. New York: Springer; 2005. p. 397–420. [Google Scholar]
- [38].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 1995;57(1):289–300. [Google Scholar]
- [39].Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Research 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9(1):559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 2018;47(D1):D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS catalog). Nucleic Acids Research 2016;45(D1):D896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nature Genetics 2012;44(7):821–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ziebarth JD, Bhattacharya A, Cui Y. Bayesian Network Webserver: a comprehensive tool for biological network modeling. Bioinformatics 2013;29(21):2801–3. [DOI] [PubMed] [Google Scholar]
- [45].Ziebarth JD, Cui Y. Precise network modeling of systems genetics data using the Bayesian network webserver. Methods in Molecular Biology 2017;1488:319–35. [DOI] [PubMed] [Google Scholar]
- [46].Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype--tissue expression (GTEx) project. Nature Genetics 2013;45(6):580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 2016;32(10):1479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genetic Epidemiology 2013;37(7):658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dehmer M, Emmert-Streib F, Graber A, Salvador A. Applied Statistics for network biology. Weinheim: Wiley Online Library; 2011. [Google Scholar]
- [51].Fang L, Zhang M, Li Y, Liu Y, Cui Q, Wang N. PPARgene: a database of experimentally verified and computationally predicted PPAR target genes. PPAR Research 2016:2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Raghunath A, Sundarraj K, Nagarajan R, Arfuso F, Bian J, Kumar AP, et al. Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biology 2018;17:297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aleksunes LM, Slitt AL, Maher JM, Dieter MZ, Knight TR, Goedken M, et al. Nuclear factor-E2-related factor 2 expression in liver is critical for induction of NAD (P) H: quinone oxidoreductase 1 during cholestasis. Cell Stress and Chaperones 2006;11(4):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhou Y, Duan S, Zhou Y, Yu S, Wu J, Wu X, et al. Sulfiredoxin-1 attenuates oxidative stress via Nrf2/ARE pathway and 2-Cys Prdxs after oxygen-glucose deprivation in astrocytes. Journal of Molecular Neuroscience 2015;55(4):941–50. [DOI] [PubMed] [Google Scholar]
- [55].Shin D, Kim EH, Lee J, Roh J-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radical Biology and Medicine 2018;129:454–62. [DOI] [PubMed] [Google Scholar]
- [56].Velázquez KT, Enos RT, Bader JE, Sougiannis AT, Carson MS, Chatzistamou I, et al. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World Journal of Hepatology 2019;11(8):619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Van Herck M, Vonghia L, Francque S. Animal models of nonalcoholic fatty liver disease—a starter’s guide. Nutrients 2017;9(10):1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Leung A, Trac C, Du J, Natarajan R, Schones DE. Persistent chromatin modifications induced by high fat diet. Journal of Biological Chemistry 2016;291(20):10446–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Damiri B, Holle E, Yu X, Baldwin WS. Lentiviral-mediated RNAi knockdown yields a novel mouse model for studying Cyp2b function. Toxicological Sciences 2011;125(2):368–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Damiri B, Baldwin WS. Cyp2b-knockdown mice poorly metabolize corn oil and are age-dependent obese. Lipids 2018;53(9):871–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Friedl A, Stoesz S, Buckley P, Gould M. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. The Histochemical Journal 1999;31(7):433–41. [DOI] [PubMed] [Google Scholar]
- [62].Guo H, Jin D, Zhang Y, Wright W, Bazuine M, Brockman DA, et al. Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes 2010;59(6):1376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Asimakopoulou A, Weiskirchen S, Weiskirchen R. Lipocalin 2 (LCN2) expression in hepatic malfunction and therapy. Frontiers in Physiology 2016;7:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Abella V, Scotece M, Conde J, Gómez R, Lois A, Pino J, et al. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 2015;20(8):565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].O’Brien KD, Chait A. Serum amyloid A: the “other” inflammatory protein. Current Atherosclerosis Reports 2006;8(1):62–8. [DOI] [PubMed] [Google Scholar]
- [66].De Beer MC, Webb NR, Wroblewski JM, Noffsinger VP, Rateri DL, Ji A, et al. Impact of serum amyloid A on high density lipoprotein composition and levels. Journal of Lipid Research 2010;51(11):3117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ouimet M, Moore KJ. A big role for small RNAs in HDL homeostasis. Journal of Lipid Research 2013;54(5):1161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gao W, He H-W, Wang Z-M, Zhao H, Lian X-Q, Wang Y-S, et al. Plasma levels of lipometabolism-related miR-122 and miR-370 are increased in patients with hyperlipidemia and associated with coronary artery disease. Lipids in Health and Disease 2012;11(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Raitoharju E, Seppälä I, Lyytikäinen L-P, Viikari J, Ala-Korpela M, Soininen P, et al. Blood hsa-miR-122–5p and hsa-miR-885–5p levels associate with fatty liver and related lipoprotein metabolism—The Young Finns Study. Scientific Reports 2016;6:38262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabolism 2006;3(2):87–98. [DOI] [PubMed] [Google Scholar]
- [71].Li J, Zhou D, Qiu W, Shi Y, Yang J-J, Chen S, et al. Application of weighted gene co-expression network analysis for data from paired design. Scientific Reports 2018;8(1):622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends in Immunology 2007;28(12):551–8. [DOI] [PubMed] [Google Scholar]
