

Summary
Individuals with PTEN hamartoma tumor syndrome (PHTS) harbor pathogenic germline PTEN variants that confer a significantly increased lifetime risk of various organ-specific cancers including second primary malignant neoplasms (SMNs). Currently, there are no reliable biomarkers that can predict individual-level cancer risk. Despite the highly promising value of cell-free DNA (cfDNA) as a biomarker for underlying sporadic cancers, the utility of cfDNA in individuals with known cancer-associated germline variants and subclinical cancers remains poorly understood. We perform ultra-low-pass whole-genome sequencing (ULP-WGS) of cfDNA from plasma samples from patients with PHTS and cancer as well as those without cancer. Analysis of cfDNA reveals that patients with PHTS and SMNs have distinct cfDNA size distribution, aberrant genome-wide fragmentation, and differential fragment end motif frequencies. Our work provides evidence that cfDNA profiles may be used as a marker for SMN risk in patients with PHTS.
Keywords: PTEN hamartoma tumor syndrome, PTEN, Cowden syndrome, cell-free DNA, cfDNA, fragmentomics, second primary malignant neoplasm, breast cancer, thyroid cancer
Graphical abstract
Highlights
-
•
Patients with PHTS and multiple primary cancers have distinct cfDNA profiles
-
•
Those with multiple cancers have decreased numbers of longer cfDNA fragments
-
•
Patients with multiple cancers have increased genome-wide cfDNA fragmentation
-
•
Differentially abundant cfDNA end motifs among patients with multiple cancers
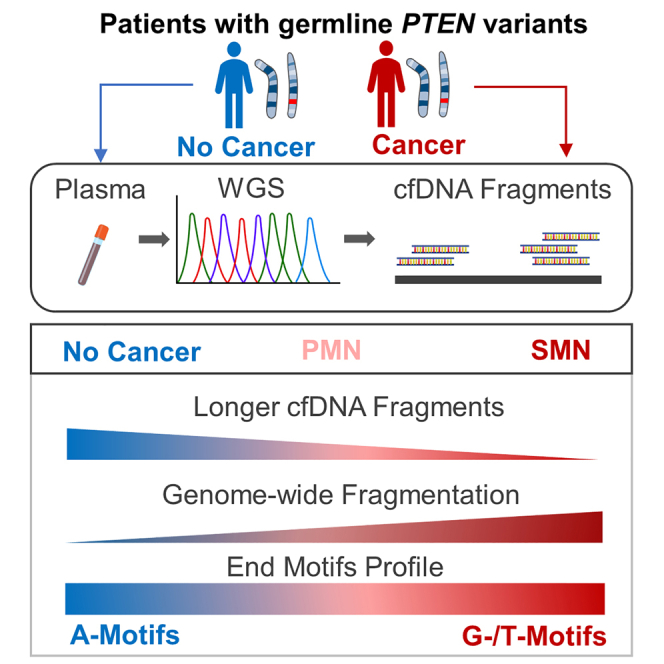
Liu et al. investigate cell-free DNA (cfDNA) fragmentomic features as a marker for cancer risk in patients with germline PTEN variants. Patients with second primary cancers exhibit a reduced proportion of oligo-nucleosome cfDNA fragments, greater genome-wide fragmentation, and increased frequency of G- and T-end motifs with concurrent decrease in A-end motifs.
Introduction
The phosphatase and tensin homolog (PTEN) tumor suppressor gene is one of the most frequently somatically mutated genes in sporadic cancers.1,2 Patients with PTEN hamartoma tumor syndrome (PHTS), an autosomal dominant hereditary cancer syndrome caused by pathogenic germline PTEN variants, have significantly elevated lifetime risks of organ-specific cancers, including breast (91%), endometrial (48%), thyroid (33%), kidney (30%), and colon cancer (17%), as well as melanoma (5%).3 Furthermore, patients with PHTS have more than a 7-fold increased risk of developing second/subsequent primary malignant neoplasms (SMNs), with at least 40% of affected individuals developing SMNs.3,4 The clinical presentation of PHTS is highly heterogeneous, with individuals with identical germline PTEN variants often exhibiting widely disparate phenotypes such as multiple primary cancers versus no cancer. Despite accurate estimates of cancer risk at the cohort level, there is currently no reliable biomarker to accurately predict which individuals with PHTS will develop malignancies.
Circulating cell-free DNA (cfDNA) are extracellular DNA fragments found in the blood and other bodily fluids.5 cfDNA is thought to be primarily released from cells undergoing apoptosis, necrosis, and potentially active secretion. The modal size of cfDNA in plasma is typically 166 bp, corresponding to the length of nucleosome-protected DNA. A portion of cfDNA in the plasma in patients with cancer originates from cancer cells, termed circulating tumor DNA (ctDNA), which exhibits distinct size and fragmentation patterns, often reflecting areas of open chromatin regions (OCRs). Cancer is typified by aberrant gene expression often linked to increased OCRs, which are more accessible to nucleases. This results in the generation of smaller and more variable DNA fragments, with the GC versus AT content reflecting signal from genomic features, including promoters, transcription start sites, repetitive elements, and heterochromatin.6,7,8 However, ctDNA levels vary between cancer types and are found in lower concentrations in early-stage cancer, limiting its utility for early cancer diagnosis.9,10
Recently, innovative approaches that comprehensively analyze the spectrum of cfDNA fragmentation patterns, known as fragmentomics, have demonstrated potential in detecting cancer with high sensitivity and specificity.11,12 The fragmentation pattern of cfDNA depends on various factors, such as nucleosomal organization, chromatin structure, gene expression, and nuclease content, resulting in characteristic signatures based on the tissue of origin. Machine-learning approaches leveraging genome-wide cfDNA fragmentation patterns show promise in identifying over 90% of patients with invasive cancer.13,14 Studies have shown that patients with various cancers have distinct cfDNA fragment end motifs compared to healthy individuals without cancer.15,16 Furthermore, by using cfDNA methylation patterns, machine-learning approaches have been used to accurately identify the tissue of origin for various sporadic cancers with high specificity.17 However, studies evaluating the utility of cfDNA in individuals with hereditary forms of cancer, particularly in the context of subclinical cancer, are lacking. This is particularly clinically useful because inherited cancer syndromes confer the highest risk of cancers as a group and are at risk for SMNs, with patients often undergoing rigorous surveillance regimens. Hence, cfDNA profiles may be useful in detecting malignancies as early as possible, especially in the premorbid phase, and/or help predict which individuals are predisposed to SMNs.
Studies have shown that PTEN not only regulates the PI3K/AKT/mTOR signaling pathway but also plays a vital role in maintaining genomic integrity and regulating DNA damage repair processes within the nucleus.18,19,20,21,22 For example, the C-terminal domain of PTEN interacts with histone H1 to promote chromatin condensation.23 Thus, patients with PHTS predisposed to cancer may have aberrant chromatin architecture characterized by increased OCRs. Furthermore, we have previously shown that patients with PHTS and cancer have distinct germline copy-number variations compared to patients with PHTS without cancer.24 Collectively, this suggests that DNA fragmentation patterns could serve as a reliable biomarker for cancer risk in patients with PHTS and could point to underlying molecular processes that may explain why only certain individuals with PHTS develop cancer.
