

Summary
A tumor ecosystem constantly evolves over time in the face of immune predation or therapeutic intervention, resulting in treatment failure and tumor progression. Here, we present a single-cell transcriptome-based strategy to determine the evolution of longitudinal tumor biopsies from liver cancer patients by measuring cellular lineage and ecology. We construct a lineage and ecological score as joint dynamics of tumor cells and their microenvironments. Tumors may be classified into four main states in the lineage-ecological space, which are associated with clinical outcomes. Analysis of longitudinal samples reveals the evolutionary trajectory of tumors in response to treatment. We validate the lineage-ecology-based scoring system in predicting clinical outcomes using bulk transcriptomic data of additional cohorts of 716 liver cancer patients. Our study provides a framework for monitoring tumor evolution in response to therapeutic intervention.
Keywords: tumor evolution, tumor ecology, tumor lineage, liver cancer, hepatocellular carcinoma, cholangiocarcinoma, single cell, tumor heterogeneity, tumor microenvironment, immunotherapy
Graphical abstract
Highlights
-
•
CASCADE is useful to monitor tumor evolution in response to immunotherapy
-
•
A malignant ecosystem can be assessed based on tumor lineage and its ecology
-
•
The tumor lineage and its ecological spaces can predict patient outcome
-
•
MAIT cells are potential contributors of treatment response in liver cancer
Revsine et al. perform single-cell transcriptome profiling of longitudinal tumor biopsies from liver cancer patients who received immunotherapy. They develop a CASCADE algorithm to classify tumors in the lineage-ecological space and further monitor tumor evolution in response to treatment, where MAIT cells are found as potential contributors of treatment response.
Introduction
A solid neoplasm constantly evolves over time as a cell community, whose fitness depends on the intrinsic ability of its tumor cells to modify their surrounding tumor microenvironment (TME) for adaptive benefits.1 Tumor cells can regulate the stromal and immune cells of the TME, which may either be resourceful or hazardous toward tumor cells depending on environmental cues. This remarkably dynamic property of a malignant ecosystem creates a diversity in tumor cells known as intratumoral heterogeneity (ITH), which is responsible for tumor progression and treatment failure, and further serves as a universal tumor prognostic biomarker.2 During tumor evolution, the continuous shaping between tumor cells and their TME defines the evolutionary trajectory of a tumor ecosystem.3,4 This intricate co-evolution mechanism creates critical challenges for classifying tumors and developing effective cancer treatments. Liver cancer is known to have extensive ITH, leading to poor treatment response and high mortality rates.5,6,7,8 Although immunotherapy has transformed the management of liver cancer, most patients still do not derive clinical benefit.9,10,11 There is an unmet need to develop consensus approaches for classifying the evolutionary and ecological features of liver tumor lesions to monitor tumor evolution and treatment response.12 In this vein, cancer cell intrinsic factors drive the evolutionary dynamics, and the ecology of a tumor lesion is defined by the TME.
The development of single-cell technologies has revolutionized our knowledge of tumor biology by providing a full landscape of cell types, cellular states, and potential mechanisms of cell-cell interactions.13,14,15 Characterization of the TME via single-cell analysis has uncovered a diversity of cell types and states in immune and stromal cells.16,17,18 Particular attention has been paid to the pro- or anti-tumor roles of the non-malignant cell types and their internal cell states. For example, studies have reported that exhausted T cells with high expression of PDCD1 keep the cytotoxic activity of T cells suppressed and provide an immune checkpoint molecule that can be targeted clinically.19,20,21 However, characterization of tumor cells at the single-cell level, especially cell clonality, has been challenging because tumor cell state heterogeneity determined by transcriptomics largely arises independently of genetic variations.22 Recent studies have demonstrated that tumor cell biodiversity may drive microenvironmental reprograming and that tumor functional clonality is a strong prognostic indicator of liver cancer,23,24,25,26 suggesting that non-genetic clonal diversity may be effective in defining tumor biology.27 Nevertheless, the extensive diversity and high plasticity of both tumor cells and non-tumor cells in a dynamic tumor ecosystem create a barrier for classifying tumor evolution. This raises the question of how best to develop a method to monitor a tumor ecosystem and its response to treatment.
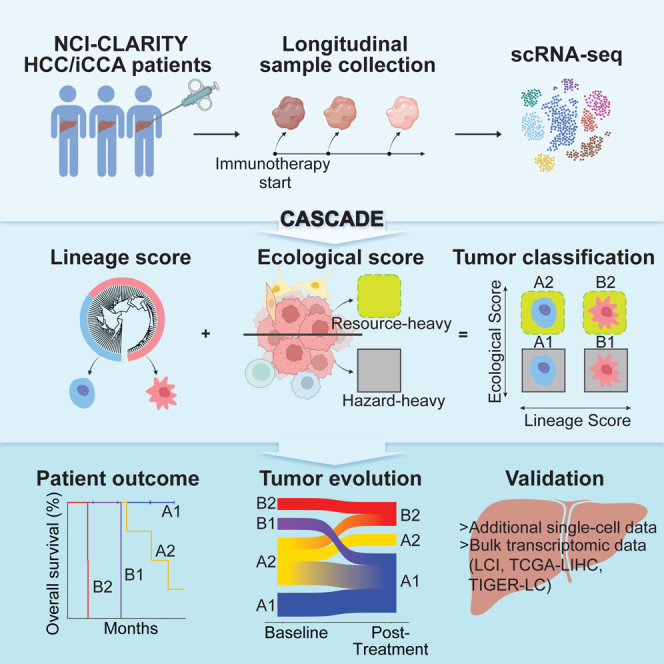
In this study, we analyzed longitudinal single-cell and bulk tissue transcriptomic data from hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA), two main clinical subtypes of liver cancer, under the NCI-CLARITY protocol (https://ccr.cancer.gov/liver-cancer-program/nci-clarity-study). We developed a bioinformatics tool, named Cancer Aggressiveness via Single-Cell Analysis During Evolution (or CASCADE), that uses tumor lineage and ecology to characterize a tumor ecosystem and its evolution in response to immunotherapy, based on the main concept from a consensus recommendation.12 We defined a lineage score based on different tumor cell states and an ecological score based on the dynamics of immune and stromal cells as being either hazardous or resourceful to tumor cells. We validate the CASCADE method in an independent single-cell cohort and four additional cohorts of bulk transcriptomic HCC and iCCA data. Collectively, we developed CASCADE as a stable platform to monitor tumor evolution in response to treatment. This method separates tumor samples into four quadrants in the lineage-ecological space, providing a framework for liver tumor classification that has potential implications in clinical practice.
