

Abstract
Key Clinical Message
Cleidocranial dysplasia (CCD) is a rare genetic skeletal disorder with only few cases reported in Africa, mostly based on clinical and radiological findings. We report the first case in Mali, caused by a novel de novo variant in the RUNX2 gene.
Abstract
Cleidocranial dysplasia (CCD) is a rare autosomal dominant skeletal dysplasia characterized by an aplastic/hypoplastic clavicles, patent sutures and fontanels, dental abnormalities and a variety of other skeletal changes. We report a novel de novo variant in the RUNX2 gene causing a severe phenotype of CCD in a Malian girl.
Keywords: cleidocranial dysplasia, de novo genetic variant, Mali, RUNX2, West Africa
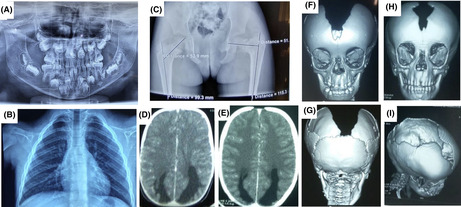
Radiological features of cleidocranial dysplasia. (A) The orthopantomogram at age 8 showing germ retention and polyodontia with supernumerary teeth; (B) Chest X‐rays showing a total absence of the clavicles and (C) Pelvic x‐rays showing a left coxa Vara lesion; Brain CT‐scan showing (D) a dilatation of the occipital horns of the lateral ventricles at the time of diagnosis and (E) 3 years later; 3D reconstruction of the CT‐scan of the skull in (F) anterior view showing patent anterior fontanel, the absence of the upper part of the frontal and the parietal bones, (G) posterior view showing the absence of the parietal bone with preservation of the occipital bone, and (H, I) progression of the cranial bone development after 3 years of evolution with a notable reduction of the bone defect.
1. INTRODUCTION
Cleidocranial dysplasia (CCD) is a rare genetic skeletal disorder with an estimated prevalence of one per million. 1 , 2 It is an autosomal dominant condition, clinically characterized by an aplastic or hypoplastic clavicles, patent sutures and fontanels, dental abnormalities, short stature, and a variety of other skeletal changes. 1 The diagnosis of CCD is established based on clinical features, radiological findings, and genetic factors. 3 , 4 , 5 CCD is caused by genetic variants in the runt‐related transcription factor 2 gene (RUNX2), located at chromosome 6p21 in approximately two‐thirds of cases. 1 , 5 Molecularly, RUNX2 gene plays a crucial role on the differentiation of stem cells into osteoblasts and bones formation. 5 Genetically diagnosed cases have been reported worldwide but only few cases were reported in the African population, mostly from South Africa where a high prevalence has been reported in the mixed ancestry population. 2 We report here the first genetically confirmed case of CCD in Mali, caused by a novel de novo variant in the RUNX2 gene.