Therefore, in this retrospective series of prospectively accrued patients with PHTS, we investigated whether plasma cfDNA profiles can be leveraged as a predictive marker of cancer risk in patients with PHTS and cancer, including those with SMNs as well as subclinical cancers, compared to those without cancer.
Results
Patient characteristics and PTEN variant spectrum
A series of 99 adult patients with confirmed germline PTEN variants were included in our study, with 49 having a cancer diagnosis and 50 without cancer. The median (interquartile range [IQR]) age at plasma draw was 46 years (36–52), with 70 females (71%) and 29 males (29%) (Table 1). The median age of first primary cancer diagnosis was 43 years (35–49). The median (IQR) Cleveland Clinic (CC) score, a semiquantitative surrogate for PHTS phenotypic burden, was 25 (15–34) with no difference between the two groups. The majority of the patients were self-reported as White (70/99, 72%). Importantly, the PTEN variant spectra were similar across individuals within the cancer and no cancer groups (Table S1). Because breast and thyroid cancers are the most common PHTS component malignancies, with thyroid cancer having the youngest age at onset, we focused on patients with plasma samples archived within 2 years of a breast (n = 35) and/or thyroid (n = 15) cancer diagnosis.
Table 1.
Baseline characteristics of patients with PHTS
| Characteristic | N | Overall, n = 99a | No cancer, n = 50a | PMN, n = 23a | SMN, n = 26a | pb |
|---|---|---|---|---|---|---|
| Gender (%) | 99 | – | – | – | – | <0.001 |
| Female | – | 70 (71) | 26 (52) | 19 (83) | 25 (96) | – |
| Male | – | 29 (29) | 24 (48) | 4 (17) | 1 (4) | – |
| Age, y | – | – | – | – | – | – |
| Study enrollment | 99 | 45 (36–52) | 41 (32–52) | 46 (38–49) | 50 (44–58) | 0.005 |
| First primary cancer diagnosis | 49 | 43 (35–49) | – | 45 (38–47) | 42 (34–50) | 0.8 |
| Race (%) | 97 | – | – | – | – | 0.7 |
| White | – | 70 (72) | 31 (62) | 19 (86) | 20 (80) | – |
| Black | – | 2 (2) | 1 (2) | 1 (5) | 0 (0) | – |
| Asian | – | 2 (2) | 2 (4) | 0 (0) | 0 (0) | – |
| Unknown | – | 12 (12) | 8 (16) | 1 (4.5) | 3 (12) | – |
| Other | – | 11 (11) | 7 (14) | 2 (10) | 2 (8) | – |
| Baseline CC score | 96 | 25 (15–34) | 23 (15–33) | 23 (13–35) | 25 (16–35) | 0.9 |
| First cancer (%) | 49 | – | – | – | – | 0.014 |
| Breast | – | 27 (55) | – | 13 (57) | 14 (54) | – |
| Thyroid | – | 14 (29) | – | 10 (43) | 4 (15) | – |
| Endometrial | – | 6 (12) | – | 0 (0) | 6 (23) | – |
| Kidney | – | 0 (0) | – | 0 (0) | 0 (0) | – |
| Colon | – | 1 (2) | – | 0 (0) | 1 (4) | – |
| Melanoma | – | 1 (2) | – | 0 (0) | 1 (4) | – |
| Second cancer (%) | 26 | – | – | – | – | <0.001 |
| Breast | – | 15 (31) | – | – | 15 (58) | – |
| Thyroid | – | 2 (4) | – | – | 2 (8) | – |
| Endometrial | – | 2 (4) | – | – | 2 (8) | – |
| Kidney | – | 2 (4) | – | – | 2 (8) | – |
| Colon | – | 1 (2) | – | – | 1 (4) | – |
| Melanoma | – | 1 (2) | – | – | 1 (4) | – |
| Other | – | 3 (6) | – | – | 3 (12) | – |
| Plasma draw timing | 49 | 7 (2–17) | – | 7 (1–15) | 7 (3–23) | 0.3 |
| Postdiagnosis | – | 9 (6–20) | – | 8.5 (6–16) | 8.5 (6–30) | – |
| Prediagnosis | – | 2 (1–3) | – | 1 (1–2) | 2.5 (2–3) | – |
IQR, interquartile range; PMN, individuals diagnosed with only a single primary malignant neoplasm; SMN, individuals diagnosed with second/subsequent primary malignant neoplasms.
n (%); median (IQR).
Pearson’s chi-squared test; Kruskal-Wallis rank-sum test; Fisher’s exact test.
Forty (82%) patients had plasma drawn after their breast or thyroid cancer diagnosis. The median time difference for patients who had plasma drawn after their cancer diagnosis was 9 months (6–20). In contrast, nine patients (9%) had plasma drawn before their cancer diagnosis. The median time difference for patients who had plasma drawn before their cancer diagnosis was 2 months (1–3). Five patients had plasma drawn before the diagnosis of their first primary malignancy, whereas four patients had plasma drawn before the diagnosis of their SMNs. Many patients had a prior or subsequent cancer diagnosis in addition to their breast and/or thyroid cancer. The most prevalent first primary malignant neoplasms (PMNs) were breast in 27 (55%), thyroid in 14 (29%), and endometrial in 6 (12%). Primary colon cancer and melanoma were observed once each. Overall, 26 (53%) patients had second primary malignant neoplasms (SMNs), with 6 patients having ≥3 malignancies. The most prevalent SMNs included breast in 15 (58%) followed by thyroid, endometrial, and kidney cancer being observed twice (15%) each.
cfDNA fragmentomic features in patients with PHTS
The series of patients with PHTS were divided into subgroups based on cancer status irrespective of plasma sample draw time. This consisted of all of the patients with SMNs (SMN, n = 26), PMNs (PMN, n = 23), and no cancer (n = 50). To investigate cfDNA profiles in patients with PHTS and subclinical cancer, we further stratified these subgroups based on plasma draw time. These subgroups consisted of the following: patients with plasma samples drawn before SMN diagnosis (pre-SMN, n = 4), patients with plasma samples drawn before PMN diagnosis (pre-PMN, n = 5), patients with plasma drawn after SMN diagnosis (post-SMN, n = 22) and patients with plasma samples drawn after PMN diagnosis (post-PMN, n = 18).
Overall, all of the groups had cfDNA fragments ranging from approximately 50 to 1,000 bp in size. We focused our analysis on fragments of 100–650 bp, which corresponds to mono- (100–250 bp), di- (251-250 bp), and tri-nucleosome-derived (451–650 bp) cfDNA fragments (Figures 1A–1I). To compare cfDNA size distributions between groups, we performed a two-sample Kolmogorov-Smirnov (KS) test. The test statistic (D) of the KS test is defined as the largest absolute difference between the cumulative frequency distributions between groups (Figure S1; Table S2). We also calculated the difference in fragment size frequency (Figure S2) and cumulative frequencies (Figure S3) to quantify the difference of proportions of fragment sizes between groups across all nucleosome fractions.
Figure 1.

Individuals with PHTS and SMNs display decreased oligo-nucleosomal cfDNA fragments
The line plot depicts the median fragment size frequency normalized by total fragment counts across the mono-, di-, and trinucleosome fractions grouped by (A–C) cancer status, (D–F) SMN status, and (G–I) SMN status and plasma draw time (see also Table S2).
