

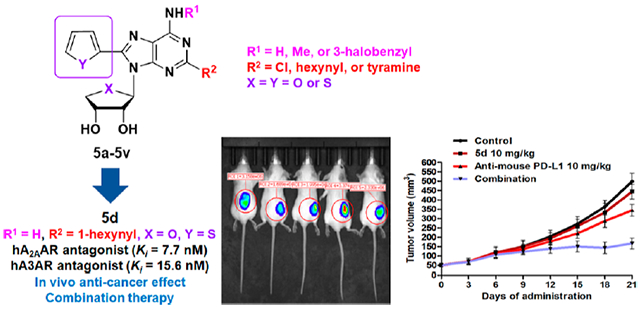
Abstract
Based on hA2AAR structures, a hydrophobic C8-heteroaromatic ring in 5′-truncated adenosine analogues occupies the subpocket tightly, converting hA2AAR agonists into antagonists while maintaining affinity toward hA3AR. The final compounds of 2,8-disubstituted-N6-substituted 4′-thionucleosides, or 4′-oxo, were synthesized from d-mannose and d-erythrono-1,4-lactone, respectively, using a Pd-catalyst-controlled regioselective cross-coupling reaction. All tested compounds completely antagonized hA2AAR, including 5d with the highest affinity (Ki,A2A = 7.7 ± 0.5 nM). The hA2AAR–5d X-ray structure revealed that C8-heteroaromatic rings prevented receptor activation-associated conformational changes. However, the C8-substituted compounds still antagonized hA3AR. Structural SAR features and docking studies supported different binding modes at A2AAR and A3AR, elucidating pharmacophores for receptor activation and selectivity. Favorable pharmacokinetics were demonstrated, in which 5d displayed high oral absorption, moderate half-life, and bioavailability. Also, 5d significantly improved the antitumor effect of anti-PD-L1 in vivo. Overall, this study suggests that the novel dual A2AAR/A3AR nucleoside antagonists would be promising drug candidates for immune-oncology.
Graphical Abstract
INTRODUCTION
Although there has been remarkable progress in anticancer therapy, cancer remains the main life-threatening disease.1 Recently, immuno-oncology agents emerged as the next generation of antitumor drugs for immune checkpoint inhibition.2 Among them, monoclonal antibodies (mAbs) have been intensively studied as immune checkpoint inhibitors.3 They block costimulatory molecules on immune cells and inhibit immune-evasion of a tumor, resulting in the reactivation of the immune system. However, mAb immune checkpoint inhibitors have some limitations such as their high cost, prolonged half-life, immune-related adverse effects, and immunogenicity.4 To overcome these drawbacks, the development of small molecules as immuno-oncology agents has been highly appealing. A number of target proteins such as toll-like receptors (TLRs), stimulators of interferon genes (STING), adenosine receptors (ARs), and so on have been investigated as the principal targets for the development of small molecule immune-oncology agents.5
ARs consist of four subtypes termed A1, A2A, A2B, and A3. Among these, A2AAR and A2BAR couple to GS protein, which activates adenylate cyclase (AC) to increase the level of cyclic AMP (cAMP), while A1AR and A3AR bind to Gi protein, inhibiting the level of cAMP. In a tumor microenvironment (TME), ecto-5′-nucleotidase (CD73) on the cell surface catalyzes the hydrolysis of extracellular adenosine-5′-monophosphate (AMP) to adenosine and phosphate, resulting in high adenosine concentrations. When adenosine is released in the tumor microenvironment, it can activate any of the four AR subtypes to control cAMP levels.6 Notably, adenosine activating the A2AAR, which is overexpressed in effector T cells and then blocks T cell receptor (TCR) signaling, abolishes the immune response.7 In addition, A2AAR also upregulates the expression of negative costimulatory molecules such as programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA4), stimulating immune evasion.8 Thus, if A2AAR is blocked, immune checkpoints by CTLA4, PD-1, or PD-L1 can be inhibited in cancer cells, giving A2AAR antagonists cancer immunotherapeutic potential. Furthermore, an A2AAR antagonist is expected to show a synergistic effect in combination with mAb immune checkpoint inhibitors.9 In fact, most of the A2AAR antagonists in clinical trials as cancer immunotherapeutic agents, are coadministered with a mAb immune checkpoint inhibitor as combination therapy.10
Based on a potent and selective A3AR agonist, 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (Cl-IB-MECA, 1a),13 we have long been interested in discovering new AR ligands with the 4′-thionucleoside skeleton. Among these, 2-chloro-N6-(3-iodobenzyl)-4′-thioadenosine-5′-N-methyluronamide (thio-Cl-IB-MECA, 1b)14 was discovered to be a more potent human (h) A3AR agonist (Ki = 0.38 ± 0.07 nM) than 1a (Ki = 1.4 ± 0.3 nM) (Chart 1). Truncation of the 5′-uronamide of 1a and 1b converted selective A3AR agonists 1a and 1b into A3AR antagonists 2a (Ki = 42.9 ± 8.9 nM) and 2b (Ki = 4.16 ± 0.5 nM).15 From this study, the amide proton of the 5′-uronamide group of 1a and 1b acted as a hydrogen-bonding donor, which was essential for the induced fit in the receptor required for A3AR agonism. Compound 2b also showed high binding affinity (Ki = 3.89 ± 1.15 nM) at the rat A3AR, indicating that it is a species-independent A3AR antagonist. These truncated nucleosides 2a and 2b were the first examples to show pure A3AR antagonism with species-independent and high binding affinity with a nucleoside skeleton,15 potentially for treatment of chronic kidney disease (CKD)16 and cancer.17 Further SAR study of 2a and 2b at the C2 and N6-positions resulted in the discovery of A2AAR agonists 3a (Ki = 63.2 ± 15 nM) and 3b (Ki = 7.19 ± 0.6 nM), in which an extended hexynyl group at the C2 position and a free amino group at the N6-position were found to act as essential pharmacophores for A2AAR binding, although they also maintained the antagonistic activity at the hA3AR.18
Chart 1.
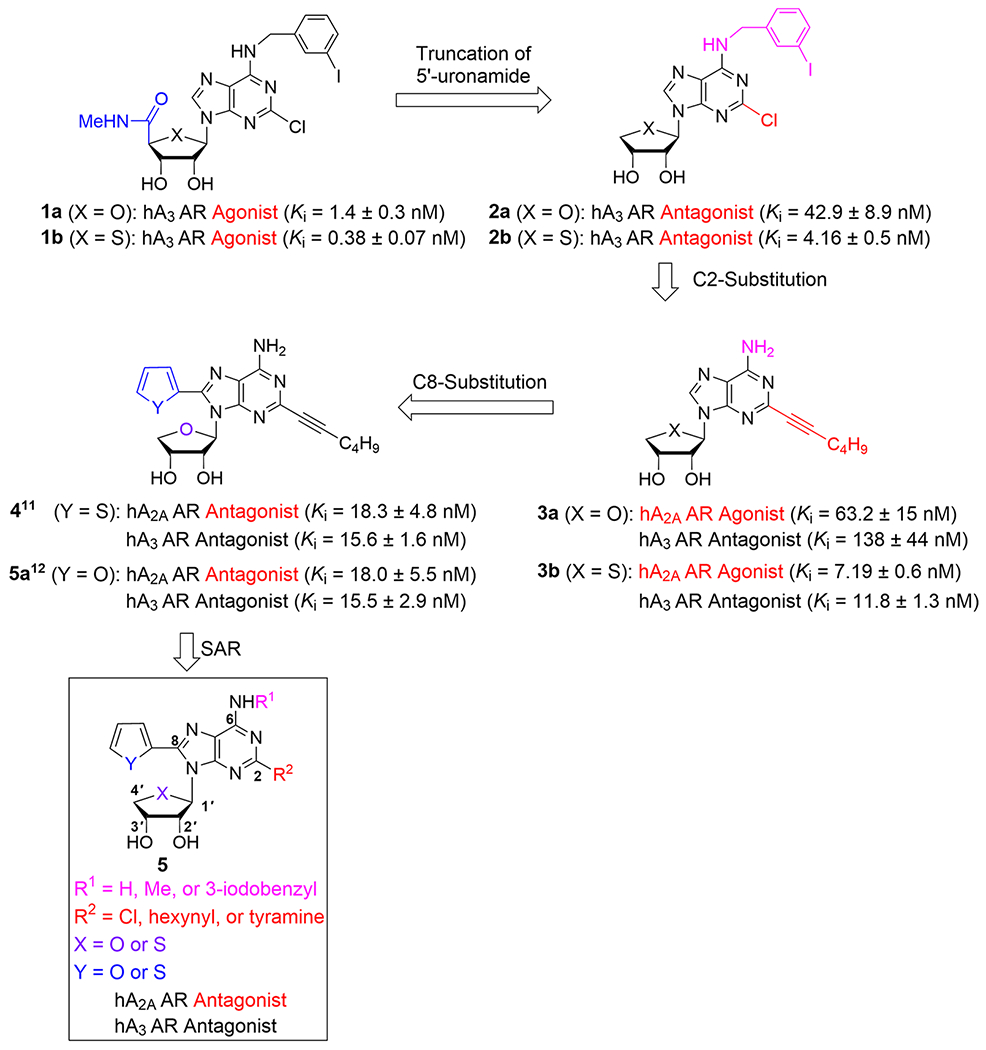
Rationale for the Design of the Target Nucleosides 5
In order to switch compound 3 to an A2AAR antagonist, we analyzed agonist- and antagonist-bound co-crystal structures of the A2AAR. As shown in Figure 1, A2AAR antagonist (ZM-241385) occupied a subpocket which was expanded by His2506.52/A2A (Figure 1A, His2506.52/A2A; Ballesteros–Weinstein residue numbering and name of the receptor are referred to as superscripts.19). Also, the predicted binding mode of 3a with hA2AAR indicated that the C8 position is located in a spacious subpocket capable of accommodating a bulky substituent (Figure 1B, yellow). Thus, if we introduce an aromatic ring at the C8 position of 3a, it was hypothesized that it induces tight binding rather than an induced fit required for receptor activation, anticipating the conversion of A2AAR agonist 3b into A2AAR antagonist 4. As expected, compound 4 was found to be a full A2AAR antagonist (Ki = 18.3 ± 4.8 nM). Recently, we reported the X-ray co-crystal structure complexed with A2AAR antagonist 4,11 confirming our hypothesis that the C8 aromatic ring of 3a tightly binds to the subpocket and serves as the key pharmacophoric feature to discriminate between agonist and antagonist.18,20 Most known A2AAR agonists possess a nucleoside structure, while all known A2AAR antagonists have heterocyclic compounds without a sugar ring.10 These results indicate that both 2′–OH and 3′–OH groups have a pivotal role for A2AAR activation but are not essential for an inactive A2AAR state.20 Namely, despite possessing a nucleoside skeleton containing both 2′–OH and 3′–OH, the compounds of this research exhibited potent A2AAR antagonism, and it is believed to be the first example.11
Figure 1.
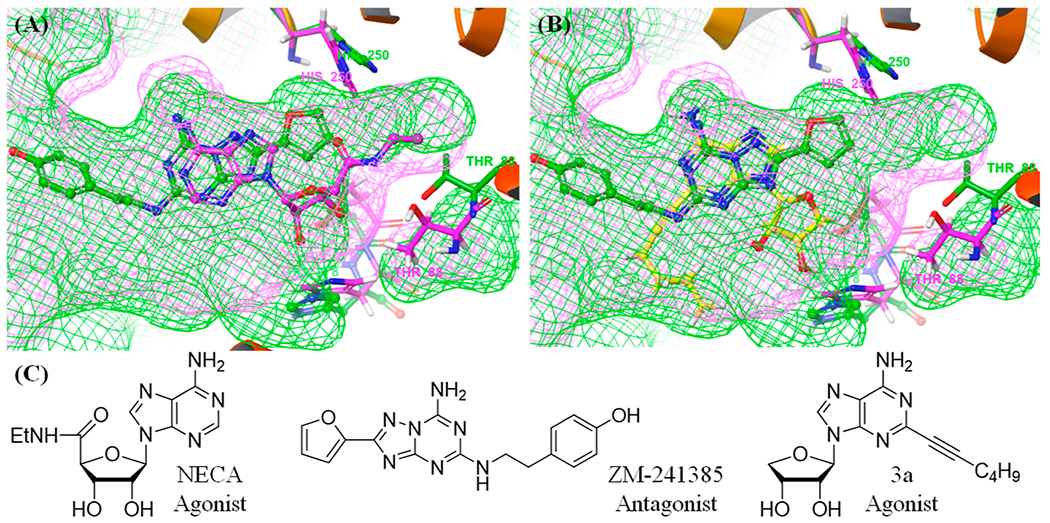
X-ray co-crystal structure of hA2AAR containing NECA (pink, PDB ID: 2YDV) and ZM-241385 (green, PDB ID: 3EML). (A) Comparison of the binding pocket for the co-crystal structures and its subpocket. (B) Predicted binding mode of 3a with hA2AAR (yellow, PDB ID: 3EML). (C) Chemical structures of the ligands.
Based on these findings, we carried out a comprehensive SAR study at the C2-, C8-, and N6-positions of 4, synthesized the truncated 4′-oxonucleoside derivatives 5a–f and measured their A2AAR binding affinities.20 Also, we synthesized the corresponding truncated 4′-thionucleosides 5g–v, based on a bioisosteric relationship between oxygen and sulfur, and evaluated them for binding affinities of the four adenosine receptor subtypes.12
Herein, we report the design, synthesis, and binding affinity of the final nucleosides 5, modified at the C2, C8, and N6-positions. We also report the pharmacokinetic characterization and in vivo antitumor effects of the most potent A2AAR antagonist in the series for its development as an immuno-oncology agent.
RESULTS AND DISCUSSION
Chemistry.
The 4′-oxonucleoside derivatives 5a–f were synthesized from d-erythrono-1,4-lactone, as shown in Scheme 1. d-Erythrono-1,4-lactone was protected as 2,3-O-acetonide under the standard conditions, which was reduced with diisobutylaluminum hydride (DIBAL-H) to afford lactol 6.15 Acetylation of lactol 6 afforded the glycosyl donor 7.15c Condensation of 7 with silylated 6-chloropurine under Vorbrüggen21 conditions in the presence of trimethylsilyl trifluoromethansulfonate (TMSOTf) as a Lewis acid yielded the protected β-nucleoside 8 as a single stereoisomer. Treatment of 8 with 1 N HCl followed by the protection of resulting diol with the t-butyldimethylsilyl (TBS) group afforded the di-O-TBS ether, which was subjected to the iodination22 at C2 and C8 positions using a freshly prepared lithium tetramethylpiperidide (LiTMP) to produce 6-chloro-2,8-diiodo-purine derivative 9. Palladium catalyst-controlled regioselective Sonogashira22–24 coupling of 9 with 1-hexyne in the presence of tetrakis(triphenylphosphine)palladium and cesium carbonate yielded C2-hexynyl derivative 10 as the major regioisomer.23 Then, another palladium-catalyzed Stille25 coupling of 10 with 2-tributylstannylfuran and–thiophene yielded the C8-furyl derivative 11a and the C8-thienyl derivative 11b, respectively, after desilylation. The regio- and stereochemistry of 11b were confirmed by X-ray crystallography (Scheme 1, please see the Supporting Information for the X-ray crystallographic details of the compound 11b). Compounds 11a and 11b were treated with ammonia, methylamine, and 3-iodobenzylamine to afford 5a–c and 5d–f, respectively.
Scheme 1. Synthesis of Truncated 2-Hexynyl-N6,8-Disubstituted-Adenosines 5a–fa.
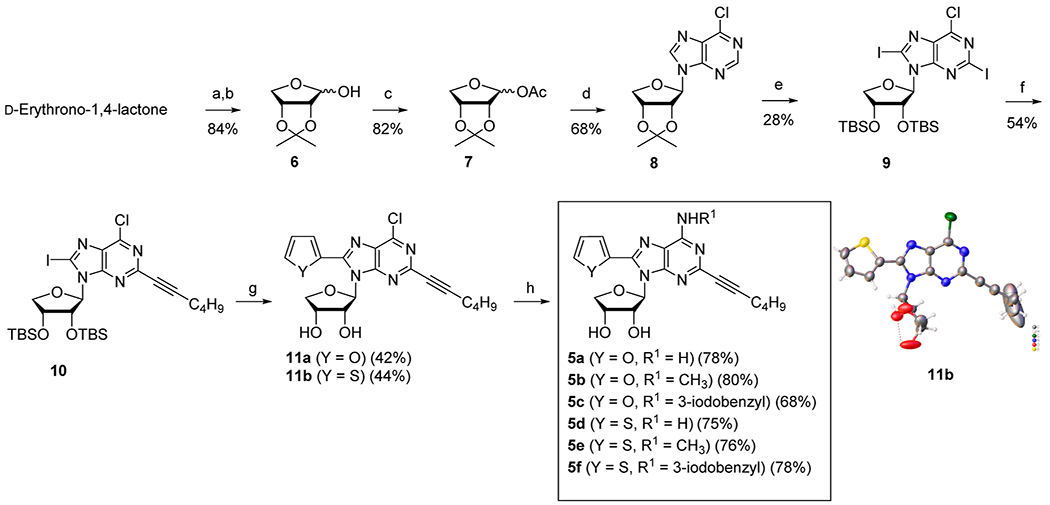
aReagents and conditions: (a) pTSA, 2,2-dimethoxypropane, DMF, 4 h, reflux; (b) DIBAL-H, toluene, −78 °C, 30 min; (c) AC2O, pyridine, rt, 3 h; (d) 6-chloropurine, HMDS, (NH4)2SO4, TMSOTf, 1,2-dichloroethane, 0 to 80 °C, 15 h; (e) (i) 1 N HCl, THF, rt, 15 h; (ii) TBSCl, imidazole, DMF, rt, 15 h; (iii) tetramethylpiperidine, I2, n-BuLi, THF, −78 °C, 4 h; (f) Pd(PPh3)4, Cs2CO3, CuI, 1-hexyne, DMF, rt, 5 h; (g) (i) Pd(PPh3)2Cl2, 2-tributylstannylfuran, THF, 70 °C, 1 h for 11a or Pd(PPh3)2Cl2, 2-tributylstannylthiophene, THF, 60 °C, 1 h for 11b; (ii) Et3N, Et3N·3HF, THF, rt, 15 h; (h) NH3/t-BuOH, 100 °C, 12 h for 5a, 5d and CH3NH2·HCl, Et3N, EtOH, rt, 24 h for 5b, 5e, and 3-iodobenzylamine·HCl, Et3N, EtOH, rt, 24 h for 5c and 5f.