Results
Single-cell transcriptome profiling of longitudinal tumor samples from liver cancer patients
We performed single-cell transcriptome profiling of longitudinal tumor samples from liver cancer patients to understand liver tumor evolution at the single-cell level. These samples are part of the NCI-CLARITY study, which seeks to understand the effectiveness of immunotherapy in liver cancer patients and develop predictors of immunotherapy response. Specifically, we analyzed fresh tumor biopsies from HCC or iCCA patients who were enrolled in clinical studies at the NIH Clinical Center (Figure S1A). We sought to develop a computational model using all longitudinal patients as a discovery cohort, then validate it with the remaining 31 patients who contributed only one sample as a validation cohort. The initial longitudinal cohort consisted of 11 patients diagnosed with HCC (n = 9) or iCCA (n = 2) (Table 1). Ten patients were treated with immunotherapy and one underwent surgical resection. Tumor biopsies were collected longitudinally for each patient, with one sample collected at baseline and between 1 and 4 samples post-treatment. Overall, 31 samples were collected across all patients. We performed single-cell RNA sequencing (scRNA-seq) of the tumor samples to generate a high-resolution transcriptomic landscape of the tumor cell communities. A total of 57,567 cells passed the initial quality controls, which included thresholds of ≥500 genes expressed and <50% mitochondrial genes detected in each single cell (Figure S1B). We further determined the malignancy of the cells by inferring large-scale chromosomal copy number variations (CNVs) based on their transcriptomes (Figures S1C and S1D). The inferred CNV profiles of the malignant cells (n = 13,042) were consistent with those found in previous studies of HCC and iCCA,28,29 demonstrating the successful separation of malignant and non-malignant cells. In addition, we found significantly higher expression of classical tumor marker genes in malignant cells compared with non-malignant cells, further supporting a confident determination of malignant cells (Figure S2A). The single-cell transcriptome data of malignant and non-malignant cells can be visualized at an online interface of scAtlasLC (https://scatlaslc.ccr.cancer.gov).
Table 1.
Clinical information of liver cancer patients with single-cell transcriptome data
| Variable | Longitudinal single-cell cohort (n = 11), no. (%) | Validation single-cell cohort (n = 31), no. (%) |
|---|---|---|
| Age (years)a | ||
| 30–39 | 0 | 2 (6.5) |
| 40–49 | 0 | 3 (9.7) |
| 50–59 | 2 (18.2) | 2 (6.5) |
| 60–69 | 5 (45.5) | 13 (41.9) |
| 70–79 | 3 (27.3) | 8 (25.8) |
| 80+ | 1 (9.1) | 3 (9.7) |
| Gender | ||
| Female | 1 (9.1) | 15 (48.4) |
| Male | 10 (90.9) | 16 (51.6) |
| Etiology | ||
| Fatty liver | 0 | 2 (6.5) |
| HBV | 2 (18.2) | 3 (9.7) |
| HCV | 5 (45.5) | 10 (32.3) |
| HBV + HDV | 1 (9.1) | 0 |
| None | 3 (27.3) | 14 (45.2) |
| No data | 0 | 2 (6.5) |
| Stage | ||
| I | 0 | 2 (6.5) |
| II | 1 (9.1) | 1 (3.2) |
| III | 2 (18.2) | 6 (19.4) |
| IV | 8 (72.7) | 20 (64.5) |
| No data | 0 | 2 (6.5) |
| Diagnosis | ||
| HCC | 9 (81.8) | 20 (64.5) |
| iCCA | 2 (18.2) | 11 (35.5) |
| Treatment | ||
| Pembrolizumab | 1 (9.1) | 5 (16.1) |
| Tremelimumab | 1 (9.1) | 1 (3.2) |
| Tremelimumab + Durvalumab | 8 (72.7) | 16 (51.6) |
| Resection only | 1 (9.1) | 6 (19.4) |
| None | 0 | 3 (9.7) |
| CA19-9 | ||
| ≤35 U/mL | 6 (54.5) | 7 (22.6) |
| >35 U/mL | 5 (45.5) | 11 (35.5) |
| No data | 0 | 13 (41.9) |
| Alpha-fetoprotein | ||
| Negative, ≤20 ng/mL | 5 (45.5) | 11 (35.5) |
| Positive, >20 ng/mL | 6 (54.5) | 13 (41.9) |
| No data | 0 | 7 (22.6) |
Age at time of baseline tissue collection used for this study.
Hierarchical clustering reveals distinct groups of malignant cells
Shared tumor cell states across patients are often observed,30 even though transcriptomic heterogeneity of tumor cells in liver cancer and other cancer types is well recognized.23,24,25,31 Consistent with previous work, intertumor heterogeneity was revealed from this study, where sample- and patient-specific clusters of malignant cells were observed in the t-distributed stochastic neighbor embedding (t-SNE) space (Figure 1A). To understand the commonalities among tumor cells across patients, we constructed a hierarchical relationship of all the malignant cells in our cohort. We identified four major branches in the hierarchical tree, with sub-branches observed within each major clade (Figure 1B). Each clade displayed different gene expression patterns (Figures 1C; Table S1). Based on the marker genes in the literature, we defined the four clades as cholangiocyte-like tumor cells (CLTCs), hepatocyte-like tumor cells (HLTCs), myeloid cell-like tumor cells (MCTCs), and mesenchymal-like tumor cells (MLTCs), respectively, for simplicity (Figures 1B–1D; Table S2). The names of these lineages refer to potential shared characteristics across member cells, such as MCTCs consistently exhibiting myeloid cell-like features. Consistently, gene set enrichment analysis revealed hepatocyte-related functional pathways (such as xenobiotic metabolism, bile acid metabolism, coagulation, and adipogenesis) in HLTCs, with additional oxidative stress-related pathways of reactive oxygen species and oxidative phosphorylation (Figure 1E). MCTCs were mainly involved in inflammatory response, with elevated TNF-α signaling via NF-κB, allograft rejection, and complement pathways. In CLTCs, the MYC targets pathway was elevated, which is related to cell proliferation and differentiation. MLTCs were enriched in cell growth and migration-related pathways, such as EMT, myogenesis, and mitotic spindle (Figure 1E). We also applied classical tumor marker genes to each of the four groups of tumors, where significantly higher expression was observed in tumor cells than non-tumor cells (Figure S2A).
Figure 1.

Determination of malignant cell groups
(A) t-SNE plot of all 13,042 malignant cells. Cells are assigned a general color by patient and a shade of that color by sample, with the lightest shade at baseline and successively darker hues for post-treatment samples. Patient IDs start with H and C to denote the clinical diagnosis of HCC and iCCA, respectively.
(B) Hierarchical relationship of all malignant cells. Inner section shows the hierarchical relationship among all cells, with colors showing the top 20 clades. Outer ring denotes the top four clades of the hierarchical tree as tumor lineages. The four interior rings indicate with matched colors the expression of the differentially expressed genes for each of the four major lineages.
(C) Violin plots of the expression of the top five differentially expressed genes by log fold change for each tumor lineage.
(D) Average expression of representative genes specific to cholangiocytes (CC), hepatocytes (HC), inflammation, or epithelial-mesenchymal transition (EMT) in malignant cells of each tumor lineage. The p values were calculated using a two-sided t test.
(E) Enriched pathways of each tumor lineage. Normalized enrichment score (NES) is shown for each pathway.
(F) Tumor lineage composition in each tumor sample with at least 15 malignant cells detected. Bars are colored by lineage: CLTC in red, HLTC in blue, MCTC in green, and MLTC in purple. See also Figures S1 and S2 and Tables S1 and S2.
