

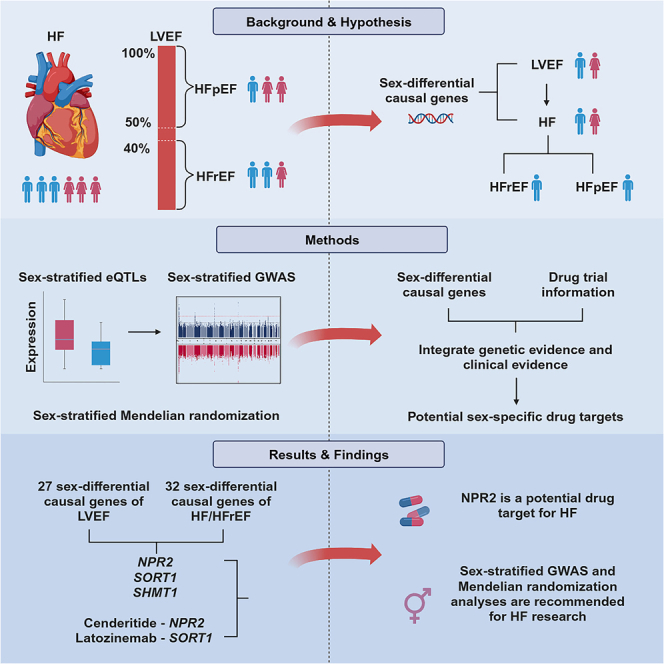
Summary
The prevalence of heart failure (HF) subtypes, which are classified by left ventricular ejection fraction (LVEF), demonstrate significant sex differences. Here, we perform sex-stratified genome-wide association studies (GWASs) on LVEF and transcriptome-wide Mendelian randomization (MR) on LVEF, all-cause HF, HF with reduced ejection fraction (HFrEF), and HF with preserved ejection fraction (HFpEF). The sex-stratified GWASs of LVEF identified three sex-specific loci that were exclusively detected in the sex-stratified GWASs. Three drug target genes show sex-differential effects on HF/HFrEF via influencing LVEF, with NPR2 as the target gene for the HF drug Cenderitide under phase 2 clinical trial. Our study highlights the importance of considering sex-differential genetic effects in sex-balanced diseases such as HF and emphasizes the value of sex-stratified GWASs and MR in identifying putative genetic variants, causal genes, and candidate drug targets for HF, which is not identifiable using a sex-combined strategy.
Keywords: heart failure, left ventricular ejection fraction, drug target, sex difference, Mendelian randomization study, genome-wide association study
Graphical abstract
Highlights
-
•
Three variants showed different associations with LVEF in women and men
-
•
Three causal genes exhibiting sex-differential effects on both LVEF and HF or HFrEF
-
•
NPR2 is likely to be a more effective drug target for HF in men
Yang et al. use sex-stratified GWASs and Mendelian randomization studies to identify sex-differential putative genetic variants, causal genes, and candidate drug targets for left ventricular ejection fraction, heart failure, and its subtypes. This study highlights the importance of considering sex-differential genetic effects in sex-balanced diseases such as heart failure.
Introduction
Heart failure (HF) is a major cause of cardiovascular morbidity, hospitalization, mortality, and healthcare expenditures,1 affecting ∼64.3 million people worldwide.2 HF can be further stratified into four subgroups based on left ventricular ejection fraction (LVEF), HF with reduced ejection fraction (HFrEF; LVEF <40%), HF with preserved ejection fraction (HFpEF; LVEF ≥50%), HF with mildly reduced ejection fraction (HFmrEF; LVEF 41%–49%), and HF with recovered ejection fraction (HFrecEF; baseline LVEF ≤40% and increase to >40%).3 Although the lifetime risk of HF is similar between males and females,4 there are profound sex differences in its two major subtypes, HFrEF and HFpEF. In the Framingham study, HFrEF is twice as common in men than in women, whereas HFpEF is the opposite.5 Due to the development of relevant drugs, such as angiotensin receptor-neprilysin inhibitors,6 the incidence rates of HF have been declining in both sexes. However, women have experienced a greater decline than men, with an overall rate change of −43% versus −29%. This can be explained by a sharper reduction in the incidence of HFrEF among women (−61%) compared to men (−29%).7
Despite the noted sex differences of HF subtypes, the underlying mechanisms remain poorly understood. Previous studies hypothesized that men’s predisposition to macrovascular coronary artery disease/myocardial infarction may contribute to the higher risk of HFrEF, whereas coronary microvascular dysfunction/endothelial inflammation may drive the elevated risk of HFpEF in women.8 In contrast, the potential sex-differential genetic regulatory mechanisms of HF and its subtypes have been less often studied. Previous genetic studies reported <50 loci associated with HF and its subtypes,9,10,11 and genome-wide association studies (GWASs) of LVEF identified six loci.12 In addition, a recent study further identified 10 putatively causal genes for HF through GWASs and Mendelian randomization (MR) analyses.13 However, these studies used sex-combined analysis strategies, in which sex-differential causal effects of genes can be masked, either in direction or magnitude.14 Adopting a sex-stratified approach may hold the key to unraveling the underlying genetic mechanisms and drug targets for HF in males and females.15
Sex-stratified GWAS and post-GWAS analyses, especially omics MR, offer practical and cost-effective ways to evaluate potential sex differences at the molecular level (e.g., transcripts).16 Omics MR uses genetic variants as instrumental variables to estimate the effect of an exposure (e.g., gene expression level) on an outcome (e.g., HF).17 The availability of sex-stratified data, including sex-stratified expression quantitative trait loci (eQTL) data,18 and sex-stratified GWASs of HF,9,11,19 has presented us with a valuable opportunity to conduct sex-stratified analyses using MR. Moreover, data from UK Biobank (UKB) enable us to generate sex-stratified GWAS results for LVEF.20
The objective of this study was to understand how genetics influence sex differences in HF, as well as in its potential intermediate phenotype, LVEF. Furthermore, we identified sex-differential causal genes whose effects on LVEF and HF and its subtypes were different between sexes, thus revealing potential drug targets that could improve LVEF levels and subsequently inform HF therapy strategies.
Results
Figure 1 illustrates the study design. First, we conducted sex-combined and sex-stratified MR analyses to estimate the causal effects of gene expression levels (represented by eQTLs) on HF. Second, sex-stratified GWAS and post-GWAS analyses were performed on LVEF. Third, the potential causal links between LVEF and HF, as well as HF subtypes, were tested using MR analyses. Fourth, we performed a male-specific MR analysis using male-only eQTLs and male-dominant HF subtype outcomes. Through the MR evidence and drug trial information, we identified shared sex-differential causal genes of LVEF and HF/HFrEF as potential drug targets.
Figure 1.

Study design
MR, mendelian randomization; TWMR analyses, transcriptome-wide MR analyses; GWAS, genome-wide association study; LVEF, left ventricular ejection fraction; HF, heart failure; HFrEF, HF with reduced ejection fraction; HFpEF, HF with preserved ejection fraction; SNP, single-nucleotide polymorphism; GTEx project, Genotype-Tissue Expression project; eQTL, expression quantitative trait loci; MVP, Million Veteran Program; GBMI, Global Biobank Meta-analysis Initiative; HERMES, Heart Failure Molecular Epidemiology for Therapeutic Targets Consortium.
Selection of instruments of gene expressions
Sex-combined and sex-stratified eQTLs (557 males and 281 females) from the Genotype-Tissue Expression (GTEx) project were selected as candidate genetic instruments for the expression levels of their respective genes.18 Our study focused on eQTLs in tissue types potentially relevant to the pathology of HF, including heart–atrial appendage, heart–left ventricle, artery–coronary, artery–aorta, artery–tibial, adipose–subcutaneous, adipose–visceral (omentum), liver, pancreas, and whole blood. Because cis-eQTLs are considered to have more direct and specific biological effects on gene expression, cis-eQTLs located within 1 Mb of the gene probe and that met the following criteria were used as instruments: (1) with F-statistics >10 to ensure sufficient instrument strength; (2) associated with the gene expression levels (p <1 × 10−5); and (3) passing the Steiger filtering to exclude reverse causal instruments.21 After selection, 15,928 (66%) eQTLs of 6,600 genes in the sex-combined population were used as exposures in the sex-combined analysis for HF. 10,592 (58%) eQTLs of 4,589 genes available in both males and females were defined as shared eQTLs and used as exposures in the sex-stratified analyses for HF and LVEF. In addition, 15,292 (84%) eQTLs of 6,565 genes in males were used as exposures in the male-specific analyses for HF subtypes (Table S1).
Selection of intermediate phenotype and primary outcomes
The primary outcomes in our study included sex-combined HF data from the HERMES (Heart Failure Molecular Epidemiology for Therapeutic Targets) Consortium10 and the Global Biobank Meta-analysis Initiative (GBMI),19 sex-stratified HF data from GBMI, and male-dominated HFrEF and HFpEF data from the Million Veteran Program (MVP).11 In addition, we investigated sex-stratified LVEF as an intermediate phenotype, with genetic associations generated through a GWAS analysis including 17,899 males and 19,467 females conducted in this study (Table S2; more details of the GWAS in later sections).
Wald’s ratio was used as the primary method to estimate putative causal effects.22
In sex-stratified MR analyses, pairwise Z score tests were applied to quantify the differences in MR estimates between males and females, with a Z score p < 0.05 suggesting sex-differential effects. MR estimates with Benjamini-Hochberg false discovery rate (FDR) <0.05 were considered to be robust evidence, and genes exhibiting such robust evidence were considered to be potential causal genes. To further strengthen the causal evidence, we evaluated linkage disequilibrium (LD) structures using the LD check method,17 which estimated the pairwise LD between each eQTL and all of the significant disease-associated GWAS variants in the cis-region. LD r2 > 0.8 was the threshold we used for the whole study.