Next, we synthesized the corresponding 4′-thionucleoside derivatives 5g–v, based on the bioisosteric relationship between oxygen and sulfur. The synthetic strategy to the 4′-thionucleoside derivatives 5g–v was to condense the glycosyl donor 16 with 6-chloro-2,8-diiodopurine 17d or 2,6-dichloro-8-iodopurine 19 under Vorbrüggen21 conditions and then apply regioselective C8-Stille25 coupling, C2-Sonogashira22–24 coupling, and N6-amination on the purine moiety. Thus, we first synthesized the glycosyl donor 16 from d-mannose according to our previously published procedure (Scheme 2).26
Scheme 2. Synthesis of the Glycosyl Donor 16a.
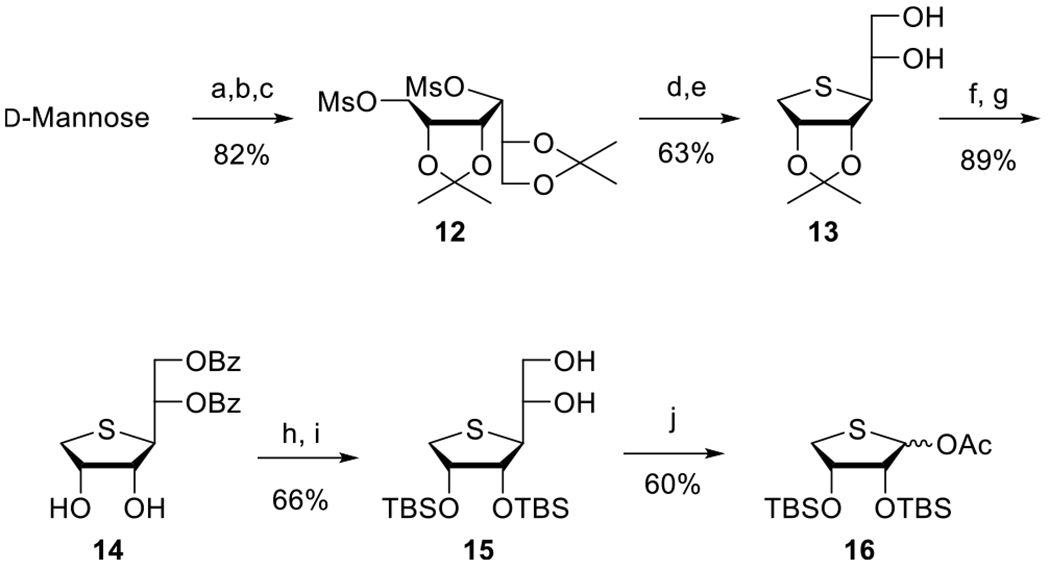
aReagents and conditions: (a) 2,2-dimethoxypropane, camphorsulfonic acid, acetone, rt, 15 h; (b) NaBH4, EtOH, rt, 2 h; (c) MsCl, Et3N, CH2Cl2, rt, 1 h; (d) Na2S, DMF, 80 °C, 15 h; (e) 60% AcOH, rt, 2 h; (f) BzCl, pyridine, CH2Cl2, rt, 18 h; (g) 80% AcOH, 70 °C, 12 h; (h) TBSCl, imidazole, DMF, rt, 18 h; (i) NaOMe, MeOH, rt, 2 h; (j) Pb(OAc)4, EtOAc, rt, 18 h.
d-Mannose was treated with 2,2-dimethoxypropane in the presence of camphorsulfonic acid to give di-O-acetonide, which was reduced with NaBH4, and the resulting diol was treated with mesyl chloride to give the dimesylate 12 in 82% yield. Treatment of 12 with Na2S in DMF at 80 °C afforded the 4-thiosugar, which was selectively hydrolyzed with 60% acetic acid to give 5,6-diol 13. As the 2,3-acetonide was problematic for removal under acidic conditions at the final step, the 2,3-O-acetonide was converted to the 2,3-di-O-TBS group as follows. Benzoylation of 13, followed by hydrolysis of 2,3-O-acetonide with 80% acetic acid, yielded 2,3-diol 14. Protection of diol 14 with a TBS group followed by the removal of the benzoyl group afforded 5,6-diol 15. Oxidative cleavage of 15 with Pb(OAc)4 yielded the glycosyl donor 16.
Then, we synthesized 6-chloro-2,8-diiodopurine 17d and 2,6-dichloro-8-iodopurine 19, which can serve as essential intermediates for selective modification at the C2, C8, and N6 positions (Scheme 3). N6-Protected 6-chloropurine (17a)27 was treated with LiTMP, prepared by treating tetramethylpiperidine with n-BuLi, followed by treatment with iodine to yield 2,8-diiodo derivative 17c.22 Because the 8-iodo position was sensitive to degradation under acidic conditions, the tetrahydropyran (THP) group of 17c was removed with CuCl2 to afford 17d. N9-Protected 2,6-dichloropurine 17b27 was treated with lithium diisopropylamide (LDA), prepared by treating i-Pr2NH with n-BuLi, followed by further treatment with iodine to give 8-iodo derivative 18. The removal of the THP group of 18 under mild acidic conditions afforded 19.
Scheme 3. Synthesis of 6-Chloro-2,8-diiodopurine 17d and 2,6-Dichloro-8-iodopurine 19a.
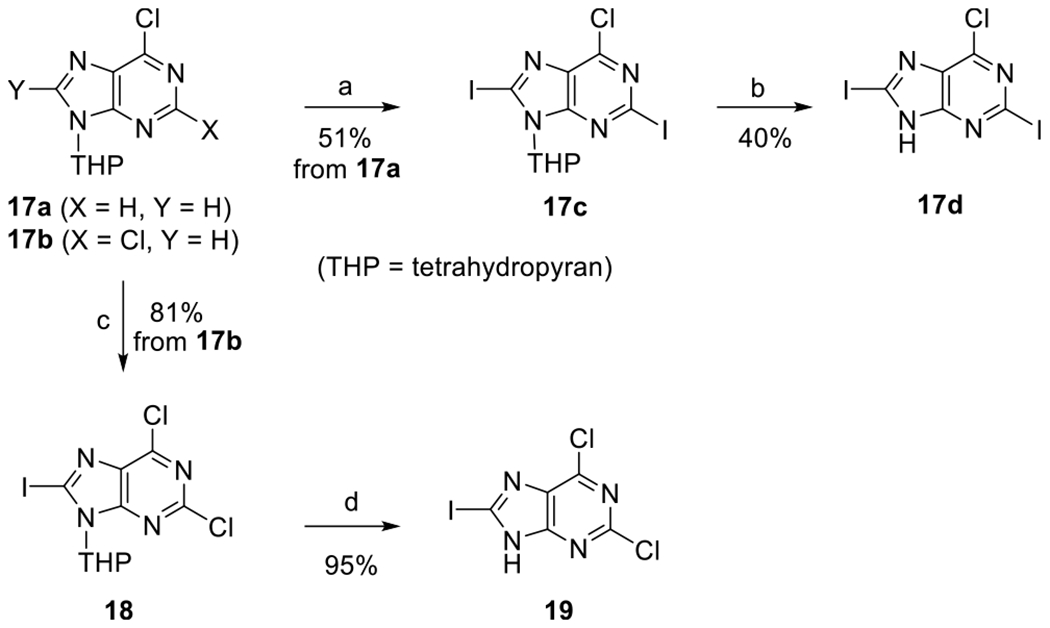
aReagents and conditions: (a) tetramethylpiperidine, I2, n-BuLi, THF, −78 °C, 4 h; (b) CuCl2, EtOH/H2O, 85 °C, 5 h; (c) i-Pr2NH, n-BuLi, I2, THF, −78 °C, 2 h; (d) PPTS, EtOH, 60 °C, 6 h.
Condensation of 16 with silylated 6-chloro-2,8-diiodopurine in the presence of TMSOTf21 as a Lewis acid yielded the protected nucleoside 20 as a single β-anomer without concomitant formation of the N7-isomer. The desired C2-selectivity was not observed in substrate 20 when we attempted regioselective Sonogashira coupling, as shown in Scheme 1. Thus, we paid attention to Stille coupling to obtain the desired product. Delightfully, we were able to regioselectively synthesize 8-furyl derivative 21a and the 8-thienyl derivative 21b regioselectively with high yield. The 2-hexynyl derivatives 22a and 22b were then synthesized via another palladium-catalyzed Sonogashira coupling of 21a and 21b with 1-hexyne, respectively, and their structures were confirmed by X-ray crystallography (Scheme 4, please see Supporting Information for the X-ray crystallographic details of the compounds 22a and 22b). N6-Substitution of 22a and 22b with ammonia, methylamine, and 3-iodobenzylamine afforded the final nucleosides 5g–i and 5j–l, respectively.
Scheme 4. Synthesis of Truncated 2-Hexynyl-N6,C8-Disubstituted-4′-Thioadenosines 5g–la.
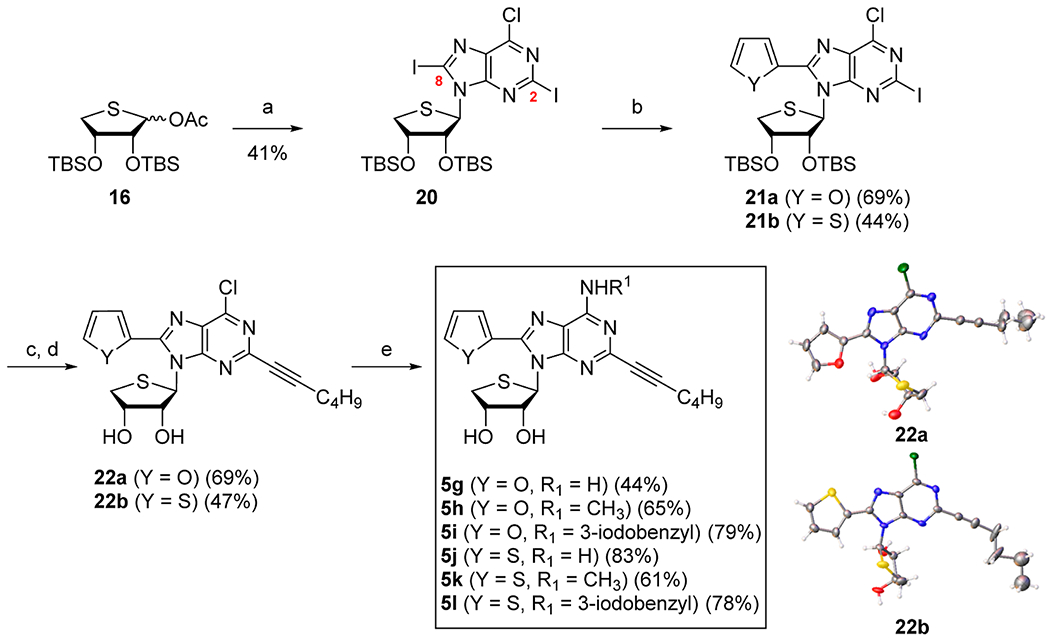
aReagents and conditions: (a) 17d, BSA, TMSOTf, CH3CN, 75 °C, 1.5 h; (b) Pd(dba)2, 2-tributylstannylfuran, DMF/PhMe, rt, 9 h for 21a or Pd(dba)2, 2-tributylstannylthiophene, DMF/PhMe, rt, 23 h for 21b; (c) Pd(PPh3)2Cl2, 1-hexyne, CuI, Cs2CO3, DMF, rt, 14 h for 22a or 16 h for 22b; (d) n-Bu4NF, THF, rt, 16 h for 22a or 50 °C, 12 h for 22b; (e) NH3/t-BuOH, 100 °C, 15 h for 5g, 5j or CH3NH2·HCl, Et3N, EtOH, 50 °C, 23 h for 5h or 28 h for 5k or 3-iodobenzylamine·HCl, Et3N, EtOH, 50 °C, 24 h for 5i or 75 °C, 20 h for 5l.
Then, we synthesized the N6,C8-disubstituted-4′-thioadenosines 5m–v with chloro or p-hydroxyphenethylamine (tyramine) groups at the C2 position because a selective A2A antagonist, 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a]-[1,3,5]triazin-5-ylamino]ethyl) phenol (ZM-241385) possessed a C2-tyramine group29 (Scheme 5). The glycosyl donor 16 was condensed with silylated 2,6-dichloro-8-iodopurine under Vorbrüggen21 conditions to afford the condensed nucleoside 23. Compound 23 was subjected to palladium-catalyzed Stille25 coupling with 2-tributylstannylfuran and -thiophene to give 8-furyl- and 8-thienyl derivatives 24a and 24b, respectively. Treatment of 24a and 24b with tetra-n-butylammonium fluoride (TBAF), followed by further treatment of resulting diol derivatives with ammonia, methylamine, and 3-iodobenzylamine afforded the N6-amino-, methylamino-, and 3-iodobenzylamino derivatives 5m–o and 5p–r, respectively. Furthermore, 2-chloro-8-furyl-N6-amino- and methylamino derivatives 5m and 5n were converted to the corresponding 2-p-hydroxyphenethylamino derivatives 5s and 5t, respectively. Similarly, 8-thienyl derivatives 5p and 5q were converted to 5u and 5v, respectively.
Scheme 5. Synthesis of Truncated 2-Chloro- and 2-p-Hydroxyphenethylamino-N6,C8-Disubstituted-4′-Thioadenosines 5m–va.
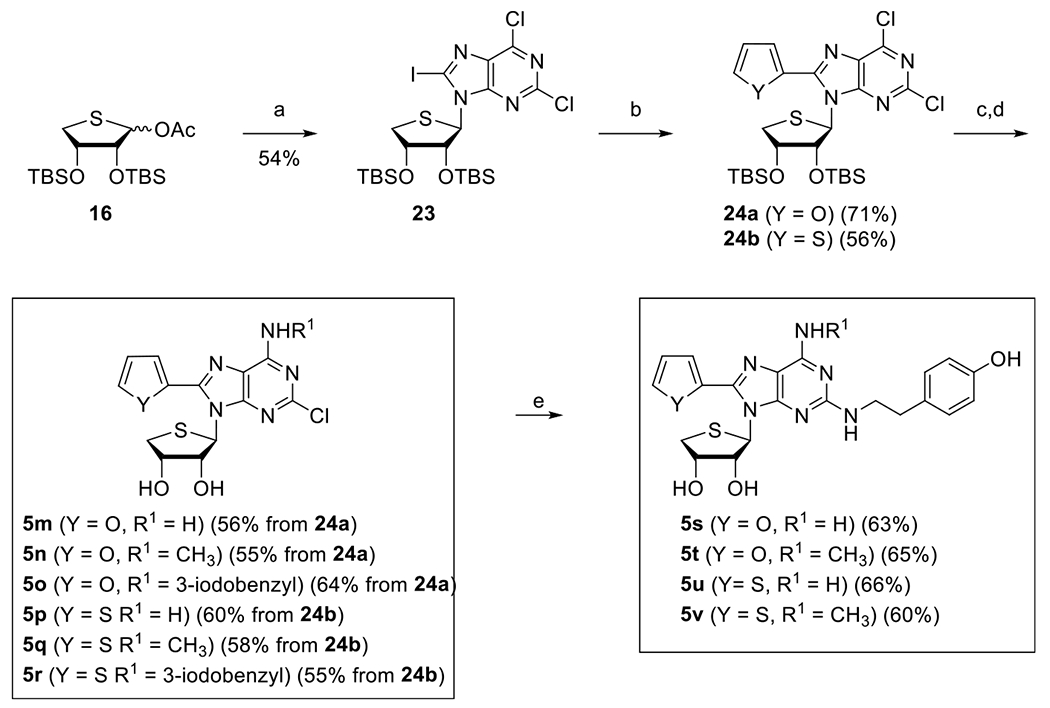
aReagents and conditions: (a) 19, BSA, TMSOTf, CH3CN, 75 °C, 1.5 h; (b) Pd(PPh3)2Cl2, 2-tributylstannylfuran, THF, 70 °C, 24 h for 24a or Pd(PPh3)2Cl2, 2-tributylstannylthiophene, THF, 70 °C, 24 h for 24b; (c) TBAF, THF, rt, 1 h; (d) NH3/t-BuOH, 120 °C, 15 h for 5m, 5p or CH3NH2·HCl, Et3N, EtOH, rt, 24 h for 5n, 5q or 3-iodobenzylamine·HCl, Et3N, EtOH, rt, 24 h for 5o, 5r; (e) tyramine, DIPEA, 1-butanol, MW, 180 °C, 2 h.
Structure–Activity Relationship Studies and Pharmacological Profiles.
Binding Affinities and cAMP Functional Data to Adenosine Receptor Subtypes.