We further determined the composition of tumor lineages in each individual tumor based on the resolved lineages of malignant cells (Figure 1F). Unsurprisingly, most of the tumors were dominated by a single lineage. Interestingly, in some cases, we did observe multiple lineages of malignant cells within the same tumor. As expected, HCC and iCCA tumors were mainly composed of HLTCs and CLTCs, respectively. However, samples from HCC patient H49 were dominated by CLTCs, indicating that tumor lineages may not always align with clinical diagnoses of liver cancer. Noticeably, tumors dominated by HLTC and MCTC lineages were clustered together in the hierarchical relationship of lineage composition profiles and were separate from samples dominated by CLTC and MLTC lineages (Figure 1F). This pattern suggests a shared tumor biology between certain lineages. Taken together, we identified four major lineages of malignant cells in HCC and iCCA, while observing extensive tumor heterogeneity.
To assess the effect of our limited sample size in the discovery cohort on the resultant tumor cell clusters, we performed the lineage pattern analysis again using a combined set of malignant cells from both the discovery and validation cohorts of 42 patients (Table 1) (n = 25,728). We observed 5 major clades in the combined hierarchical tree, showing a strong concordance with the originally defined clusters (Figure S2B). Almost all cells classified as HLTC, CLTC, and MLTC in the discovery cohort fell into clusters 1, 3, and 4, respectively, while almost all MCTCs were grouped into clusters 2 and 5. All clusters from the combined tumor cell set were comprised of at least 10 patients besides the MLTC-based cluster 4, which included 6 patients but was dominated by a single sample. Given the high similarity between our two lineage analyses and the heterogeneity of patients in our clusters, we concluded that our lineages were stable and representative of the tumor cell landscape. We therefore used the lineages originally developed from our discovery cohort in all downstream analyses as this strategy can test the stability of CASCADE to model tumor evolution. We also performed analysis of tumor cells from HCC and iCCA separately to demonstrate the stability of the four tumor lineages (Figure S2C). Consistently, there is a high concordance of tumor cell lineage types identified between a combined HCC/iCCA and HCC (or iCCA) alone.
Tumor microenvironments vary among tumors
Characterizing the TME can deepen our understanding of intrinsic tumor biology. Accordingly, we performed t-SNE and clustering analyses on the non-malignant cells in our cohort to determine the landscape of the TME of the tumor samples (Figure 2A). Based on the marker genes specifically expressed in each cluster, we identified T and NK cells, B and plasma cells, cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), tumor-associated endothelial cells (TECs), hepatocytes, and cholangiocytes. We then determined cell subtypes within each major cell type to better characterize the dynamics of cellular states (Figure S3). To this end, we first separated B cells (CD19+CD38−) from plasma cells (CD19−CD38+) based on clustering analysis (Figure 2B). Within the T and NK cell population, we defined multiple cell subtypes, including CD4+ or CD8+ memory T cells (CD69+), regulatory T cells (FOXP3+), central memory CD4+ cells (IL7R+), CD8+ effector cells (GZMH+), and CD8+ effector memory cells (GZMK+), as well as tissue-resident NK cells (CD160+) and circulatory NK cells (GNLY+) (Figure 2C). Similarly, subtypes of CAFs (LUM+ inflammatory, MYH11+ vascular, and APOA1+ hepatocyte-like), TAMs (monocytes, M1 macrophages, and M2 macrophages), and TECs were determined based on the differentially expressed genes in each identified cluster (Figures 2D–2F).
Figure 2.

The landscape of the TME
(A‒F) t-SNE plots of all non-malignant cells (A) (n = 44,525), B and plasma cells (B) (n = 1,914), T and NK cells (C) (n = 30,020), CAFs (D) (n = 1,029), TAMs (E) (n = 4,702), and TECs (F) (n = 2,762).
(G) Hierarchical relationship of the TME composition in each tumor sample with at least 15 non-malignant cells detected. The two major clades are colored, with clade 1 in blue and clade 2 in red.
(H) Relative abundance of each non-malignant cell subtype in each clade of the TME in (G).
(I) Composition of immune and stromal cells in each clade of the TME in (G). See also Figures S3 and S4 and Table S3.
We further determined the TME composition of each individual tumor based on the resolved TME landscape. Hierarchical clustering analysis of the TME compositions revealed two distinct clades among the tumors (Figure 2G). Clade 1 was relatively homogeneous, with nearly all constituent samples composed of large populations of CD8+GZMK+ effector memory T cells and either CD4+KLRB1+ T cells or CD4+IL7R+ central memory T cells. Other elevated non-malignant cell types in this clade included CD8+GZMH+ effector T cells, B cells, NK cells, and mucosal-associated invariant T (MAIT) cells. Clade 2 was more heterogeneous, with distinct subclades of samples showing elevated populations of CD4+/CD8+CD69+ memory T cells, LUM+ inflammatory CAFs, ANGPT2+ TECs, monocytes, and M2 macrophages. Tumors in clade 2 also tended to have higher proportions of MKI67+ proliferative T cells and VWF+ TECs (Figure 2H; Table S3). Although immune cells dominated the TME across all tumors in our cohort, stromal cells were nearly four times more prevalent in clade 2 than clade 1 (Figure 2I). To account for differences in sample number between patients, we also performed this analysis using only the baseline sample for each patient and saw consistent clustering (Figure S4). This stark difference in the TME composition of the two clades indicates that the tumors involved in our study faced distinct environmental conditions.
Modeling a tumor in the context of its microenvironment
Since a tumor and its microenvironment continuously interact with and shape each other, modeling the two elements together as a tumor ecosystem may better reflect the biology underlying tumor cell biodiversity. While the importance of considering both elements has been increasingly noted by the cancer biology field, difficulties remain as how best to model them. In a consensus statement formulated by a group of experts in the fields of cancer evolution and ecology, a theoretical framework was proposed to classify tumors using an Evo-index (evolutionary; intratumoral heterogeneity and its change over time) and an Eco-index (ecological; hazards and resources to tumor cells, where hazards are harmful to tumor cells while resources are beneficial to them).12 This conceptual framework motivated us to develop a computational model based on the principles of an Evo- and Eco-index using our single-cell profiles generated from liver tumors. To characterize tumor cells, we developed a lineage score, which was calculated based on the expression of genes associated with the CLTC, MLTC, HLTC, and MCTC lineages (see STAR Methods). A low lineage score represents an HLTC/MCTC-enriched lineage, whereas a high score indicates a CLTC/MLTC-enriched lineage. Meanwhile, the TME is represented by an ecological score based on resources and hazards to tumor cells. Here, resources and hazards were determined using the cell types associated with the two clades of the TME hierarchical clustering (see STAR Methods), where clade 1 represents a hazard-heavy TME and a low ecological score while clade 2 represents a resource-heavy TME and a high ecological score (Figure 2G). We combined the lineage and ecological scores to characterize the tumor ecosystem and named the method CASCADE (Figure 3A).
Figure 3.

Modeling a tumor ecosystem using lineage and ecological scores
(A) Graphical representation of using lineage and ecological scores to model a tumor ecosystem.
(B) Projection of the baseline samples from each of the 11 patients in the discovery single-cell cohort to the lineage-ecological space.
(C) Biological features of the tumors in each quadrant of the lineage-ecological space. The heatmap on the left shows the average expression of the top positive and negative differentially expressed genes in a quadrant. Each section of the heatmap, denoting differentially expressed genes for a quadrant (from top to bottom A1, A2, B1, and B2), has a corresponding plot on the right showing the top significantly enriched hallmark pathways for those genes. Pathways are ordered by high to low normalized enrichment score (NES). Only upregulated pathways are shown here.