Test for MR assumptions
To assess the relevance assumption, we examined the strength of each genetic variant with F-statistics. Only instruments with F-statistics >10, which indicated that weak instrument bias is unlikely to bias the MR estimates of this study, were selected.23 The exchangeability assumption cannot be definitively proven, but the inherent attributes of human genetics make it not susceptible to conventional confounders such as age, sex, and environmental risk factors.24 Although factors such as population stratification may introduce confounding in the outcome,25 and the utilization of only lead cis-eQTLs for target genes in each tissue limited the detection of potential bias,24,26 we should note that this study focused exclusively on homogeneous ethnic populations of Europeans, which avoid bias due to population admixture. Concerning the exclusion restriction assumption, we conducted LD check analysis to assess the LD structure and mitigate bias resulting from pleiotropy.24 In addition, we applied Steiger filtering to test the directionality of exposure-outcome effects. Only SNPs that explain more of the variance in the exposure than it does the variance in the outcome were selected in this study. By doing this, we could address potential reverse causality.21
Sex-combined and sex-differential causal effects of gene expression levels on HF
To start with, we conducted sex-combined and sex-stratified MR analyses to assess the potential causal effects of gene expression levels on HF. Using sex-combined gene expression levels (GTEx) and two HF outcome datasets (HERMES and GBMI), we yielded 11,083 and 9,835 pairs of gene-HF MR estimates, respectively. After conducting meta-analyses with the fixed-effects model to obtain the meta-analyzed MR results derived from two HF outcome datasets, we identified 140 potential causal genes with robust signals (FDR<0.05) and 89 causal genes showing LD checked evidence with HF (Table S5).
Then, using sex-stratified gene expression (GTEx) and HF outcome (GBMI) data, we further estimated the effects of the 140 candidate causal genes identified in the above sex-combined MR analysis in males and females separately. We observed seven robust signals (FDR <0.05) in males and one in females, whereas no significant sex difference was found among these 140 genes (Table S6). Furthermore, to comprehensively identify the sex-differential causal genes in HF, we extended the sex-stratified MR analyses to all shared eQTLs in males and females. By combining these sex-stratified eQTLs with HF GWASs, we yielded 6,555 pairs of gene-HF MR estimates in males and females for the following comparisons (Table S7).
Pairwise Z score tests were applied to quantify the differences of gene-HF MR estimates in the two sexes. 5.0% (328 of 6,555) of the gene-HF pairs showed nonoverlapped effect estimates in males and females (pairwise Z score p < 0.05; Table S7). When further narrowing down the analysis to gene-HF pairs with pairwise Z score p < 0.01, we identified 47 genes exhibiting different effects on HF in males and females. Among these, 24 of the causal genes showed robust MR evidence (FDR <0.05), and 12 of them showed evidence of LD checked (Table S8). The above MR results indicated substantial sex differences in the causal effects of gene expression levels on HF.
MR estimates were simultaneously influenced by expression regulations (eQTL data) and genetic associations of HF (outcome GWAS data). To explore the reasons behind the sex-differential causal effects of genes on HF, we compared the expression regulations and genetic associations of 6,555 pairs of gene-HF MR estimates in males and females (Table S9). Spearman correlation coefficients observed a very high correlation in genetic associations of eQTLs (Spearman correlation [ρ] = 0.97, p < 0.001) but an extremely low correlation in genetic associations of HF (ρ = 0.10, p < 0.001) between males and females (Figure 2). As a validation analysis, we performed MR using sex-combined eQTL data as exposures and male- or female-only HF GWAS summary statistics as outcomes separately. We observed that the MR results were very similar regardless of whether sex-stratified or sex-combined eQTLs were used (ρ = 0.99; Figure S1). This further confirmed that the sex-differential MR estimates were mainly driven by the outcome data used.
Figure 2.

Scatterplots of gene expression regulations and SNP associations of LVEF and HF in males and females
(A) The relationship of genetic effects of eQTLs between males and females.
(B) SNP associations of LVEF between males and females.
(C) SNP associations of HF between males and females.
The x axis represents estimates in males and the y axis represents estimates in females. The gray smoothing lines and shaded regions around them represent the overall trend and 95% confidence intervals (CIs) of the estimates. Ρ calculated by Spearman correlation methods were used to quantify the correlations. Only cis-eQTLs that are closely associated with genes (with p < 1.0 × 10−5) and have sufficient power (F-statistics ≥10) were included in MR analyses and depicted.
Sex-stratified GWAS and post-GWAS analyses of LVEF
Sex differences were reported in two HF subtypes, HFrEF, and HFpEF, which were stratified by LVEF. We hypothesized that LVEF may play a crucial role in explaining the sex differences. Therefore, we conducted a sex-stratified GWAS of LVEF using data from the UKB (details about methods of GWASs were provided in the STAR Methods), which included 17,899 males and 19,467 females of European ancestry. The GWAS identified six genetic variants robustly associated with LVEF in six loci (p < 5 × 10−8; Figure 3). Among these loci, BAG3, CLCNKA, and CDKN1A showed genetic signals in both males and females, and their associations with LVEF or other LV traits have been reported in previous GWASs.12,27,28 The remaining three loci were sex-specific loci, which were either found in males (FHOD3 and LSM3) or females (PTK2). These three loci have not been found in previous GWASs that used the same data as ours,12 which indicates that these sex-specific loci may be hidden in sex-combined GWASs and only be identifiable in sex-stratified GWASs. The female-specific variant, rs4073554, was close to the PTK2 gene (β of T allele = 0.33, SE = 0.06, p = 6.9 × 10−9, effect allele frequency = 0.48; Figure 3). This variant is a common intron variant that exhibited a remarkable ranking score (1a) and a high probability score (0.95) using RegulomeDB.29 Specifically, this variant overlaps with the chromatin accessibility peak and transcription factor footprints for heart tissue and was predicted to be within the active enhancer region of relevant tissues such as the heart and vessel. The above evidence indicated that the female-specific variant within the PTK2 is likely to be a functional variant for cardiovascular tissues.
Figure 3.

Manhattan plot of sex-stratified genome-wide LVEF associations
Sex-stratified GWAS was conducted using the BOLT-LMM method (n = 17,899 in males, n = 19,467 in females). The x axis represents the genomic position and the y axis represents the strength of association as represented by −log10 (p value). Suggestive associations at a significance level of p < 1 × 10−5 are indicated by the black line and genome-wide significance at p < 5 × 10−8 is indicated by the red line. Independent genome-wide significant variants are annotated with the nearest gene(s). Male-specific candidate genes are denoted by blue boxes, female-specific candidate genes are denoted by red boxes, and candidate genes found in both sexes are denoted by green boxes.
To assess the proportion of phenotypic variance explained by the common genetic variation, we estimated the SNP heritability (h2) of LVEF. The SNP heritability of LVEF was similar between males (h2 = 0.17, SE = 0.04) and females (h2 = 0.19, SE = 0.03). The bivariate LD score30 using SNPs across the whole genome identified comparable genetic correlations between LVEF and HF in males (rg = −0.39) and females (rg = −0.37), and a strong positive genetic correlation between LVEF in males versus females (rg = 0.92, SE = 0.13, p = 4.66 × 10−12; Table S10). However, when we restricted the comparisons to sex-stratified eQTLs from GTEx, the Spearman correlation between SNP-LVEF associations in males and females was largely attenuated (ρ = 0.13, p < 0.001), suggesting a higher sex disparity of these genetic variants influencing the expression of genes.
To investigate potential sex-differential causal effects of gene expression levels on LVEF, we undertook MR analyses by combining sex-stratified shared eQTLs (GTEx) and LVEF data (this study) and obtained 7,931 pairs of gene-LVEF MR estimates in both males and females (Table S11). We identified 41 genes exhibiting sex-differential effects on LVEF using pairwise Z score tests (p < 0.01; Table S12). Among these 41 genes, 27 were potential causal genes with robust MR signals (FDR <0.05), and 11 of them showed LD checked evidence (Table S12). Among the causal genes, four genes, OPRL1, CLEC12A, CLEC12B, and CLEC1B, exhibited robust causal effects on LVEF in both males and females (Table S13).
Sex-stratified causal links between LVEF and HF
We hypothesized that LVEF plays an intermediate role of causal genes on HF, and sex differences in HF can be partially explained by sex differences in LVEF. Using four SNPs robustly (with p < 5 × 10−8) and independently (filtered by LD clumping with r2 < 0.001) associated with LVEF in males and two SNPs in females as instruments (Table S14), we found strong MR evidence to support the effect of upregulated LVEF on a reduced risk of HF in both males (inverse variance weighted [IVW] odds ratio [OR] = 0.90, 95% confidence interval [CI] = 0.85–0.94, p = 1.86 × 10−5 per percent change unit increase of LVEF) and females (IVW OR = 0.84, 95% CI = 0.78–0.90, p = 2.70 × 10−7). The direction of effects was consistent across the five MR sensitivity methods, including IVW, weighted median, weighted mode, simple mode, and MR-Egger (Table S15).
Male-specific causal links between LVEF and HFrEF and causal genes of HFrEF
Due to a more pronounced decline in the incidence of HFrEF among women compared to men, women experienced a greater reduction in the overall incidence of HF.7 Therefore, we aimed to find drug target genes for HF, with a focus on increasing LVEF and preventing HFrEF for males. We performed male-specific MR analyses to examine the causal links between LVEF and two HF subtypes (HFrEF and HFpEF; data from MVP, which comprised 92% male participants). Using the four male-specific LVEF variants as instruments, MR analysis revealed a robust effect of higher LVEF on the reduced risk of HFrEF (IVW OR = 0.82, 95% CI = 0.78–0.86, p = 1.64 × 10−15) in males, but not on HFpEF in males, which fit with our expectations (Figure 4; Table S15). Furthermore, we performed male-specific MR using male-only eQTLs (GTEx) and a GWAS of HFrEF (MVP). We identified 6,254 gene-HFrEF MR estimates, 8 of which showed robust MR signals (FDR <0.05), and 2 genes showed LD checked evidence with HFrEF (Table S16). In addition, to evaluate the effects of genes on HFpEF for reference, we performed MR analysis by combing male-only eQTLs and HFpEF, which identified 10 putative causal genes, with 9 showing LD checked evidence with HFrEF (Table S17).
Figure 4.

Sex-stratified MR to estimate the causal effects of genetically determined LVEF levels on risks of HF/HF subtypes
GWAS of LVEF is from the present study, whereas sex-stratified GWAS of HF is from GBMI, and GWASs of male-dominant HFrEF and HFpEF are from MVP. Inverse variance weighted 2-sample MR method was performed. The odds ratio (OR) and 95% CI are for the risk of HF and its subtypes per percent change in the LVEF levels. The squares are the causal estimates on the OR scale, and the whiskers represent the 95% CI for these ORs.