The binding affinities of all final nucleosides 5a–v at the four subtypes of the hARs were measured using standard radioligands and membrane preparations.29 The hA1AR and hA3AR were expressed in Chinese hamster ovary (CHO) cells, and the hA2AAR and hA2BAR were expressed in human embryonic kidney (HEK)-293 cells. [3H](−)-N6-2-Phenylisopropyladenosine (25, R-PIA), [3H]2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamido-adenosine (26, CGS21680), 1,3-[3H]-dipropyl-8-cyclopentylxanthine (27, DPCPX), and [125I]N6-(4-amino-3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (28, I-AB-MECA) were used for the hA1AR, hA2AAR, hA2BAR, and hA3AR, respectively. The percent inhibition of radioligand binding at 10 μM was reported in cases of weak binding. Nonspecific binding was defined using 5′-N-ethylcarboxamidoadenosine (29, NECA).
As shown in Table 1, the addition of a hydrophobic furan or thiophene ring at the C8 position of 3 preserved considerable AR affinity in general and at the A2AAR, specifically. The furan substitution exhibited comparable hA2AAR binding affinity with the thiophene substitution, although the orientation of thiophene can be stabilized as a preferred binding pose by the 1,4-N…S noncovalent sulfur interaction between thiophene and N7 nitrogen.30 It implies that the interaction between hA2AAR and the heteroatom of the C8-aromatic ring is not crucial for binding with the receptor. As the C2 position is appended with a rigid and hydrophobic hexyne substituent (5a–5l), the addition of a hydrophobic group at the N6-position decreased hA2AAR binding affinity in the following order: R1 = H > CH3 > 3-iodobenzyl. This pattern indicated that a free amino group is essential for appropriate hydrogen bonding in the hA2AAR, which matches our previous findings.18 In the case of the binding affinities of C2-hexyne compounds for hA3AR, the 4′-thionucleoside derivatives (5g–5l) could not tolerate the bulky N6-3-iodobenzylamine, as opposed to the 4′-oxonucleoside derivatives (5a–5f) and the series of compounds with C8–H substitution.18b On the other hand, 2-Cl or 2-tyramine compounds (5m–5v) favored bulkier N6-substituents than free amines. These results indicate that the tight binding by both bulky C8-aromatic ring and rigid C2-hexyne can contract the binding pocket near N6-position. In the N6-NH2 derivatives, the C2-hexyne compounds maintained substantial binding affinity at the hA2A and hA3ARs, but 2-Cl or 2-tyramine substitution decreased the binding affinities at both receptors dramatically. It is surprising in that a C2-tyramine substitution28 was reported to increase the binding affinity at the hA2AAR. In general, 4′-oxonucleoside derivatives exhibited better binding affinities at the hA2AAR and hA3AR than the corresponding 4′-thionucleoside derivatives, among which 5d exhibited the best binding affinity at the hA2AAR (Ki = 7.7 ± 0.5 nM) along with good binding affinity at the hA3AR (Ki = 15.6 ± 1.6 nM). Among tested 4′-thionucleoside derivatives, compound 5g exhibited the highest binding affinity (Ki = 13.2 ± 0.2 nM at hA2AAR) with high selectivity (Ki,5g = 119 ± 39 nM at hA3AR). These findings provide an essential clue leading to the discovery of selective nucleoside A2AAR ligands.
Table 1.
Binding Affinities of Compounds 5a–v at Four Subtypes of the hARs
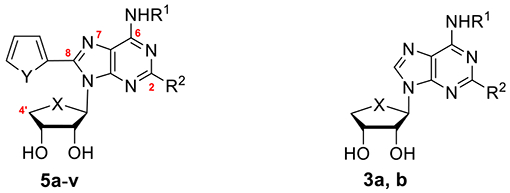
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| compd no. |
X |
Y |
R1 |
R2 |
Ki value (nM ± SEMa or % displacement at 10 μMb) |
|||
| hA1AR | hA2AAR | hA2BAR | hA3AR | |||||
|
| ||||||||
| 5a | O | O | H | 1-hexyne | 183 ± 28 | 18.0 ± 5.5 | 327 ± 32 | 15.5 ± 2.9 |
| 5b | O | O | CH3 | 1-hexyne | 3030 ± 50 | 50.5 ± 12.1 | 76 ± 1% | 2.9 ± 0.7 |
| 5c | O | O | 3-I-Bn | 1-hexyne | 13 ± 1% | 579 ± 53 | 4 ± 2% | 199 ± 20 |
| 5d = 4 | O | S | H | 1-hexyne | 392 ± 99 | 7.7 ± 0.5 | 834 ± 11 | 15.6 ± 1.6 |
| 5e | O | S | CH3 | 1-hexyne | 2310 ± 600 | 126 ± 18 | 52 ± 1% | 2.9 ± 0.5 |
| 5f | O | S | 3-I-Bn | 1-hexyne | 2580 ± 460 | 513 ± 153 | 2 ± 3% | 8.9 ± 4.8 |
| 5g | S | O | H | 1-hexyne | 46 ± 1% | 13.2 ± 0.2 | 1973 ± 251 | 119 ± 39 |
| 5h | S | O | CH3 | 1-hexyne | 17 ± 9% | 159 ± 17 | 41 ± 2% | 53.5 ± 16.9 |
| 5i | S | O | 3-I-Bn | 1-hexyne | 11 ± 7% | 5 ± 5% | 3 ± 2% | 2 ± 8% |
| 5j | S | S | H | 1-hexyne | 494 ± 122 | 61.4 ± 4.1 | 27 ± 2% | 150 ± 6 |
| 5k | S | S | CH3 | 1-hexyne | 11 ± 3% | 2820 ± 1080 | 11 ± 2% | 206 ± 26 |
| 5l | S | S | 3-I-Bn | 1-hexyne | 1 ± 0.2% | 2 ± 2% | 3 ± 3% | 12 ± 5% |
| 5m | S | O | H | Cl | 3890 ± 650 | 140 ± 10 | 53 ± 3% | 855 ± 157 |
| 5n | S | O | CH3 | Cl | 49 ± 1% | 752 ± 67 | 46 ± 1% | 94.6 ± 45.2 |
| 5o | S | O | 3-I-Bn | Cl | 4900 ± 1070 | 336 ± 108 | 51 ± 1% | 89.7 ± 20.4 |
| 5p | S | S | H | Cl | 1980 ± 140 | 459 ± 46 | 62 ± 3% | 294 ± 80 |
| 5q | S | S | CH3 | Cl | 26 ± 4% | 955 ± 704 | 17 ± 1% | 138 ± 10 |
| 5r | S | S | 3-I-Bn | Cl | 50 ± 2% | 720 ± 9 | 17 ± 2% | 137 ± 28 |
| 5s | S | O | H | tyramine | 46 ± 3% | 112 ± 6 | 227 ± 41 | 1650 ± 100 |
| 5t | S | O | CH3 | tyramine | 5330 ± 340 | 468 ± 28 | 713 ± 88 | 445 ± 148 |
| 5u | S | S | H | tyramine | 8050 ± 910 | 946 ± 94 | 608 ± 81 | 3010 ± 1140 |
| 5v | S | S | CH3 | tyramine | 47 ± 2% | 917 ± 59 | 579 ± 135 | 106 ± 27 |
| 3ac | S | H | 1-hexyne | 39 ± 10% | 7.19 ± 0.6 | N.D.d | 11.8 ± 1.3 | |
| 3bc | O | H | 1-hexyne | 740 ± 430 | 63.2 ± 15 | N.D.d | 138 ± 44 | |
All binding experiments were performed using adherent CHO cells and HEK293 cells stably transfected with cDNA encoding the appropriate hAR binding was carried out using 1 nM 25, 10 nM 26, 25 nM 27, and 0.2 nM 28 as radioligands for A1, A2A, A2B, and A3ARs, respectively. Values are expressed as mean ± SEM, n ± 3–4 (outliers eliminated), and are normalized against a nonspecific binder, 29.
When a percent value is shown, it refers to the percent inhibition of a specific radioligand binding at 10 μM, with nonspecific binding defined using 10 μM of 29.
Ref 18.
Not determined.
Three synthesized compounds were evaluated in functional cAMP assays in hA2AAR- or hA3AR-expressing CHO cell lines. As shown in Table 2, no receptor activation at the hA2AAR at 10 μM concentration for compounds 5a, 5d, and 5g was observed, demonstrating that they act as hA2AAR full antagonists. Among them, the compound 5d demonstrated the highest potency, and its KB value was calculated to be 25.8 nM at the hA2AAR using full agonist NECA (29) (Table S1). Similarly, compounds 5a, 5d, and 5g inhibited hA3AR activation at 10 μM, and the KB value of 5d for hA3AR was calculated to be 107.4 nM, also indicating that it is a full hA3AR antagonist. It should be noted that the addition of a hydrophobic furan or thiophene at the C8 position could convert A2AAR agonists into A2AAR antagonists successfully by inhibiting the interaction for receptor inactivation.
Table 2.
cAMP Functional Assay Data at hA2AAR and hA3AR Expressed in CHO Cellsa
| compound no. | % activation, A2A cAMP assay | % activation, A3 cAMP assay |
|---|---|---|
| 5a | 0.3 ± 0.3 | 16 ± 4 |
| 5d | 0.8 ± 0.3 | 4 ± 4 |
| 5g | 0.05 ± 0.1 | 11 ± 5 |
At a concentration of 10 μM, unless noted.
Molecular Docking Studies.
In order to elucidate the effect of 4′-thioribose, a homology modeling study was performed using the co-crystal structure of A2AAR containing 5d (Figure 2, PDB ID: 8CU6). Comparison of the binding sites of hA2AAR and hA3AR revealed two characteristic differences that can induce variation in binding affinity between the two receptors: the expanded binding pocket by Ser2476.52/A3 (blue arrow), and the reduced ribose-binding pocket by Leu903.32/A3 of hA3AR (red arrow) (Figure 2A). Molecular docking was then performed for 4′-thionucleoside 5j and 4′-oxonucleoside 5d. Docking with the homology model of hA3AR showed that the binding pose of 5d was similar to that of hA2AAR, whereas 5j exhibited a reverse-oriented binding pose due to steric clashes between 4′-thioribose and Leu903.32/A3 (Figure 2B). In hA2AAR, although there were minor steric clashes between Ile2747.39/A2A/His2787.43/A2A and 5j, the ligand could still retain its forward-oriented binding pose due to the smaller residue Val843.32/A2A compared to Leu903.32/A3 (red dashed line, Figure 2C,D).
Figure 2.
Comparison of the crystal structure of hA2AAR (PDB ID: 8CU6) and the homology model of A3AR. (A) Binding pocket of the co-crystal structure of hA2AAR (orange), and the homology model of A3AR. (green) (B) Predicted binding mode of 5d (cyan) and 5j (magenta) for hA3AR. (C) Predicted binding mode of 5j (lime) and (D) co-crystal structure of 5d (white) with hA2AAR. The red dashed line represents steric clashes, and the yellow dashed line represents hydrogen bonding.
Overall, the molecular docking analysis suggests that steric clashes between the sugar moiety and the receptors can be a key factor in the binding of both hA2AAR and hA3AR. Although the difference in the bulkiness of ribose can be considered subtle, as observed in the X-ray crystal structure of 11b [C(4′)–O: 1.39 Å in ribose] and 22b [C(4′)–S: 1.82 Å in 4′-thioribose], the results showed it was significant enough to result in several-fold changes in binding affinities and even flip the binding mode in hA3AR (Figure 2B).
ADME Evaluation of Compound 5d.
We examined the druggability of the most potent compound 5d primarily through in vitro ADME assays to determine its potential as a preclinical candidate. Our results showed that compound 5d did not significantly inhibit five types of CYP isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) and hERG K+ channels (Tables S2 and S3).
We also conducted an in vivo pharmacokinetic study of 5d in mice, which revealed proper distribution (Vss = 0.69 L/kg) and moderate plasma clearance (CL = 60.5 mL/min/kg). After oral administration, compound 5d was rapidly absorbed (Tmax = 0.25 h) with moderate bioavailability (F = 22.81%) and half-life (t1/2 = 1.99 h) (Table 3).
Table 3.
In Vivo Pharmacokinetic Properties of Compound 5d
| PK parameter |
route of dosing |
|
|---|---|---|
| i.v. (n = 3) | p.o. (n = 3) | |
| dose (mg/kg) | 2.00 | 10.00 |
| AUC0–t (h·ng/mL) | 550.95 | 626.55 |
| Cmax (ng/mL) | 1984.94 | 1241.79 |
| Tmax (h) | 0.08 | 0.25 |
| CL ((mL/min)/kg) | 60.5 | |
| Vss (L/kg) | 0.69 | |
| t1/2 (h) | 0.16 | 1.99 |
| F (%) | 22.81 | |
Synergistic Antitumor Activity of Compound 5d with the Immune Checkpoint Inhibitor in 4T1-Luc Cells Implanted in Mouse Xenograft Models.
We conducted an in vivo test to investigate the antitumor effect of compound 5d and its potential synergistic effect with an immune checkpoint inhibitor. 4T1-Luc tumor-bearing mice were intraperitoneally treated with compound 5d (10 mg/kg), antimouse PD-L1 (10 mg/kg), or a combination of both treatments thrice per week for 21 days. As shown in Figure 3A–D, the single administration of compound 5d resulted in a weak antitumor effect, with a TGI of 15%. However, coadministration of 5d with antimouse PD-L1 demonstrated a substantial increase in TGI up to 77%, while the single administration of antimouse PD-L1 exhibited only 43% of TGI. No adverse effects or body weight loss were observed. In addition, bioluminescence imaging of mice was conducted on the final day of the experiment to precisely quantify the tumor mass. Consistent with the tumor volume decrease, the group of mice treated with combination therapy exhibited a remarkable reduction in signal intensity from the tumor (Figure 3E). Taken together, these results demonstrate that compound 5d effectively improves the antitumor potential of the immune checkpoint inhibitor anti-PD-L1.
Figure 3.
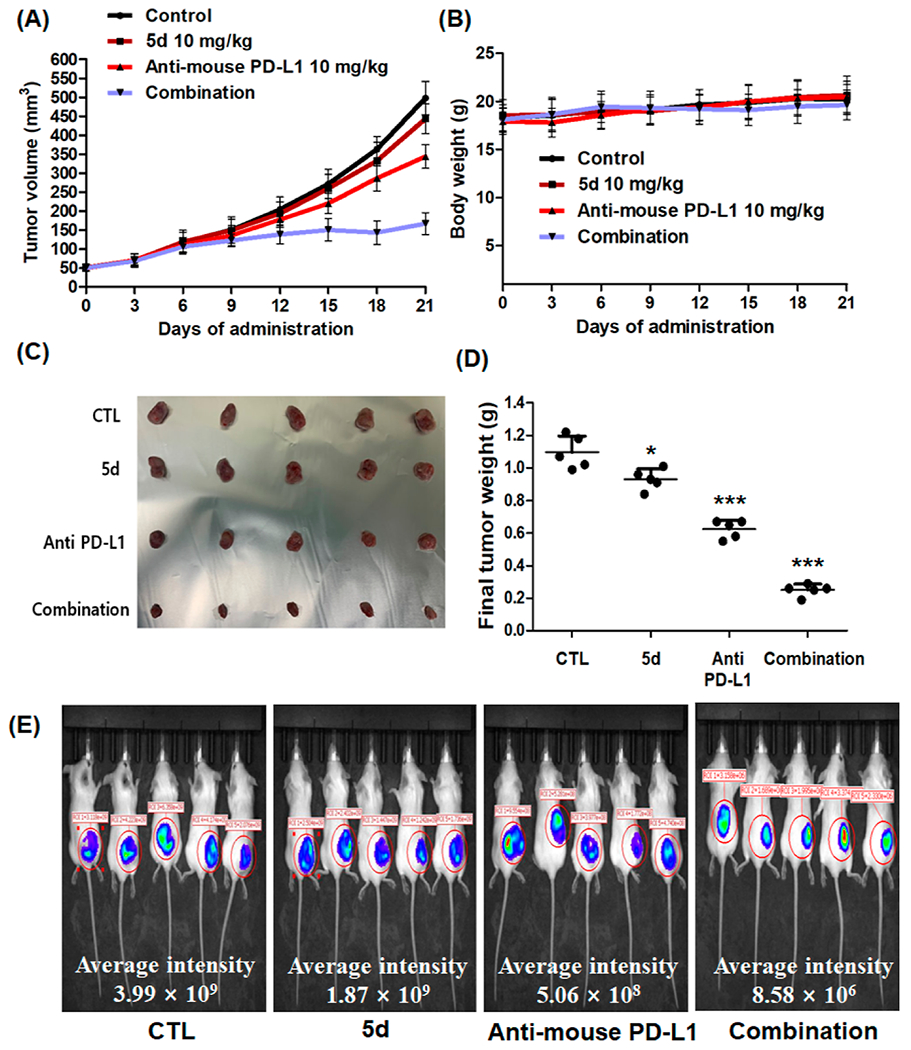
In vivo antitumor activity of the compound 5d and its synergistic effect with immune checkpoint inhibitor. Mice bearing 4T1-Luc tumors were treated thrice per week with vehicle (DMSO/cremophor/normal saline = 5:5:90), 5d (10 mg/kg, i.p.), anti-PD-L1 (10 mg/kg, i.p.), or combination of 5d (10 mg/kg, i.p.), and anti-PD-L1 (10 mg/kg, i.p.). (A) Tumor volume and (B) body weight of the mice bearing 4T1-Luc tumors during administration. (C) Image of excised tumors from the mice and (D) their tumor weight. (E) Luciferase imaging (IVIS) of the mice on the final day of the experiment. Average signal intensity was measured by PerkinElmer Living Image software. *P < 0.05, **P < 0.01, ***P < 0.001 vs the control.