(D and E) Kaplan-Meier plots of the 11 liver cancer patients (D) or only HCC patients (E) from the quadrants in (B). The p value was calculated using the log rank test for trend.
(F) Forest plot of hazard ratio in an additional HCC/iCCA single-cell cohort (sc) (n = 20) and three bulk HCC cohorts of LCI (n = 239), TCGA (n = 363), and TIGER-LC (n = 62). Quadrant B2 was used as the reference group for hazard ratio calculation. Bars show 95% confidence interval. See also Figures S5 and S6 and Tables S4 and S5.
We first used CASCADE to model the tumor ecosystems of baseline samples derived from liver cancer patients in our study. We found that tumors fell into four quadrants, i.e., A1, A2, B1, and B2, in the lineage-ecological space (Figure 3B). Based on the definitions of the lineage and ecological scores, A1 indicates an HLTC/MCTC-enriched lineage with a hazard-heavy TME, A2 represents an HLTC/MCTC-enriched lineage with a resource-heavy TME, B1 refers to a CLTC/MLTC-enriched lineage with a hazard-heavy TME, and B2 denotes a CLTC/MLTC-enriched lineage with a resource-heavy TME. We next performed differential gene expression analysis (Table S4) and gene set enrichment analysis between quadrants. Genes enriched in quadrant A1 were related to immune response (inflammatory, interferon α and γ responses) and cell proliferation (TNF-α and KRAS signaling), as well as xenobiotic metabolism (Figure 3C). Genes enriched in quadrant A2 were similarly associated with immune response and cell growth, but also had a much higher enrichment in metabolic pathways (oxidative phosphorylation, fatty and bile acid metabolism, peroxisome). Quadrant B1 only showed enrichment for mTORC1 signaling and glycolysis pathways. Finally, quadrant B2 was enriched in pathways related to cell differentiation (EMT, myogenesis), growth (angiogenesis, IL2-STAT5 and TGF-β signaling), and the cell cycle (G2M checkpoint, P53). We noticed that both iCCA tumors fell into B2, while HCC tumors were spread out over the other three quadrants (Figure 3B). Interestingly, patients in the four quadrants had significantly different overall survival, with quadrants A1, A2, B1, and B2 yielding the best to the worst patient outcomes in that order (Figure 3D). The same trend was observed when analyzing HCC samples separately (Figure 3E). We also developed a third metric, the tumor score, as the product of the lineage and ecological scores (Figure S5A). We found varying tumor scores among patients, with higher tumor scores related to poorer patient outcomes (Figuress S5B‒S5D). These results suggest that CASCADE can reflect the biology of a tumor ecosystem by capturing the characteristics of a tumor cell population and its TME.
We further validated CASCADE in additional single-cell data of 31 HCC and iCCA patients treated at the NIH clinical center as the validation cohort. We calculated lineage and ecological scores using the same procedure as before. Similar to our discovery cohort, tumors fell into four quadrants, with A1 having the best overall survival and B2 having the worst (Figures 3F and S6A). We further increased the power of our validation by analyzing three additional independent cohorts of HCC patients with bulk transcriptome data: the LCI (Liver Cancer Institute) cohort (n = 239), TCGA-LIHC (The Cancer Genome Atlas Liver Hepatocellular Carcinoma Collection) cohort (n = 363), and the TIGER-LC (Thailand Initiative in Genomics and Expression Research for Liver Cancer) cohort (n = 62). We used the top differentially expressed genes from our discovery single-cell dataset to calculate lineage and ecological scores for samples in the three bulk cohorts, since cell types and lineages cannot be well determined directly in bulk data (see STAR Methods; Table S5). We observed consistent trends of quadrant-related overall survival in all the three cohorts (Figure 3F). We also found that patient survival outcomes could be predicted using the tumor score metric (Figures S6B‒S6E). These results further suggest that CASCADE can be successfully utilized to evaluate a tumor ecosystem linked to survival outcomes.
Application of CASCADE in tracing tumor evolution
To study tumor evolution in response to treatment, we investigated the composition of tumor cell lineage and the TME for all the longitudinal samples of each individual patient in our single-cell study. Tumor lineages tended to stay relatively stable within each patient, while TME compositions underwent dynamic changes over time (Figure 4). For example, patient H08 was dominated by an HLTC lineage during tumor evolution, with just a small proportion of tumor cells of the MCTC lineage occurring in the biopsy collected at 76 weeks after treatment (Figure 4G). However, dramatic changes could be observed when examining the TME of this patient, with a TME that started with a large proportion of CD4+CD69+ memory T cells and later shifted to dominant populations of CD4+IL7R+ central memory T cells and CD8+GZMK+ effector memory T cells (Figure 4G). Noticeably, although relatively rare in our cohort, we did occasionally observe lineage shifts during tumor evolution. For example, for the first 30 weeks after being enrolled for treatment, patient H34 had a homogeneous HLTC lineage and a mostly consistent TME with large populations of LUM+ inflammatory CAFs, monocytes, VWF+ endothelial cells, and CD4+CD69+ memory T cells. However, by the last biopsy collection time point at 97 weeks, the tumor lineage had completely shifted to MLTC, accompanied by a dramatically different TME with expanded proportions of LUM+ inflammatory CAFs, M2 macrophages, and NK cells (Figure 4K). Across our cohort, we did not observe any relationship between tumor lineage/TME and the alpha-fetoprotein (AFP) in HCC patients or cancer antigen 19-9 (CA 19-9) in iCCA patients.
Figure 4.

The evolutionary landscape of the tumor and the TME
(A‒K) The composition of tumor cells and the TME during tumor evolution for patients H73 (A), H85 (B), H77 (C), H68 (D), C26 (E), C46 (F), H08 (G), H58 (H), H01 (I), H49 (J), and H34 (K). Levels of alpha-fetoprotein (AFP) or cancer antigen 19-9 (CA 19-9) are indicated for HCC and iCCA patients, respectively. Not every sample has both detectable malignant and non-malignant single cells. The baseline sample for patient C46 only has five malignant cells and is excluded from our analyses but is shown here for reference.
We then modeled the complex evolutionary trajectories of the tumor cells and TMEs by applying CASCADE to the longitudinal samples of our initial discovery cohort of 11 patients (n = 23 samples). Only samples with both detectable malignant and non-malignant cells were included in this analysis. We observed that most of the samples were distributed in quadrants A1, A2, and B2 in the lineage-ecological space, with only a single sample located in B1 (Figure 5A). This could be the result of a low sample size or an indication that the B1 quadrant is biologically rare. We further determined the patterns of tumor evolution by evaluating quadrant changes for each individual patient (n = 8 patients with both baseline and post-treatment samples available from Figure 5A). Interestingly, all patients that started in A1 or B2 ended in the same quadrant during tumor evolution, while those starting in A2 or B1 largely migrated elsewhere (Figure 5B). Remarkably, patients with both start and end in A1 had a stable disease, whereas those with a start and end in B2 experienced disease progression upon treatment. Patients who started in A2 had mixed treatment responses depending on the quadrants they ended up with. Since A1 and B2 represent the least and the most aggressive tumors, respectively, these patterns of tumor evolution seem not to be stochastic, but rather embedded in the tumor.
Figure 5.

Application of CASCADE in modeling tumor evolution
(A) Projection of longitudinal samples (n = 23) from the 11 patients in the single-cell discovery cohort into the lineage-ecological space. Only samples with both detectable malignant and non-malignant cells were included in this plot.