Individuals with PHTS and SMNs display decreased oligo-nucleosomal cfDNA fragments
Overall, we found that cfDNA size distribution between patients with and without cancer was statistically significant (D = 0.21, p = 7.54E−11), with notable differences in the di- and trinucleosome fractions (D = 0.19, p = 0.002 and D = 0.53, p < 2.2E−16, respectively) (Figures 1A–1C; Table S2). These differences appeared to be primarily driven by patients with SMNs (Figures 1D–1F; Table S2). The SMN group had significantly different cfDNA size distributions compared to the PMN group (D = 0.20, p = 8.64E−10) and those without cancer (D = 0.24, p = 9.56E−14), specifically within the di- (D = 0.20, p = 0.001; D = 0.23, p = 8.01E−05) and trinucleosome fractions (D = 0.59, p < 2.2E−16; D = 0.50, p < 2.2E−16). When stratifying by plasma draw time, the pre-SMN group was significantly different compared to all of the groups (pre-PMN, D = 0.25, p = 8.66E−15; post-PMN, D = 0.13, p = 9.66E−05; post-PMN, D = 0.27, p < 2.2E−16; no cancer, D = 0.28, p < 2.2E−16), with differences between the pre-SMN and post-SMN groups driven within the trinucleosome fraction (D = 0.34, p = 3.6E−10) (Figures 1G–1I; Table S2).
The SMN group had approximately 5% fewer fragments cumulatively within the di- and trinucleosome fractions compared to those without cancer (Figures S2 and S3). Although not statistically significant, the SMN group also had approximately 6% more fragments ≤175 bp within the mononucleosome fraction compared to those without cancer. Similarly, those in the pre-SMN group also had approximately 5% fewer fragments in the di- and trinucleosome fractions. Notably, the pre-SMN group had approximately 8% more fragments ≤175 bp within the mononucleosome fraction, albeit not statistically significant.
We also compared whether major peaks for each nucleosome fraction were different between groups (Table S3). Although none of the comparisons were statistically significant, there were notable patterns. Patients with SMNs had shorter major peaks of the di- (340 versus 360 bp, p = 0.067) and trinucleosome (540 versus 555 bp, p = 0.12) fractions compared to patients without cancer. Interestingly, the pre-SMN group had even shorter major peaks of the di- (333 versus 360 bp, p = 0.053) and trinucleosome (532 versus 555 bp, p = 0.08) fractions relative to those without cancer. In contrast, no differences between major peaks were observed when comparing the post-SMN and post-PMN group with those without cancer.
SMN status correlates with increased genome-wide cfDNA fragmentation
We subsequently evaluated genome-wide cfDNA fragmentation patterns using 5-Mb bins by SMN status. The median fragment ratios of the mono- (p = 0.06), di- (p = 0.07), and trinucleosome (p = 0.11) were not significant in overall testing. However, on pairwise comparison, the SMN group had higher median fragment ratios than those without cancer (0.21 versus 0.18, p = 0.03; 0.17 versus 0.14, p = 0.02; 0.38 versus 0.23, p = 0.03) across the mono-, di-, and trinucleosome fractions, respectively (Figures 2A–2C; Table S3). Upon differentiation based on plasma draw time, we observe that the pre-SMN group had even higher fragment ratios compared to those without cancer within the di- (0.19 versus 0.14, p = 0.036) and trinucleosome (0.41 versus 0.23, p = 0.017) fractions (Figures 3A–3C; Table S3). Despite the lack of statistical significance, the mononucleosome fraction had higher fragment ratios in the pre-SMN group compared to those without cancer (0.23 versus 0.18, p = 0.16).
Figure 2.

SMN status correlates with increased genome-wide cfDNA fragmentation
The median fragment ratio (defined as the ratio of short to long fragments) plotted in 5-Mb genomic bins for patients with PHTS stratified by SMN status within the (A) mononucleosome, (B) dinucleosome, and (C) trinucleosome fractions (see also Table S3).
Figure 3.

Individuals with PHTS and pre-SMN display more pronounced genome-wide cfDNA fragmentation
The median fragment ratio (defined as the ratio of short to long fragments) plotted in 5-Mb genomic bins for patients with PHTS stratified by SMN status and plasma draw time within the (A) mononucleosome, (B) dinucleosome, and (C) trinucleosome fractions (see also Table S3).
We also created three separate multivariable logistic regression models to assess the strength of correlation between SMN status and the median cfDNA fragment ratio associated with each nucleosome fraction while adjusting for age at plasma draw and CC score (i.e., phenotypic burden) (Table 2). We observed that a 0.1 increase in fragment ratio is significantly associated with a 2.37 (95% confidence interval [CI], 1.09–7.27; p = 0.025) and 3.03 (95% CI, 1.07–15.7; p = 0.029)-fold increase in the odds of being diagnosed with SMN for the mono- and dinucleosome fractions, respectively. Fragment ratios for the trinucleosome fraction were no longer statistically significant results after controlling for covariates.
Table 2.
Multivariable logistic regression: Elevated mono- and dinucleosome cfDNA fragments associated with SMN diagnosis
| Variables | Model 1 |
Model 2 |
Model 3 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | p | OR | 95% CI | p | OR | 95% CI | p | |
| Age at draw | 1.08 | 1.03–1.15 | 0.001 | 1.08 | 1.03–1.15 | <0.001 | 1.08 | 1.03–1.14 | <0.001 |
| CC score | 1.01 | 0.96–1.05 | 0.8 | 1.00 | 0.95–1.05 | >0.9 | 1.0 | 0.95–1.04 | 0.8 |
| Ratio1 | 2.37 | 1.09–7.27 | 0.025 | – | – | – | – | – | – |
| Ratio2 | – | – | – | 3.03 | 1.07–15.7 | 0.029 | – | – | – |
| Ratio3 | – | – | – | – | – | – | 1.40 | 0.96–2.10 | 0.078 |
CC, Cleveland Clinic; CI, confidence interval; OR, odds ratio; ratio1, mononucleosome-associated fragment ratios; ratio2, dinucleosome-associated fragment ratios; ratio3, trinucleosome-associated fragment ratios.
To investigate the contribution of metadata (i.e., age and CC score) on classification performance, we first constructed a model containing only metadata. We performed leave-one-out cross-validation, yielding an area under the receiver operating characteristic curve (AUC) of 0.68, with age being the primary driver for classification. We subsequently performed likelihood-ratio tests comparing models with and without fragment ratios that demonstrated that there was a modest but significant improvement in classification performance using fragment ratios derived from the mono- (p = 0.018) and dinucleosome (p = 0.024) fractions, with AUCs of 0.71 and 0.70, respectively (Figure S4).
Finally, we performed a genome-wide correlation analysis of fragment ratios for each sample compared to the median fragment ratio profile of PHTS patients without cancer. Individual samples from the SMN, PMN, and no cancer group weakly correlated to the median fragmentation profile of patients without cancer, with median correlation coefficients ranging between 0.43 and 0.59 (Table S3). Notably, the SMN group (0.54 versus 0.59, p = 0.004) and the PMN group (0.57 versus 0.59, p = 0.044) had significantly lower median correlations compared to those without cancer within the trinucleosome fraction.
Although no significant differences in median correlation coefficients were observed in any nucleosome fraction between the pre-SMN group to those without cancer, both the post-SMN (0.55 versus 0.59, p = 0.007) and pre-PMN (0.51 versus 0.59, p = 0.011) groups displayed greater discordance, specifically within the trinucleosome fraction, when compared to those without cancer (Table S3). However, the statistically significant difference observed in the pre-PMN group may be an artifact of GC correction because the difference was nonsignificant (p = 0.96) when the comparison was made without GC correction.