CONCLUSIONS
A series of truncated 4’-thio- and 4’-oxonucleoside derivatives substituted at C2, C8, and N6-positions were synthesized from d-mannose and d-erythrono-1,4-lactone, respectively. Functional groups such as C2-hexynyl and C8-aryl groups were introduced by palladium catalyst-controlled regioselective cross-coupling.22 From this study, it was discovered that the introduction of hydrophobic rings such as furan and thiophene at the C8 position successfully converted an A2AAR agonist into an A2AAR antagonist. The X-ray co-crystal structure of hA2AAR complexed with antagonist 5d,11 revealed that the subpocket was occupied by heteroaromatic rings, inducing tight binding that can abolish receptor activation. Additionally, the C8 substituent caused the ribose ring to rotate from its canonical orientation, disrupting a crucial interaction for receptor activation. Also, it was elucidated that the C2-hexynyl group formed favorable hydrophobic interactions in a relatively large hydrophobic pocket of the A2AAR.
4’-Oxonucleoside derivatives, 5a–5f, showed better binding affinities than 4’-thionucleosides. Among them, compound 5d was discovered as the best hA2AAR antagonist (Ki = 7.7 ± 0.5 nM) along with a high antagonistic binding affinity at the hA3AR (Ki = 15.6 ± 1.6 nM). Among the 4’-thionucleoside derivatives, 5g–5v, the 4’-thionucleoside 5g exhibited the highest binding affinity at hA2AAR (Ki = 13.2 ± 0.2 nM), along with a ninefold stronger binding affinity than hA3AR (Ki = 119 ± 39 nM). According to the molecular docking study, the steric clashes between 4′-thioribose and Leu903.32/A3 weaken the binding with A3AR, flipping the binding pose of the ligand.
The most potent compound 5d also showed good druggability and an acceptable pharmacokinetic profile suitable for a preclinical candidate. Furthermore, 5d exhibited a potent in vivo antitumor effect as a combination therapy with the immune checkpoint inhibitor, anti-PD-L1, without any adverse effects and body weight loss.
A2AAR antagonists are known to be highly associated with cancer immunotherapy activity. On the other hand, existing research demonstrates that the modulation of A3AR can exhibit anticancer effects through distinct mechanisms, such as the induction of cell cycle arrest and inhibition of HIF-1α expression.17,31 Hence, further investigation into the anticancer effects of compound 5d will be conducted to elucidate its potential additive effect achieved through the modulation of A3AR.
To the best of our knowledge, this is the first SAR study of A2AAR antagonists with a nucleoside skeleton. The characteristics of the nucleoside moiety can provide beneficial effects such as species independence and pharmacokinetic properties, which are favorable for preclinical and clinical trials, as in the case of the A3AR antagonist with a nucleoside skeleton.15a,b Thus, the novel nucleoside dual A2AAR/A3AR antagonist 5d can be a promising drug candidate for combination therapy with immune checkpoint inhibitors.
EXPERIMENTAL SECTION
General Procedures.
The study employed various scientific techniques to measure and analyze the physical and chemical properties of synthesized compounds. NMR spectra (1H/13C) were obtained by Jeol JNM-ECZ 400s (400 MHz/100 MHz), Bruker AV500 (500 MHz/125 MHz), or Bruker AV800 (800 MHz/200 MHz) and reported as chemical shifts in parts per million (δ) relative to solvent peaks, with coupling constants (J) expressed in hertz (Hz). Melting points were measured using a Barnstead Electrothermal instrument, while optical rotations were obtained using a Jasco-P2000 instrument. Elemental analyses (C, H, and N) were conducted to assess compound purity, and HPLC (Agilent 1260 infinity series) was also used to determine the purities of the representative compounds using a binary solvent system [0.01 M KH2PO4 buffer in H2O/MeCN] and an Agilent Zorbax Eclipse XDB-C18 column [5.0 μm, 4.6 mm i.d. × 250 mm]. The HPLC purity was more than 95% purity. High-resolution mass spectrometry spectra were obtained using fast atom bombardment (FAB) and electrospray ionization (ESI) methods. Silica gel 60 (230–400 mesh) was used for flash column chromatography, and all solvents were purified and dried before use using standard techniques. Commercially available materials were used without purification unless otherwise stated.
Synthesis and Characterization of the Compounds.
The compound 5d was synthesized in the previous paper through the intermediates 6–10, 11b.11 The glycosylic donor 1626 and the diiodinated purine 17d23 were prepared by a known procedure.
(2R,3R,4R)-2-(6-Chloro-8-(furan-2-yl)-2-(hex-1-yn-1-yl)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (11a). Stille Coupling.
To a solution of 10 (530 mg, 0.77 mmol) in anhydrous THF (38 mL) were added 2-(tributylstannyl)furan (0.48 mL, 1.53 mmol) and bis-(triphenylphosphine)palladium(II) dichloride (107 mg, 0.15 mmol) at room temperature under N2. After being refluxed with stirring for 1 h, the reaction mixture was cooled to room temperature and evaporated. The residue was purified by column chromatography (silica gel, hexane/EtOAc, 30/1 to 15/1) to give the intermediate.
TBS Deprotection.
To a cooled (0 °C) solution of the above-generated intermediate in anhydrous THF (30 mL) were dropwise added triethylamine (0.50 mL, 3.83 mmol) and trimethylamine-trihydrofluoride (0.68 mL, 3.83 mmol) at room temperature under N2. After being stirred at room temperature for 15 h, the reaction mixture was evaporated. The residue was purified by column chromatography (silica gel, hexane/EtOAc, 1/1) to give 11a (130 mg, 42%) as white solid: mp 184 °C; (c 0.11, MeOH); UV (MeOH) λmax 225, 239, 331 nm; 1H NMR (400 MHz, MeOD) δ 7.93 (dd, J = 1.6, 0.8 Hz, 1H), 7.49 (dd, J = 4.0, 1.2 Hz, 1H), 6.77 (dd, J = 3.6, 1.6 Hz, 1H), 6.52 (d, J = 6.4 Hz, 1H), 5.40 (dd, J = 6.4, 4.8 Hz, 1H), 4.68 (dd, J = 9.6, 3.2 Hz, 1H), 4.50–4.54 (m, 1H), 4.03 (dd, J = 9.6, 1.6 Hz, 1H), 2.51 (t, J = 6.8 Hz, 2H), 1.63–1.69 (m, 2H), 1.50–1.59 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, MeOD): δ 154.9, 150.9, 150.2, 148.9, 147.3, 144.9, 132.7, 119.2, 114.4, 93.0, 92.2, 81.4, 77.2, 76.2, 73.7, 32.1, 23.9, 20.3, 14.7; HRMS (FAB): found 403.1165 [calcd for C19H20ClN4O4+ (M + H)+ 403.1173]; Anal. Calcd for C19H19ClN4O4: C, 56.65; H, 4.75; N, 13.91. Found: C, 56.91; H, 4.35; N, 13.77.
General Procedure for N6-Amination for the Preparation of 5a–5f.
To a solution of 11a or 11b (1.00 equiv) in EtOH (0.20 M) or t-BuOH (0.20 M) were dropwise added appropriate amines (1.50 equiv to excess) at room temperature under N2. After being stirred at the provided temperature for 12–24 h, the reaction mixture was evaporated. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH, 20/1) to give 5a–5f.
(2R,3R,4R)-2-(6-Amino-8-(furan-2-yl)-2-(hex-1-yn-1-yl)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (5a).
11a was dissolved in NH3/t-BuOH (0.20 M) and stirred for 12 h at 100 °C in a steel bomb. Yield = 78%; white solid; mp 194 °C; (c 2.16, MeOH); UV (MeOH) λmax 237, 318 nm; 1H NMR (400 MHz, MeOD): δ 7.82 (dd, J = 1.6, 0.4 Hz, 1H), 7.19 (dd, J = 3.4, 0.4 Hz, 1H), 6.70 (dd, J = 3.5, 1.7 Hz, 1H), 6.31 (d, J = 6.3 Hz, 1H), 5.44 (dd, J = 6.2, 4.8 Hz, 1H), 4.63 (dd, J = 9.6, 3.5 Hz, 1H), 4.47–4.48 (m, 1H), 3.98 (dd, J = 9.6, 1.3 Hz, 1H), 2.44 (t, J = 7.1 Hz, 2H), 1.60–1.63 (m, 2H), 1.49–1.54 (m, 2H), 0.97 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, MeOD): δ 156.7, 152.0, 147.7, 146.7, 144.8, 144.7, 119.8, 115.5, 113.1, 91.4, 88.1, 81.6, 76.1, 75.0, 72.9, 31.5, 23.1, 19.4, 13.9; HRMS (FAB): found 384.1666 [calcd for C19H22N5O4+ (M + H)+ 384.1672]; Anal. Calcd for C19H21N5O4: C, 59.52; H, 5.52; N, 18.27. Found: C, 59.21; H, 5.32; N, 18.56.
(2R,3R,4R)-2-(8-(Furan-2-yl)-2-(hex-1-yn-1-yl)-6-(methylamino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (5b).
11a and methylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 24 h at room temperature. Yield = 80%; white solid; mp 201 °C; (c 0.09, MeOH); UV (MeOH) λmax 244, 325 nm; 1H NMR (800 MHz, DMSO-d6): δ 7.99 (d, J = 0.8 Hz, 1H), 7.97 (br s, 1H), 7.09 (d, J = 3.4 Hz, 1H), 6.76 (dd, J = 3.0, 1.5 Hz, 1H), 6.11 (d, J = 6.6 Hz, 1H), 5.43 (d, J = 6.5 Hz, 1H), 5.20 (d, J = 3.8 Hz, 1H), 5.17 (q, J = 12.1, 5.5 Hz, 1H), 4.43 (dd, J = 9.2, 3.2 Hz, 1H), 4.30 (d, J = 2.9 Hz, 1H), 3.84 (d, J = 9.2 Hz, 1H), 2.94 (br s, 3H), 2.44 (t, J = 7.0 Hz, 2H), 1.51–1.57 (m, 2H), 1.41–1.47 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (200 MHz, DMSO-d6): δ 154.2, 149.7, 145.7, 145.3, 130.8, 130.0, 129.6, 128.2, 119.0, 88.8, 85.6, 81.8, 74.2, 72.5, 70.5, 29.9, 27.0, 21.5, 17.9, 13.4; HRMS (FAB): found 398.1817 [calcd for C20H24N5O4+ (M + H)+ 398.1817]; Anal. Calcd for C20H23N5O4: C, 60.44; H, 5.83; N, 17.62. Found: C, 60.84; H, 5.45; N, 17.35.
(2R,3R,4R)-2-(8-(Furan-2-yl)-2-(hex-1-yn-1-yl)-6-((3-iodobenzyl)-amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (5c).
11a and 3-iodobenzylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 24 h at room temperature. Yield = 68%; pale yellow solid; mp 215 °C; (c 0.055, MeOH); UV (MeOH) λmax 229, 325 nm; 1H NMR (400 MHz, MeOD): δ 7.81–7.82 (m, 2H), 7.61 (d, J = 8.0 Hz, 1H), 7.42 (dd, J = 7.6, 0.4 Hz, 1H), 7.19 (dd, J = 3.2, 0.4 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 6.70 (dd, J = 3.6, 1.6 Hz, 1H), 6.33 (d, J = 6.8 Hz, 1H), 5.46 (dd, J = 6.4, 4.8, Hz, 1H), 4.77 (br s, 2H), 4.65 (dd, J = 9.6, 3.6 Hz, 1H), 4.48–4.50 (m, 1H), 3.99 (dd, J = 9.6, 1.2 Hz, 1H), 2.46 (t, J = 7.2 Hz, 2H), 1.60–1.66 (m, 2H), 1.51–1.56 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 150.3, 145.59, 145.54, 145.52, 143.3, 142.3, 142.1, 135.9, 135.3, 130.4, 126.71, 126.73, 113.7, 112.1, 94.6, 89.1, 86.0, 74.3, 73.0, 70.5, 40.0, 29.8, 27.8, 21.4, 17.9, 13.4; HRMS (FAB): found 600.1109 [calcd for C26H27IN5O4+ (M + H)+ 600.1108]; Anal. Calcd for C26H26IN5O4: C, 52.10; H, 4.37; N, 11.68. Found: C, 52.15; H, 4.32; N, 11.70.
(2R,3R,4R)-2-(6-Amino-2-(hex-1-yn-1-yl)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (5d).11
The compound was synthesized by the reported procedure. HPLC purity: 98.73%.
(2R,3R,4R)-2-(2-(Hex-1-yn-1-yl)-6-(methylamino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (5e).
11b and methylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 24 h at room temperature. Yield = 76%; white solid; mp 216 °C; (c 0.01, MeOH); UV (MeOH) λmax 239, 325 nm; 1H NMR (400 MHz, MeOD): δ 7.71 (dd, J = 5.1, 0.9 Hz, 1H), 7.65 (dd, J = 3.6, 0.9 Hz, 1H), 7.23 (dd, J = 5.0, 3.6 Hz, 1H), 6.11 (d, J = 6.4 Hz, 1H), 5.50 (dd, J = 6.3, 4.8 Hz, 1H), 4.63 (dd, J = 9.6, 3.5 Hz, 1H), 4.46 (t, J = 4.6 Hz, 1H), 3.97 (dd, J = 6.0, 1.2 Hz, 1H), 3.08 (br s, 3H), 2.45 (t, J = 7.2 Hz, 2H), 1.61–1.65 (m, 2H), 1.50–1.55 (m, 2H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (200 MHz, DMSO-d6): δ 154.2, 149.7, 145.7, 145.3, 130.8, 130.0, 129.6, 128.2, 119.0, 88.8 (d, J = 7.9 Hz), 85.6, 81.8, 79.1, 74.2 (d, J = 10.5 Hz), 72.6 (d, J = 20.9 Hz), 70.5 (d, J = 21.0 Hz), 29.9, 21.5, 17.9, 13.4; HRMS (FAB): found 414.1605 [calcd for C20H24N5O3S+ (M + H)+ 414.1600]; Anal. Calcd for C20H23N5O3S: C, 58.09; H, 5.61; N, 16.94. Found: C, 57.99; H, 5.86; N, 16.67.
(2R,3R,4R)-2-(2-(Hex-1-yn-1-yl)-6-((3-iodobenzyl)amino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (5f).
11b and 3-iodobenzylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 24 h at room temperature. Yield = 78%; white solid; mp 225 °C; (c 0.06, MeOH); UV (MeOH) λmax 231, 326 nm; 1H NMR (400 MHz, DMSO-d6): δ 8.55 (br s, 1H), 7.88 (dd, J = 5.1, 0.7 Hz, 1H), 7.74 (s, 1H), 7.58–7.60 (m, 2H), 7.35 (d, J = 6.4 Hz, 1H), 7.29 (dd, J = 4.8, 3.7 Hz, 1H), 7.11 (t, J = 7.7 Hz, 1H), 5.97 (d, J = 6.8 Hz, 1H), 5.51 (d, J = 6.5 Hz, 1H), 5.22–5.24 (m, 2H), 4.41 (dd, J = 9.3, 3.3 Hz, 1H), 3.83 (d, J = 8.9 Hz, 1H), 2.42 (t, J = 7.1 Hz, 2H), 1.52–1.56 (m, 2H), 1.42–1.46 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 153.6, 150.2, 145.8, 145.5, 142.5, 136.1, 135.4, 130.8, 130.5, 130.2, 129.8, 128.3, 126.8, 118.9, 94.7, 89.0, 81.8, 74.4, 70.6, 42.3, 29.9, 21.5, 18.0, 13.5; HRMS (FAB): found 616.0887 [calcd for C26H27IN5O3S+ (M + H)+ 616.0879]; Anal. Calcd for C26H26IN5O3S: C, 50.74; H, 4.26; N, 11.38. Found: C, 50.75; H, 4.30; N, 11.15.
2,6-Dichloro-8-iodo-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (18).
To a cooled (−78 °C) solution of 17b (1.27 g, 4.65 mmol) in anhydrous THF (30 mL) was dropwise added LDA (14.0 mL, 1.0 M in THF/hexanes, 14.0 mmol) under N2, and the reaction mixture was stirred at the same temperature for 1 h. After adding iodine (5.90 g, 23.25 mmol) in THF (10 mL), the reaction mixture was stirred for 1 h. The reaction mixture was quenched with saturated aqueous Na2S2O3 (50 mL), diluted with EtOAc (50 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed successively with H2O and saturated brine, dried over anhydrous MgSO4, filtered, and evaporated. The residue was purified by column chromatography (silica gel, hexanes/EtOAc, 2/1) to give 18 (1.50 g, 81%) as a brown solid: 1H NMR (800 MHz, CDCl3): δ 5.65 (dd, J = 11.6, 2.4 Hz, 1H), 4.16–4.20 (m, 1H), 3.72 (td, J = 11.6, 2.0 Hz, 1H), 3.03 (ddd, J = 24.0, 12.8, 4.4 Hz, 1H), 2.11–2.19 (m, 1H), 1.57–1.93 (m, 4H); 13C NMR (200 MHz, CDCl3): δ 153.4, 152.6, 150.0, 133.2, 106.4, 86.8, 69.2, 28.7, 24.4, 23.1; HRMS (FAB): found 398.9273 [calcd for C10H10Cl2IN4O+ (M + H)+ 398.9283]; Anal. Calcd for C10H9Cl2IN4O: C, 30.10; H, 2.27; N, 14.04. Found: C, 30.37; H, 1.91; N, 14.01.
2,6-Dichloro-8-iodopurine (19).