Evidence synthesis and HF/HFrEF drug targets identification
Through the above MR analyses, we first found 140 causal genes in sex-combined HF, and 7 of them showed robust MR evidence in males and 1 of them showed robust MR evidence in females. Second, we found 24 sex-differential causal genes of HF in males or females. Third, we identified 8 male-specific causal genes of HFrEF. Taken together, we found 32 sex-differential causal genes for HF/HFrEF and 27 sex-differential causal genes for LVEF. To pinpoint causal genes playing sex-differential roles on HF/HFrEF via LVEF, we identified genes exhibiting robust MR evidence (FDR <0.05) for both LVEF and HF. Considering the potential tissue-specific effects of gene expression regulations,31 we initially focused on shared genes that demonstrated causal effects in the same tissue. As a result, we identified 2 causal genes, SORT1 (sortilin 1) and NPR2 (natriuretic peptide receptor 2), that showed sex-differential causal effects on HF/HFrEF and LVEF (Figures 5 and 6; Table S18). Then, to extensively examine shared causal genes between LVEF and HF/HFrEF, we expanded the search to genes showing MR evidence in different tissues and identified SHMT1 (Serine hydroxymethyltransferase 1) as a putative causal gene for both HF and LVEF in females.
Figure 5.

Sex-stratified TWMR to estimate the causal effects of gene expression levels on levels of LVEF and risks of HF/HF subtypes
Wald’s ratio method was applied. The effects on LVEF were reported as effect sizes (β) with 95% CIs. Effects of per 1-SD change in gene expression level on binary outcomes (HF, HFrEF, and HFpEF) were represented as ORs with 95% CIs. The squares are the causal estimates on the β scale for LVEF levels, or OR scale for binary outcomes, and the whiskers represent the 95% CIs.∗ The instrument of NPR2, rs78920801, was not available in the male-specific HF GWAS data. We attempted to use a proxy genetic variant in high LD with this eQTL (r2 > 0.8 in the 1000 Genomes data for the relevant population32) instead. However, no variant met this criterion.
Figure 6.

Shared causal genes between LVEF and HF/HFrEF identified in this study
Two genes in red (NPR2 and SORT1) were shared causal genes between LVEF and HF/HFrEF with eQTLs from the same tissue. The remaining gene in black (SHMT1) was a shared causal gene with eQTLs from different tissues.
For these three prioritized causal genes/targets, NPR2, SORT1, and SHMT1, we integrated their MR evidence with drug trial information from OpenTargets33 and the Therapeutic Target Database34 to assess the potential of these causal genes as viable drug targets. First, genetically predicted higher expression levels of NPR2 showed an effect on increased LVEF levels (β = 0.65, 95% CI = 0.23–1.07, p = 2.39 × 10−3) and reduced risk of HFrEF (OR = 0.87, 95% CI = 0.81–0.93, p = 4.57 × 10−5), but not associated with risk of HFpEF in males. For MR results in females, the expression levels of NPR2 showed little evidence to support its effect on LVEF or HF. Its effect on HF in both sexes was naturally neutralized after combining the differential effects in males and females, which partly explained why previous sex-combined studies were not able to identify the association of NPR2 on HF (genetic score from OpenTargets = 0; Table 1). The drug trial information further supported the protective effect of NPR2 activation on HF and LVEF. Cenderitide, also known as CD-NP, is a dual agonist targeting both NPR2 and NPR1.35 It has entered phase 1 and 2 and proof-of-concept clinical studies that aimed to evaluate its efficacy in treating HF (clinicaltrials.gov: NCT00839007, NCT00620308, NCT02359227, and NCT01750905).
Table 1.
Drug target validation
| MR results in this study | Open targets | Therapeutic target database | Clinical trial | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Causal gene | Outcomes | Effect sizesa | p | Overall score | Genetic score | Drug | Action | Indication | Phase |
| NPR2 | Male-stratified LVEF | 0.65 (0.23–1.07) |
2.39E−3 | 0.01 | 0.00 | Cenderitide | Agonist | Heart failure | 2 (NCT02359227) |
| Female-stratified LVEF | −0.09 (−0.36 to 0.17) |
4.86E−1 | |||||||
| Male-dominated HFrEF | 0.87 (0.81–0.93) |
4.57E−5 | |||||||
| OPRL1 | Male-stratified LVEF | −0.32 (−0.61 to −0.04) |
2.75E−2 | 0.00 | 0.00 | SER-100 | Agonist | 2 (NCT00283361) | |
| Female-stratified LVEF | 0.32 (0.08–0.56) |
8.12E−3 | |||||||
| ADORA2B | Sex-combined HF | 0.97 (0.95–0.99) |
4.49E−4 | 0.02 | 0.00 | Tonapofylline | Inhibitor | 3 (NCT00709865) | |
| KCNH2 | Sex-combined HF | 1.03 (1.02–1.05) |
2.21E−4 | 0.50 | 0.00 | Vesnarinone | Inhibitor | Approved | |
| SORT1 | Male-stratified LVEF | −0.05 (−0.17 to 0.16) |
3.85E−1 | 0.06 | 0.09 | Latozinemab (AL001) | Monoclonal antibody | Frontotemporal dementia | 2 (NCT03987295) |
| Female-stratified LVEF | 0.18 (0.06–0.29) |
2.20E−3 | |||||||
| Male-dominated HFrEF | 0.95 (0.93–0.97) |
3.64E−7 | |||||||
| CLEC12A | Male-stratified LVEF | −0.16 (−0.28 to −0.05) |
6.33E−3 | 0.00 | 0.00 | CD123/CLL1 CAR-T cells | CAR-T cell-therapy (dual specific) | Acute myeloid leukemia | 2/3 (NCT03631576) |
| Female-stratified LVEF | 0.17 (0.03–0.30) |
1.46E−2 | |||||||
| CETP | Sex-combined HF | 1.05 (1.02–1.08) |
4.36E−4 | 0.20 | 0.32 | Evacetrapib | Inhibitor | Cardiovascular disease | 3 (NCT02227784) |
| EDNRA | Sex-combined HF | 0.94 (0.91–0.97) |
3.16E−5 | 0.34 | 0.00 | Macitentan | Modulator | Cardiovascular disease | Approved |
| VKORC1 | Sex-combined HF | 1.02 (1.01–1.03) |
3.76E−4 | 0.50 | 0.00 | Warfarin | Inhibitor | Atrial fibrillation | Approved |
| ALDH2 | Sex-combined HF | 0.91 (0.87–0.95) |
1.27E−5 | 0.07 | 0.05 | ALD-401 | Modulator | Cerebrovascular ischemia | 2 (NCT01273337) |
| FP-045 | Agonist | Cardiovascular disease | 1 | ||||||
NPR2 and SORT1 were causal genes that showed sex-differential effects on HF/HFrEF and LVEF in this study.
Effect sizes are represented by ORs and 95% CI for binary outcomes, including HF, HFrEF and HFpEF; for the continuous outcome, LVEF, the effect sizes are β coefficients and 95% CI.
Second, genetically predicted higher expression levels of SORT1 exhibited effects on reducing the risk of HF in both males and females, although the evidence was comparatively weaker in females. Meanwhile, elevated expression levels of these two genes were associated with increased LVEF levels solely in females, whereas no such correlation was observed in males. Drug targeting SORT1 for the treatment of frontotemporal dementia is currently under clinical development (phase 3 clinical trial, clinicaltrials.gov: NCT04374136), and the design of drugs targeting SORT1 and clinical trials evaluating their effects on HF are awaited.
Third, we identified SHMT1 as a putative causal gene that showed effects on both LVEF and HF in females, with MR evidence in different tissues. Our findings revealed that genetically predicted higher expression levels of SHMT1 were linked to an increased risk of HF and elevated levels of LVEF in females, whereas little effect was found in males. We did not find a developed drug targeting SHMT1.
Furthermore, this study identified three causal genes related to LVEF or HF as potential drug targets: ADORA2B, OPRL1, and KCNH2. KCNH2 is already targeted by an approved HF drug, vesnarinone,36 which is an inhibitor of KCNH2. This aligned with our finding that a lower expression level of KCNH2 was associated with a lower risk of HF. In addition, we discovered five other causal genes, CLEC12A, CETP, EDNRA, VKORC1, and ALDH2, which were already targeted by existing drugs, four of which were used in the treatment of cardiovascular diseases (Table 1).
Discussion
In this study, we comprehensively investigated the sex-differential putative causal genes of HF/HF subtypes and LVEF. Although previous sex-stratified genetic studies recognized HF as a sex-balanced disease,14,31 we observed that 5.0% of the tested genes exhibit sex-differential effects on HF, and these differences can be attributed to sex-differential genetic associations of HF. Sex-stratified GWASs identified three loci for LVEF; two loci were only identified in males (FHOD3 and LSM3), and one locus was found in females only (PTK2). After detecting causal links between LVEF and HF/HF subtypes, especially HFrEF in males, we looked for sex-differential causal genes shared between LVEF and HF/HFrEF. Among 32 sex-differential causal genes for HF/HFrEF and 27 sex-differential causal genes for LVEF, 3 shared causal genes, NPR2, SORT1, and SHMT1, may play their causal roles on HF through LVEF. Among these three genes, SORT1 has been previously reported in GWASs of HF,10 whereas NPR2 and SHMT1 were not. Integrating the MR evidence with drug trial information, we prioritized two sex-differential genes as candidate drug targets for HF. NPR2, the target gene of HF drug Cenderitide, showed robust MR evidence on LVEF and HFrEF in males but much weaker in females. SORT1 showed effects on LVEF only in females and showed effects on HF in both sexes, where SORT1 is the target gene for the drug latozinemab. In addition, among a total of 202 causal genes of LVEF, HF, and its subtypes identified in this study, except for NPR2 and SORT1, there were 8 drug targets for existing drugs, including 3 HF drugs and 4 other cardiovascular drugs. Our study highlighted the importance of considering sex-differential genetic effects even in sex-balanced diseases such as HF and emphasized the value of sex-stratified GWAS and MR analyses in identifying genetic variants, putative causal genes, and candidate drug targets for HF, which may not be identifiable using a sex-combined strategy.