2. CASE PRESENTATION
A 20‐month‐old girl was seen in the Neurology Department of the Teaching Hospital of Point “G”, Bamako, Mali for right body side muscle weakness. She was born from a normal pregnancy and a non‐incidental birth. The obstetrical ultrasounds of the three trimesters revealed no abnormalities. There is no consanguinity between her healthy parents (aged 55 and 31 years) and she is the third and youngest child of a sibship with two healthy sisters (Figure 1A). Her mother did not report any teratogenic drugs intake during pregnancy, but noticed a strange softness of the whole upper part of the head of her newborn and was reassured by the healthcare providers that time will address the issue as the child grows up. Four months later, she developed muscle weakness in the right‐side of the body and started walking at 10 months age. Her pediatrician referred her to a rehabilitation center where she underwent physical therapy. With the persisting hemiparesis, parents decided to come to our clinic for a better care. In clinical examination she weighed 9 kg, the head circumference was 43 cm, and facial deformity characterized by a frontal bossing with midline depression and hypertelorism and midfacial hypoplasia were noted (Figure 1B,C). At the age of eight, she was 116 cm with a standard deviation of third percentile (short stature for age). There was a depression in the upper parietal and frontal posterior region measuring 21 × 15 cm but completely recovered with healthy skin and hair. She was cooperative and did not have a language or behavioral disorder. There was a right hemiparesis with a motor strength rated 3/5 in upper and lower limbs, but walked without assistance. The tendon reflexes were brisk in the right side with extensor plantar responses (positive Babinski sign). There were no other neurological anomalies. She had dental anomalies including generalized delayed teeth and unilateral maxillary endo‐alveoli associated with mandibular prognathism (Figure 1D–F). The orthopantomogram confirmed germ retention and polyodontia with supernumerary teeth (Figure 2A). Ophthalmological examination found a right exotropia with no visual impairment. ENT examination did not find any hearing impairment. In addition, cardiac and abdominal ultrasounds were normal. Blood chemistries including total cell count, serum creatinine, alkaline phosphatase and ions were normal except phosphorus levels that were slightly high at 1.84 mmol/L (normal range 0.87–1.50 mmol/L). Spine and hands X‐rays were unremarkable, while chest and pelvic X‐rays revealed bilateral aplastic clavicles and a left coxa Vara deformation (Figure 2B,C). Brain CT‐scan showed a dilatation of the occipital horns of the lateral ventricles that persisted after the 3 years of evolution (Figure 2D,E). The three‐dimensional CT (3D CT) reconstruction of the skull bones showed patent anterior fontanel, the absence of the posterior part of the frontal and the upper part of the parietal bones with normal facial bones and the skull base (Figure 2F,G). Three years later a reduction of the soft tissue from 21 × 15 cm to 14.5 × 10 cm was noticed with a notable progression of bone development (Figure 2H,I). These clinical and radiographic features were consistent with CCD. Sanger sequencing using primers for all the exons of the RUNX2 gene identified a novel heterozygote nonsense de novo variant at position c.925C>T, resulting in a premature stop codon of the protein at residue 309 in exon 6 (c.925C>T; p.Gln309X) in the patient (II.3), segregating with the phenotype in the family (Figure 3A–C). Furthermore, the residue Gln309 is well conserved across a wide range of species (Figure 3D). This variant (p.Gln309*) is absent from public SNP databases including 1000 Genomes, gnomAD/ExAC browser: 0.000000, and dbSNP. This variant (c.925C>T; p.Gln309*) in RUNX2 is predicted to be pathogenic by several in silico prediction tools and according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Table 1).
FIGURE 1.
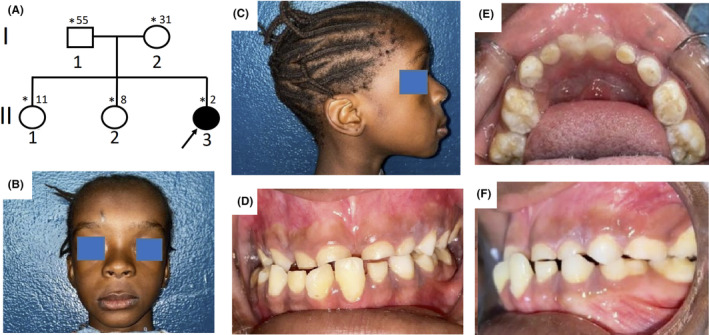
Phenotypic characteristics of cleidocranial dysplasia. (A) Pedigree of the family showing a sporadic case, the black arrow indicates the proband and asterisks those seen in clinic; (B, C) Photos of the patient showing a bossing of the forehead, hypertelorism, midline depression and midfacial hypoplasia; (D–F) Dental images of the patient showing a generalized delayed teething and unilateral maxillary endo‐alveoli associated with mandibular prognathism.
FIGURE 2.