To a solution of 18 (3.0 g, 7.52 mmol) in anhydrous EtOH (50.0 mL, 0.15 M) was added PPTS (378 mg, 1.50 mmol) at room temperature under N2. After being heated at 60 °C for 6 h, the reaction mixture was quenched with triethylamine (1.0 mL) and evaporated. The residue was purified by column chromatography (silica gel, hexanes/EtOAc, 1/1) to give 19 (2.25 g, 95%) as a yellow solid: 13C NMR (200 MHz, MeOD): δ 163.1, 152.6, 147.0, 134.2, 115.1; HRMS (FAB): found 314.8683 [calcd for C5H2Cl2IN4+ (M + H)+ 314.8683]; Anal. Calcd for C5HCl2IN4: C, 19.07; H, 0.32; N, 17.79. Found: C, 19.07; H, 0.31; N, 17.71.
9-((2R,3R,4S)-3,4-Bis((tert-butyldimethylsilyl)oxy)-tetrahydrothiophen-2-yl)-6-chloro-2,8-diiodo-9H-purine (20).
To a stirred suspension of 2-chloro-8-iodo-6-chloropurine (17d) (581 mg, 1.84 mmol) in CH3CN (10.0 mL) was dropwise added BSA (2.40 mL, 9.83 mmol) at room temperature under N2, and the mixture was heated at 60 °C until obtaining clear brown solution. To the reaction mixture was quickly added 16 (500 mg, 1.23 mmol) in CH3CN (5.0 mL) and TMSOTf (0.22 mL, 1.23 mmol), and the mixture was stirred at 75 °C for 1.5 h. The reaction mixture was cooled to room temperature, quenched with saturated NaHCO3 solution (30 mL), and diluted with EtOAc (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed successively with H2O (50 mL), dried over anhydrous MgSO4, filtered, and evaporated. The residue was purified by column chromatography (silica gel, hexanes/EtOAc, 30/1) to give 20 (569.0 mg, 41%) as brown syrup: 1H NMR (400 MHz, CDCl3): δ 6.01 (d, J = 7.2 Hz, 1H), 5.26 (dd, J = 7.2, 2.8 Hz, 1H), 4.55 (dd, J = 5.6, 2.4 Hz, 1H), 3.54 (dd, J = 11.2, 3.2 Hz, 1H), 2.86 (dd, J = 11.2, 2.4 Hz, 1H), 0.98 (s, 9H), 0.74 (s, 9H), 0.17 (s, 3H), 0.16 (s, 3H); 13C NMR (100 MHz, MeOD): δ 154.7, 149.9, 136.2, 118.2, 113.3, 95.1, 76.9, 76.7, 75.0, 27.2, 27.1, 19.8, 19.5; HRMS (FAB): found 752.9854 [calcd for C21H36ClI2N4O2SSi2+ (M + H)+ 752.9876]; Anal. Calcd for C21H35ClI2N4O2SSi2: C, 33.49; H, 4.68; N, 7.44. Found: C, 33.87; H, 4.31; N, 7.11.
General Procedure for Stille Coupling for the Preparation of 21a and 21b.
To a stirred solution of 20 (1.00 equiv) in DMF/toluene (v/v = 1/1, 0.040 M) was added bis(dibenzylideneacetone)-palladium(0) (0.10 equiv), followed by 2-(tributylstannyl)furan (1.05 equiv, for 21a), or 2-(tributylstannyl)thiophene (1.05 equiv, for 21b) dropwise at room temperature under N2. After being stirred for 9 h (for 21a) or 23 h (for 21b) at the same condition, the reaction mixture was quenched with saturated NaHCO3 solution and diluted with Et2O. The layers were separated, and the aqueous layer was extracted with Et2O. The combined organic layers were washed successively with H2O and saturated brine, dried over anhydrous MgSO4, filtered, and evaporated. The residue was purified by column chromatography to give 21a, or 21b.
9-((2R,3R,4S)-3,4-Bis((tert-butyldimethylsilyl)oxy)-tetrahydrothiophen-2-yl)-6-chloro-8-(furan-2-yl)-2-iodo-9H-purine (21a).
21a was isolated by column chromatography (silica gel, hexanes/EtOAc, 100/3 to 25/2). Yield = 69%; yellow oil; (c 1.21, MeOH); UV (MeOH) λmax 326 nm; 1H NMR (400 MHz, MeOD): δ 7.89 (dd, J = 0.9, 0.7 Hz, 1H), 7.46 (dd, J = 0.9, 2.8 Hz, 1H), 6.79 (dd, J = 3.6, 1.6 Hz, 1H), 6.72 (d, J = 7.8 Hz, 1H), 5.44 (dd, J = 7.8, 2.7 Hz, 1H), 4.61 (q, J = 2.4 Hz, 1H), 3.61 (dd, J = 11.2, 3.0, Hz, 1H), 2.87 (dd, J = 11.2, 2.1 Hz, 1H), 1.00 (s, 9h), 0.63 (s, 9H), 0.20 (s, 3H), 0.17 (s, 3H), −0.07 (s, 3H), −0.54 (s, 3H); 13C NMR (200 MHz, MeOD): δ 155.4, 150.8, 149.7, 148.8, 145.0, 134.0, 119.3, 117.2, 114.5, 80.0, 76.2, 66.5, 37.6, 27.1, 26.8, 19.8, 19.4, −3.4, −3.4, −3.5, −4.6. HRMS (ESI): found 693.1027 [calcd for C25H38ClIN4O3SSi2+ (M + H)+ 692.0936].
9-((2R,3R,4S)-3,4-Bis((tert-butyldimethylsilyl)oxy)-tetrahydrothiophen-2-yl)-6-chloro-2-iodo-8-(thiophen-2-yl)-9H-purine (21b).
21b was isolated by column chromatography (silica gel, hexanes/EtOAc, 40/1). Yield = 44%; yellow oil; (c 0.18, MeOH); UV (MeOH) λmax 330 nm; 1H NMR (400 MHz, MeOD): δ 7.91 (dd, J = 5.1, 0.9 Hz, 1H), 7.81 (dd, J = 3.7, 0.9 Hz, 1H), 7.29 (dd, J = 5.1, 3.7 Hz, 1H), 6.43 (d, J = 8.3 Hz, 1H), 5.53 (dd, J = 8.3, 2.8 Hz, 1H), 4.60 (q, J = 2.5 Hz, 1H), 3.60 (dd, J = 11.5, 2.8 Hz, 1H), 2.87 (dd, J = 11.5, 1.8 Hz, 1H), 0.94 (s, 9H), 0.63 (s, 9H), 0.19 (s, 3H), 0.15 (s, 3H), −0.06 (s, 3H), −0.44 (s, 3H); 13C NMR (200 MHz, MeOD): δ 153.66, 151.63, 149.67, 132.37, 131.48, 131.33, 129.33, 128.28, 115.19, 77.73, 73.98, 64.29, 36.03, 25.83, 25.68, 18.17, 17.93, −4.20, −4.36, −4.47, −5.14. HRMS (ESI): found 709.0781 [calcd for C25H38ClIN4O2S2Si2+ (M + H)+ 709.0781].
General Procedure for Sonogashira Coupling and Desilylation for the Preparation of 22a and 22b. Sonogashira Coupling.
To a stirred solution of 21a (1.00 equiv, for 22a) or 21b (1.00 equiv, for 22b) in DMF (0.10 M) were added bis(triphenylphosphine)-palladium(II) dichloride (0.10 equiv), copper(I) iodide (0.20 equiv), and cesium carbonate (1.5 equiv), followed by 1-hexyne (1.05 equiv) dropwise at room temperature under N2. After being stirred for 14 h (for 22a) or 16 h (for 22b) at the same condition, the reaction mixture was quenched with H2O and diluted with Et2O. The layers were separated, and the aqueous layer was extracted with Et2O. The combined organic layers were washed successively with H2O and saturated brine, dried over anhydrous MgSO4, filtered, and evaporated. The residue was purified by column chromatography to give the coupling products.
TBS Deprotection.
To a solution of coupling product (1.00 equiv) in THF (0.10 M) was added the mixture of tetrabutylammoniumfluoride (1.0 M in THF) and acetic acid (v/v = 10/1, 2.50 equiv) at room temperature under N2. After being stirred for 16 h (for 22a) or 12 h (for 22b) at 50 °C under N2, the reaction mixture was quenched by H2O, and diluted with EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous MgSO4, filtered, and evaporated. The residue was purified by column chromatography to give 22a or 22b.
(2R,3R,4S)-2-(6-Chloro-8-(furan-2-yl)-2-(hex-1-yn-1-yl)-9H-purin-9-yl)tetrahydro-thiophene-3,4-diol (22a). Sonogashira Coupling.
The coupling product was isolated by column chromatography (silica gel, hexanes/EtOAc, 100/3 to 50/2). Yield = 73%; yellow oil; (c 0.84, MeOH); UV (MeOH) λmax 330 nm; 1H NMR (400 MHz, MeOD): δ 7.89 (d, J = 1.8 Hz, 1H), 7.46 (dd, J = 3.6, 0.8 Hz, 1H), 6.79 (dd, J = 3.4, 1.6 Hz, 1H), 6.72 (d, J = 7.8 Hz, 1H), 5.48 (dd, J = 7.5, 3.0 Hz, 1H), 4.66 (q, J = 2.6 Hz, 1H), 3.63 (dd, J = 11.2, 3.0 Hz, 1H), 2.88 (dd, J = 11.4, 2.3 Hz, 1H), 2.53 (t, J = 6.9 Hz, 2H), 1.64–1.71 (m, 2H), 1.51–1.60 (m, 2H), 1.00 (s, 9H), 1.00 (t, J = 7.2 Hz, 3H), 0.20 (s, 3H), 0.17 (s, 3H), −0.07 (s, 3H), −0.55 (s, 3H); 13C NMR (200 MHz, MeOD): δ 154.9, 151.2, 150.4, 148.7, 147.0, 145.2, 133.0, 119.1, 114.5, 92.5, 81.5, 79.9, 76.3, 66.5, 37.5, 32.1, 27.1, 26.8, 23.9, 20.4, 19.8, 19.4, 14.8, −3.4, −3.5, −3.6, −4.7. HRMS (ESI): found 647.2664 [calcd for C31H48ClN4O3SSi2+ (M + H)+ 646.2596].
TBS Deprotection.
22a was isolated by column chromatography (silica gel, CH2Cl2/MeOH, 50/1). Yield = 94%; yellow solid; (c 0.13, MeOH); UV (MeOH) λmax 330 nm; 1H NMR (400 MHz, MeOD): δ 7.95 (d, J = 1.4 Hz, 1H), 7.51 (d, J = 3.7 Hz, 1H), 6.79 (dd, J = 3.7, 1.8 Hz, 1H), 6.61 (d, J = 7.8 Hz, 1H), 5.61 (dd, J = 7.8, 3.7 Hz, 1H), 4.59 (q, J = 2.8 Hz, 1H), 3.74 (dd, J = 11.5, 3.2 Hz, 1H), 2.97 (dd, J = 11.5, 1.8 Hz, 1H), 2.52 (t, J = 6.9 Hz, 2H), 1.63–1.70 (m, 2H), 1.50–1.60 (m, 2H), 1.00 (t, J = 7.1 Hz, 3H); 13C NMR (200 MHz, MeOD): δ 154.9, 151.0, 150.4, 148.8, 146.9, 145.0, 133.0, 118.9, 114.5, 92.2, 81.5, 79.2, 75.5, 67.7, 37.8, 32.2, 23.9, 20.3, 14.7. HRMS (FAB): found 419.0944 [calcd for C19H20ClN4O3S+ (M + H)+ 419.0945].
(2R,3R,4S)-2-(6-Chloro-2-(hex-1-yn-1-yl)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydro-thiophene-3,4-diol (22b). Sonogashira Coupling.
The coupling product was isolated by column chromatography (silica gel, hexanes/EtOAc, 100/3 to 50/2). Yield = 64%; colorless oil; (c 0.84, MeOH); 1H NMR (400 MHz, MeOD): δ 7.91 (dd, J = 5.3, 1.1 Hz, 1H), 7.83 (dd, J = 3.7, 0.9 Hz, 1H), 7.30 (dd, J = 5.3, 3.9 Hz, 1H), 6.44 (d, J = 8.2 Hz, 1H), 5.57 (dd, J = 8.0, 3.0 Hz, 1H), 4.64 (q, J = 2.4 Hz, 1H), 3.64 (dd, J = 11.4, 3.2 Hz, 1H), 2.88 (dd, J = 11.4, 2.3 Hz, 1H), 2.54 (t, J = 7.1 Hz, 2H), 1.64–1.72 (m, 2H), 1.51–1.60 (m, 2H), 1.01 (t, J = 7.3 Hz, 3H), 0.94 (s, 9H), 0.63 (s, 9H), 0.19 (s, 3H), 0.15 (s, 3H), −0.06 (s, 3H), −0.47 (s, 3H); 13C NMR (200 MHz, MeOD): δ 155.1, 154.7, 151.2, 147.0, 133.8, 133.3, 132.9, 131.1, 130.1, 92.5, 81.5, 79.5, 75.9, 66.3, 37.2, 32.1, 27.1, 26.8, 23.9, 20.3, 19.7, 19.4, 14.7, −3.4, −3.4, −3.6, −4.5. HRMS (ESI): found 663.2450 [calcd for C31H48ClN4O2S2Si2+ (M + H)+ 663.2440].
TBS Deprotection.
22b was isolated by column chromatography (silica gel, CH2Cl2/MeOH, 50/1). Yield = 73%; white solid; (c 0.52, MeOH); UV (MeOH) λmax 326 nm; 1H NMR (400 MHz, MeOD): δ 7.90 (dd, J = 3.7, 0.9 Hz, 1H), 7.88 (dd, J = 5.1, 1.4 Hz, 1H), 7.32 (dd, J = 5.1, 3.7 Hz, 1H), 6.33 (d, J = 7.8 Hz, 1H), 5.65 (q, J = 3.8 Hz, 1H), 4.56 (td, J = 3.4, 1.7 Hz, 1H), 3.73 (dd, J = 11.5, 3.7 Hz, 1H), 2.96 (dd, J = 11.5, 1.8 Hz, 1H), 2.52 (t, J = 6.9 Hz, 2H), 1.63–1.70 (m, 2H), 1.50–1.59 (m, 2H), 1.00 (t, J = 7.1 Hz, 3H); 13C NMR (200 MHz, MeOD): δ 155.2, 154.6, 150.9, 146.9, 133.7, 133.4, 132.9, 131.2, 130.3, 92.1, 81.5, 78.9, 75.3, 67.5, 37.6, 32.2, 23.9, 20.3, 14.7. HRMS (FAB): found 435.0717 [calcd for C19H20ClN4O3S+ (M + H)+ 435.0711].
General Procedure for N6-Amination for the Preparation of 5g–5l.
To a solution of 22a or 22b (1.00 equiv) in EtOH (0.20 M) or t-BuOH (0.20 M) were dropwise added appropriate amines (1.50 equiv to excess) at room temperature under N2. After being stirred at the provided temperature for 12–28 h, the reaction mixture was evaporated. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH, 20/1) to give 5g–5l.
(2R,3R,4S)-2-(6-Amino-8-(furan-2-yl)-2-(hex-1-yn-1-yl)-9H-purin-9-yl)tetrahydro-thiophene-3,4-diol (5g).
22a was dissolved in NH3/t-BuOH (0.20 M) and stirred for 15 h at 100 °C in a steel bomb. Yield = 44%; white solid; mp 220 °C; (c 0.05, MeOH); UV (MeOH) λmax 238, 320 nm; 1H NMR (400 MHz, MeOD): δ 7.85 (d, J = 1.8 Hz, 1H), 7.23 (d, J = 3.2 Hz, 1H), 6.72 (q, J = 1.7 Hz, 1H), 6.41 (d, J = 7.8 Hz, 1H), 5.65 (dd, J = 3.2, 1.8 Hz, 1H), 4.56 (td, J = 3.4, 1.8 Hz, 1H), 3.73 (dd, J = 11.4, 3.7 Hz, 1H), 2.92 (dd, J = 11.4, 1.8 Hz, 1H), 2.46 (t, J = 6.9 Hz, 2H), 1.60–1.68 (m, 2H), 1.49–1.58 (m, 2H), 0.99 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 155.68, 150.44, 145.49, 145.48, 143.53, 142.14, 118.89, 113.36, 112.20, 85.70, 81.64, 75.98, 72.14, 63.88, 35.65, 29.93, 21.51, 17.97, 13.48. HRMS (FAB): found 400.1443 [calcd for C19H22N5O3S+ (M + H)+ 400.1441]; HPLC purity: 99.50%.
(2R,3R,4S)-2-(8-(Furan-2-yl)-2-(hex-1-yn-1-yl)-6-(methylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5h).
22a and methylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 23 h at 50 °C. Yield = 65%; white solid; mp 210 °C; (c 0.06, MeOH); UV (MeOH) λmax 244, 326 nm; 1H NMR (400 MHz, MeOD): δ 7.82 (s, 1H), 7.19 (d, J = 3.7 Hz, 1H), 6.70 (dd, J = 3.4, 1.6 Hz, 1H), 6.40 (d, J = 7.8 Hz, 1H), 5.65 (dd, J = 7.6, 3.4 Hz, 1H), 4.56 (q, J = 2.8 Hz, 1H), 3.73 (dd, J = 11.5, 3.7 Hz, 1H), 3.10 (s, 3H), 2.92 (dd, J = 11.5, 1.4 Hz, 1H), 2.47 (t, J = 6.9 Hz, 2H), 1.62–1.69 (m, 2H), 1.49–1.59 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H); 13C NMR (200 MHz, MeOD): δ 157.08, 148.45, 147.27, 145.83, 144.92, 121.48, 115.62, 113.89, 88.49, 82.80, 79.02, 75.52, 66.78, 37.49, 32.38, 23.94, 20.35, 14.76. HRMS (FAB): found 414.1600 [calcd for C20H24N5O3S+ (M + H)+ 414.1607]; HPLC purity: 99.00%.