Although omics MR studies integrating QTLs data (i.e., eQTLs) with GWASs have been widely used to pinpoint potential causal genes for complex traits and diseases,17,37,38,39,40 the availability of sex-stratified omics MR studies has only recently emerged.16 Most of these studies have focused on phenotypes exhibiting notable sex differences, such as testosterone levels and dementia.41,42,43,44 In previous sex-stratified genomic studies that systematically screened phenotypes with sex differences,14,31 HF was considered to be a phenotype without sex differences due to its similar SNP heritability between males and females. This may partly lead to the lack of further discoveries (e.g., omics MR study) on the sex differences in HF. However, such similarity in heritability for binary traits, such as HF, may arise due to the limited power to detect sex differences in the currently available cohorts.16 Interestingly, our findings reveal that 5.0% of the tested genes exhibit sex-differential causal effects on HF. Such sex-differential MR estimates were driven by different genetic associations of HF between males and females (ρ = 0.10) rather than that of eQTLs (ρ = 0.97). This observation aligns with findings from a previous MR analysis using sex-stratified eQTL data as exposure.42
Previous studies have validated the effectiveness of evaluating the causal effects of genes on intermediate phenotypes to inform causal pathways and drug targets for cardiovascular diseases.13,45,46,47 However, the application of a sex-aware approach in this field was scarce. Considering that LVEF stratified HF into subtypes with significant sex disparities, we hypothesized that sex-differential causal relationships between genes and LVEF levels may contribute to the sex disparities in HF subtypes. We attempted to inform the mechanism of HF by the following three sex-stratified analyses. First, by conducting a sex-stratified GWAS on LVEF, we found that although the heritability (0.17 in males and 0.19 in females) and genetic correlation (rg = 0.92) of LVEF were highly similar between males and females, our GWAS identified three sex-specific LVEF loci. This further highlights the need to go beyond heritability and genetic correlation analyses and indicates that sex-specific loci may be masked in sex-combined GWASs and only identifiable in sex-stratified GWASs. One of the findings, PTK2, encodes FAK (focal adhesion kinase) and has been reported to be associated with other LVEF-related phenotypes, including LV diastolic function measurement and atrial fibrillation.48,49 In line with our findings, animal research yielded evidence of sex-specific neurological effects of FAK.50,51 Second, sex-stratified MR analysis for LVEF revealed four sex-differential genes—OPRL1, CLEC12A, CLEC12B, and CLEC1B—that exhibited strong causal associations with LVEF in both sexes. Intriguingly, these genes had opposite effects on LVEF levels in males and females. In females, higher expression levels of these genes were related to increased LVEF levels, whereas in males, it led to decreased LVEF. Third, sex-stratified MR analysis identified causal genes showing sex-differential effects on HF, and three of these genes also affect LVEF levels in a sex-biased manner. Among these shared genes, higher expression levels of NPR2 showed a positive association with LVEF levels in males and a negative association with risk of HFrEF in males. Notably, these effects were not observed in females, suggesting that NPR2 may exclusively affect LVEF levels and the risk of HFrEF in males. Taken together, the sex-stratified method outperformed the common sex-combined analysis in identifying genes that affect LVEF and HF in a sex-specific manner and potentially explain causal pathways in HF.
In this study, NPR2, SORT1, and SHMT1 were identified as three prioritized drug targets. However, the validation of the drug targets should not rely on only genetic evidence but, more important, on evidence derived from experiments and clinical trials. NPR2 encodes the primary receptor for C-type natriuretic peptide (CNP), a hormone involved in cardioprotective actions mediated by NPR2/cGMP signaling.52,53,54 Existing experimental evidence showed that CNP has antiproliferative and antihypertrophic effects, and activating NPR2 with CNP was effective in improving ventricular contractility and reducing the risk of HF and other related cardiovascular conditions in mice.55,56,57,58 In contrast, inhibiting NPR2 in mice led to precursors to HF, including deteriorated left ventricular systolic function.55,59,60 Furthermore, a drug targeting NPR2, Cenderitide, has been evaluated in clinical studies with patients with acute and chronic HF (NCT00839007 and NCT00620308) after myocardial infarction (NCT02071602), and in stable patients with left ventricular assist devices (NCT01750905). In a recent randomized trial involving 18 HF patients with LVEF <40%, Cenderitide showed a safety profile and pharmacological effects, compared with placebo.61 This further supports NPR2 prioritization as a candidate drug target for HFrEF prevention. Furthermore, a next-generation drug called CRRL408, which was designed based on Cenderitide, is currently in its preclinical investigational drug-enabling program.62 This clinical trial evidence aligns well with our MR evidence, in which our results further indicate that NPR2 could have better efficacy on HFrEF in males through increasing LVEF.
SORT1 was previously identified in a sex-combined GWAS of HF conducted by the HERMES Consortium.10 Building upon this study, we further investigated the sex-differential associations between SORT1 expression levels and LVEF and HF/HF subtypes. Our MR evidence indicates that the drug targeting this cluster may be more effective in the increase of LVEF levels in females and have protective effects on HF in both sexes and HFrEF in males. However, the relationship between SORT1 and HFrEF risk in females remains to be elucidated. Preclinical studies have identified the crucial role of SORT1 in cardiovascular phenotypes.63,64,65,66,67,68,69,70 However, the direction of the causal effects of SORT1 on these phenotypes remains inconsistent.71,72 To pave the way for the development of cardiovascular drugs targeting SORT1, additional experimental and clinical evidence is required to fully understand the role of SORT1 in the development of cardiovascular diseases, including HF. SORT1 has emerged as a potential therapeutic target for AL001, a monoclonal antibody that blocks the receptor encoded by SORT1.73 A phase 3 clinical trial is in progress to assess the effectiveness of AL001 in treating frontotemporal dementia (NCT04374136). However, our findings, together with the existing evidence mentioned above, raise concerns about the potential side effects of AL001 on cardiovascular health.
In addition, we found that expression levels of SHMT1 were associated with elevated LVEF levels and increased risk of HF in females. However, the causal effects were observed in different tissues and should be interpreted with caution. SHMT1 regulates key reactions in folate-mediated one-carbon metabolism.74 The functions of SHMT1 in the development of HF or other cardiovascular diseases have been poorly investigated.75,76 Mimosine, a nonprotein amino acid, attenuates the transcription of SHMT1 and was reported to have an antifibrotic effect.77 However, it was unclear whether this antifibrotic activity can benefit heart diseases in vivo.78 Further studies are needed to investigate the effects of SHMT1 on LVEF and HF, particularly in females.
Furthermore, among the causal genes for LVEF, HF, and its subtypes identified in this study, three served as existing drug targets for HF. Our findings offer additional genetic evidence that may help explain the observed drug effects in clinical trials. First, an approved HF drug, vesnarinone, inhibited the expression of KCNH2. This aligned with our finding that lower expression levels of KCNH2 were associated with a reduced risk of HF. Second, SER100 was an agonist targeting OPRL1 that was evaluated in a phase 2 clinical trial (NCT00283361). However, the development of SER100 as a drug candidate for HF was terminated due to its adverse dose-limiting decrease in systolic blood pressure.79 Our study revealed the sex-differential effects of OPRL1 on LVEF. Specifically, we found a positive association between OPRL1 expression levels and LVEF in females and a negative relationship in males. Third, a lower expression level of ADORA2B was related to an increased risk of HF in this study; however, an HF drug, tonapofylline, assessed in clinical trials was an inhibitor targeting ADORA2B. Notably, the phase 3 clinical trial failed to demonstrate the efficacy of intravenous tonapofylline treatment for acute HF,80 although another trial suggested that oral agents were beneficial in increasing sodium excretion in stable HF patients.81
Limitations of the study
Our study has some limitations. First, the outcome data we used were derived from case-control GWASs. Therefore, the causal genes identified in this study represent potential targets for disease prevention rather than treatment. Further investigations conducted with HF progression data are needed to validate the treatment effects of drugs targeting these genes. However, despite this limitation, one of three gene targets, NPR2, is an HF drug target. In addition, expression levels of several other HF treatment targets (i.e., KCNH2) were found to be associated with the risk of HF. This suggests that there may be some mechanistic overlaps between HF prevention and treatment targets. Second, due to a lack of other types of omics data, our analyses were restricted to transcriptome data and GWAS data. Studies incorporating other types of large-scale molecular QTL datasets are needed to better discover disease mechanisms and identify potential drug targets.82 Third, this study included only participants of European ancestry, which limits the generalizability of the findings to non-European ancestry. Although multiancestry MR comparisons are of great importance,37,38 data in non-European ancestries are still lacking, which highlights the importance of well-powered genetic studies in non-European ancestries. Fourth, the eQTL data used in our study were derived from relatively small sample sizes with 838 individuals, and only 1 instrument for target genes in each tissue was available. This may limit the statistical power and precision of the MR results. However, we prioritized using GWASs with larger sample sizes and cases as outcomes to minimize the impact of this limitation. Fifth, we used eQTLs data with more males than females and sex-combined and sex-stratified HF data with more HF cases in males than in females. This sample size imbalance may affect the statistical power of our MR analyses, potentially weakening evidence of correlations between gene expression levels (e.g., SORT1) and HF/HFrEF risk in females compared to males. In an ideal situation, we could conduct GWASs after matching the sample size between males and females. However, our limited access to summary statistics prevented us from taking this approach. Sixth, limited by a lack of female-only data, causal genes of HF subtypes identified in this study were male specific, and their effects in females remain unknown. This constrained our investigation of the mechanism of HF. However, a paucity of female data is a common problem in HF research,8 and such knowledge gaps can only be addressed with sex-balanced or female-only studies that have increased representation of females.
In conclusion, our findings indicate that the sex differences in sex-balanced diseases, such as HF, can still be partly explained by genetics. Meanwhile, sex-stratified GWASs have the potential to reveal genetic loci (e.g., PTK2 for LVEF in females). These findings highlight the importance of considering sex-stratified disease GWAS associations when examining the genetic basis of sex differences in diseases. Furthermore, we identified two genes, NPR2 and SORT1, that are causally linked to both LVEF and HF/HFrEF, exhibiting sex-differential effects. These genes are potential drug targets for the prevention of HF and HFrEF by increasing LVEF levels in specific sexes. Further clinical and experimental research is warranted to validate and explore the feasibility and effectiveness of targeting these genes for HF prevention and treatment. The analytical model used in this study can be generalized to other research on complex diseases without noted sex differences.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| eQTLs | Genotype-Tissue Expression (GTEx) project18 | www.gtexportal.org/home/ |
| GWAS of sex-combined HF | Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES) Consortium10 Global Biobank Meta-analysis Initiative (GBMI)19 |
www.nature.com/articles/s41467-019-13690-5 www.globalbiobankmeta.org/ |
| GWAS of sex-stratified HF | Global Biobank Meta-analysis Initiative (GBMI)19 | www.globalbiobankmeta.org/ |
| GWAS of sex-stratified LVEF | UK Biobank20 This study |
www.ukbiobank.ac.uk/ |
| GWAS of male-dominate HFrEF and HFpEF | Million Veteran Program (MVP)11 | www.nature.com/articles/s41467-022-35323-0 |
| Software and algorithms | ||
| MR models | Hemani et al.83 | github.com/MRCIEU/TwoSampleMR |
| LD check | Zhao et al.38 | github.com/globalbiobankmeta/multi-ancestry-pwmr |
| LDSC | Ning et al.84 | github.com/bulik/ldsc |
| R | Open source | https://www.r-project.org/ |
| Other | ||
| Variant Effect Predictor (VEP) | McLaren et al.85 | useast.ensembl.org/ |
| Therapeutic Target Database | Zhou et al.34 | db.idrblab.net |
| OpenTarget | Koscielny et al.33 | https://platform.opentargets.org/ |
| ClinicalTrials | Open source | https://clinicaltrials.gov/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Yu Xu (jane.yuxu@gmail.com).