Individuals with PHTS and SMNs display distinct end motif profiles
Subsequently, to comprehensively characterize cfDNA fragment end profiles, we calculated the frequencies for each the first 4-nt sequence (4-mer end motif) at each 5′ fragment end of cfDNA molecules. Out of the possible 256 4-mer end motifs, we identified 35 end motifs that differed significantly, specifically when comparing patients with SMN to those without cancer (Figure 4A; Table S4). These findings remained statistically significant even after correcting for multiple comparisons using the Benjamini-Hochberg procedure. No differences in end motif frequencies were observed between the PMN group and those without cancer.
Figure 4.

Individuals with PHTS and SMNs display distinct end motif profiles
(A) Heatmap displaying Z score transformed of the significantly differentially abundant 4-mer motif frequencies. The color gradient reflects the number of SDs from the mean frequency. Darker shades indicate lower relative abundance, whereas lighter shades suggest higher relative abundance. (B and C) Boxplot of the 2-mer and 1-mer motif frequencies stratified by SMN status. (D and E) Boxplot of 2-mer and 1-mer end motif frequencies stratified by SMN status and plasma draw time. Motif frequencies are relative to the total number of end motifs across all of the nucleosome fractions. The center line in each boxplot represents the median. Each box extends from the 25th to the 75th percentile (IQR) (see also Table S4).
The top three most significantly increased 4-mer end motifs include GAAG (0.54% versus 0.44%, p = 0.014), CAGG (0.78% versus 0.66%, p = 0.012), and GGGA (0.50% versus 0.42%, p = 0.013) (Table S4). In contrast, CATT (0.82% versus 1.0%, p = 0.013), CTCT (0.69% versus 0.79%, p = 0.014), and ATCG (0.040% versus 0.045%, p = 0.009) were the three most significantly decreased 4-mer end motifs. We observed similar patterns when stratifying by SMN status and plasma draw time, although only nine and four 4-mer end motifs differed significantly when comparing the pre-SMN group and post-SMN to those without cancer, respectively, likely due to the reduced sample size.
As part of an exploratory analysis, we performed pairwise comparisons of all 4-mer end motifs irrespective of whether they reached overall significance. In this analysis, we observed an interesting trend among patients with SMN compared to those without cancer. Specifically, the most differentially increased end motifs in patients with SMN were C-end motifs, including CAAG (0.75% versus 0.59%, p = 0.013), CAGA (0.79% versus 0.64%, p = 0.012), and CCAG (1.35% versus 1.09%, p = 0.01) (Table S5). Similarly, we observed even more pronounced increases in C-end motifs in the pre-SMN group, including CCAG (1.51% versus 1.09%, p = 0.01), CAAG (0.78% versus 0.59%, p = 0.013), CAGA (0.83% versus 0.64%, p = 0.012), CCCA (1.6% versus 1.29%, p = 0.013), and CAGG (0.84% versus 0.66%, p = 0.018).
To examine broader trends, we also calculated the frequency of both 2-mer and 1-mer end motifs. Overall, patients with SMNs exhibited a decreased frequency of A-end motifs (24% versus 26%, p = 0.033), specifically in AC- and AT-end motifs, and increased frequency of T-end motifs (20% versus 19%, p = 0.008), notably TA- and TG-end motifs (Figures 4B and 4C). Although not statistically significant, we also observed an increased frequency of G-end motifs (23% versus 22%, p = 0.051), particularly GA- and GG-end motifs. Although similar patterns were observed in the pre-SMN group, C-end motifs (32% versus 32%, p = 0.053), specifically CA- and CC-end motifs appear uniquely increased in this subgroup, consistent with our 4-mer end motif results (Figures 4D and 4E).
Discussion
Overall, the results from our retrospective cohort study have highlighted that patients with PHTS and SMNs have distinct plasma cfDNA profiles (i.e., size distribution, fragmentation profile, and fragment end profiles) compared to patients with PHTS and PMNs, as well as those without cancer. Our findings provide strong supporting evidence that cfDNA fragmentomic features can be leveraged as a potential marker of individual SMN risk in PHTS and suggest conducting a confirmatory prospective study to validate our findings.
Interestingly, all cfDNA fragmentomic features (i.e., size distribution, fragmentation profile, and fragment end profiles) were more pronounced in those with plasma drawn before their cancer diagnosis. This is likely because patients with plasma drawn before their cancer diagnosis likely had active cancer, whereas patients with plasma drawn after their cancer diagnosis may include those who had undergone cancer treatment. The latter may suggest that these plasma samples, following cancer treatment, contain low to no tumor content. Although this raises concerns that differences in cfDNA profiles may be confounded by different cancer treatment modalities (i.e., surgery, radiation, and chemotherapy), close examination of cancer subgroups stratified by plasma draw time suggests that treatment effects are negligible. Notably, the pre-PMN group (i.e., treatment-naive individuals) exhibited subtle differences in all cfDNA fragmentomic features compared to the post-PMN (i.e., likely treated) and no cancer group. Moreover, the post-PMN group were nearly identical to the no cancer group. Taken together, this suggests that the treatment effects on cfDNA profiles were minimal.
The differential distribution of specific cfDNA fragment sizes in the SMN group compared to the no cancer group explains the significantly higher genome-wide fragment ratios within the mono- and dinucleosome fractions, even after controlling for age at plasma draw and phenotypic burden. Consistent with our findings, Sanchez et al. reported that healthy individuals and patients with sporadic metastatic colon cancer had similar mononucleosome-associated peaks, but significantly different dinucleosome-associated peaks—approximately 30 bp shorter in cancer patients.25 In addition, they found that patients with sporadic colorectal cancer had an increased proportion of fragments below 160 bp, and that the mutant allele frequency was positively correlated with the proportion of shorter cfDNA fragments. Other studies have also similarly found that the proportion of ctDNA is positively correlated with the proportion of shorter cfDNA fragments.7,26,27 These findings parallel our observations, even in our germline series, with the pre-SMN group (i.e., those who likely have active cancer) having the highest proportion of short fragments below 175 bp, which we suspect contains enrichment of ctDNA. In addition, Mouliere et al. and Ganesamoorthy et al. provide evidence that ctDNA is enriched within the dinucleosome fraction in early- and late-stage tumors, respectively.27,28 Ganesamoorthy et al. investigated plasma samples with low tumor content and did not detect enrichment in cfDNA fragments <150 bp, but reported significant enrichment of tumor-derived fragments within the dinucleosome fraction.28
As expected, genome-wide correlation analysis demonstrated that the fragmentation profiles of both cancer subgroups (i.e., SMN and PMN) correlated weakly with the median fragmentation profiles of the individuals without cancer. However, contrary to our expectations based on prior work in sporadic cancers, fragmentation profiles among individuals without cancer also weakly correlated with one another, suggesting high intragroup variability.13 We speculate that patients with PHTS, irrespective of cancer status, have baseline genomic instability due to PTEN dysfunction, which may result in more heterogeneous cfDNA fragmentation profiles between individuals, thus limiting the reliability of comparing genome-wide correlation analyses in this patient population. Specifically, PTEN interacts with histone H1 via its C2 domain, thereby promoting chromatin condensation.23 Thus, patients with PHTS likely display irregular nucleosome eviction and chromatin decondensation that is potentially more pronounced in patients with SMN, which may be an important early event in the development of SMNs. Increased chromatin decondensation could lead to greater nuclease accessibility and, consequently, increased cfDNA fragmentation. We recognize the importance of validating these findings to better discern potential technical effects from genuine biological differences.