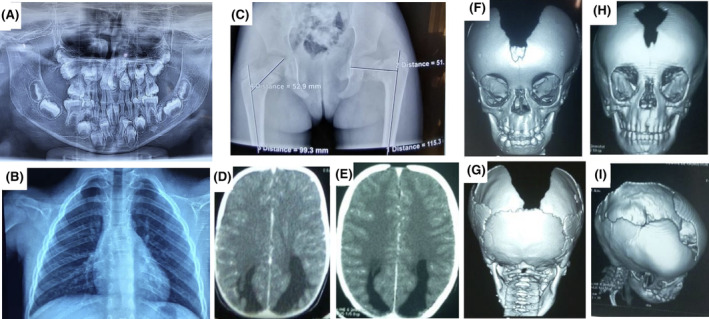
Radiological features of cleidocranial dysplasia. (A) The orthopantomogram at age 8 showing germ retention and polyodontia with supernumerary teeth; (B) Chest X‐rays showing a total absence of the clavicles and (C) Pelvic x‐rays showing a left coxa Vara lesion; Brain CT‐scan showing (D) a dilatation of the occipital horns of the lateral ventricles at the time of diagnosis and (E) 3 years later; 3D reconstruction of the CT‐scan of the skull in (F) anterior view showing patent anterior fontanel, the absence of the upper part of the frontal and the parietal bones, (G) posterior view showing the absence of the parietal bone with preservation of the occipital bone, and (H, I) progression of the cranial bone development after 3 years of evolution with a notable reduction of the bone defect.
FIGURE 3.
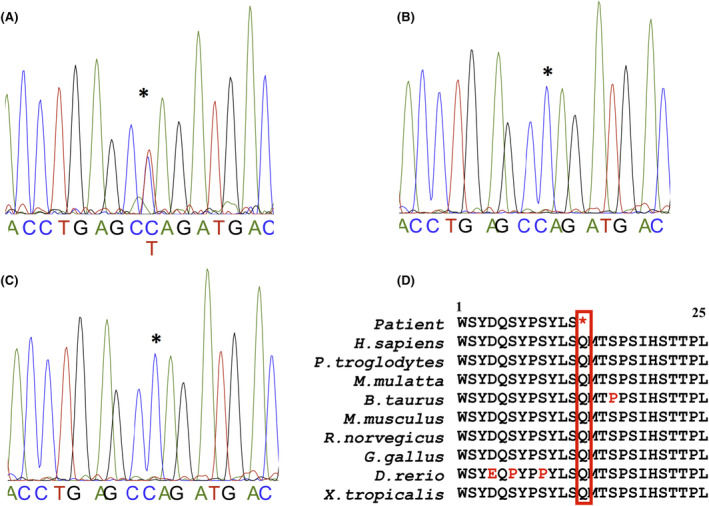
Genetic data. Chromatograms showing (A) the nucleotide “C” to “T” change in the proband and (B, C) the wildtype sequence in the unaffected parents and relatives; (D) portion of the highly conserved region of the protein spanning the Gln309 residue across a wide range of species. “*” Indicates the position of the changed amino acid.
TABLE 1.
Description of the pathogenicity of the variant c.925C>T (p.Gln309*) in the RUNX2 gene.
| Tools | Scores | Indicative prediction |
|---|---|---|
| ACMG classification | PVS1, PS2, PM2, PP1 | Pathogenic |
| CADD | 40 | Pathogenic |
| BayesDel addAF | 0.625 | Pathogenic |
| BayesDel noAF | 0.66 | Pathogenic |
| EIGEN | 1.1911 | Pathogenic |
| EIGEN PC | 1.0745 | Pathogenic |
| FATHMM‐MKL | 0.98 | Pathogenic |
| LRT | 0 | Deleterious |
Over the 6 years of follow up, the disease course was marked by two episodes of generalized tonic and clonic seizures that did not necessitate antiepileptic medication. The first episode occurred in a febrile context related to malaria infection and the second was unspecified. No other abnormalities were noted. Parents were reassured about the possible bone development progression but were advised to protect the patient from activities that may lead head injury and to follow‐up every 6 months.