Materials availability
This study did not generate unique reagents.
Data and code availability
The summary statistics of the present sex-stratified GWAS analysis on left ventricular ejection fraction (LVEF) will be deposited to the IEU Open GWAS database (https://gwas.mrcieu.ac.uk/) upon publication of the article. The data can be accessed using the GWAS IDs 'ieu-b-5120' and 'ieu-b-5121. Code for reproducing the study is available at GitHub repository (https://github.com/geneinmylife/MR-project-Heart-failure). Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
Experimental model and subject details
This study did not use any experimental models or enroll human subjects.
Method details
Study design
The current study consists of four parts, as shown in Figure 1. (1) to identify causal genes for heart failure (HF) and explore potential sex differences in gene-HF associations, we conducted sex-combined and sex-stratified omics Mendelian Randomization (MR) analyses. A pairwise Z score test was used to quantify the sex differences in HF and find sex-differential causal genes. To understand the underlying reasons for sex differences in HF, we calculated Spearman correlations between gene expression regulations and genetic effects on HF between males and females. (2) to comprehend the sex differences in left ventricular ejection fraction (LVEF), we conducted a GWAS analysis for LVEF by employing data from the cardiac magnetic resonance (CMR) imaging sub-study of UK Biobank (UKB) and stratified the samples by sex. Post-GWAS analyses included Spearman correlation calculation, estimation of heritability and genetic correlations, as well as sex-stratified MR analyses applying the same analytical pipeline described above for HF. (3) To investigate the sex-differential causal effects of LVEF on HF, we conducted two-sample MR with sex-stratified LVEF and HF data. Besides, we analyzed the causal effects of LVEF on two subtypes of HF, HF with reduced ejection fraction (HFrEF), and HF with preserved ejection fraction (HFpEF) using male-dominate data. (4) Based on evidence gathered from the previous steps, we assumed that certain genes may play causal roles in HF/HFrEF through their effects on LVEF. Therefore, we identified shared causal genes between these two phenotypes. This MR evidence was incorporated with drug trial information to find potential drug targets. Details about the study participants and methods are provided below.
Data resources
Table S2 provides full details of the data sources and sample size.
Study exposures
In the current analysis, expression quantitative trait loci (eQTLs) were used as instruments to evaluate the causal role of genes on outcomes. The eQTL data were obtained from version 8 of the Genotype-Tissue Expression (GTEx) project, which consisted of 838 individuals (557 males and 281 females, sex was classified based on sex chromosomes).18 Our study focused on eQTLs in tissue types potentially relevant to the pathology of HF. The evaluated tissue types included: Heart – Atrial Appendage, Heart – Left Ventricle, Artery – Coronary, Artery – Aorta, Artery – Tibial, Adipose – Subcutaneous, Adipose – Visceral (Omentum), Liver, Pancreas, and Whole Blood. The tissue selection process was informed of biological mechanisms known to be related to HF.13,86,87
The following steps were applied to select eQTL instruments: (1) we focused on cis-eQTLs, which are located within 1 Mb of the associated probe, to minimize associations potentially driven by horizontal pleiotropy; (2) only eQTLs that were closely associated with genes (with p < 1.0 × 10−5) were selected; (3) we assessed the strength of the genetic predictors for each variant using F-statistics. We considered single nucleotide polymorphisms (SNPs) with F-statistics ≥10 as having sufficient power and included them in our analysis; (4) we used Steiger filtering to identify SNPs that explain more of the variance in the exposure than it does the variance in the outcome, and only eQTLs with the Steiger filter flag as TRUE were selected. By implementing these rigorous selection criteria, we aimed to ensure the reliability and validity of the instrumental variables used in our analysis.
Primary outcomes
The primary outcomes were HF and its two subtypes: HFrEF and HFpEF. We obtained sex-combined GWAS summary statistics of HF from 1) the Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES) Consortium,10 which includes 26 studies of European ancestry; and 2) Global Biobank Meta-analysis Initiative (GBMI).19 GBMI is a global biobank that includes genetic and health data from approximate 2.6 million participants, from which sex-stratified HF data were obtained. To analyze the subtypes of HF, specifically HFrEF and HFpEF, we utilized GWAS data from the Million Veteran Program (MVP).11 MVP is a large biobank linked to Veterans Affairs (VA) electronic health record databases. MVP dataset primarily consists of male participants (92%), and most of the HF cases were males (96.9%), the GWAS data from MVP were specifically used as male-dominate data.
Intermediate phenotypes
There are marked sex biases in the landscape of the two major subtypes of HF, HFrEF, and HFpEF, which are categorized based on LVEF. Therefore, in the current analysis, the sex-stratified LVEF was used as an intermediate phenotype. We performed sex-stratified GWAS of LVEF, using data from the CMR imaging sub-study of UK Biobank (UKB).20 The UKB is a large prospective cohort that initially recruited around half a million participants resident in the UK at an age of 40–69 years in its initial recruitment visit. As a sub-study of UKB, CMR imaging was performed in Cheadle, United Kingdom, on a clinical wide bore 1.5 Tesla scanner according to a protocol reported before.88 At the time of the current analysis, data on LVEF from 39,695 individuals were available.
Before conducting two-sample MR analyses to test the causal links between LVEF and HF and its two subtypes, we performed the following criteria to select instruments that were strongly and independently associated with LVEF: (1) instruments were required to be associated with LVEF at genome-wide significance level (p < 5.0 × 10−8); (2) instruments with F-statistics lower than 10 were excluded to ensure sufficient instrument strength; (3) linkage disequilibrium (LD) clumping was applied to remove instruments strongly correlated with each other (r2 < 0.001) in 1Mb region.
Identify sex-combined and sex-stratified causal genes associated with HF
First, we employed sex-combined MR analyses to identify potential causal genes of HF, thus informing the subsequent follow-up investigation about whether these genes exhibited sex-differential effects. To increase statistical power, we conducted sex-combined MR analyses separately using HF outcome datasets from GBMI and HERMES, and then synthesized the results obtained from these two datasets using meta-analyses with fixed-effects model. FDR was applied among all MR estimates within each tissue respectively. Genes with robust MR signals (FDR<0.05) in this sex-combined MR analysis were further tested in sex-stratified MR analyses. LD check were performed for these potential causal genes of HF.
Second, to examine sex differences in HF, we conducted sex-stratified MR analyses in three steps: 1) identified genes with instruments proxy their expression levels in both sexes, and performed MR on HF separately for males and females; 2) calculated Spearman correlations to quantify the differences in all pairs of MR estimates shared between the two sexes; 3) conducted pairwise Z score tests to quantify differences in each pair of MR estimates shared by males and females. A p value of pairwise z-scores <0.05 indicated non-overlapping MR estimates between males and females.
Using Spearman correlation coefficient and the percentage of non-overlapping MR estimates, we detected sex differences in HF. To reveal the underlying reasons for these differences, we calculated the correlations between gene expression regulations (eQTL data) and genetic associations with HF (GWAS data) for all gene-HF pairs between males and females. Furthermore, to validate the findings that sex differences in HF were driven by sex-stratified GWAS associations of HF, we performed MR again with sex-combined eQTL data and male- or female-only HF GWAS summary statistics, separately. Subsequently, we calculated correlations between these MR estimates and those derived from sex-stratified MR (i.e., the MR estimates from sex-combined eQTLs and male-only HF GWAS, and the MR estimates from male-only eQTLs and male-only HF GWAS).
To extensively investigate genes with sex-differential causal effects on HF, we selected MR estimates with significant sex-differential effects (defined as a pairwise Z score p value < 0.01), and then applied an FDR correction to exclude genes that showed weak association in both sexes. For causal genes with robust evidence (FDR<0.05), LD check was further conducted to evaluate the LD structure. This approach allowed us to pinpoint genes with sex-differential effects on LVEF and HF, aligning with the primary focus of our investigation.
Identify sex-stratified causal genes associated with LVEF
The same statistical model used in sex-stratified MR analyses for HF was applied to identify the sex-differential putative causal genes of LVEF.
Identify causal relationships between LVEF and HF/HF subtypes
To identify potential causal links between LVEF and HF, we performed a two-sample MR analysis. In this analysis, LVEF was the exposure variable, and the risk of HF/HF subtypes was evaluated as the outcomes.
Identify male-specific causal genes on HF subtypes
Male-specific MR was performed in male-only eQTLs and male-dominant GWAS of HFrEF and HFpEF. We used an FDR threshold of 0.05 for all MR estimates within each tissue respectively, and then LD check for validation. Furthermore, the causal effects of genes on HFpEF were tested for reference.
Identify drug targets for HF/HF subtypes
Based on previous studies that identified cardiovascular drug targets among shared causal genes between disease-relevant traits and diseases,45,46,47 we investigated the overlap between sex-differential causal genes of LVEF and HF/HFrEF to explore potential drug targets. We assumed that these shared genes could be potential drug targets with sex-specific effects on LVEF and HF, so we conducted an extensive search for drug information on all the causal genes identified in this study, with a particular focus on these shared causal genes. To gather drug information and clinical annotations for the potential target genes, two primary sources were utilized: Therapeutic Target Database (TTD)34 and OpenTarget.33 TTD provides valuable information about therapeutic protein and nucleic acid targets, the specific diseases they target, and the drugs associated with these targets. The OpenTarget recorded the relationships between genes and diseases, and rated evidence score for each category (e.g., genetic association score) (Table 1). Details of the clinical trials for potential targets were further verified by checking Clinialtrials.gov.