We have also demonstrated that cfDNA fragment end motif profiling is a potential approach for identifying patients with PHTS and SMNs. We show that 35 4-mer end motifs were significantly associated with SMNs, even after multiple comparison correction. Patients with SMNs displayed a decreased frequency of A-end motif and increased G- and T-end motifs. Caspase-dependent activation of DNA nucleases is a hallmark of apoptosis. Aberrant nuclease activity may also contribute to distinct aberrant fragment end profiles in patients with PHTS and SMNs. PTEN plays an important role in regulating the apoptotic threshold, and is a known substrate of caspase-3 via the C-terminal tail.29 Studies have found that a subset of PTEN variants in patients with PHTS displays diminished caspase-3 cleavage.30 Furthermore, PTEN expression has been shown to be correlated with caspase-3 expression.31 PTEN-mediated apoptosis is orchestrated by its lipid phosphatase activity and results in caspase activation.32 The apoptotic nuclease DNA fragmentation factor B plays a key role in DNA fragmentation of chromatin into oligo-nucleosomes with a preference for generating A-end motifs over T-motifs.8 This increased frequency of A-end motifs, concurrent decrease in T-end motifs, and decreased proportion of oligo-nucleosome cfDNA fragments may suggest that patients with PHTS and SMNs may have impaired PTEN-mediated apoptosis resulting in aberrant nuclease activity.
Interestingly, 4-mer C-end motifs—particularly those starting with CA, CC, and CG—were the most increased end motifs in patients with SMN, and even more so in the pre-SMN group, despite the lack of significance in overall testing. In contrast to our observations, sporadic cancers typically demonstrate a decrease in C-end motif frequencies, accompanied by concurrent increase in A-end motifs. Prior studies observed that patients with hepatocellular carcinoma had consistent decreases in 4-mer CC-end motifs likely due to the downregulation of DNASE1L3,15 a nuclease responsible for producing CC-end motifs and digesting oligonucleosomes.8,33 Similarly, another study investigating 2-mer end motif profiles of sporadic renal cell carcinoma and colorectal cancer observed consistent decreases in CC-, CA-, and CT-end motifs while concurrently noting increases in AA- and AC-end motifs.16
Prior studies have shown that normal plasma cfDNA fragment end profiles are characterized by an over- and underrepresentation of C- and A-end motif fragments, respectively, corresponding with the distribution of nucleosome-occupied and open-chromatin regions.6,8,34 The observation that C- and A-end motif frequencies are in the opposite direction in patients with PHTS with SMNs compared to sporadic cancers may hint at differences in underlying pathophysiology. The underrepresentation of C-end motifs in sporadic cancers appears to be driven primarily by aberrations in nuclease expression. In contrast, we postulate that the enrichment of C-end motifs in patients with PHTS and SMNs reflect increased chromatin decondensation, likely originating from cancer-derived cfDNA (i.e., ctDNA). The increased frequency of C-motifs may be a distinct marker of active SMN in patients with PHTS. However, studies with larger sample sizes are required to validate this finding.
The presence of disparate phenotypes, such as multiple primary cancers versus no cancer, among individuals with identical germline PTEN variants suggests additional phenotypic modifiers in PHTS. An interesting observation, in the context that treatment effects are likely minimal on cfDNA profiles, is that both the post-SMN (i.e., those with likely treated cancer) and the pre-SMN group (i.e., those with likely active cancer) shared similar but less pronounced differences in cfDNA fragmentomic features relative to those without cancer. These observations may suggest that patients with PHTS and SMNs have distinct baseline genetic or molecular differences. Individuals predisposed to SMNs may harbor PTEN variants that exhibit more pronounced PTEN nuclear dysfunction. Patients with Cowden syndrome with the K289E allele of PTEN demonstrating altered PTEN ubiquitination leading to nuclear exclusion of PTEN have been described.35 It is also possible that those at risk of SMNs possess genetic modifiers that further impair PTEN function. Indeed, our group has previously shown that patients with PHTS harboring germline variants in genes encoding for the subunits of the mitochondrial complex II (SDHx) have an increased risk of breast and thyroid cancer that surpasses the risk mediated by PTEN variants alone.36,37 Further investigation is warranted to better understand modifier genes related to SMN risk.
Typical for rare diseases such as PHTS, one limitation of this study is the relatively small sample size for each phenotype group, despite our series being the largest globally to our knowledge. This limits our ability to perform in-depth analyses such as examining whether cancer-type specific cfDNA fragmentomic features exist in individuals with PHTS and SMNs. It is noteworthy that the pre-SMN group, despite having a small sample size (n = 4), exhibited the most pronounced differences in cfDNA fragmentomic features. This finding suggests a large effect size, underscoring the significance of the observed differences. Furthermore, we did not adjust for multiple comparisons due to the exploratory nature of the analysis and limited sample size. However, it is worth highlighting that our cfDNA fragment size distribution and 4-mer end motif analysis results were particularly robust and would remain significant after correction with the Benjamini-Hochberg procedure.
Ultra-low-pass whole-genome sequencing also limits our ability to gain a deeper understanding of the biological mechanisms that contribute to SMN risk. We attempted to characterized cfDNA fragmentation hotspots—namely OCRs and gene regulatory elements—using a computation method known as CRAG.38 However, despite pooling samples from both the cancer and no cancer group to increase coverage, we could only identify hotspots after merging samples into a singular dataset for each group, precluding us from conducting meaningful statistical comparisons. Finally, another limitation is the absence of an independent series to ascertain our findings. Given the rarity of PHTS, suitable external datasets and independent samples do not exist. We acknowledge the need for validation through a prospectively collected, independent series of patients with PHTS, paired with controls who have PHTS but no cancer, and are rigorously matched for other features such as age, gender, and clinical characteristics.
Our findings suggest potential cfDNA fragmentomic features associated with SMN risk in patients with PHTS, which require further validation to determine their clinical utility for risk stratification in this patient population. Patients with PHTS and SMNs are characterized by the enrichment of shorter cfDNA fragments between 100 and 175 bp, decreased proportion of cfDNA fragments in the di- and trinucleosome fractions, and increased genome-wide fragmentation (i.e., ratio of short to long fragments) in both the mono- and dinucleosome fractions. Furthermore, fragment end profiles are characterized by the concurrent decrease in A-end motifs and increase in G- and T-end motifs in patients with PHTS and SMNs. Increased frequency of C-end motifs appears to be unique to patients who had plasma drawn before their SMN diagnosis, a potential marker for active cancer. Analysis of cfDNA profiles in patients with PHTS and PMN or those with a strong family history of SMN may be beneficial. The presence of the aforementioned cfDNA fragmentomic features in a patient with PHTS would suggest the need for closer cancer surveillance for SMNs. This may facilitate earlier cancer detection and intervention leading to better clinical outcomes as well as prevent unnecessary high-risk cancer surveillance and prophylactic surgeries in this vulnerable population.