3. DISCUSSION
CCD is a rare spectrum of autosomal dominant skeletal dysplasia caused by mutations in the RUNX2 gene. To date, more than 200 different mutations have been reported with missense variants being the commonest type. 5 Although CCD is mostly familial, some CCD cases occur sporadically caused by de novo mutations. 5 Classically, CCD manifestations may include aplastic or hypoplastic clavicles, patent sutures and fontanels and dental abnormalities. Dental abnormalities are one of the main features of CCD and are reported in about 93.5% of all cases like in the case of our patient. 6 Also, some phenotype–genotype correlation has been reported about the dental abnormalities. 7 Thus, when the variant is located in the runt domain of the protein, dental symptoms are commonly present when compared to none runt domain mutations as seen in this patient. 7 , 8 In addition, only 10% of the CCD patients appear with bilateral clavicles aplasia confirming the severity of the case we report here caused by early stop codon in the RUNX2 protein. 1 Another particularity presented by this patient is the severe parietal bone dysplasia which has been reported in only few cases. 9 , 10 Furthermore, our patient had a focal neurological deficit and seizures but no intellectual disability. A case with neurological symptoms were previously reported but attributed to secondary complications such as brain injuries, infections or hemorrhage. 11 Brain CT‐scan showed the absence of the upper part of the frontal and the parietal bones and a dilatation of the occipital horns of the lateral ventricles which may be related to focal atrophy. Although, the brain lesions found on the brain CT‐scan did not explain entirely the neurological symptoms, other unrevealed malformations or lesions from perinatal related trauma could be the possible explanation in the case of our patient.
CCD cases reported in the African population were mainly based on the clinical presentation and radiological features. 2 , 12 Only few genetically diagnosed cases were reported in the African population. 10 , 13 We identified a novel de novo stop mutation in the RUNX2 gene associated with CCD, which causes the RUNX2 protein to end its translation earlier than expected leading to shortened and probably non‐functional RUNX2 protein to be expressed.
4. CONCLUSION
This study reports the first genetically confirmed case of CCD caused by a novel de novo variant in the RUNX2 gene in the Malian population and one of the rare cases in Africa. It highlights the phenotypic variability and expands the genetic spectrum and epidemiology of this rare disease.
5. ETHICAL COMPLIANCE
This study was conducted in full compliance of the declaration of Helsinki and the study protocol has been approved by the institutional Ethics Committee of Faculty of Medicine and Dentistry of the University of Sciences, Techniques and Technologies of Bamako, Mali. Written consent and assent forms were obtained from all participants before enrollment and share of data including photographs.
AUTHOR CONTRIBUTIONS
Lassana Cissé: Conceptualization; investigation; validation; writing – original draft; writing – review and editing. Abdoulaye Yalcouyé: Investigation; validation; writing – review and editing. Kadidia Oumar Touré: Investigation; validation; writing – review and editing. Youlouza Coulibaly: Investigation; validation; writing – review and editing. Alassane Baneye Maiga: Investigation; validation; writing – review and editing. Salia Bamba: Investigation; validation; writing – review and editing. Dramane Diallo: Investigation; validation; writing – review and editing. Salimata Diarra: Investigation; validation; writing – review and editing. Abdoulaye Taméga: Investigation; validation; writing – review and editing. Oumou Traoré: Investigation; validation; writing – review and editing. Mahamadou Kotioumbé: Investigation; validation; writing – review and editing. Moussa Aly Sangaré: Validation; writing – review and editing. Hamidou Oumar Ba: Investigation; validation; writing – review and editing. Assiatou Simaga: Investigation; validation; writing – review and editing. Fatogoma Issa Koné: Investigation; validation; writing – review and editing. Oumar Samassekou: Supervision; validation; writing – review and editing. Amadou Koné: Investigation; supervision; validation; writing – review and editing. Cheick Oumar Guinto: Supervision; validation; writing – review and editing. Guida Landouré: Conceptualization; funding acquisition; investigation; supervision; validation; writing – original draft; writing – review and editing.