(2R,3R,4S)-2-(8-(Furan-2-yl)-2-(hex-1-yn-1-yl)-6-((3-iodobenzyl)-amino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5i).
22a and 3-iodobenzylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 24 h at 50 °C. Yield = 79%; white solid; mp 238 °C; (c 0.07, MeOH); UV (MeOH) λmax 246, 325 nm; 1H NMR (400 MHz, DMSO-d6): δ 8.62 (s, 1H), 8.03 (d, J = 1.4 Hz, 1H), 7.75 (s, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.14 (d, J = 3.2 Hz, 1H), 7.11 (d, J = 7.8 Hz, 1H), 6.79 (dd, J = 3.2, 1.8 Hz, 1H), 6.20 (d, J = 7.8 Hz, 1H), 5.47 (d, J = 6.4 Hz, 1H), 5.38–5.41 (m, 2H), 4.63 (s, 2H), 4.38 (s, 1H), 3.48 (dd, J = 11.2, 3.4 Hz, 1H), 2.81 (d, J = 10.5 Hz, 1H), 2.44 (t, J = 7.1 Hz, 2H), 1.51–1.59 (m, 2H), 1.40–1.49 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 153.75, 149.88, 145.60, 145.33, 143.51, 142.50, 142.26, 136.05, 135.45, 130.51, 126.78, 119.32, 113.51, 112.26, 94.74, 86.10, 81.90, 76.12, 72.15, 63.94, 42.26, 35.69, 29.91, 21.56, 18.06, 13.53. HRMS (FAB): found 616.0879 [calcd for C26H27IN5O3S+ (M + H)+ 616.0876]; HPLC purity: 95.75%.
(2R,3R,4S)-2-(6-Amino-2-(hex-1-yn-1-yl)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydro-thiophene-3,4-diol (5j).
22b was dissolved in NH3/t-BuOH (0.20 M) and stirred for 15 h at 100 °C in a steel bomb. Yield = 83%; white solid; mp 197 °C; (c 0.15, MeOH); UV (MeOH) λmax 236, 319 nm; 1H NMR (400 MHz, DMSO-d6): δ 7.88 (d, J = 4.1 Hz, 1H), 7.64 (d, J = 3.2 Hz, 1H), 7.45 (s, 2H), 7.31 (dd, J = 5.1, 3.7 Hz, 1H), 6.05 (d, J = 7.8 Hz, 1H), 5.43–5.49 (d, J = 6.8 Hz, 1H), 5.49 (d, J = 6.8 Hz, 1H), 5.46 (qd, J = 7.2, 2.8 Hz, 1H), 5.35 (d, J = 3.7 Hz, 1H), 4.37 (d, J = 1.8 Hz, 1H), 3.47 (dd, J = 11.0, 3.2 Hz, 1H), 2.80 (d, J = 10.6 Hz, 1H), 2.43 (t, J = 7.1 Hz, 2H), 1.50–1.58 (m, 2H), 1.39–1.48 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H); 13C NMR (200 MHz, DMSO-d6): δ 155.46, 150.78, 145.56, 145.29, 130.90, 129.99, 128.97, 128.21, 118.73, 85.60, 81.63, 75.58, 71.92, 63.94, 35.56, 29.89, 21.48, 17.94, 13.44. HRMS (FAB): found 416.1219 [calcd for C19H22N5O2S2+ (M + H)+ 416.1215]; HPLC purity: 99.56%.
(2R,3R,4S)-2-(2-(Hex-1-yn-1-yl)-6-(methylamino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5k).
22b and methylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 28 h at 50 °C. Yield = 61%; white solid; mp 238 °C; (c 0.04, DMSO); UV (MeOH) λmax 242, 326 nm; 1H NMR (400 MHz, DMSO-d6): δ 7.89 (s, 1H), 7.87 (dd, J = 5.0, 0.9 Hz, 1H), 7.64 (dd, J = 3.7, 0.9 Hz, 1H), 7.31 (dd, J = 5.0, 3.7 Hz, 1H), 6.06 (d, J = 8.2 Hz, 1H), 5.51 (d, J = 6.4 Hz, 1H), 5.42–5.46 (m, 1H), 5.37 (d, J = 3.2 Hz, 1H), 4.37 (d, J = 1.4 Hz, 1H), 3.48 (dd, J = 11.2, 3.4 Hz, 1H), 2.92 (s, 3H), 2.81 (dd, J = 11.2, 1.1 Hz, 1H), 2.45 (t, J = 7.3 Hz, 2H), 1.53–1.60 (m, 2H), 1.40–1.49 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (200 MHz, MeOD): δ 154.32, 149.77, 145.37, 145.27, 130.96, 129.96, 128.91, 128.24, 119.31, 85.72, 81.97, 75.70, 71.95, 63.95, 35.59, 29.93, 27.08, 21.59, 18.03, 13.47. HRMS (FAB): found 430.1372 [calcd for C20H24N5O2S2+ (M + H)+ 430.1371]; HPLC purity: 99.17%.
(2R,3R,4S)-2-(2-(Hex-1-yn-1-yl)-6-((3-iodobenzyl)amino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5l).
22b and 3-iodobenzylamine hydrochloride (1.50 equiv) were dissolved in EtOH (0.20 M) and stirred for 20 h at 75 °C. Yield = 78%; white solid; mp 236 °C; (c 0.11, DMSO); UV (MeOH) λmax 248, 326 nm; 1H NMR (400 MHz, DMSO-d6): δ 8.53 (s, 1H), 7.89 (d, J = 5.0 Hz, 1H), 7.76 (s, 1H), 7.65 (d, J = 3.2 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.36 (d, J = 5.5 Hz, 1H), 7.31 (t, J = 4.1 Hz, 1H), 7.12 (t, J = 7.8 Hz, 1H), 6.07 (d, J = 8.2 Hz, 1H), 5.52 (d, J = 5.0 Hz, 1H), 5.44 (t, J = 3.0 Hz, 1H), 5.38 (s, 1H), 4.62 (s, 2H), 4.37 (s, 1H), 3.47 (dd, J = 11.0, 2.7 Hz, 1H), 2.81 (d, J = 11.0 Hz, 1H), 2.44 (t, J = 7.1 Hz, 2H), 1.52–1.59 (m, 2H), 1.40–1.49 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 153.59, 150.29, 145.80, 145.23, 142.58, 136.12, 135.49, 130.92, 130.55, 130.21, 129.13, 128.39, 126.87, 119.23, 94.77, 86.12, 81.96, 75.77, 72.01, 64.06, 54.98, 42.32, 35.67, 29.95, 21.62, 18.10, 13.58. HRMS (FAB): found 632.0653 [calcd for C26H27IN5O2S2+ (M + H)+ 632.0651].
9-((2R,3R,4S)-3,4-Bis((tert-butyldimethylsilyl)oxy)-tetrahydrothiophen-2-yl)-6-chloro-2,8-diiodo-9H-purine (23).
To a stirred solution of 2,6-dichloro-8-iodopurine (19) (581 mg, 1.84 mmol) in CH3CN (10.0 mL) was dropwise added BSA (2.4 mL, 9.83 mmol) at room temperature under N2, and the reaction mixture was heated at 60 °C until obtaining a clear solution. To the reaction mixture was dropwise added 16 (500 mg, 1.23 mmol) in CH3CN (5.0 mL) and TMSOTf (0.22 mL, 1.23 mmol). After being stirred at 75 °C for 1.5 h, the reaction mixture was cooled to room temperature and evaporated. The residue was purified by column chromatography (silica gel, hexanes/EtOAc, 10/1) to give 23 (400 mg, 54%) as a colorless syrup: (c 37.53, MeOH); UV (MeOH) λmax 229, 286 nm; 1H NMR (400 MHz, DMSO-d6): δ 5.98 (d, J = 7.2 Hz, 1H), 5.08 (dd, J = 7.2, 2.8 Hz, 1H), 4.64 (dd, J = 5.2, 2.8 Hz, 1H), 3.43 (dd, J = 11.2, 3.2 Hz, 1H), 2.78 (dd, J = 11.6, 2.8 Hz, 1H), 0.91 (s, 9H), 0.62 (s, 9H), 0.11 (s, 3H), 0.09 (s, 3H), −0.1 (s, 3H), −0.53 (s, 3H); 13C NMR (200 MHz, DMSO-d6): δ 152.9, 150.5, 148.3, 133.5, 128.2, 77.1, 73.3, 66.5, 35.4, 25.6, 25.2, 17.7, 17.3, −4.6, −4.7, −4.7, −5.8; HRMS (FAB): found 661.0459 [calcd for C21H36Cl2IN4O2SSi2+ (M + H)+ 661.0451]; Anal. Calcd for C21H35ClI2N4O2SSi2: C, 33.49; H, 4.68; N, 7.44. Found: C, 33.11; H, 5.08; N, 7.19.
General Procedure for Stille Coupling for the Preparation of 24a and 24b.
To a solution of 23 (2.90 g, 4.38 mmol) in anhydrous THF (0.10 M) was added bis(triphenylphosphine)palladium(II) dichloride (0.10 equiv), 2-(tributylstannyl)furan (1.50 equiv, for 24a), or 2-(tributylstannyl)thiophene (1.50 equiv, for 24b) at room temperature under N2. After being heated reflux with stirring for 24 h, the reaction mixture was cooled to room temperature and evaporated. The residue was purified by column chromatography to give 24a or 24b.
9-((2R,3R,4S)-3,4-Bis((tert-butyldimethylsilyl)oxy)-tetrahydrothiophen-2-yl)-2,6-dichloro-8-(furan-2-yl)-9H-purine (24a).
24a was isolated by column chromatography (silica gel, hexanes/EtOAc, 20/1). Yield = 71%; colorless oil; (c 0.045, MeOH); UV (MeOH) λmax 322 nm; 1H NMR (400 MHz, CDCl3): δ 7.66–7.69 (m, 1H), 7.38 (d, J = 3.2 Hz, 1H), 6.65–6.66 (m, 2H), 5.32 (dd, J = 6.8, 2.8 Hz, 1H), 4.56–4.60 (m, 1H), 3.57 (dd, J = 10.8, 3.2 Hz, 1H), 2.86 (dd, J = 11.2, 3.2 Hz, 1H), 0.96 (s, 9H), 0.65 (s, 9H), 0.16 (s, 3H), 0.14 (s, 9H), −0.11 (s, 3H), −0.52 (s, 3H); 13C NMR (150 MHz, MeOD): δ 155.3, 152.8, 151.5, 149.7, 148.0, 144.3, 132.6, 118.6, 113.8, 79.3, 75.4, 65.8, 36.8, 26.4, 26.0, 19.0, 18.6, −4.2, −4.2, −4.3, −5.5; HRMS (FAB): found 601.1652 [calcd for C25H39Cl2N4O3SSi2+ (M + H)+ 601.1651]; Anal. Calcd for C25H38Cl2N4O3SSi2: C, 49.90; H, 6.37; N, 9.31. Found: C, 49.94; H, 6.36; N, 9.37.
9-((2R,3R,4S)-3,4-Bis((tert-butyldimethylsilyl)oxy)-tetrahydrothiophen-2-yl)-2,6-dichloro-8-(thiophen-2-yl)-9H-purine (24b).
24b was isolated by column chromatography (silica gel, hexanes/EtOAc, 40/1). Yield = 56%; colorless oil; (c 0.10, DMSO); UV (MeOH) λmax 253, 315 nm; 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 4.4 Hz, 1H), 7.65 (d, J = 5.2 Hz, 1H), 7.19 (dd, J = 5.2, 4.0 Hz, 1H), 6.40 (d, J = 7.6 Hz, 1H), 5.39 (dd, J = 7.6, 2.8 Hz, 1H), 4.52–4.56 (m, 1H), 3.58 (dd, J = 10.8, 2.4 Hz, 1H), 2.84 (dd, J = 10.8, 2.4 Hz, 1H), 0.92 (s, 9H), 0.65 (s, 9H), 0.14 (s, 3H), 0.12 (s, 3H), −0.11 (s, 3H), −0.46 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 154.4, 152.4, 151.9, 151.0, 135.8, 131.5, 131.4, 129.5, 128.3, 77.8, 74.0, 64.4, 36.0, 25.9, 25.7, 18.2, 18.0, −4.2, −4.3, −4.4, −5.2; HRMS (FAB): found 617.1452 [calcd for C25H39Cl2N4O2S2Si2+ (M + H)+ 617.1451]; Anal. Calcd for C25H38Cl2N4O2S2Si2: C, 48.60; H, 6.20; N, 9.07. Found: C, 48.66; H, 6.24; N, 9.02.
General Procedure for Desilylation and N6-Amination for the Synthesis of 5m–5r.
To a solution of 24a or 24b (1.00 equiv) in anhydrous THF (0.10 M) was dropwise added TBAF (2.50 equiv) at room temperature. The reaction mixture was stirred at the same temperature for 1 h and evaporated. The residue was purified by column chromatography (silica gel, hexanes/EtOAc, 5/1 to 3/1) to give a diol intermediate. To a solution of the diol intermediate (1.00 equiv) in EtOH (0.20 M) or t-BuOH (0.20 M) were dropwise added appropriate amines (1.50 equiv to excess) at room temperature under N2. After being stirred at the provided temperature for 15–24 h, the reaction mixture was evaporated. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH, 20/1) to give 5m–5r.
(2R,3R,4S)-2-(6-Amino-2-chloro-8-(furan-2-yl)-9H-purin-9-yl)-tetrahydrothiophene-3,4-diol (5m).
Yield = 56%; pale yellow solid; mp 241 °C; (c 0.045, MeOH); UV (MeOH) λmax 242, 305 nm; 1H NMR (400 MHz, MeOD): δ 7.82 (d, J = 1.8 Hz, 1H), 7.20 (d, J = 2.7 Hz, 1H), 6.70 (dd, J = 3.7, 1.8 Hz, 1H), 6.37 (d, J = 7.8 Hz, 1H), 5.54 (dd, J = 7.3, 3.6 Hz, 1H), 4.54 (dd, J = 5.4, 3.6 Hz, 1H), 3.67 (dd, J = 11.4, 3.2 Hz, 1H), 2.91 (dd, J = 11.4, 1.8 Hz, 1H); 13C NMR (150 MHz, MeOD): δ 158.6, 155.5, 153.5, 147.4, 145.5, 145.2, 120.4, 115.8, 113.9, 79.1, 75.5, 67.0, 37.5; HRMS (FAB): found 354.0416 [calcd for C13H13ClN5O3S+ (M + H)+ 354.0428]; Anal. Calcd for C13H12ClN5O3S: C, 44.13; H, 3.42; N, 19.80. Found: C, 43.91; H, 3.14; N, 19.95.
(2R,3R,4S)-2-(2-Chloro-8-(furan-2-yl)-6-(methylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5n).
Yield = 55%; white solid; mp 224 °C; (c 0.06, MeOH); UV (MeOH) λmax 245, 310 nm; 1H NMR (400 MHz, MeOD): δ 7.80 (d, J = 1.9 Hz, 1H), 7.16 (d, J = 3.1 Hz, 1H), 6.68 (dd, J = 3.7, 1.8 Hz, 1H), 6.36 (d, J = 7.3 Hz, 1H), 5.54 (dd, J = 7.8, 3.6 Hz, 1H), 4.54 (dd, J = 5.0, 3.1 Hz, 1H), 3.67 (dd, J = 11.4, 3.6 Hz, 1H), 3.07 (br s, 3H), 2.91 (dd, J = 11.4, 2.2 Hz, 1H); 13C NMR (200 MHz, MeOD): δ 157.8, 155.8, 152.3, 147.2, 145.7, 144.6, 121.2, 115.6, 113.8, 79.1, 75.5, 66.9, 37.5, 28.4; HRMS (FAB): found 368.0587 [calcd for C14H15ClN5O3S+ (M + H)+ 368.0584]; Anal. Calcd for C14H14ClN5O3S: C, 45.72; H, 3.84; N, 19.04. Found: C, 45.75; H, 4.06; N, 19.39.
(2R,3R,4S)-2-(2-Chloro-8-(furan-2-yl)-6-((3-iodobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5o).
Yield = 64%; white solid; mp 219 °C; (c 0.13, MeOH); UV (MeOH) λmax 248, 311 nm; 1H NMR (800 MHz, DMSO-d6): δ 9.06 (t, J = 6.0 Hz, 1H), 8.03 (s, 1H), 7.75 (s, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.36 (d, J = 7.6 Hz, 1H), 7.12–7.14 (m, 2H), 6.79 (s, 1H), 6.17 (d, J = 7.8 Hz, 1H), 5.48 (d, J = 6.2 Hz, 1H), 5.35 (br s, 1H), 5.30 (br s, 1H), 4.57–4.63 (m, 2H), 4.38 (s, 1H), 3.46 (dd, J = 11.3, 3.4 Hz, 1H), 2.80 (d, J = 11.2 Hz, 1H); 13C NMR (200 MHz, DMSO-d6): δ 154.5, 152.5, 150.5, 145.6, 143.1, 142.1, 141.7, 136.0, 135.5, 130.5, 126.8, 118.9, 113.5, 112.2, 94.7, 76.2, 72.0, 64.1, 42.5, 35.6; HRMS (FAB): found 569.9866 [calcd for C20H18ClIN5O3S+ (M + H)+ 569.9864]; Anal. Calcd for C20H17ClIN5O3S: C, 42.16; H, 3.01; N, 12.29. Found: C, 42.55; H, 2.97; N, 12.61.