Quantification and statistical analysis
MR analyses
MR allows for the simultaneous use of multiple SNPs as instruments and multiple gene expression traits as exposures.89 The analysis was conducted based on three key assumptions of MR: (1) relevance: the genetic predictors have robust associations with the exposure; (2) exchangeability: the association of instruments with exposures and outcomes is not confounded; (3) exclusion restriction: the instruments are only related to the outcome through the exposure being studied. Findings were reported according to the STROBE-MR (Strengthening the Reporting of Mendelian Randomization Studies) guidelines.90
For MR analysis aimed at identifying causal genes, the Wald’s ratio method was applied to calculate the MR estimates for all instruments, as only one instrument was available for target genes in each tissue. To establish multiple testing-adjusted thresholds and discover gene-outcome associations with robust signals, we applied a Benjamini-Hochberg FDR of 0.05. For MR analysis aimed at identifying causal links between LVEF and HF and HF subtypes, we utilized Wald’s ratio method for exposures with only one instrument, and the inverse variance weighted (IVW) method for exposures with two or more instruments.91 To ensure the robustness of associations, we further performed sensitivity analyses using weighted median, MR-Egger, simple mode, and weighted mode methods.
We reported the effects of per 1-standard deviation (SD) change in gene expression level on binary outcomes (HF, HFrEF, and HFpEF) as odds ratios (ORs) with 95% confidence intervals (CIs). The effects on LVEF were reported as effect sizes with 95% CIs. All ethical approvals for these analyses can be found in the original studies.
LD check
For causal genes showing robust MR evidence (FDR<0.05), to further distinguish causality from confounding by LD, we evaluated the LD structure. As gene expression association lacks sufficient SNP coverage in the test region (only top hits were provided from GTEx), we conducted the ‘LD check’ analysis17 to evaluate the LD. We calculated LD r2 between each eQTL and all variants in the disease-associated region (1Mb window) with GWAS p < 1 × 10−3, and r2 of more than 0.8 between the eQTL and any of the outcome variants was considered as evidence for LD checked.
GWAS and post-GWAS study of LVEF
Sex-stratified GWAS of LVEF (continuous variable, UKB data-field ID 22420) was performed among 17,899 men and 19,467 women of European ancestry in UKB separately. To maximize sample sizes, these two GWAS were conducted using BOLT-LMM,92 a linear mixed model to account for both relatedness and population stratification, following the in-house UKB GWAS pipeline.93 Genotyping array (UKB data-field ID 22000) was included in BOLT-LMM as a covariate to account for the potential batch effect, and we assumed an additive genetic effect on LVEF for each genetic variant. Full details of BOLT-LMM were described elsewhere.94 This analysis has been conducted using the UKB Resources under application number 15825.
Manhattan plots of genome-wide associations were depicted. To estimate the proportion of LVEF that could be attributed to common variants, we estimated heritability (h2) with linkage disequilibrium core regression (LDSC, v.1.0.1).30 Besides, we employed LDSC to calculate the genetic correlation between male- and female-stratified HF and LVEF, as well as between male-only LVEF and HFrEF or HFpEF. These included the following steps: (1) reformatting summary statistics; (2) filtering for the HapMap3 SNPs with corresponding allele; (3) assessing genetic correlation. The LD scores from the European 1000 Genomes Project dataset were referenced.32
Acknowledgments
J.Z. is supported by the Academy of Medical Sciences (AMS) Springboard Award, the Wellcome Trust, the Government Department of Business, Energy and Industrial Strategy, and the British Heart Foundation and Diabetes UK (SBF006\1117). This work was supported by grants from the National Key Research and Development Program of China (2022YFC2505203). J.L., M.X., G.N., and Y.B. are supported by the National Natural Science Foundation of China (82088102, 81970691, 81970728, 81930021, and 81941017), the Shanghai Clinical Research Center for Metabolic Diseases (19MC1910100), and the Shanghai Municipal Government (22Y31900300). J.L., M.X., Y.X., T.W., M.L., Z.Z., S.W., H.L., G.N., W.W., and Y.B. are members of the Innovative Research Team of High-level Local Universities in Shanghai. G.N. and Y.B. are supported by the Shanghai Shenkang Hospital Development Center (SHDC12019101, SHDC2020CR1001A, and SHDC2020CR3064B). Y.X. is supported by the National Top Young Talents program. Qian Yang works in a unit that is supported by the University of Bristol and UK Medical Research Council (MC_UU_00032/05). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank GTEx, HERMES Consortium, GBMI, and MVP for sharing their summary-level data. The graphic abstract was generated using www.biorender.com.
Author contributions
Y.X., J.Z., and Y.B. designed the study, wrote the research plan, and interpreted the results. Qianqian Yang undertook the main, replication, and sensitivity MR analyses. Qian Yang conducted the sex-stratified GWAS of LVEF. Qianqian Yang wrote the first draft of the manuscript, with critical comments from J.Z., Qian Yang, and Y.V.S. and revision from all authors. Y.X. is the guarantor. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Declaration of interests
The authors declare no competing interests.
Published: January 17, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101382.
Contributor Information
Weiqing Wang, Email: wqingw61@163.com.
Yufang Bi, Email: byf10784@rjh.com.cn.
Jie Zheng, Email: jie.zheng@bristol.ac.uk.
Yu Xu, Email: jane.yuxu@gmail.com.
Supplemental information
References
- 1.Virani S.S., Alonso A., Aparicio H.J., Benjamin E.J., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Cheng S., Delling F.N., et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143:e254–e743. doi: 10.1161/CIR.0000000000000950. [DOI] [PubMed] [Google Scholar]
- 2.GBD 2017 Disease and Injury Incidence and Prevalence Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. 2018;392:1789–1858. doi: 10.1016/S0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heidenreich P.A., Bozkurt B., Aguilar D., Allen L.A., Byun J.J., Colvin M.M., Deswal A., Drazner M.H., Dunlay S.M., Evers L.R., et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e876–e894. doi: 10.1161/CIR.0000000000001062. [DOI] [PubMed] [Google Scholar]
- 4.Motiejūnaitė J., Akiyama E., Cohen-Solal A., Maggioni A.P., Mueller C., Choi D.J., Kavoliūnienė A., Čelutkienė J., Parenica J., Lassus J., et al. The association of long-term outcome and biological sex in patients with acute heart failure from different geographic regions. Eur. Heart J. 2020;41:1357–1364. doi: 10.1093/eurheartj/ehaa071. [DOI] [PubMed] [Google Scholar]
- 5.Lloyd-Jones D.M., Larson M.G., Leip E.P., Beiser A., D'Agostino R.B., Kannel W.B., Murabito J.M., Vasan R.S., Benjamin E.J., Levy D., Framingham Heart Study Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106:3068–3072. doi: 10.1161/01.cir.0000039105.49749.6f. [DOI] [PubMed] [Google Scholar]
- 6.McMurray J.J.V., Packer M., Desai A.S., Gong J., Lefkowitz M.P., Rizkala A.R., Rouleau J.L., Shi V.C., Solomon S.D., Swedberg K., et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014;371:993–1004. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 7.Gerber Y., Weston S.A., Redfield M.M., Chamberlain A.M., Manemann S.M., Jiang R., Killian J.M., Roger V.L. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 2015;175:996–1004. doi: 10.1001/jamainternmed.2015.0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam C.S.P., Arnott C., Beale A.L., Chandramouli C., Hilfiker-Kleiner D., Kaye D.M., Ky B., Santema B.T., Sliwa K., Voors A.A. Sex differences in heart failure. Eur. Heart J. 2019;40:3859–3868c. doi: 10.1093/eurheartj/ehz835. [DOI] [PubMed] [Google Scholar]
- 9.Levin M.G., Tsao N.L., Singhal P., Liu C., Vy H.M.T., Paranjpe I., Backman J.D., Bellomo T.R., Bone W.P., Biddinger K.J., et al. Genome-wide association and multi-trait analyses characterize the common genetic architecture of heart failure. Nat. Commun. 2022;13:6914. doi: 10.1038/s41467-022-34216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah S., Henry A., Roselli C., Lin H., Sveinbjörnsson G., Fatemifar G., Hedman Å.K., Wilk J.B., Morley M.P., Chaffin M.D., et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 2020;11:163. doi: 10.1038/s41467-019-13690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joseph J., Liu C., Hui Q., Aragam K., Wang Z., Charest B., Huffman J.E., Keaton J.M., Edwards T.L., Demissie S., et al. Genetic architecture of heart failure with preserved versus reduced ejection fraction. Nat. Commun. 2022;13:7753. doi: 10.1038/s41467-022-35323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tadros R., Francis C., Xu X., Vermeer A.M.C., Harper A.R., Huurman R., Kelu Bisabu K., Walsh R., Hoorntje E.T., Te Rijdt W.P., et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 2021;53:128–134. doi: 10.1038/s41588-020-00762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasooly D., Peloso G.M., Pereira A.C., Dashti H., Giambartolomei C., Wheeler E., Aung N., Ferolito B.R., Pietzner M., Farber-Eger E.H., et al. Genome-wide association analysis and Mendelian randomization proteomics identify drug targets for heart failure. Nat. Commun. 2023;14:3826. doi: 10.1038/s41467-023-39253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernabeu E., Canela-Xandri O., Rawlik K., Talenti A., Prendergast J., Tenesa A. Sex differences in genetic architecture in the UK Biobank. Nat. Genet. 2021;53:1283–1289. doi: 10.1038/s41588-021-00912-0. [DOI] [PubMed] [Google Scholar]