Limitations of the study
Key limitations to this study primarily relate to factors that constrain our statistical power. A significant challenge in studying rare diseases such as PHTS is the relatively small sample size, which limited our ability to identify cancer-type specific cfDNA fragmentomic features. In addition, the variability in the timing of plasma samples collection relative to cancer diagnosis may have led to the inclusion of samples from patients who have undergone cancer treatment (i.e., plasma containing little to no ctDNA contributing to the total cfDNA pool), further limiting our statistical power. Moreover, the observed differences in cfDNA profiles may also be confounded by the effects of cancer treatment, such as surgery, radiation, or chemotherapy. The use of ultralow coverage whole-genome sequencing also precluded us from performing more detailed analyses, such as characterizing OCRs and gene regulatory elements, which may have offered additional functional insights underlying SMN risk. Finally, due to the rarity of PHTS, the absence of external independent datasets for validation emphasizes the preliminary nature of our findings.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Plasma samples from patients with PHTS with and without cancer | Genomic Medicine Biorepository, Cleveland Clinic | N/A |
| Critical commercial assays | ||
| QIAamp Circulating Nucleic Acid Kit | QIAGEN | Cat #55114 |
| NEBNext Ultra DNA Library Prep Kit for Illumina | New England BioLabs | Cat #E7370L |
| cfDNA extraction and Ultra-low pass WGS | Broad Institute | http://genomics.broadinstitute.org/products/liquid-biopsy-sequencing |
| Deposited data | ||
| De-identified cfDNA fragment files | This paper | https://doi.org/10.5281/zenodo.8422534 |
| Software and algorithms | ||
| R (v.4.2.3) | CRAN | https://www.r-project.org/; RRID: SCR_003005 |
| R Studio | Posit | https://posit.co/products/open-source/rstudio/; RRID: SCR_000432 |
| Original code | This paper | https://doi.org/10.5281/zenodo.10372575 |
| SAMtools (v.1.16.1) | Danecek et al.39 | http://www.htslib.org/; RRID: SCR_002105 |
| BEDtools (v.2.29.0) | Quinlan and Hall40 | https://bedtools.readthedocs.io/en/latest/content/installation.html; RRID: SCR_006646 |
| ggplot2 (v.3.4.1) | Wickham et al.41 | https://ggplot2.tidyverse.org/; RRID: SCR_014601 |
| pROC (1.18.4) | Robin et al.42 | https://cran.r-project.org/web/packages/pROC/index.html; RRID:SCR_001905 |
| pheatmap (v.1.0.2) | Kolde43 | https://cran.r-project.org/web/packages/pheatmap/index.html; RRID: SCR_016418 |
| Other | ||
| ENCODE Blacklist Regions | Amemiya et al.44 | https://github.com/Boyle-Lab/Blacklist/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Charis Eng (engc@ccf.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
De-identified fragment files mapped to hg19 and individual summary statistics have been deposited at Zenodo.org and are publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
All original code deposited at GitHub and is publicly available as the date of publication. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Patients and participants
A total of 6,923 patients were prospectively accrued in a study approved by the institutional review board (IRB) of the Cleveland Clinic under IRB protocol 8458 from 2005 through 2020. Eligible patients met at least the relaxed operational diagnostic criteria of the International Cowden Consortium, defined as full diagnostic criteria minus one feature, having macrocephaly plus neurodevelopmental disorder and/or penile freckling, or presence of a known pathogenic germline PTEN variant. All neoplasia diagnoses are documented by pathology (first choice), other objective (e.g., imaging) reports, clinical note or death certificate. For each consented patient, we review medical records, including pedigrees, clinical genetic testing reports, and clinical notes associated with genetics evaluations, and/or genetic-counseling visits. After undergoing germline PTEN variation and deletion analysis, 611 patients were found to have germline PTEN variants. Accrued patient biospecimens including peripheral blood and plasma are stored and managed at the Genomic Medicine Biorepository (GMB) at the Cleveland Clinic (Cleveland, OH, USA) using standard protocols.
To identify potential participants, we searched the GMB for patients with confirmed germline PTEN variants with archived plasma samples that were drawn within approximately two years of a breast (N = 35) and/or thyroid (N = 15) cancer diagnosis. We focused on breast and thyroid cancers as they are of the most common component malignancies in PHTS, with thyroid cancer having the youngest age at onset.3 In total, 100 samples were identified including 50 PHTS patients without cancer and 50 PHTS patients with cancer. One sample from the cancer group was excluded due to insufficient cfDNA for sequencing. De-identified patient demographic (e.g., age, gender, race, etc.) and clinical data are found in Table S2. Summarized characteristics were summarized in Table 1 in the Results section.”
Method details
Sample collection and DNA isolation
Following receipt by the Genomic Medicine Biorepository, plasma samples were inventoried and immediately stored at −80°C until DNA extraction. cfDNA was isolated from all plasma samples using the Circulating Nucleic Acids Kit (Cat #55114, Qiagen, Hilden, Germany). A volume of 1 mL of plasma was used for cfDNA extraction. Plasma samples were sent to the Broad Institute for cfDNA extraction, library preparation and ultra-low pass whole genome sequencing (ULP-WGS).
Sequencing library preparation
Next-generation sequencing libraries from cfDNA were prepared using 5–50 ng of DNA using the NEBNext DNA Library Prep Kit for Illumina (Cat #E7370L, New England Biolabs, Ipswich, MA, USA). The quality and concentration of cfDNA and generated genomic libraries were examined using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Whole genome sequencing data processing
Whole genome libraries were sequenced using 100-bp paired-end runs on Illumina NovaSeq 6000 at 0.1-0.3X coverage per genome. Processing of raw data, including demultiplexing and masking of dual-index adaptor sequences, was performed using Illumina CASAVA (Consensus Assessment of Sequence and Variation) software. Trimmed reads were aligned to human reference genome version hg19. Read pairs with MAPQ score <30, PCR duplicates, secondary alignments, and unmapped reads were removed prior to further downstream analysis using SAMtools (v.1.16.1).39 Filtered bam files were sorted using SAMtools followed by summarizing paired-end reads as fragments with the fragment start, end, strand alignment, insert size, and insert GC content as BEDPE files using BEDTools (v.2.29.0), specifically the ‘bedtools bamtobed’ command.40 Reads overlapping with the ENCODE blacklist regions44 were excluded using the ‘bedtools subtract’ command.