(2R,3R,4S)-2-(6-Amino-2-chloro-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5p).
Yield = 60%; white solid; mp 235 °C; (c 0.205, MeOH); UV (MeOH) λmax 246, 303 nm; 1H NMR (600 MHz, MeOD): δ 7.73 (dd, J = 5.0, 0.9 Hz, 1H), 7.71 (dd, J = 3.6, 0.9 Hz, 1H), 7.25 (dd, J = 4.9, 3.6 Hz, 1H), 6.20 (d, J = 7.3 Hz, 1H), 5.56 (dd, J = 7.8, 3.6 Hz, 1H), 4.52–4.53 (m, 1H), 3.67 (dd, J = 11.4, 3.1 Hz, 1H), 2.91 (dd, J = 11.4, 1.8 Hz, 1H); 13C NMR (200 MHz, MeOD): δ 158.5, 155.3, 153.9, 148.7, 132.2, 131.77, 131.74, 130.0, 120.4, 78.8, 75.4, 66.9, 37.5; HRMS (FAB): found 370.0194 [calcd for C13H13ClN5O2S2+ (M + H)+ 370.0199]; Anal. Calcd for C13H12ClN5O2S2: C, 42.23; H, 3.29; N, 18.95. Found: C, 42.61; H, 3.01; N, 18.91.
(2R,3R,4S)-2-(2-Chloro-6-(methylamino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5q).
Yield = 58%; white solid; mp 236 °C; (c 0.10, MeOH); UV (MeOH) λmax 249, 311 nm; 1H NMR (400 MHz, MeOD): δ 7.71 (dd, J = 5.0, 1.3 Hz, 1H), 7.68 (dd, J = 3.9, 0.9 Hz, 1H), 7.24 (dd, J = 5.0, 3.6 Hz, 1H), 6.19 (d, J = 7.3 Hz, 1H), 5.56 (dd, J = 7.3, 3.2 Hz, 1H), 4.52–4.53 (m, 1H), 3.67 (dd, J = 11.4, 3.6 Hz, 1H), 3.06 (br s, 3H), 2.90 (dd, J = 11.4, 1.8 Hz, 1H); 13C NMR (200 MHz, MeOD): δ 160.3, 157.8, 155.8, 152.8, 148.1, 132.5, 131.5, 129.9, 121.0, 78.8, 75.4, 66.9, 37.4, 28.4; HRMS (FAB): found 384.0349 [calcd for C14H15ClN5O2S2+ (M + H)+ 384.0356]; Anal. Calcd for C14H14ClN5O2S2: C, 43.80; H, 3.68; N, 18.24. Found: C, 43.81; H, 3.60; N, 17.99.
(2R,3R,4S)-2-(2-Chloro-6-((3-iodobenzyl)amino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5r).
Yield = 55%; white solid; mp 207 °C; (c 0.12, MeOH); UV (MeOH) λmax 252, 310 nm; 1H NMR (6 00 MHz, MeOD): δ 7.79 (br s, 1H), 7.71 (d, J = 5.0 Hz, 1H), 7.69 (d, J = 3.6 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.24 (dd, J = 5.0, 3.7 Hz, 1H), 7.09 (t, J = 7.8 Hz, 1H), 6.20 (d, J = 7.8 Hz, 1H), 5.57 (dd, J = 7.8, 3.6 Hz, 1H), 4.66–4.74 (m, 2H), 4.51–4.53 (m, 1H), 3.67 (dd, J = 11.4, 3.6 Hz, 1H), 2.90 (dd, J = 11.4, 1.8 Hz, 1H); 13C NMR (200 MHz, DMSO-d6): δ 154.4, 152.3, 150.9, 145.6, 141.8, 136.1, 135.5, 130.5, 130.4, 130.2, 129.2, 128.2, 126.8, 118.8, 94.7, 75.9, 71.8, 64.1, 42.5, 35.6; HRMS (FAB): found 585.9620 [calcd for C20H18ClIN5O2S2+ (M + H)+ 585.9635]; Anal. Calcd for C20H17ClIN5O2S2: C, 41.00; H, 2.92; N, 11.95. Found: C, 41.40; H, 2.50; N, 11.55.
General Procedure for N2-Amination for the Synthesis of 5s-5v.
To a solution of 5m, 5n, 5p, and 5q in anhydrous 1-butanol (0.10 M) was dropwise added N,N-diisopropylethylaminetriethylamine (10.00 equiv) followed by tyramine (5.00 equiv) at room temperature. After being heated with microwave at 180 °C for 2 h, the reaction mixture was cooled to room temperature and evaporated. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH, 25/1) to give 5s–5v.
(2R,3R,4S)-2-(6-Amino-8-(furan-2-yl)-2-((4-hydroxyphenethyl)-amino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5s).
Yield = 63%; yellow solid; mp 232 °C; (c 0.10, MeOH); UV (MeOH) λmax 228, 324 nm; 1H NMR (800 MHz, MeOD): δ 7.75 (d, J = 1.2 Hz, 1H), 7.05 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 3.2 Hz, 1H), 6.69–6.71 (m, 2H), 6.66 (dd, J = 3.4, 1.8 Hz, 1H), 6.27 (d, J = 7.2 Hz, 1H), 5.72–5.75 (m, 1H), 4.53 (dd, J = 5.6, 3.3 Hz, 1H), 3.59 (dd, J = 11.5, 3.6 Hz, 1H), 3.56 (t, J = 7.2 Hz, 2H), 2.87 (dd, J = 11.5, 2.2 Hz, 1H), 2.83 (t, J = 7.2 Hz, 2H); 13C NMR (200 MHz, DMSO-d6): δ 158.7, 156.0, 155.4, 154.8, 144.3, 144.2, 138.0, 132.1, 130.3, 130.0, 129.3, 115.0, 114.9, 111.7, 111.3, 79.1, 78.9, 75.7, 72.6, 35.7, 34.5; HRMS (FAB): found 455.1439 [calcd for C21H23N6O4S+ (M + H)+ 455.1435]; Anal. Calcd for C21H22N6O4S: C, 55.49; H, 4.88; N, 18.49. Found: C, 55.51; H, 4.85; N, 18.51.
(2R,3R,4S)-2-(8-(Furan-2-yl)-2-((4-hydroxyphenethyl)amino)-6-(methylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5t).
Yield = 65%; pale yellow solid; mp 262 °C; (c 8.67, MeOH); UV (MeOH) λmax 219, 287, 327 nm; 1H NMR (800 MHz, MeOD): δ 7.72 (d, J = 1.2 Hz, 1H), 7.05 (d, J = 8.4 Hz, 2H), 6.99 (d, J = 3.1 Hz, 1H), 6.70–6.71 (m, 2H), 6.63 (dd, J = 3.4, 1.8 Hz, 1H), 6.26 (d, J = 7.2 Hz, 1H), 5.73 (br s, 1H), 4.53 (dd, J = 3.2, 5.6 Hz, 1H), 3.60 (dd, J = 11.4, 3.5 Hz, 1 H), 3.58 (s, 1H), 3.57 (s, 1H), 3.02 (br s, 3H), 2.87 (dd, J = 11.5, 2.1 Hz, 1H), 2.83 (t, J = 7.4 Hz, 2H); 13C NMR (200 MHz, DMSO-d6): δ 158.7, 155.4, 155.3, 155.2, 151.4, 144.4, 144.2, 137.6, 130.0, 129.3, 115.0, 114.3, 111.7, 111.1, 75.8, 72.6, 63.9, 62.7, 43.1, 35.7, 34.6, 26.8; HRMS (FAB): found 469.1670 [calcd for C22H25N6O4S+ (M + H)+ 469.1658]; Anal. Calcd for C22H24N6O4S: C, 56.40; H, 5.16; N, 17.94. Found: C, 56.12; H, 5.00; N, 17.86.
(2R,3R,4S)-2-(6-Amino-2-((4-hydroxyphenethyl)amino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5u).
Yield = 66%; pale yellow solid; mp 165 °C; (c 0.185, MeOH); UV (MeOH) λmax 231, 324 nm; 1H NMR (800 MHz, DMSO-d6): δ 9.15 (s, 1H), 7.74 (dd, J = 5.0, 0.9 Hz, 1H), 7.48 (dd, J = 3.4, 0.7 Hz, 1H), 7.24 (dd, J = 5.1, 3.6 Hz, 1H), 7.02 (d, J = 7.9 Hz, 2H), 6.79 (br s, 2H), 6.68–6.69 (m, 2H), 6.39 (t, J = 5.7 Hz, 1H), 5.98 (d, J = 7.6 Hz, 1H), 5.62 (br s, 1H), 5.46 (br s, 1H), 5.26 (br s, 1H), 4.36 (s, 1H), 3.44 (d, J = 10.9 Hz, 1H), 3.41 (dd, J = 14.8, 5.4 Hz, 2H), 2.78 (dd, J = 11.2, 1.7 Hz, 1H), 2.73–2.75 (m, 2H); 13C NMR (200 MHz, DMSO-d6): δ 158.6, 155.9, 155.4, 152.7, 141.1, 132.0, 130.1, 129.38, 129.35, 128.3, 127.9, 127.6, 115.08, 115.05, 113.8, 72.5, 63.9, 62.7, 43.1, 35.7, 34.6; HRMS (FAB): found 471.1268 [calcd for C21H23N6O3S2+ (M + H)+ 471.1273]; Anal. Calcd for C21H22N6O3S2: C, 53.60; H, 4.71; N, 17.86. Found: C, 53.41; H, 4.68; N, 17.94.
(2R,3R,4S)-2-(2-((4-Hydroxyphenethyl)amino)-6-(methylamino)-8-(thiophen-2-yl)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (5v).
Yield = 60%; pale yellow solid; mp 195 °C; (c 3.38, MeOH); UV (MeOH) λmax 219, 331 nm; 1H NMR (800 MHz, DMSO-d6): δ 9.15 (s, 1H), 7.73 (dd, J = 5.1, 0.9 Hz, 1H), 7.47 (d, J = 2.9 Hz, 1H), 7.33 (br s, 1H), 7.24 (dd, J = 5.1, 3.6 Hz, 1H), 7.02 (d, J = 8.2 Hz, 2H), 6.68 (d, J = 8.4 Hz, 2H), 6.54 (br s, 1H), 5.98 (d, J = 7.6 Hz, 1H), 5.62 (br s, 1H), 5.44 (br s, 1H), 5.25 (s, 1H), 4.36 (s, 1H), 3.41–3.45 (m, 3H), 2.89 (br s, 2H), 2.75–2.79 (m, 3H); 13C NMR (200 MHz, DMSO-d6): δ 158.6, 155.4, 155.0, 151.9, 140.7, 132.1, 130.1, 130.1, 129.3, 128.2, 127.9, 127.4, 115.0, 115.0, 114.1, 75.6, 72.5, 63.9, 43.1, 35.7, 34.6, 26.8; HRMS (FAB): found 485.1432 [calcd for C22H25N6O3S2+ (M + H)+ 485.1430]; Anal. Calcd for C22H24N6O3S2: C, 54.53; H, 4.99; N, 17.34. Found: C, 54.31; H, 4.67; N, 17.36.
Molecular Docking Study.
The crystal structures of A2AAR containing the A2AAR agonist (NECA) or antagonists (LJ-4517, ZM-241385) were used for the molecular docking study. (PDB ID: 2YDV, 3EML, 8CU6). The preparation of ligands and proteins and homology modeling of A3AR with the crystal structure of A2AAR (PDB ID: 8CU6) were performed by Maestro 13.0 (Schrödinger, LLC, New York, NY, 2021). The grid generation and ligand docking were progressed in a Glide docking program in Maestro 13.0, and the predicted binding mode was calculated as XP-precision.
Pharmacological Assays.
Cell Culture and Membrane Preparation.
HEK293 cells expressing recombinant hA1AR, hA2AAR, or A3AR were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. Cells were collected by scraping into ice-cold PBS buffer. After homogenization and suspension, cells were centrifuged at 1000g or 10 min, and the pellet was discarded. The suspension was then recentrifuged at 20,000g for 60 min at 4 °C. The resultant pellets were resuspended in buffer containing 3 units/mL adenosine deaminase (from bovine spleen, Worthington Biochemical Corp., Lakewood, NJ) and incubated at 37 °C for 30 min. The aliquots of membrane preparation were kept at −80 °C until further use.
Binding Assays at hA1AR and hA2AAR.
For binding to hA1AR, 50 μL of increasing concentrations of a test ligand and 50 μL of 25 (1.0 nM, PerkinElmer, Boston, MA) were incubated with membrane preparations (40 μg/tube) at 25 °C for 60 min in 50 mM Tris·HCl buffer (pH 7.4) containing 10 mM MgCl2 in a total assay volume of 200 μL. Nonspecific binding was determined using 10 μM of 29. For hA2AAR binding, membrane preparations (20 μg/tube) were incubated at 25 °C for 60 min with a final concentration of 26 (5 nM, American Radiolabeled Chemicals, Inc., St. Louis, MO) in a mixture containing 50 μL of increasing concentrations of a test ligand and 200 μL of 50 mM Tris·HCl, pH 7.4, containing 10 mM MgCl2. 29 (10 μM) was used to define nonspecific binding. The reaction was terminated by filtration with GF/B filters. Filters were placed in scintillation vials containing 5 mL of Hydrofluor scintillation liquid and counted using a PerkinElmer Tricarb 2810TR liquid scintillation counter.
Binding Assays at hA2BAR.
Adenosin A2B receptor competition binding experiments were carried out in a multiscreen GF/C 96-well plate. In each well was incubated 25 μg of membranes from the Euroscreen HEK-A2B cell line and prepared in our laboratory (lot: A009/14–02–2020, protein concentration = 5254.8 μg/mL), 25 nM of 27 (140 Ci/mmol, 1 mCi/mL, PerkinElmer NET974001MC) and compounds studied and standard. Nonspecific binding was determined in the presence of 1000 μM of 29 (Sigma E2397). The reaction mixture (Vt: 250 μL/well) was incubated at 25 °C for 30 min, 200 μL was transferred to GF/C 96-well plate (Millipore, Madrid, Spain) pretreated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 5 mM, bacitracin 100 μg/μL, adenosine deaminase 2 U/mL, pH = 6.5), which was then filtered and washed 4 times with 250 μL wash buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 5 mM, pH = 6.5) before measuring in a microplate β scintillation counter (MicroBeta Trilux, PerkinElmer, Madrid, Spain).
Binding Assay at hA3AR.
Each tube in the binding assay contained 100 μL of membrane suspension (10 μg protein), 50 μL of 28 (0.2 nM, PerkinElmer, Boston, MA), and 50 μL of increasing concentrations of the test ligands in Tris·HCl buffer (50 mM, pH 8.0) containing 10 mM MgCl2. Nonspecific binding was determined using 10 μM of 29. The mixtures were incubated at 25 °C for 60 min. Binding reaction was terminated by filtration through Whatman GF/B filters using a MT-24 cell harvester (Brandel, Gaithersburg, MD, USA). Filters were washed 3 times with 9 mL of ice-cold buffer. Filters were counted using a PerkinElmer Cobra II γ-counter.
Cyclic AMP Accumulation Assay.
Human adenosine receptor functional experiments were carried out in the CHO-A2A cell line and the CHO-A3 cell line for hA2AAR and hA3AR, respectively. The day before the assay, the cells are seeded on the 96 well culture plate (Falcon 353072). The cells are washed with wash buffer [Dulbecco’s modified Eagle’s medium nutrient mixture F-12 ham (Sigma D8062), 25 mM Hepes; pH = 7.4]. Wash buffer is replaced by incubation buffer [Dulbecco’s modified Eagle’s medium nutrient mixture F-12 ham (Sigma D8062), 25 mM Hepes, 30 μM rolipram (Sigma R6520); pH = 7.4]. Test compounds are added and incubated at 37 °C for 15 min. Afterward, 1 μM of 29 (Sigma E2387) is added and incubated at 37 °C for 15 min. Also, for hA3AR, FSK (Sigma F3917) is added and incubated at 37 °C for 5 min. After incubation, the amount of cAMP is determined using the cAMP Biotrak Enzymeimmunoassay (EIA) System Kit (GE Healthcare RPN225).
Statistical Analysis.
Binding and functional parameters were calculated using Prism 5.0 software (GraphPAD, San Diego, CA, USA). IC50 values obtained from competition curves were converted to Ki values using the Cheng–Prusoff equation.32 Data were expressed as mean ± standard error of the mean.
Pharmacokinetic Assay.
For pharmacokinetic studies, CrljOr-i:CD1 male mice were obtained as weanlings of 6 weeks of age upon arrival (provided by DBL Co., Ltd.). This study was reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) of Chaon Co., Ltd. (IACUC number: CE21077). Mice weighed about 31.4 g; animals were 7 weeks old at the start of dosing. For the single pharmacokinetics (i.v.: 2 mg/5 mL/kg; oral: 10 mg/10 mL/kg) of compound 5d in mice, the dosing solution was 5% DMSO/40% PEG 400/55% DW (v/v/v). This study was conducted using a cross-over (n = 3) design. Blood samples (0.1 mL) were collected from the retro-orbital plexus at 5, 15, and 30 min and 1, 2, 4, 8, 12, and 24 h postdose for the i.v. group and 15, 30 min, and 1, 2, 4, 8, 12, and 24 h postdose for the oral group. The collected blood samples were placed in heparinized tubes (5 IU/mL). Plasma was obtained by centrifugation at 5000 rpm for 5 min at 4 °C. Aliquots of plasma (30–40 μL) were transferred to polypropylene tubes and stored at −15 °C until analysis. LC–MS/MS was used to determine the concentration of the compound 5d at plasma. To measure the concentration of compound 5d at plasma, selectivity, linearity, and stability in plasma (at 10 °C for 4 h) were confirmed. In vivo PK data were analyzed using Phoenix WinNonlin software (Phoenix WinNonlin 8.3). A noncompartment model analysis was performed to obtain the following parameters: AUC0–t, C0 or Cmax, Tmax, clearance (CL), volume of distribution (Vss), half-life (t1/2), and bioavailability (F).