(B) Sankey plot of transitions in the lineage-ecological space between the baseline and the final post-treatment sample of each patient in the single-cell discovery cohort (n = 8). Patients with only one sample in (A) are not included in this plot.
(C) Projection of longitudinal samples (n = 75) from liver cancer patients in the NCI CLARITY retrospective cohort into the lineage-ecological space. Patients may have more than one baseline or post-treatment sample.
(D) Sankey plot of transitions in the lineage-ecological space between the baseline and the final post-treatment sample of each patient in the NCI CLARITY retrospective cohort (n = 32 patients). Only patients with at least two samples are included here.
(E) Kaplan-Meier plot of the patients in the NCI CLARITY retrospective cohort based on quadrants of the baseline samples (A1, n = 13 patients; A2, n = 5; B1, n = 6; B2, n = 8). The p value was calculated using the log rank test for trend.
(F) Forest plot of the hazard ratio in all patients from the NCI CLARITY retrospective cohort (n = 32). Patients are grouped by their lineage-ecological quadrant at baseline (left) (A1, n = 13 patients; A2, n = 5; B1, n = 6; B2, n = 8) and their final follow-up time point (right) (A1, n = 13 patients; A2, n = 6; B1, n = 5; B2, n = 8). Quadrant B2 was used as the reference group for hazard ratio calculation. Bars show 95% confidence interval. ∗p < 0.05 for baseline A1 or A2 compared with baseline B2; p < 0.05 for follow-up A2 compared with follow-up B2; p > 0.05 for follow-up A1 or B2 compared with follow-up B2.
(G) Forest plot of the hazard ratio in HCC patients from the NCI CLARITY retrospective cohort (n = 18). Patients are grouped by their lineage-ecological quadrant at baseline (left) (A1, n = 11 patients; A2, n = 5; B1, n = 1; B2, n = 1) and their final follow-up time point (right) (A1, n = 9 patients; A2, n = 6; B1, n = 1; B2, n = 2). Quadrant B2 was used as the reference group for hazard ratio calculation. Bars show 95% confidence interval. p < 0.05 for baseline A1 or A2 compared with B2; p > 0.05 for baseline B1 compared with baseline B2; p < 0.05 for follow-up A2 compared with follow-up B2; p > 0.05 for follow-up A1 or B1 compared with follow-up B2. See also Figures S7 and S8 and Table S5.
Applying CASCADE to a prospective cohort of liver cancer patients
To validate the findings of tumor evolution in our single-cell cohort, we applied CASCADE to bulk transcriptomic data of longitudinal samples from the NCI CLARITY retrospective cohort,32 which consists of primary liver cancer patients undergoing immunotherapy treatment at multiple cancer centers in the United States. We determined the lineage and ecological scores of 75 longitudinal samples collected from 32 patients (18 HCC and 14 CCA). We found that tumors were distributed over all four lineage-ecological quadrants, including B1, a region with little representation in our discovery data (Figure 5C). This suggests that the lack of B1 samples in our discovery single-cell cohort may be due to a small sample size. Consistent with the findings from the discovery cohort, samples that started in A1 or B2 mostly ended in the same quadrant, while those that started in B1 tended to migrate elsewhere (Figure 5D). However, we found that all but one of the samples located in A2 at baseline ended in A2 as well, which is different from the observations in the single-cell discovery cohort where A2 samples tended to migrate. These results further suggest that patterns of tumor evolution might be embedded in the tumor, while also revealing a more complex picture of quadrant changes. We next performed survival analysis of all patients in the CLARITY retrospective cohort using their baseline samples. We found a significant trend of differing survival outcomes in the four lineage-ecological quadrants (Figure 5E). The results are generally consistent with the findings from our discovery cohort, with A1 and A2 having better outcomes than B1 and B2. Here, unlike in our discovery cohort, patients in A2 have the best overall survival, although this trend may be driven by a small number of samples. Multivariate analysis revealed that the CASCADE classifier is independent of age, gender, and race/ethnicity (Figure S7A). We also performed survival analysis of HCC and iCCA separately in the validation cohort. We found that, consistent with general clinical responses, more A1 and A2 cases were identified in HCC than iCCA while a majority of iCCA belonged to B1 and B2 quadrants (Figures S7B and S7C). While a significant difference in survival among the CASCADE subtypes of HCC was observed, iCCA only showed a trend, but statistically not significant (Figures S7B and S7C).
We next wanted to investigate how our CASCADE quadrant classification system compared with other published signatures of liver cancer subtypes. Within our prospective cohort, we calculated sample labels using CASCADE, CLARITY, TCGA, Hoshida, Yamashita, and Lee signatures (Figure S8) with the same strategy described previously.33 Quadrants A1 and A2 tended to align with different subtypes than quadrants B1 and B2; the former mainly matching C1 and C2 from CLARITY, iC2 from TCGA, S3 from Hoshida, MH subtype of Yamashita, and cluster-B of Lee, while the latter matched CLARITY C3 and C4, TCGA iC1, Hoshida S1, Yamashita HpSC subtype, and Lee cluster-A. Quadrant A2 showed a bit more similarity to B1/B2 than did A1, with slightly higher levels of alignment with CLARITY C3 and TCGA iC1. Quadrants B1 and B2 also had modest differences due to the elevation of TCGA iC3 and Hoshida S2 in B2 samples. Overall, the separation of samples in our prospective cohort using CASCADE quadrants matched some of the clusters using other published HCC signatures.
To better understand the effect of patients migrating across quadrants during treatment, we analyzed patient survival among the prospective cohort using their quadrant at baseline and in their final post-treatment samples. Among all patients, survival by their final sample followed a similar pattern as at baseline, with B1 and B2 having much worse outcomes than A1 and A2 (Figure 5F). This trend was consistent when looking at HCC patients only (Figure 5G). In the overall cohort and in HCC only, survival differences between quadrants were slightly more pronounced at baseline than at the final time point. For example, in HCC, quadrant A1 was associated with a significant 90% hazard reduction compared with B2 at baseline, but only an 80% non-significant hazard reduction at the last follow-up (Figure 5G). These results suggest that survival outcomes are embedded in a patient at baseline.
Immune cell types are associated with clinical outcomes
We compared the TME composition to identify immune cell subtypes differentially enriched among the CASCADE-defined quadrants (Figure 6A). Among nine immune/stromal cell subtypes elevated in A1 (the quadrant with the best patient outcomes) we found that many of them individually showed little to no impact on patient survival in our discovery cohort. However, we found substantial survival differences between patients based on whether they had detectable MAIT cell populations. Strikingly, we found perfect survival in four patients with MAIT cells and poor survival in the remaining seven patients without these cells (Figure 6B). Noticeably, the identified MAIT cells constituted a distinct cluster among all T cells in the t-SNE space, indicating a different transcriptomic profile (Figure 6C). Among the differentially expressed genes between MAIT cells and all other T cells, we found six upregulated genes (Figure 6D) that overlapped with MAIT-related gene signatures.34 We used these six genes (SLC4A10, ZBTB16, NCR3, KLRB1, TMIGD2, LST1) as a MAIT cell signature to determine MAIT levels in four bulk cohorts and found significant association with patient survival in each, where higher MAIT levels corresponded to improved overall survival of liver cancer patients (Figures 6E–6H). Hallmark pathway analysis revealed only TNF-α signaling via NF-κB to be significantly enriched in the MAIT-related genes. Consistently, we found a significant correlation between MAIT cells and TNF-α gene expression in the CLARITY retrospective cohort (Figure 6I). These results are consistent with our recent findings on the role of MAIT dysfunction in HCC35 and the model that a MAIT cell subset with elevated TNF signaling may be anti-tumorigenic in HCC,36 providing further confidence about the utility of the CASCADE algorithm.