- 15.Sotomi Y., Hikoso S., Nakatani D., Mizuno H., Okada K., Dohi T., Kitamura T., Sunaga A., Kida H., Oeun B., et al. Sex Differences in Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2021;10 doi: 10.1161/JAHA.120.018574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khramtsova E.A., Wilson M.A., Martin J., Winham S.J., He K.Y., Davis L.K., Stranger B.E. Quality control and analytic best practices for testing genetic models of sex differences in large populations. Cell. 2023;186:2044–2061. doi: 10.1016/j.cell.2023.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng J., Haberland V., Baird D., Walker V., Haycock P.C., Hurle M.R., Gutteridge A., Erola P., Liu Y., Luo S., et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 2020;52:1122–1131. doi: 10.1038/s41588-020-0682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliva M., Muñoz-Aguirre M., Kim-Hellmuth S., Wucher V., Gewirtz A.D.H., Cotter D.J., Parsana P., Kasela S., Balliu B., Viñuela A., et al. The impact of sex on gene expression across human tissues. Science. 2020;369 doi: 10.1126/science.aba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W., Kanai M., Wu K.H.H., Rasheed H., Tsuo K., Hirbo J.B., Wang Y., Bhattacharya A., Zhao H., Namba S., et al. Global Biobank Meta-analysis Initiative: Powering genetic discovery across human disease. Cell Genom. 2022;2 doi: 10.1016/j.xgen.2022.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O'Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemani G., Tilling K., Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13 doi: 10.1371/journal.pgen.1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawlor D.A., Harbord R.M., Sterne J.A.C., Timpson N., Davey Smith G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 23.Burgess S., Thompson S.G. Bias in causal estimates from Mendelian randomization studies with weak instruments. Stat. Med. 2011;30:1312–1323. doi: 10.1002/sim.4197. [DOI] [PubMed] [Google Scholar]
- 24.Sanderson E., Glymour M.M., Holmes M.V., Kang H., Morrison J., Munafò M.R., Palmer T., Schooling C.M., Wallace C., Zhao Q., Smith G.D. Mendelian randomization. Nat. Rev. Methods Primers. 2022;2 doi: 10.1038/s43586-021-00092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brumpton B., Sanderson E., Heilbron K., Hartwig F.P., Harrison S., Vie G.Å., Cho Y., Howe L.D., Hughes A., Boomsma D.I., et al. Avoiding dynastic, assortative mating, and population stratification biases in Mendelian randomization through within-family analyses. Nat. Commun. 2020;11:3519. doi: 10.1038/s41467-020-17117-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmer T.M., Lawlor D.A., Harbord R.M., Sheehan N.A., Tobias J.H., Timpson N.J., Davey Smith G., Sterne J.A.C. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat. Methods Med. Res. 2012;21:223–242. doi: 10.1177/0962280210394459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aung N., Vargas J.D., Yang C., Cabrera C.P., Warren H.R., Fung K., Tzanis E., Barnes M.R., Rotter J.I., Taylor K.D., et al. Genome-Wide Analysis of Left Ventricular Image-Derived Phenotypes Identifies Fourteen Loci Associated With Cardiac Morphogenesis and Heart Failure Development. Circulation. 2019;140:1318–1330. doi: 10.1161/CIRCULATIONAHA.119.041161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer H.V., Dawes T.J.W., Serrani M., Bai W., Tokarczuk P., Cai J., de Marvao A., Henry A., Lumbers R.T., Gierten J., et al. Genetic and functional insights into the fractal structure of the heart. Nature. 2020;584:589–594. doi: 10.1038/s41586-020-2635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong S., Zhao N., Spragins E., Kagda M.S., Li M., Assis P., Jolanki O., Luo Y., Cherry J.M., Boyle A.P., Hitz B.C. Annotating and prioritizing human non-coding variants with RegulomeDB v.2. Nat. Genet. 2023;55:724–726. doi: 10.1038/s41588-023-01365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y., Shan Y., Zhang W., Lee A.M., Li F., Stranger B.E., Huang R.S. Deciphering genetic causes for sex differences in human health through drug metabolism and transporter genes. Nat. Commun. 2023;14:175. doi: 10.1038/s41467-023-35808-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.1000 Genomes Project Consortium. Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koscielny G., An P., Carvalho-Silva D., Cham J.A., Fumis L., Gasparyan R., Hasan S., Karamanis N., Maguire M., Papa E., et al. Open Targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 2017;45:D985–D994. doi: 10.1093/nar/gkw1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y., Zhang Y., Zhao D., Yu X., Shen X., Zhou Y., Wang S., Qiu Y., Chen Y., Zhu F. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2023 doi: 10.1093/nar/gkad751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lisy O., Huntley B.K., McCormick D.J., Kurlansky P.A., Burnett J.C., Jr. Design, synthesis, and actions of a novel chimeric natriuretic peptide: CD-NP. J. Am. Coll. Cardiol. 2008;52:60–68. doi: 10.1016/j.jacc.2008.02.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumori A., Shioi T., Yamada T., Matsui S., Sasayama S. Vesnarinone, a new inotropic agent, inhibits cytokine production by stimulated human blood from patients with heart failure. Circulation. 1994;89:955–958. doi: 10.1161/01.cir.89.3.955. [DOI] [PubMed] [Google Scholar]
- 37.Zheng J., Zhang Y., Zhao H., Liu Y., Baird D., Karim M.A., Ghoussaini M., Schwartzentruber J., Dunham I., Elsworth B., et al. Multi-ancestry Mendelian randomization of omics traits revealing drug targets of COVID-19 severity. EBioMedicine. 2022;81 doi: 10.1016/j.ebiom.2022.104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H., Rasheed H., Nøst T.H., Cho Y., Liu Y., Bhatta L., Bhattacharya A., Hemani G., Davey Smith G., Brumpton B.M., et al. Proteome-wide Mendelian randomization in global biobank meta-analysis reveals multi-ancestry drug targets for common diseases. Cell Genom. 2022;2 doi: 10.1016/j.xgen.2022.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt A.F., Finan C., Gordillo-Marañón M., Asselbergs F.W., Freitag D.F., Patel R.S., Tyl B., Chopade S., Faraway R., Zwierzyna M., Hingorani A.D. Genetic drug target validation using Mendelian randomisation. Nat. Commun. 2020;11:3255. doi: 10.1038/s41467-020-16969-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt A.F., Bourfiss M., Alasiri A., Puyol-Anton E., Chopade S., van Vugt M., van der Laan S.W., Gross C., Clarkson C., Henry A., et al. Druggable proteins influencing cardiac structure and function: Implications for heart failure therapies and cancer cardiotoxicity. Sci. Adv. 2023;9 doi: 10.1126/sciadv.add4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y., Gagliano Taliun S.A. Lipid-lowering drug targets and Parkinson's disease: A sex-specific Mendelian randomization study. Front. Neurol. 2022;13 doi: 10.3389/fneur.2022.940118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porcu E., Claringbould A., Weihs A., Lepik K., BIOS Consortium. Völker U., Völker U., Santoni F.A., Teumer A., Franke L., et al. Limited evidence for blood eQTLs in human sexual dimorphism. Genome Med. 2022;14:89. doi: 10.1186/s13073-022-01088-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell A.S., Rosoff D.B., Mavromatis L.A., Jung J., Wagner J., Lohoff F.W. Comparing the Relationships of Genetically Proxied PCSK9 Inhibition With Mood Disorders, Cognition, and Dementia Between Men and Women: A Drug-Target Mendelian Randomization Study. J. Am. Heart Assoc. 2022;11 doi: 10.1161/JAHA.122.026122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang P., Hou Y., Tu W., Campbell N., Pieper A.A., Leverenz J.B., Gao S., Cummings J., Cheng F. Population-based discovery and Mendelian randomization analysis identify telmisartan as a candidate medicine for Alzheimer's disease in African Americans. Alzheimers Dement. 2023;19:1876–1887. doi: 10.1002/alz.12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L., Peters J.E., Prins B., Persyn E., Traylor M., Surendran P., Karthikeyan S., Yonova-Doing E., Di Angelantonio E., Roberts D.J., et al. Systematic Mendelian randomization using the human plasma proteome to discover potential therapeutic targets for stroke. Nat. Commun. 2022;13:6143. doi: 10.1038/s41467-022-33675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swerdlow D.I., Preiss D., Kuchenbaecker K.B., Holmes M.V., Engmann J.E.L., Shah T., Sofat R., Stender S., Johnson P.C.D., Scott R.A., et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385:351–361. doi: 10.1016/S0140-6736(14)61183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt A.F., Swerdlow D.I., Holmes M.V., Patel R.S., Fairhurst-Hunter Z., Lyall D.M., Hartwig F.P., Horta B.L., Hyppönen E., Power C., et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105. doi: 10.1016/S2213-8587(16)30396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roselli C., Chaffin M.D., Weng L.C., Aeschbacher S., Ahlberg G., Albert C.M., Almgren P., Alonso A., Anderson C.D., Aragam K.G., et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wild P.S., Felix J.F., Schillert A., Teumer A., Chen M.H., Leening M.J.G., Völker U., Großmann V., Brody J.A., Irvin M.R., et al. Large-scale genome-wide analysis identifies genetic variants associated with cardiac structure and function. J. Clin. Invest. 2017;127:1798–1812. doi: 10.1172/JCI84840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia C., Lovins C., Malone H.M., Keasey M.P., Hagg T. Female-specific neuroprotection after ischemic stroke by vitronectin-focal adhesion kinase inhibition. J. Cerebr. Blood Flow Metabol. 2022;42:1961–1974. doi: 10.1177/0271678X221107871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Speert D.B., Konkle A.T.M., Zup S.L., Schwarz J.M., Shiroor C., Taylor M.E., McCarthy M.M. Focal adhesion kinase and paxillin: novel regulators of brain sexual differentiation? Endocrinology. 2007;148:3391–3401. doi: 10.1210/en.2006-0845. [DOI] [PubMed] [Google Scholar]
- 52.Kuhn M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016;96:751–804. doi: 10.1152/physrev.00022.2015. [DOI] [PubMed] [Google Scholar]
- 53.Špiranec K., Chen W., Werner F., Nikolaev V.O., Naruke T., Koch F., Werner A., Eder-Negrin P., Diéguez-Hurtado R., Adams R.H., et al. Endothelial C-Type Natriuretic Peptide Acts on Pericytes to Regulate Microcirculatory Flow and Blood Pressure. Circulation. 2018;138:494–508. doi: 10.1161/CIRCULATIONAHA.117.033383. [DOI] [PubMed] [Google Scholar]
- 54.Becker J.R., Chatterjee S., Robinson T.Y., Bennett J.S., Panáková D., Galindo C.L., Zhong L., Shin J.T., Coy S.M., Kelly A.E., et al. Differential activation of natriuretic peptide receptors modulates cardiomyocyte proliferation during development. Development. 2014;141:335–345. doi: 10.1242/dev.100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langenickel T.H., Buttgereit J., Pagel-Langenickel I., Lindner M., Monti J., Beuerlein K., Al-Saadi N., Plehm R., Popova E., Tank J., et al. Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc. Natl. Acad. Sci. USA. 2006;103:4735–4740. doi: 10.1073/pnas.0510019103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian H., Froese A., Jönsson P., Schmidt H., Gorelik J., Nikolaev V.O. Distinct submembrane localisation compartmentalises cardiac NPR1 and NPR2 signalling to cGMP. Nat. Commun. 2018;9:2446. doi: 10.1038/s41467-018-04891-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dickey D.M., Flora D.R., Bryan P.M., Xu X., Chen Y., Potter L.R. Differential regulation of membrane guanylyl cyclases in congestive heart failure: natriuretic peptide receptor (NPR)-B, Not NPR-A, is the predominant natriuretic peptide receptor in the failing heart. Endocrinology. 2007;148:3518–3522. doi: 10.1210/en.2007-0081. [DOI] [PubMed] [Google Scholar]