To account for GC-related coverage bias, we performed a fragment-level GC adjustment for each sample by applying a locally weighted scatterplot smoothing (LOESS) regression analysis with a default span setting of 0.75.45 To account for potential differences in GC effects on coverage by fragment length, LOESS regression was performed separately for each nucleosome fraction. For each sample, we counted the total number of fragments binned by GC content from 0 to 1. We filtered out the bottom 5th and 95th percentile of fragments due to increased variance of the LOESS regression model at the tails of the GC content distribution. A scalar correction factor for each GC stratum was calculated from the ratio of corrected to uncorrected counts to obtain GC-adjusted fragment counts.
cfDNA Fragment size distribution analysis
Fragment size frequencies were obtained by normalizing the total number of fragments for each sample by the total fragment counts across all the mono-, di-, and tri-nucleosome fractions (100–650 bp). Longer fragment sizes, especially those >600 bp, tend to exhibit lower R2 read quality and characteristics sequencing errors when using Illumina paired-end sequencing.46 In our dataset, fragments >650 bp, representing larger oligo-nucleosomes, accounted for less than 1–4% of all fragments. Taken together, we chose to exclude fragments >650 bp to minimize potential biases arising from technical challenges and to focus on fragments that are likely more clinically relevant based on their prevalence. We then calculated the median frequency for each fragment size in each group. Fragment size distribution was visualized using R package ggplot2.41
Genome-wide fragmentation pattern analysis
To investigate fragment size and coverage in a position-dependent manner, fragments were assigned to 5-Mb adjacent, non-overlapping bins using the ‘bedtools intersect’ tool. Bins with an average GC content <0.3 and an average mappability <0.9 were excluded, leaving 473 bins spanning approximately 2.4 GB of the genome as previously described.13,14 Fragment sizes corresponding with mono- (100–250 bp), di- (251–450 bp), and tri-nucleosomes (451–650 bp) were included for downstream analysis. For each 5-Mb bin, we calculated the ratios of the number of short (mono, 100–150 bp; di, 251–300; tri, 451–500 bp) and long (mono, 151–250 bp; di, 301–450 bp; tri, 501–650 bp) fragments across each nucleosome fraction to obtain genome-wide fragmentation profiles for each sample. Genome-wide fragmentation profiles were visualized using the R package ggplot2. We also performed genome-wide correlation analysis of fragment ratios as described previously.13 Briefly, we calculated the Spearman correlation coefficients for each genomic bin by comparing each individual sample to the median fragment ratio profiles of patients with PHTS without cancer.
Multivariable logistic regression was performed to investigate the association between median cfDNA fragment ratios and second primary malignant neoplasm (SMN) diagnosis while controlling for relevant covariates. Age at plasma draw and the Cleveland Clinic (CC) score, a semi-quantitative measure of phenotypic burden,47 were chosen as covariates that could influence cfDNA fragmentation profiles. Due to limited sample size, and that only one patient with SMN was male, we did not include gender as a covariate in our models. Leave-one-out cross-validation (LOOCV) was utilized to assess model performance. Individual samples were sequentially used the validation set, while the remaining samples formed the training set. The procedure was repeated until each sample had been used as a validation set once. The predicted probabilities from each iteration were compiled and use to calculate and visualize the area under the receiver operating characteristics (AUROC) curve using the R package pROC42 and ggplot2 respectively.
Fragment end motif analysis
From the BEDPE files summarizing paired-end reads, we generated two distinct BED files containing the first four nucleotides (i.e., 4-mer end motif) from the 5′ end of each paired-end read. Nucleotide sequences were extracted using the ‘bedtools getfasta’ function. For each individual sample, the two BED files corresponding to each read were then concatenated into a single BED file. Subsequently, we then calculated the frequency of 4-mer end motif frequencies across all nucleosome fractions. To calculate 2-mer end motif frequencies, we repeated this procedure, generating BED files containing only the first two nucleotides. The 1-mer end motifs were then subsequently derived from these 2-mer end motif frequencies. To visualize differentially abundant end motifs, a heatmap of Z score transformed 4-mer end motif frequencies was generated using the R package pheatmap.43 Additionally, boxplots of the 2-mer and 1-mer end motifs was visualized using the R package ggplot2.
Quantification and statistical analysis
Statistical analysis and data visualization was performed using R (v.4.2.3). We utilized the two-sample Kolmogorov-Smirnov (KS) test to compare if the cumulative frequency distributions between groups were sampled from different distributions. Additionally, the difference in fragment size frequency (ΔF) and cumulative frequency distributions (ΔCF) relative to patients without cancer were calculated and visualized. The Kruskal-Wallis test and Wilcoxon Rank-Sum test were used to compare medians of continuous variables for multiple groups and pairwise comparisons, respectively. As an exploratory analysis, we performed post-hoc pairwise comparisons between subgroups using the Wilcoxon rank-sum test if the Kruskal-Wallis test did not yield statistical significance. This was done to identify any potentially clinically relevant differences between subgroups. A p value less than 0.05 was considered as the threshold for statistical significance. Benjamini-Hochberg procedure was used to calculate adjusted p-values to correct for multiple hypothesis testing.
Acknowledgments
This study was supported, in part, by the VeloSano Pilot Award (to C.E.). L.Y. and A.D. are Ambrose Monell Cancer Genomic Medicine Fellows. A.D. is funded, in part, by a PTEN Research Foundation Young Investigator Award (to C.E.) and an American Association of Neurological Surgeons Award (to A.D.). C.E. is the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic. We thank all patients and families who contributed to this study. We thank the Genomic Medicine Biorepository of the Cleveland Clinic Genomic Medicine Institute and our database and clinical research teams.
Author contributions
Conceptualization, D.L., L.Y., and C.E.; methodology, D.L., A.D., Y.N., and L.Y.; formal analysis, D.L.; investigation, D.L and L.Y.; resources, C.E.; writing – original draft, D.L.; writing – review & editing, L.Y. and C.E.; visualization, D.L.; supervision, A.D., Y.N., L.Y., and C.E.; funding acquisition, C.E.
Declaration of interests
The authors declare no competing interests.
Published: January 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101384.
Supplemental information
References
- 1.Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S.I., Puc J., Miliaresis C., Rodgers L., McCombie R., et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 2.Steck P.A., Pershouse M.A., Jasser S.A., Yung W.K., Lin H., Ligon A.H., Langford L.A., Baumgard M.L., Hattier T., Davis T., et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 3.Yehia L., Plitt G., Tushar A.M., Joo J., Burke C.A., Campbell S.C., Heiden K., Jin J., Macaron C., Michener C.M., et al. Longitudinal analysis of cancer risk in children and adults with germline PTEN variants. JAMA Netw. Open. 2023;6:e239705. doi: 10.1001/jamanetworkopen.2023.9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ngeow J., Stanuch K., Mester J.L., Barnholtz-Sloan J.S., Eng C. Second malignant neoplasms in patients with Cowden syndrome with underlying germline PTEN mutations. J. Clin. Oncol. 2014;32:1818–1824. doi: 10.1200/JCO.2013.53.6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wan J.C.M., Massie C., Garcia-Corbacho J., Mouliere F., Brenton J.D., Caldas C., Pacey S., Baird R., Rosenfeld N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 6.Fu Y., Sinha M., Peterson C.L., Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouliere F., Robert B., Arnau Peyrotte E., Del Rio M., Ychou M., Molina F., Gongora C., Thierry A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS One. 2011;6:e23418. doi: 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han D.S.C., Ni M., Chan R.W.Y., Chan V.W.H., Lui K.O., Chiu R.W.K., Lo Y.M.D. The biology of cell-free DNA fragmentation and the roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020;106:202–214. doi: 10.1016/j.ajhg.