In Vivo Tumor Xenograft Model.
All animal experiments were conducted according to the guidelines approved by the Seoul National University Institutional Animal Care and Use Committee (IACUC; permission number: SNU-190408–5). Female mice (BALB/c, aged 4–5 weeks, weighing 18 g) were purchased from the Central Laboratory Animal, Inc. (Seoul, Korea), and they were housed under pathogen-free conditions with a 12 h light–dark schedule. 4T1-Luc cells were then injected subcutaneously into the flanks of mice (1 × 106 cells in 200 μL of 50:50 PBS/Matrigel), and tumors were allowed to grow for 7 days until their volume reached approximately 50 mm3. The mice were randomized into vehicle control and treatment groups (n = 5). Vehicle control (DMSO/cremophor/normal saline = 5:5:90), 5d (10 mg/kg body weight), antimouse PD-L1 (10 mg/kg body weight), or a combination of 5d (10 mg/kg body weight) and antimouse PD-L1 (10 mg/kg body weight) was administered intraperitoneally (i.p.) 3 times per week for 21 days; mice were then euthanized 1 week later. Tumors were excised, weighed, and frozen for further biochemical analysis. Tumor volume was measured using an electronic caliper according to the following formula: tumor volume (mm3) = 3.14 × length × width × height/6. Toxicity was evaluated based on body weight loss. Bioluminescence images of mice were acquired on the final day of the experiment. Mice were anesthetized, positioned in the IVIS (PerkinElmer, Waltham, MA, USA), and imaged 15 min after injection of d-luciferin (150 mg/kg, Gold Biotechnology) resuspended in PBS.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2022R1A6A1A0304624712), the Mid-Career Researcher Programs (NRF-2021R1A2B5B0200154413 and 2021M3E5E308084321) of the National Research Foundation of Korea (NRF), Korea, and in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (ZIADK031117).
ABBREVIATIONS
- AUC
area under the curve
- AR
adenosine receptor
- cAMP
cyclic adenosine-5′-monophosphate
- BSA
bis(trimethylsilyl)-acetamide
- CGS21680
2-[p-(2-carboxyethyl)-phenylethylamino]-5′-N-ethylcar-boxamidoadenosine
- CHO
Chinese hamster ovary
- CKD
chronic kidney disease
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyladenosine
- CYP
cytochrome P450
- DIBAL-H
diisobutylaluminum hydride
- HEK
human embryonic kidney
- hERG
human ether-a-go-go related gene
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- IVIS
in vivo imaging system
- LDA
lithium diisopropylamide
- LiTMP
lithium tetramethylpiperidide
- mAb
monoclonal antibody
- NECA
5′-N-ethylcarboxamidoadenosine
- PBS
phosphate buffered saline
- PD-L1
programmed death-ligand 1
- PKC
protein kinase C
- PLC
phospholipase C
- PPTS
pyridinium p-toluenesulfonate
- pTSA
para-toluene sulfonic acid
- R-PIA
(−)-N6-2-phenylisopropyl adenosine
- SAR
structure—activity relationship
- TBAF
tetra-n-butylammonium fluoride
- TBS
t-butyldimethylsilyl
- thio-Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyl–4′-thioadenosine
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- Tris-HCl
tris(hydroxymethyl)aminomethane hydrochloride
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00806.
Additional in vitro data, X-ray crystallography data, HPLC data of representative final compounds, and 1H and 13C NMR copies of intermediates and all final compounds 5a–5v (PDF)
Coordinate file for the docking model of compound 3a to A2AAR, coordinate file for the docking model of compound 5a and 5j to A2AAR, and coordinate file for the homology docking model of compound 5a and 5j to A3AR (ZIP)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.3c00806
The authors declare no competing financial interest.
Contributor Information
Gibae Kim, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
Xiyan Hou, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; College of Life Science, Dalian Minzu University, Dalian 116600, People’s Republic of China.
Woong Sub Byun, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; Department of Chemical and Systems Biology, Chem-H and Stanford Cancer Institute, Stanford School of Medicine, Stanford University, Stanford, California 94305, United States.
Gyudong Kim, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; College of Pharmacy & Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Republic of Korea.
Dnyandev B. Jarhad, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea
Grim Lee, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
Young Eum Hyun, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
Jinha Yu, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, South Korea.
Chang Soo Lee, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
Shuhao Qu, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
Eugene Warnick, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, and Digestive and Kidney Disease, National Institutes of Health, Bethesda, Maryland 20892, United States.
Zhan-Guo Gao, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, and Digestive and Kidney Disease, National Institutes of Health, Bethesda, Maryland 20892, United States.
Ji Yong Kim, Future Medicine Company Limited, Seoul 06665, Republic of Korea.
Seunghee Ji, HK Inno.N Corporation, Seoul 04551, Republic of Korea.
Hyunwoo Shin, HK Inno.N Corporation, Seoul 04551, Republic of Korea.
Jong-Ryoul Choi, HK Inno.N Corporation, Seoul 04551, Republic of Korea.
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, and Digestive and Kidney Disease, National Institutes of Health, Bethesda, Maryland 20892, United States
Hyuk Woo Lee, Future Medicine Company Limited, Seoul 06665, Republic of Korea.
Sang Kook Lee, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
Lak Shin Jeong, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; Future Medicine Company Limited, Seoul 06665, Republic of Korea.
REFERENCES
- (1).Sung H; Ferlay J; Siegel RL; Laversanne M; Soerjomataram I; Jemal A; Bray F Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J. Clin 2021, 71, 209–249. [DOI] [PubMed] [Google Scholar]
- (2).(a) Waldman AD; Fritz JM; Lenardo MJ A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol 2020, 20, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Darvin P; Toor SM; Sasidharan Nair V; Elkord E Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med 2018, 50 (12), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Marin-Acevedo JA; Kimbrough EO; Lou Y Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol 2021, 14, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun 2020, 11, 3801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johnson DB; Nebhan CA; Moslehi JJ; Balko JM Immune-checkpoint inhibitors: long-term implications of toxicity. Nat. Rev. Clin. Oncol 2022, 19, 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Liu C; Seeram NP; Ma H Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: a review. Cancer Cell Int. 2021, 21, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dhanak D; Edwards JP; Nguyen A; Tummino PJ Small-Molecule Targets in Immuno-Oncology. Cell Chem. Biol 2017, 24, 1148–1160. [DOI] [PubMed] [Google Scholar]
- (6).Ohta A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol 2016, 7, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Haskó G; Linden J; Cronstein B; Pacher P Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discovery 2008, 7 (9), 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sevigny CP; Li L; Awad AS; Huang L; McDuffie M; Linden J; Lobo PI; Okusa MD Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J. Immunol 2007, 178 (7), 4240–4249. [DOI] [PubMed] [Google Scholar]
- (9).(a) Leone RD; Lo YC; Powell JD A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J 2015, 13, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mediavilla-Varela M; Castro J; Chiappori A; Noyes D; Hernandez DC; Allard B; Stagg J; Antonia SJ A Novel Antagonist of the Immune Checkpoint Protein Adenosine A2a Receptor Restore Tumor Infiltrating Lymphocyte Activity in the Context of the Tumor Microenvironment. Neoplasia 2017, 19 (7), 530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yu F; Zhu C; Xie Q; Wang Y Adenosine A2A Receptor Antagonists for Cancer Immunotherapy. J. Med. Chem 2020, 63 (21), 12196–12212. [DOI] [PubMed] [Google Scholar]
- (11).Shiriaeva A; Park D; Kim G; Lee Y; Hou X; Jarhad DB; Kim G; Yu J; Hyun YE; Kim W; Gao ZG; Jacobson KA; Han GW; Stevens RC; Jeong LS; Choi S; Cherezov V GPCR Agonist-to-Antagonist Conversion: Enabling the Design of Nucleoside Functional Switches for the A2A Adenosine Receptor. J. Med. Chem 2022, 65 (17), 11648–11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lee HW; Son SH; Kang JY; Jang HJ; Ahn SY; Kim JW; Byun JS; Ji SH; Kim SH; Choi JR Adenosine derivative having antagonistic action on A2a and A3 adenosine receptors and method for preparing same. WO 2022203427 A1, September 29, 2022.
- (13).Kim HO; Ji X.-d.; Siddiqi SM; Olah ME; Stiles GL; Jacobson KA 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3-adenosine receptors. J. Med. Chem 1994, 37, 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Jeong LS; Jin DZ; Kim HO; Shin DH; Moon HR; Gunaga P; Chun MW; Kim Y-C; Melman N; Gao Z-G; Jacobson KA N6-Substituted D-4′-thioadenosine-5′-methyluronamides: potent and selective agonists at the human A3 adenosine receptor. J. Med. Chem 2003, 46, 3775–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeong LS; Lee HW; Jacobson KA; Kim HO; Shin DH; Lee JA; Gao Z-G; Lu C; Duong HT; Gunaga P; Lee SK; Jin DZ; Chun MW; Moon HR Structure-activity relationships of 2-chloro-N6-substituted-4′-thioadenosine-5′-methyluronamides as highly potent and selective agonists at the human A3 adenosine receptor. J. Med. Chem 2006, 49, 273–281. [DOI] [PubMed] [Google Scholar]; (c) Choi WJ; Lee HW; Kim HO; Chinn M; Gao Z-G; Patel A; Jacobson KA; Moon HR; Jung YH; Jeong LS Design and synthesis of N6-substituted-4′-thioadenosine-5′-uronamides as potent and selective human A3 adenosine receptor agonists. Bioorg. Med. Chem 2009, 17, 8003–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Jeong LS; Choe SA; Gunaga P; Kim HO; Lee HW; Lee SK; Tosh DK; Patel A; Palaniappan KK; Gao Z-G; Jacobson KA; Moon HR Discovery of a new nucleoside template for human A3 adenosine receptor ligands: D-4′-thioadenosine derivatives without 4′-hydroxymethyl group as highly potent and selective antagonists. J. Med. Chem 2007, 50, 3159–3162. [DOI] [PubMed] [Google Scholar]; (b) Jeong LS; Pal S; Choe SA; Choi WJ; Jacobson KA; Gao Z-G; Klutz AM; Hou X; Kim HO; Lee HW; Lee SK; Tosh DK; et al. Structure-activity relationships of truncated D- and L-4′-thioadenosine derivatives as species- independent A3 adenosine receptor antagonists. J. Med. Chem 2008, 51, 6609–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pal S; Choi WJ; Choe SA; Heller CL; Gao Z-G; Chinn M; Jacobson KA; Hou X; Lee SK; Kim HO; Jeong LS Structure-activity relationships of truncated adenosine derivatives as highly potent and selective human A3 adenosine receptor antagonists. Bioorg. Med. Chem 2009, 17, 3733–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Choi WJ; Park JG; Yoo SJ; Kim HO; Moon HR; Chun MW; Jung YH; Jeong LS Syntheses of D- and L-Cyclopentenone derivatives using ring-closing metathesis: Versatile intermediates for the synthesis of D- and L-carbocyclic nucleosides. J. Org. Chem 2001, 66, 6490–6494. [DOI] [PubMed] [Google Scholar]
- (16).Lee J; Hwang I; Lee JH; Lee HW; Jeong LS; Ha H The selective A3AR antagonist LJ-1888 ameliorates UUO-induced tubulointerstitial fibrosis. Am. J. Pathol 2013, 183, 1488–1497. [DOI] [PubMed] [Google Scholar]
- (17).Kim H; Kang JW; Lee S; Choi WJ; Jeong LS; Yang Y; Hong JT; Yoon DY A3 Adenosine receptor antagonist, truncated Thio-Cl-IB-MECA, induces apoptosis in T24 human bladder cancer cells. Anticancer Res. 2010, 30, 2823–2830. [PubMed] [Google Scholar]
- (18).(a) Hou X; Kim HO; Alexander V; Kim K; Choi S; Park S; Lee JH; Yoo LS; Gao Z; Jacobson KA; Jeong LS Discovery of a new human A2A adenosine receptor agonist, truncated 2-hexynyl-4′-thioadenosine. ACS Med. Chem. Lett 2010, 1, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hou X; Majik MS; Kim K; Pyee Y; Lee Y; Alexander V; Chung HJ; Lee HW; Chandra G; Lee JH; Park S.-g.; Choi WJ; Kim HO; Phan K; Gao Z-G; Jacobson KA; Choi S; Lee SK; Jeong LS Structure—activity relationships of truncated C2- or C8-substituted adenosine derivatives as dual acting A2A and A3 adenosine receptor ligands. J. Med. Chem 2012, 55, 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ballesteros JA; Weinstein H [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- (20).(a) Xu F; Wu H; Katritch V; Han GW; Jacobson KA; Gao Z-G; Cherezov V; Stevens RC Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lebon G; Warne T; Edwards PC; Bennett K; Langmead CJ; Leslie AGW; Tate CG Agonist-bound adenosine A2A receptor structure reveal common features of GPCR activation. Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Carpenter B; Lebon G Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol 2017, 8, 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vorbrüggen H; Ruh-Pohlenz C Synthesis of nucleosides. Org. React 2004, 55, 1–630. [Google Scholar]
- (22).Ibrahim N; Chevot F; Legraverend M Regioselective Sonogashira cross-coupling reactions of 6-chloro-2,8-diiodo-9-THP-9H-purine with alkyne derivatives. Tetrahedron Lett. 2011, 52, 305–307. [Google Scholar]
- (23).Kim G; Lee G; Kim G; Seo Y; Jarhad DB; Jeong LS Catalyst-controlled Regioselctive Sonogashira Coupling of 9-Sub-stituted-6-chloro-2,8-diiodopurines. Org. Chem. Front 2022, 9, 5536–5543. [Google Scholar]
- (24).(a) Sonogashira K Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem 2002, 653, 46–49. [Google Scholar]; (b) Chinchilla R; Najera C The Sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]
- (25).Stille JK The palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles. Angew. Chem., Int. Ed. Engl 1986, 25, 508–524. [Google Scholar]
- (26).Qu S; Mulamoottil VA; Nayak A; Ryu S; Hou X; Song J; Yu J; Sahu PK; Zhao LX; Choi S; Lee SK; Jeong LS Design, synthesis, and anticancer activity of C8-substituted-4′-thionucleosides as potential HSP90 inhibitors. Bioorg. Med. Chem 2016, 24, 3418–3428. [DOI] [PubMed] [Google Scholar]
- (27).(a) Robins RK; Godefroi EF; Taylor EC; Lewis LR; Jackson A Purine nucleosides. I. The synthesis of certain 6-substituted-9-(tetrahydro-2-pyranyl)-purines as models of purine deoxynucleosides. J. Am. Chem. Soc 1961, 83, 2574–2579. [Google Scholar]; (b) Cassidy F; Olsen RK; Robins RK Aromaticity in heterocyclic system V. Bromination of certain purines, pyrrolo[3,2-d]pyrimidines and pyrazolo[4,3-d]pyrimidines (1). J. Heterocycl. Chem 1968, 5, 461–465. [Google Scholar]
- (28).Ongini E; Dionisotti S; Gessi S; Irenius E; Fredholm BB Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonist at human adenosine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol 1999, 359, 7–10. [DOI] [PubMed] [Google Scholar]
- (29).Nayak A; Chandra G; Hwang I; Kim K; Hou X; Kim HO; Sahu PK; Roy KK; Yoo J; Lee Y; Cui M; Choi S; Moss SM; Phan K; Gao Z-G; Ha H; Jacobson KA; Jeong LS Synthesis and anti-renal fibrosis activity of conformationally locked truncated 2-hexynyl-N6-substituted-(N)-methano carbanucleosides as A3 adenosine receptor antagonists and partial agonists. J. Med. Chem 2014, 57, 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Beno BR; Yeung KS; Bartberger MD; Pennington LD; Meanwell NA A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem 2015, 58, 4383–4438. [DOI] [PubMed] [Google Scholar]; (b) It also could be observed in the crystal structures of 11b, 22a, and 22b.
- (31).(a) Merighi S; Benini A; Mirandola P; Gessi S; Varani K; Leung E; MacLennan S; Baraldi PG; Borea PA A3 Adenosine Receptors Modulate Hypoxia-Inducible Factor-1A Expression in Human A375 Melanoma Cells. Neoplasia 2005, 7, 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flamant L; Notte N; Ninane N; Raes M; Michiels C Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast cancer cells under hypoxia. Mol. Cancer 2010, 9, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Merighi S; Benini A; Mirandola P; Gessi S; Varani K; Leung E; Maclennan S; Borea PA A3 Adenosine Receptor Activation Inhibits Cell Proliferation via Phosphatidylinositol 3-Kinase/Akt-dependent Inhibition of the Extracellular Signal-regulated Kinase 1/2 Phosphorylation in A375 Human Melanoma Cells. J. Biol. Chem 2005, 280 (20), 19516–19526. [DOI] [PubMed] [Google Scholar]; (d) Brambilla R; Cattabeni F; Ceruti S; Barbieri D; Franceschi C; Kim YC; Jacobson K; Klotz KN; Lohse M; Abbracchio M Activation of the A3 adenosine receptor affects cell cycle progression and cell growth. Naunyn-Schmiedeberg’s Arch. Pharmacol 2000, 361, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yung-Chi C; Prusoff WH Relationship between the inhibition constant (Ki and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.