- [73].Duval C, Chinetti G, Trottein F, Fruchart J-C, Staels B. The role of PPARs in atherosclerosis. Trends in Molecular Medicine 2002;8(9):422–30. [DOI] [PubMed] [Google Scholar]
- [74].Moller D, Berger J. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. International Journal of Obesity 2003;27(S3):S17. [DOI] [PubMed] [Google Scholar]
- [75].Ruchat S-M, Rankinen T, Weisnagel S, Rice T, Rao D, Bergman R, et al. Improvements in glucose homeostasis in response to regular exercise are influenced by the PPARG Pro12Ala variant: results from the HERITAGE Family Study. Diabetologia 2010;53(4):679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Heikkinen S, Argmann C, Feige JN, Koutnikova H, Champy M-F, Dali-Youcef N, et al. The Pro12Ala PPARγ 2 variant determines metabolism at the gene-environment interface. Cell Metabolism 2009;9(1):88–98. [DOI] [PubMed] [Google Scholar]
- [77].Freedman BD, Lee E-J, Park Y, Jameson JL. A dominant negative peroxisome proliferator-activated receptor-γ knock-in mouse exhibits features of the metabolic syndrome. Journal of Biological Chemistry 2005;280(17):17118–25. [DOI] [PubMed] [Google Scholar]
- [78].Argmann C, Dobrin R, Heikkinen S, Auburtin A, Pouilly L, Cock T-A, et al. Pparγ 2 is a key driver of longevity in the mouse. PLoS Genetics 2009;5(12):e1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lee YK, Park JE, Lee M, Hardwick JP. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Research 2018;2(4):209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liss KH, Finck BN. PPARs and nonalcoholic fatty liver disease. Biochimie 2017;136:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator-activated receptor γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. Journal of Biological Chemistry 2003;278(36):34268–76. [DOI] [PubMed] [Google Scholar]
- [82].Gong J, Sun Z, Li P. CIDE proteins and metabolic disorders. Current Opinion in Lipidology 2009;20(2):121–6. [DOI] [PubMed] [Google Scholar]
- [83].Tanaka N, Takahashi S, Matsubara T, Jiang C, Sakamoto W, Chanturiya T, et al. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. Journal of Biological Chemistry 2015;290(5):3092–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J, et al. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PloS one 2008;3(8):e2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. The Journal of Clinical Investigation 2008;118(8):2808–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhou Z, Toh SY, Chen Z, Guo K, Ng CP, Ponniah S, et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nature Genetics 2003;35(1):49. [DOI] [PubMed] [Google Scholar]
- [87].Magnusson B, Gummesson A, Glad CA, Goedecke JH, Jernås M, Lystig TC, et al. Cell death–inducing DFF45-like effector C is reduced by caloric restriction and regulates adipocyte lipid metabolism. Metabolism 2008;57(9):1307–13. [DOI] [PubMed] [Google Scholar]
- [88].Karbowska J, Kochan Z. Intermittent fasting up-regulates Fsp27/Cidec gene expression in white adipose tissue. Nutrition 2012;28(3):294–9. [DOI] [PubMed] [Google Scholar]
- [89].Zhu R, Wang Y, Zhang L, Guo Q. Oxidative stress and liver disease. Hepatology Research 2012;42(8):741–9. [DOI] [PubMed] [Google Scholar]
- [90].Vomund S, Schäfer A, Parnham M, Brüne B, von Knethen A. Nrf2, the master regulator of anti-oxidative responses. International Journal of Molecular Sciences 2017;18(12):2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhang Q, Pi J, Woods CG, Andersen ME. A systems biology perspective on Nrf2-mediated antioxidant response. Toxicology and Applied Pharmacology 2010;244(1):84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, et al. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. Journal of Pharmacology and Experimental Therapeutics 2008;325(2):655–64. [DOI] [PubMed] [Google Scholar]
- [93].da Costa RM, Rodrigues D, Pereira CA, Silva JF, Alves JV, Lobato NS, et al. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Frontiers in Pharmacology 2019;10:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yoo S-E, Chen L, Na R, Liu Y, Rios C, Van Remmen H, et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radical Biology and Medicine 2012;52(9):1820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gaikwad A, Long DJ, Stringer JL, Jaiswal AK. In vivo role of NAD (P) H: quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. Journal of Biological Chemistry 2001;276(25):22559–64. [DOI] [PubMed] [Google Scholar]
- [96].Rockman MV. Reverse engineering the genotype–phenotype map with natural genetic variation. Nature 2008;456(7223):738–44. [DOI] [PubMed] [Google Scholar]
- [97].Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. A primer on learning in Bayesian networks for computational biology. PLoS Computational Biology 2007;3(8):e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Both phenotype and liver transcriptome data of the BXD mice used in this study were uploaded to our GeneNetwork (http://www.genenetwork.org) and systems-genetics (https://www.systems-genetics.org/) websites [33,34].
The phenotype data can be accessed with the following GN accession number for HFD: 17,562, 17,572, 17,576, 17,602, 17,604, 17,788, 17,790, 17,800, 17,802, 17,804, 17,806, 17,808, 17,810, and 17,812; and for CD: 17,561, 17,571, 17,575, 17,601, 17,787, 17,789, 17,799, 17,801, 17,803, 17,807, 17,809, and 17,811.
Raw microarray data is available on GEO (https://www.ncbi.nlm.nih.gov/geo/)) under the identifier GSE60149. The normalized data is available on GeneNetwork under the “BXD” group and “Liver mRNA” type with the identifier “EPFL/LISP BXD CD Liver Affy Mouse Gene 1.0 ST (August 18) RMA” for the control diet cohort, and “EPFL/LISP BXD HFD Liver Affy Mouse Gene 1.0 ST (Aug18) RMA” for the high-fat diet cohort.