Figure 6.

MAIT cells drive survival differences between CASCADE quadrants
(A) Heatmap of the abundance of each cell subtype in the TME in each CASCADE quadrant from our discovery single-cell cohort.
(B) Overall survival of liver cancer patients by MAIT cell level in the discovery single-cell cohort (n = 11). Samples were divided by whether they had any detectable MAIT cells or not. The p value was calculated using the log rank test.
(C) t-SNE plot of all T cells (n = 30,020).
(D) Volcano plot of differentially expressed genes between MAIT cells and all other T cells. Red lines denote thresholds for significance; the horizontal line is located at an adjusted p value of 0.05 and the two vertical lines are located at a log2 fold change of ±0.5. Red dots, highly upregulated genes; blue dots, downregulated genes. Genes that overlap with MAIT markers are labeled.
(E‒H) Kaplan-Meier plots of hazard ratio by MAIT levels in four cohorts of bulk transcriptomic data: CLARITY-retrospective (E) (n = 92), LCI (F) (n = 239), TCGA (G) (n = 363), and TIGER-LC (H) (n = 62). Each cohort is dichotomized by high/low MAIT levels based on gene expression. The p values were calculated using the log rank test.
(I) Scatterplot of MAIT signature gene expression and TNF-α in all samples of the CLARITY retrospective cohort (n = 141 samples). The blue line shows a trend line fit using a linear model; the R and p values for correlation trend are indicated.
Discussion
Despite decades of progress in understanding cancer,37,38 our knowledge of the dynamics and evolution of tumor ecosystems remains limited. It is still unclear how tumor cells continuously survive and evolve under innate immune surveillance by an adverse TME. Moreover, we still do not fully understand what drives treatment response and resistance. These challenges limit our ability to understand the mechanisms of tumor progression and to develop effective treatment strategies. Recently, the development of cutting-edge technologies such as scRNA-seq has led to groundbreaking findings by resolving the tumor at single-cell resolution, uncovering a full cellular landscape of the tumor ecosystem. However, with this powerful increase in resolution, a new challenge has emerged to gain a comprehensive understanding of such an intricate tumor landscape. HCC and iCCA remain difficult to treat due to a multitude of factors including a highly complex tumor cell community. Vast inter- and intratumor heterogeneity make it extremely challenging to identify driver events that may serve as potential therapeutic targets.28,39,40 In addition, the TME, especially during treatment, continuously interacts with tumor cells to shape tumor biology.41,42 Monitoring such a complex tumor ecosystem requires the development of a unified classification system for clinicians and researchers that can be used for better prognostic and predictive assessments of tumor behavior, as recommended by a consensus conference of experts in the fields of cancer evolution and ecology.12
In this study, we developed a tool called CASCADE using single-cell profiles derived from liver cancer patients treated with immunotherapy. CASCADE reduces the complex architecture of a tumor and its microenvironment into a simple and prognostic framework. It leverages shared types of malignant cells to determine a lineage score and hazards and resources in the TME to determine an ecological score, which together classify tumors into four distinct quadrants in the lineage-ecological space. Tumors in quadrant A1 had a restrictive TME and a relatively passive tumor cell population, which resulted in a limited tumor with the best patient outcomes. By contrast, tumors in quadrant B2 had highly proliferative tumor cells and a TME that enabled their growth, leading to the worst overall survival. Quadrant A2 had mixed patient outcomes. In this quadrant the TME was lenient, but the tumor cell population lacked highly proliferative characteristics and seemed unable to take full advantage of its favorable environment. Finally, quadrant B1 also had mixed patient outcomes with its proliferative tumor cells but inhibitory TME. Patients in this quadrant also showed mixed outcomes intermediate between those of A1 and B2. We applied CASCADE to several additional cohorts of single-cell and bulk transcriptomic data derived from HCC or iCCA patients and observed consistent results. This suggests that the CASCADE method may be applicable for classifying tumor ecosystems. Using CASCADE as a classification model, we identified a MAIT subset with elevated TNF signaling to be associated with improved overall survival of patients with liver tumors, a feature that was stable across all cohorts. These results are consistent with the hypothesis that MAIT cells play an important role in tumor immunity.35,43
A major strength of the current study is the utilization of single-cell transcriptomic data from biopsies both at baseline and at different time points following treatment. CASCADE is therefore able to model tumor evolution in response to treatment. We found that patients who started in quadrants A1 and B2 tended to stay there through the course of treatment, while those who started in A2 or B1 often migrated elsewhere. Given that quadrants A1 and B2 represented the best and worst states for patient outcome, respectively, it is reasonable to think that an evolutionary trajectory is embedded in the tumor. We anticipate that tracking patient migration across quadrants during treatment may help clinicians monitor the effectiveness of the treatment in real time. However, we could not conclusively find that changes in quadrant over time reflected changes in patient disease status; more research with larger cohort sizes is needed to clarify this point. Instead, the prognostic features classified by CASCADE are most clearly present in baseline biopsies. CASCADE’s two-metric approach may lend a more complete picture of the dynamics underlying a tumor cell community, revealing both the state of the tumor cells and the degree of response from the immune system, which could help identify patients in advance who would respond to immunotherapy. Patients in quadrant A1 at baseline are potentially most receptive to immunotherapy, while those in B2 may benefit from other avenues of treatment.
Taken together, we developed CASCADE to model tumor cells and the TME in concert to fully capture their joint dynamics. The lineage and ecological scores serve as two metrics to characterize the whole tumor ecosystem in defining a tumor’s evolutionary behavior. The defined quadrants may provide a framework for liver tumor classification to potentially guide treatment. We anticipate that application of this framework to other cancer types in a more generalized way may further benefit patient care.