FUNDING INFORMATION
This work was funded by the National Institute of Neurological Disorders and Stroke, (grant/award number: U01HG007044).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
This work has been supported by the funds U01HG007044 from the National Institute of Neurological Disorders and Stroke, USA, administered by the National Human Genome Research Institute as part of the H3Africa initiative. We thank UCRC for sequencing support and we are deeply grateful to patient and her parents for participating in this study.
Cissé L, Yalcouyé A, Touré KO, et al. A novel de novo variant in the RUNX2 gene causes cleidocranial dysplasia in a Malian girl. Clin Case Rep. 2024;12:e8551. doi: 10.1002/ccr3.8551
DATA AVAILABILITY STATEMENT
Data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet. 1999;36(3):177‐182. [PMC free article] [PubMed] [Google Scholar]
- 2. Roberts T, Stephen L, Beighton P. Cleidocranial dysplasia: a review of the dental, historical, and practical implications with an overview of the south African experience. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115(1):46‐55. doi: 10.1016/j.oooo.2012.07.435 [DOI] [PubMed] [Google Scholar]
- 3. Hermann NV, Hove HD, Jørgensen C, et al. Prenatal 3D ultrasound diagnostics in cleidocranial dysplasia. Fetal Diagn Ther. 2009;25(1):36‐39. doi: 10.1159/000195634 [DOI] [PubMed] [Google Scholar]
- 4. Adhikari A, Shrestha S, Bhattarai P, et al. Cleidocranial dysplasia–a case report of incidentally found and lately diagnosed disorder. Clin Case Reports. 2022;10:e06440. doi: 10.1002/ccr3.6440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Motaei J, Salmaninejad A, Jamali E, et al. Molecular genetics of cleidocranial dysplasia. Fetal Pediatr Pathol. 2021;40(5):442‐454. doi: 10.1080/15513815.2019.1710792 [DOI] [PubMed] [Google Scholar]
- 6. McNamara CM, O'Riordan BC, Blake M, Sandy JR. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography. Dentomaxillofac Radiol. 1999;28:89‐97. doi: 10.1038/sj/dmfr/4600417 [DOI] [PubMed] [Google Scholar]
- 7. Bufalino A, Paranaíba LMR, Gouvêa AF, et al. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Dis. 2012;18:184‐190. doi: 10.1111/j.1601-0825.2011.01862.x [DOI] [PubMed] [Google Scholar]
- 8. Ma D, Wang X, Guo J, Zhang J, Cai T. Identification of a novel mutation of RUNX2 in a family with supernumerary teeth and craniofacial dysplasia by whole‐exome sequencing: a case report and literature review. Medicine (Baltimore). 2018;97:e11328. doi: 10.1097/MD.0000000000011328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cunningham ML, Seto ML, Hing AV, Bull MJ, Hopkin RJ, Leppig KA. Cleidocranial dysplasia with severe parietal bone dysplasia: C‐terminal RUNX2 mutations. Birth Defects Res A Clin Mol Teratol. 2006;76:78‐85. [DOI] [PubMed] [Google Scholar]
- 10. Cardoso BM, Dupont J, Castanhinha S, et al. Cleidocranial dysplasia with severe parietal bone dysplasia: a new (p.Val124Serfs) RUNX2 mutation. Clin Dysmorphol. 2010;19(3):150‐152. doi: 10.1097/MCD.0b013e32833593a1 [DOI] [PubMed] [Google Scholar]
- 11. Ma Y, Zhao F, DanYu D. Cleidocranial dysplasia syndrome with epilepsy: a case report. BMC Pediatr. 2019;19(1):97. doi: 10.1186/s12887-019-1472-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmad AG, Osman M, Awadalkreem F. Full‐mouth rehabilitation of a patient with cleidocranial dysplasia using immediately loaded basal implant‐supported fixed prostheses: a case report. Int J Surg Case Rep. 2019;65:344‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salem Y, Omar Y, Dorsaf S, Sonia M, Samir B, Olfa B. The Pierre Marie‐Sainton syndrome: report of a family. J Oral Health Craniofac Sci. 2019;4:12‐14. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.