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phallen J., Sausen M., Adleff V., Leal A., Hruban C., White J., Anagnostou V., Fiksel J., Cristiano S., Papp E., et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017;9:eaan2415. doi: 10.1126/scitranslmed.aan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen J.D., Li L., Wang Y., Thoburn C., Afsari B., Danilova L., Douville C., Javed A.A., Wong F., Mattox A., et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y. At the dawn: cell-free DNA fragmentomics and gene regulation. Br. J. Cancer. 2022;126:379–390. doi: 10.1038/s41416-021-01635-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thierry A.R. Circulating DNA fragmentomics and cancer screening. Cell Genom. 2023;3:100242. doi: 10.1016/j.xgen.2022.100242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cristiano S., Leal A., Phallen J., Fiksel J., Adleff V., Bruhm D.C., Jensen S.Ø., Medina J.E., Hruban C., White J.R., et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathios D., Johansen J.S., Cristiano S., Medina J.E., Phallen J., Larsen K.R., Bruhm D.C., Niknafs N., Ferreira L., Adleff V., et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 2021;12:5060. doi: 10.1038/s41467-021-24994-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang P., Sun K., Peng W., Cheng S.H., Ni M., Yeung P.C., Heung M.M.S., Xie T., Shang H., Zhou Z., et al. Plasma DNA end motif profiling as a fragmentomic marker in cancer, pregnancy and transplantation. Cancer Discov. 2020;10:664–673. doi: 10.1158/2159-8290.CD-19-0622. [DOI] [PubMed] [Google Scholar]
- 16.Zhitnyuk Y.V., Koval A.P., Alferov A.A., Shtykova Y.A., Mamedov I.Z., Kushlinskii N.E., Chudakov D.M., Shcherbo D.S. Deep cfDNA fragment end profiling enables cancer detection. Mol. Cancer. 2022;21:26. doi: 10.1186/s12943-021-01491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein E.A., Richards D., Cohn A., Tummala M., Lapham R., Cosgrove D., Chung G., Clement J., Gao J., Hunkapiller N., et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021;32:1167–1177. doi: 10.1016/j.annonc.2021.05.806. [DOI] [PubMed] [Google Scholar]
- 18.Yehia L., Ngeow J., Eng C. PTEN-opathies: From biological insights to evidence-based precision medicine. J. Clin. Invest. 2019;129:452–464. doi: 10.1172/JCI121277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weng L.-P., Brown J.L., Eng C. PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum. Mol. Genet. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]
- 20.Shen W.H., Balajee A.S., Wang J., Wu H., Eng C., Pandolfi P.P., Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 21.He J., Kang X., Yin Y., Chao K.S.C., Shen W.H. PTEN regulates DNA replication progression and stalled fork recovery. Nat. Commun. 2015;6:7620. doi: 10.1038/ncomms8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G., Li Y., Wang P., Liang H., Cui M., Zhu M., Guo L., Su Q., Sun Y., McNutt M.A., Yin Y. PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 2015;25:1189–1204. doi: 10.1038/cr.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z.H., Zhu M., Yang J., Liang H., He J., He S., Wang P., Kang X., McNutt M.A., Yin Y., Shen W.H. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep. 2014;8:2003–2014. doi: 10.1016/j.celrep.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yehia L., Seyfi M., Niestroj L.M., Padmanabhan R., Ni Y., Frazier T.W., Lal D., Eng C. Copy number variation and clinical outcomes in patients with germline PTEN mutations. JAMA Netw. Open. 2020;3:e1920415. doi: 10.1001/jamanetworkopen.2019.20415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez C., Roch B., Mazard T., Blache P., Dache Z.A.A., Pastor B., Pisareva E., Tanos R., Thierry A.R. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight. 2021;6:e144561. doi: 10.1172/jci.insight.144561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diehl F., Li M., Dressman D., He Y., Shen D., Szabo S., Diaz L.A., Goodman S.N., David K.A., Juhl H., et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA. 2005;102:16368–16373. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mouliere F., Chandrananda D., Piskorz A.M., Moore E.K., Morris J., Ahlborn L.B., Mair R., Goranova T., Marass F., Heider K., et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018;10:eaat4921. doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ganesamoorthy D., Robertson A.J., Chen W., Hall M.B., Cao M.D., Ferguson K., Lakhani S.R., Nones K., Simpson P.T., Coin L.J.M. Whole genome deep sequencing analysis of cell-free DNA in samples with low tumour content. BMC Cancer. 2022;22:85. doi: 10.1186/s12885-021-09160-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torres J., Rodriguez J., Myers M.P., Valiente M., Graves J.D., Tonks N.K., Pulido R. Phosphorylation-regulated cleavage of the tumor suppressor PTEN by caspase-3. J. Biol. Chem. 2003;278:30652–30660. doi: 10.1074/jbc.M212610200. [DOI] [PubMed] [Google Scholar]
- 30.Torices L., Mingo J., Rodríguez-Escudero I., Fernández-Acero T., Luna S., Nunes-Xavier C.E., López J.I., Mercadillo F., Currás M., Urioste M., et al. Functional analysis of PTEN variants of unknown significance from PHTS patients unveils complex patterns of PTEN biological activity in disease. Eur. J. Hum. Genet. 2023;31:568–577. doi: 10.1038/s41431-022-01265-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X.-F., Xin Y., Mao L.-L. Clinicopathological significance of PTEN and caspase-3 expressions in breast cancer. Chin. Med. Sci. J. 2008;23:95–102. doi: 10.1016/s1001-9294(09)60019-5. [DOI] [PubMed] [Google Scholar]
- 32.Yuan X.-J., Whang Y.E. PTEN sensitizes prostate cancer cells to death receptor-mediated and drug-induced apoptosis through a FADD-dependent pathway. Oncogene. 2002;21:319–327. doi: 10.1038/sj.onc.1205054. [DOI] [PubMed] [Google Scholar]
- 33.Han D.S.C., Lo Y.M.D. The nexus of cfDNA and nuclease biology. Trends Genet. 2021;37:758–770. doi: 10.1016/j.tig.2021.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Song L., Zhang Z., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.-K., Sheffield N.C., Gräf S., Huss M., Keefe D., et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trotman L.C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N.P., Carver B.S., Cordon-Cardo C., Erdjument-Bromage H., et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ni Y., Zbuk K.M., Sadler T., Patocs A., Lobo G., Edelman E., Platzer P., Orloff M.S., Waite K.A., Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ni Y., He X., Chen J., Moline J., Mester J., Orloff M.S., Ringel M.D., Eng C. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum. Mol. Genet. 2012;21:300–310. doi: 10.1093/hmg/ddr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X., Zheng H., Fu H., Dillehay McKillip K.L., Pinney S.M., Liu Y. CRAG: de novo characterization of cell-free DNA fragmentation hotspots in plasma whole-genome sequencing. Genome Med. 2022;14:138. doi: 10.1186/s13073-022-01141-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickham H. Springer-Verlag New York; 2016. ggplot2: Elegant Graphics for Data Analysis.https://ggplot2.tidyverse.org [Google Scholar]
- 42.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolde R. 2019. Pheatmap: Pretty Heatmaps.https://cran.r-project.org/web/packages/pheatmap/index.html [Google Scholar]
- 44.Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE blacklist: Identification of problematic regions of the genome. Sci. Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y., Speed T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan G., Opitz L., Schlapbach R., Rehrauer H. Long fragments achieve lower base quality in Illumina paired-end sequencing. Sci. Rep. 2019;9:2856. doi: 10.1038/s41598-019-39076-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan M.-H., Mester J., Peterson C., Yang Y., Chen J.-L., Rybicki L.A., Milas K., Pederson H., Remzi B., Orloff M.S., Eng C. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011;88:42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
De-identified fragment files mapped to hg19 and individual summary statistics have been deposited at Zenodo.org and are publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
All original code deposited at GitHub and is publicly available as the date of publication. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze reported in this paper is available from the lead contact upon request.