- 58.Soeki T., Kishimoto I., Okumura H., Tokudome T., Horio T., Mori K., Kangawa K. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2005;45:608–616. doi: 10.1016/j.jacc.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 59.Blaser M.C., Wei K., Adams R.L.E., Zhou Y.Q., Caruso L.L., Mirzaei Z., Lam A.Y.L., Tam R.K.K., Zhang H., Heximer S.P., et al. Deficiency of Natriuretic Peptide Receptor 2 Promotes Bicuspid Aortic Valves, Aortic Valve Disease, Left Ventricular Dysfunction, and Ascending Aortic Dilatations in Mice. Circ. Res. 2018;122:405–416. doi: 10.1161/CIRCRESAHA.117.311194. [DOI] [PubMed] [Google Scholar]
- 60.Buttgereit J., Shanks J., Li D., Hao G., Athwal A., Langenickel T.H., Wright H., da Costa Goncalves A.C., Monti J., Plehm R., et al. C-type natriuretic peptide and natriuretic peptide receptor B signalling inhibits cardiac sympathetic neurotransmission and autonomic function. Cardiovasc. Res. 2016;112:637–644. doi: 10.1093/cvr/cvw184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kawakami R., Lee C.Y.W., Scott C., Bailey K.R., Schirger J.A., Chen H.H., Benike S.L., Cannone V., Martin F.L., Sangaralingham S.J., et al. A Human Study to Evaluate Safety, Tolerability, and Cyclic GMP Activating Properties of Cenderitide in Subjects With Stable Chronic Heart Failure. Clin. Pharmacol. Ther. 2018;104:546–552. doi: 10.1002/cpt.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sangaralingham S.J., Kuhn M., Cannone V., Chen H.H., Burnett J.C. Natriuretic peptide pathways in heart failure: further therapeutic possibilities. Cardiovasc. Res. 2023;118:3416–3433. doi: 10.1093/cvr/cvac125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M., et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel K.M., Strong A., Tohyama J., Jin X., Morales C.R., Billheimer J., Millar J., Kruth H., Rader D.J. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ. Res. 2015;116:789–796. doi: 10.1161/CIRCRESAHA.116.305811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goettsch C., Hutcheson J.D., Aikawa M., Iwata H., Pham T., Nykjaer A., Kjolby M., Rogers M., Michel T., Shibasaki M., et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Invest. 2016;126:1323–1336. doi: 10.1172/JCI80851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kjolby M., Andersen O.M., Breiderhoff T., Fjorback A.W., Pedersen K.M., Madsen P., Jansen P., Heeren J., Willnow T.E., Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metabol. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Strong A., Ding Q., Edmondson A.C., Millar J.S., Sachs K.V., Li X., Kumaravel A., Wang M.Y., Ai D., Guo L., et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J. Clin. Invest. 2012;122:2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hartiala J.A., Han Y., Jia Q., Hilser J.R., Huang P., Gukasyan J., Schwartzman W.S., Cai Z., Biswas S., Trégouët D.A., et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. Heart J. 2021;42:919–933. doi: 10.1093/eurheartj/ehaa1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 71.Mitok K.A., Keller M.P., Attie A.D. Sorting through the extensive and confusing roles of sortilin in metabolic disease. J. Lipid Res. 2022;63 doi: 10.1016/j.jlr.2022.100243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goettsch C., Kjolby M., Aikawa E. Sortilin and Its Multiple Roles in Cardiovascular and Metabolic Diseases. Arterioscler. Thromb. Vasc. Biol. 2018;38:19–25. doi: 10.1161/ATVBAHA.117.310292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jackson S., Yeh F.L., Ward M., Rhinn H., Huang J.Y., Pappalardo J.L., Liao Y., Hagey M., Long H., Paul R. Six months interim analysis of the phase 2 study of AL001 in frontotemporal dementia patients carrying a granulin mutation. Alzheimer's Dementia. 2021;17 [Google Scholar]
- 74.Macfarlane A.J., Perry C.A., McEntee M.F., Lin D.M., Stover P.J. Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apc(min)-mediated intestinal cancer risk. Cancer Res. 2011;71:2098–2107. doi: 10.1158/0008-5472.CAN-10-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lim U., Peng K., Shane B., Stover P.J., Litonjua A.A., Weiss S.T., Gaziano J.M., Strawderman R.L., Raiszadeh F., Selhub J., et al. Polymorphisms in cytoplasmic serine hydroxymethyltransferase and methylenetetrahydrofolate reductase affect the risk of cardiovascular disease in men. J. Nutr. 2005;135:1989–1994. doi: 10.1093/jn/135.8.1989. [DOI] [PubMed] [Google Scholar]
- 76.Wang J., Gu J., Huang Y., Fang Y., Lin J. The association between serine hydroxymethyl transferase 1 gene hypermethylation and ischemic stroke. Bosn. J. Basic Med. Sci. 2021;21:454–460. doi: 10.17305/bjbms.2020.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perry C., Sastry R., Nasrallah I.M., Stover P.J. Mimosine attenuates serine hydroxymethyltransferase transcription by chelating zinc. Implications for inhibition of DNA replication. J. Biol. Chem. 2005;280:396–400. doi: 10.1074/jbc.M410467200. [DOI] [PubMed] [Google Scholar]
- 78.Crowe B., Poynter J.A., Manukyan M.C., Wang Y., Brewster B.D., Herrmann J.L., Abarbanell A.M., Weil B.R., Meldrum D.R. Pretreatment with intracoronary mimosine improves postischemic myocardial functional recovery. Surgery. 2011;150:191–196. doi: 10.1016/j.surg.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 79.Kantola I., Scheinin M., Gulbrandsen T., Meland N., Smerud K.T. Safety, Tolerability, and Antihypertensive Effect of SER100, an Opiate Receptor-Like 1 (ORL-1) Partial Agonist, in Patients With Isolated Systolic Hypertension. Clin. Pharmacol. Drug Dev. 2017;6:584–591. doi: 10.1002/cpdd.330. [DOI] [PubMed] [Google Scholar]
- 80.Cleland J.G., Coletta A.P., Clark A.L. Clinical trials update from the European Society of Cardiology Heart Failure Meeting 2010: TRIDENT 1, BENEFICIAL, CUPID, RFA-HF, MUSIC, DUEL, handheld BNP, phrenic nerve stimulation, CHAMPION and CABG with CRT study. Eur. J. Heart Fail. 2010;12:883–888. [Google Scholar]
- 81.Greenberg B., Thomas I., Banish D., Goldman S., Havranek E., Massie B.M., Zhu Y., Ticho B., Abraham W.T. Effects of multiple oral doses of an A1 adenosine antagonist, BG9928, in patients with heart failure: results of a placebo-controlled, dose-escalation study. J. Am. Coll. Cardiol. 2007;50:600–606. doi: 10.1016/j.jacc.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 82.Sadler M.C., Auwerx C., Deelen P., Kutalik Z. Multi-layered genetic approaches to identify approved drug targets. Cell Genom. 2023;3 doi: 10.1016/j.xgen.2023.100341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hemani G., Zheng J., Elsworth B., Wade K.H., Haberland V., Baird D., Laurin C., Burgess S., Bowden J., Langdon R., et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7 doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ning Z., Pawitan Y., Shen X. High-definition likelihood inference of genetic correlations across human complex traits. Nat. Genet. 2020;52:859–864. doi: 10.1038/s41588-020-0653-y. [DOI] [PubMed] [Google Scholar]
- 85.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aday A.W., Matsushita K. Epidemiology of Peripheral Artery Disease and Polyvascular Disease. Circ. Res. 2021;128:1818–1832. doi: 10.1161/CIRCRESAHA.121.318535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taylor K., Davey Smith G., Relton C.L., Gaunt T.R., Richardson T.G. Prioritizing putative influential genes in cardiovascular disease susceptibility by applying tissue-specific Mendelian randomization. Genome Med. 2019;11:6. doi: 10.1186/s13073-019-0613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Petersen S.E., Matthews P.M., Francis J.M., Robson M.D., Zemrak F., Boubertakh R., Young A.A., Hudson S., Weale P., Garratt S., et al. UK Biobank's cardiovascular magnetic resonance protocol. J. Cardiovasc. Magn. Reson. 2016;18:8. doi: 10.1186/s12968-016-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Porcu E., Rüeger S., Lepik K., eQTLGen Consortium, BIOS Consortium. Santoni F.A., Reymond A., Kutalik Z. Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nat. Commun. 2019;10:3300. doi: 10.1038/s41467-019-10936-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Skrivankova V.W., Richmond R.C., Woolf B.A.R., Yarmolinsky J., Davies N.M., Swanson S.A., VanderWeele T.J., Higgins J.P.T., Timpson N.J., Dimou N., et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021;326:1614–1621. doi: 10.1001/jama.2021.18236. [DOI] [PubMed] [Google Scholar]
- 91.Burgess S., Zuber V., Valdes-Marquez E., Sun B.B., Hopewell J.C. Mendelian randomization with fine-mapped genetic data: Choosing from large numbers of correlated instrumental variables. Genet. Epidemiol. 2017;41:714–725. doi: 10.1002/gepi.22077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loh P.R., Tucker G., Bulik-Sullivan B.K., Vilhjálmsson B.J., Finucane H.K., Salem R.M., Chasman D.I., Ridker P.M., Neale B.M., Berger B., et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 2015;47:284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dashti H.S., Jones S.E., Wood A.R., Lane J.M., van Hees V.T., Wang H., Rhodes J.A., Song Y., Patel K., Anderson S.G., et al. Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat. Commun. 2019;10:1100. doi: 10.1038/s41467-019-08917-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Loh P.R., Kichaev G., Gazal S., Schoech A.P., Price A.L. Mixed-model association for biobank-scale datasets. Nat. Genet. 2018;50:906–908. doi: 10.1038/s41588-018-0144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary statistics of the present sex-stratified GWAS analysis on left ventricular ejection fraction (LVEF) will be deposited to the IEU Open GWAS database (https://gwas.mrcieu.ac.uk/) upon publication of the article. The data can be accessed using the GWAS IDs 'ieu-b-5120' and 'ieu-b-5121. Code for reproducing the study is available at GitHub repository (https://github.com/geneinmylife/MR-project-Heart-failure). Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.