Limitations of the study
This study has several limitations. First, the cohort size is small and treatment regimens for its patients are complex. Therefore, we were not able to test the tool for single agents vs. agents in combination. To determine the stability of tumor cell lineage, we combined tumor cells from both discovery and validation cohorts and found that tumor cell lineage is stable and insensitive to the total cell numbers used for clustering. Second, a recent study indicates that immunotherapy is more effective in HCC patients with viral hepatitis than those with NASH etiology.44 This study was not able to verify these results and determine its utility in patients with different etiologies, such as viral hepatitis- vs. non-viral hepatitis-related etiologies, again due to a small cohort size. Third, our conclusion was based on retrospective analysis of patients with tumor biopsies enrolled from our clinical trials. Furthermore, while our initial analysis was based on combined cells from both HCC and iCCA following the rationale that tumor evolution regardless of tumor types should follow the same principle for establishing a successful tumor colony, only two iCCA cases were included in the initial discovery cohort. To determine the reliability and specificity of the method, we also analyzed HCC and iCCA separately for defining tumor cell lineage and found consistent data among two different approaches. We also performed HCC and iCCA separately in the validation cohort. Consistent with the clinical responses of HCC and iCCA to immunotherapy,21 we found more A1 and A2 HCC cases than iCCA while most iCCA cases belong to B1 and B2 quadrants. While these results are encouraging, the study was based on only a few cases. A clinical utility of this tool will need to be validated prospectively in our ongoing NCI-CLARITY study (ClinicalTrials Identifier: NCT0414514), which aims to enroll a large cohort of patients with HCC and iCCA and monitor them in response to immunotherapy with longitudinal biopsies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Patient-derived biopsy tissues | NIH/NCI Clinical Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 Medium | Gibco | Cat# 11875 |
| MACS Tissue Storage Solution | Miltenyi Biotech | Cat# 130-100-008 |
| Cell Culture Freezing Medium | Gibco | Cat# 12648-010 |
| 1X Red Blood Cell Lysis Buffer | Invitrogen (Thermo-Fisher) | Cat# 00-4333-57 |
| C Tubes | Miltenyi Biotech | Cat# 130-093-237 |
| MACS Strainers (70μm) | Miltenyi Biotech | Cat# 130-095-823 |
| Critical commercial assays | ||
| Tumor Dissociation Kit, Human | Miltenyi Biotech | Cat# 130-095-929 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v1 & v2 | 10x Genomics | Cat# PN-120237 |
| Chromium Single Cell A Chip Kit | 10x Genomics | Cat# PN-120236 |
| Chromium Single Cell G Chip Kit | 10x Genomics | Cat# PN-1000127 |
| Chromium i7 Multiplex Kit | 10x Genomics | Cat# PN-120262 |
| Deposited data | ||
| scRNA-seq data | This paper | GEO: GSE229772 |
| scRNA-seq data | Ma et al. 24 | GEO: GSE151530 |
| LCI (HCC) | Roessler et al. 45 | GEO: GSE14520 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = gse14520) |
| TCGA (HCC) | TCGA (https://portal.gdc.cancer.gov) | TCGA-LIHC |
| TIGER-LC (HCC) | TIGER-LC | GEO: GSE76297 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76297) |
| Software and algorithms | ||
| Cell Ranger version 6.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Seurat version 4.1.0 | Satija Lab | https://satijalab.org/seurat/ |
| R version 3.4.3 | The R Project for Statistical Computing | https://www.r-project.org/ |
| GSEA version 4.1.0 | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| InferCNV | GitHub | https://github.com/broadinstitute/inferCNV |
| GraphPad Prism version 9 | GraphPad Software, San Diego, CA, USA | https://www.graphpad.com/ |
| CASCADE | This paper | https://doi.org/10.5281/zenodo.10424656 |
| Other | ||
| GentleMACS Dissociator | Miltenyi Biotech | 130-093-235 |
| Sequencer NextSeq 500 | Illumina | N/A |
| Sequencer HiSeq 4000 | Illumina | N/A |
| Sequencer NovaSeq 6000 | Illumina | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lichun Ma (lichun.ma@nih.gov).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The scRNA-seq data are available through the Gene Expression Omnibus (accession number GSE229772). The data can also be visualized at scAtlasLC (https://scatlaslc.ccr.cancer.gov).
-
•
Original code of CASCADE is available at https://github.com/MaLab621/CASCADE (https://doi.org/10.5281/zenodo.10424656). Any additional code is available upon reasonable request.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Human sample collection
Fresh HCC or iCCA tumor specimens were collected from primary liver cancer patients from the NIH clinical center as part of the NCI CLARITY study. In the discovery cohort, 11 liver cancer patients (HCC, 9; iCCA, 2) were involved. In total, 31 longitudinal samples were collected from the patients with between 2 and 5 samples from each one. The validation cohort consists of 31 cases (HCC, 20; iCCA, 11). A total of 26 males and 16 females were included. Tissue acquisition was performed with informed consent from patients and was further approved by the ethics committee of the National Institutes of Health. Detailed clinical information of the patients was summarized in Table 1.
Method details
Single-cell library preparation
Single cell suspensions were prepared from patient biopsies or resected tumor tissues as described in our previous work.23,24 In short, tumor tissues were stored in a tissue storage buffer (Miltenyi Biotec, Catalog #130-100-008) immediately after surgical removal and were digested with a Tumor Dissociation Kit (Miltenyi Biotec, Catalog #130-095-929) in Tumor Dissociation C Tubes (Miltenyi Biotec, Catalog #130-093-237). Single cells were then captured either directly or from cryopreserved cell stocks, following the protocol of the Chromium Next GEM Chip G Single Cell Kit (10x Genomics, PN-1000127). The libraries were prepared with the Chromium Next GEM Single Cell 3′ Kit (10x Genomics, PN-1000269) following the kit instruction manuals. Libraries were then pooled and sequenced with Illumina HiSeq 4000/NovaSeq 6000 platforms with a targeting sequencing depth of 50,000 raw reads per cell. Afterward, demultiplexing with bcl2fastq, reads alignment, tagging, and counting using Cell Ranger were performed. All samples yielded a high number of reads ranging from 100 to 500 million and with >95% Q30 bases in barcodes.
scRNA-seq data processing
Raw gene counts from different samples were normalized to the same sequencing depth using Cell Ranger (version 6.0.1). Afterward, the raw counts were used to construct a Seurat object, which kept all genes expressed in at least 0.1% of cells. We filtered out the cells with fewer than 500 detected genes or with at least 50% of their detected genes being mitochondrial. A total of 57, 567 cells passed this initial quality control. We further performed log-normalization (scale factor = 10,000), variable feature selection (k = 2,000, vst method), scaling, and principal component analysis using the variable features. All processing steps were done using the Seurat package (version 4.1.0) in R (version 4.1.3).
CNV estimation
We determined the malignancy of the cells by inferring the CNV using transcriptomic profiles. The exact procedure has been defined in the paper of our previous study.23 In short, we calculated CNV profiles and a CNV score for all cells, then a correlation score between the two. Cells in the bottom 40% of CNV score and CNV correlation score were defined as reference cells. We then re-calculated CNV profiles, CNV score, and CNV correlation score for all samples compared to our reference population of non-malignant cells. Finally, cells above a CNV score threshold of the 70th percentile and a CNV correlation score threshold of 0.4 were designated as malignant. The same cutoffs were applied to all samples for consistency. After labeling the malignancy of all cells, samples with ≥15 malignant cells were included in the analysis of malignant cells. Only newly collected samples (n = 13) in this study were involved in the CNV analysis to distinguish malignant cells and non-malignant cells. The CNV of other samples can be found in our previous publications.23,24 In total, there are 13,042 malignant cells and 44,525 non-malignant cells in this longitudinal single-cell dataset.
Grouping of malignant cells
We selected the top 200 most variable genes from each sample within the malignant cell population and combined them into a set of variable malignant cell genes (k = 2,422). These top variable genes were selected using Seurat’s vst method with default parameters, which identifies genes with the highest variance after normalizing with the global mean-variance fit among the single-cell data. We chose to use a combined set of the top variable genes from each sample to avoid over-representing samples with a larger number of cells. We then performed a hierarchical clustering of all malignant cells (n = 13,042) based on the selected variable genes using a correlation distance method and a complete linkage. Four major clades were identified from hierarchical tree as tumor cell groups.
Gene set enrichment analysis
We extracted the top differentially expressed genes between malignant cells of each lineage to perform gene set enrichment analysis. Genes were selected if they were expressed in at least 25% of cells and had an adjusted p value <0.05 and an average logfold change ≥ 0.25. Both positive and negative markers were selected for each lineage. Pathway analysis was performed on the hallmark gene sets of the Molecular Signatures Database (Human MSigDB v7.5.1) using FGSEA (version 1.22.0) with 10,000 permutations and an adjusted p value cutoff <0.05. Pathways were validated using GAGE (version 2.46.1) with a q value cutoff <0.05. The enrichment of each pathway was indicated by a normalized enrichment score. For pathway analysis of the lineage-ecological quadrants, the same procedure was used except for the GAGE validation. We calculated the top differentially expressed genes (expressed in at least 0.25% of cells, adjusted p value <0.05, average logfold change ≥ 0.25, positive and negative markers) between the four quadrants among malignant cells and each major type of nonmalignant cells (T cells, B cells, CAFs, TAMs, TECs) separately. We then pooled all the differentially expressed genes for each quadrant, keeping the instance with the highest logfold change for duplicates. Pathway analysis was again performed using the MSigDB procedure described above.
Identification of non-malignant cell types
We extracted all non-malignant cells (n = 44,525) from the whole single-cell population using the InferCNV method described above. We selected the top 2,000 most variable genes using Seurat’s vst method, scaled the data, and performed PCA dimensionality reduction. The top 20 PCs were used for a t-SNE analysis and to find neighbors in a shared nearest neighbor graph using Seurat’s FindNeighbors method. We then ran Seurat’s FindClusters method with a resolution of 0.8 (the default), which identified distinct clusters of single cells. These cell clusters were annotated using known marker genes as T cells (CD2, CD3D, CD3E, CD3G), B cells (BLNK, CD79A, FCRL5, SLAMF7), CAFs (COL1A2, COL3A1, COL6A1, DCN, FAP, PDPN), TAMs (CD14, CD163, CD68, CSF1R), TECs (CDH5, ENG, PECAM1, VWF), Cholangiocytes (CD24, EPCAM, KRT19), and Hepatocytes (ALB, CYP2A6, HNF4A, TF, TTR). For each major cell type, we determined its subtypes using this same general procedure. We first separated out cells of that type, then identified variable features, scaled the data, ran PCA, identified clusters using a nearest neighbors approach, and labeled them using differentially expressed marker genes. We used 10 PCs and a resolution of 0.1 to find clusters for all major cell types except for T cells, for which we used a resolution of 1.5 due to the large number of cells and the complex landscape of T cell subtypes.
Relative abundance of TME subtypes
We performed hierarchical clustering (correlation distance, complete linkage) on all tumor samples based on the cell subtype composition of their TMEs. The top 2 clades in the resulting hierarchical tree represent distinct patterns of the TME composition in the samples. Clade 1 TMEs were mainly composed of immune cells, with a high occurrence of cytotoxic GZMH+ and GZMK+ CD8 T cells. By contrast, clade 2 TMEs contained many more CAFs, TECs, and M2 macrophages, with a much higher overall proportion of stromal cells. We therefore hypothesized that clade 1 is a hazard-heavy TME to the tumors while clade 2 is a resource-heavy TME. We calculated the relative abundance of each cell subtype, or its weight Ws, as
where Hs is the average incidence of that subtype in the hazard-heavy clade and Rs is the average incidence of that subtype in the resource-heavy clade (Table S3). These values range from 0, where a subtype is evenly represented in each clade, to 1, where a subtype is entirely present in one clade or the other.
Ecological score
The ecological or Eco-index describes the conditions facing a tumor within its microenvironment. It is composed of a resource score and a hazard score, both of which rely on the subtype labels and weights defined in the above Relative Abundance of TME Subtypes section. For each sample, we calculated a hazard and a resource score as the weighted composition of all hazard or resource types in that sample’s TME. We finally defined the eco score of a sample as its resource score divided by the sum of its resource and hazard scores. The hazard score H is defined as
where Cs is the percent composition of a subtype in a sample’s TME and Ws is the previously defined weight for that subtype, for all subtypes in the set of hazardous subtypes SH. The resource score follows the same formula
for all subtypes in the set of resourceful subtypes SR. We calculated a hazard and resource score for each sample, then computed the final ecological score E as
for a given sample. This metric ranges from 0 to 1 with an implied uniform distribution and natural mean of 0.5. A value above 0.5 denotes a resource-heavy TME and a value below 0.5 denotes a hazard-heavy TME.
Lineage score
We determined a lineage score of each tumor to reflect the features of malignant cells. We calculated the differentially expressed genes (DEGs, expressed in at least 25% of cells, logfold change ≥ 0.25, positive markers only) between each of the 4 tumor lineages (Table S1). This gene set was further reduced to the top DEGs (adjusted p value <0.05, logfold change ≥ 2) to pick up only the strongest signal differentiating each lineage. We calculated the average expression of these genes (k = 316) in each of the samples to yield an expression score for each lineage. We only used average expression of all tumor cells in each tumor to avoid variation of tumor cell numbers among cases. We then calculated the final lineage score L as
where CLTC is the average expression of the top CLTC DEGs in a sample, MLTC is the average expression of the top MLTC DEGs in a sample, and so on. We grouped the CLTC and MLTC lineages together because the hierarchical analysis of lineage compositions showed that samples with mostly CLTC and MLTC lineages clustered together while separating from those with mostly HLTC and MCTC lineages. In addition, both showed an elevation in pathways related to cell growth and proliferation. This metric ranges from 0 to 1 with an implied uniform distribution and natural mean of 0.5. A value above 0.5 denotes a more CLTC or MLTC lineage type and a value below 0.5 denotes a more HLTC or MCTC type.
Tumor score
Tumor score is calculated as the product of the lineage and ecological scores,
for a given sample. This metric is the product of two uniform random variables that range from 0 to 1, and therefore also ranges from 0 to 1. However, unlike the other two metrics it does not assume a uniform distribution. The lineage and eco scores both have a mathematically meaningful balance point at 0.5; as their product, the tumor score has a theoretical balance point at 0.25.
CASCADE scores in bulk transcriptomic data
Since the method in calculating lineage and ecological scores cannot be directly applied to bulk transcriptomic data, We therefore used the single-cell dataset to curate gene sets representing high and low ecological scores as well as high and low lineage scores, which we termed high-E, low-E, high-L, and low-L, respectively (Table S5). First, based on the top DEGs (adjusted p val <0.05, logfold change ≥ 2, positive markers only) between the tumor lineages derived from single-cell data, we combined the CLTC and MLTC DEGs as a high-L related gene set and the HLTC and MCTC DEGs as low-L related genes. The high-L genes represent samples with a high lineage score and vice versa. Next, for each major cell type in the single-cell data (T cells, B cells, CAFs, TAMs, TECs), we calculated the DEGs (adjusted p val <0.05, logfold change ≥ 1, positive markers only) for cells of that type between samples with high and low ecological scores (using the natural mean of 0.5 to distinguish high and low ecological score values). We pooled DEGs across the major cell types from samples with high ecological scores to generate the high-E genes and those from samples with low ecological scores to generate the low-E genes. We used different logfold change thresholds for the ecological and lineage DEGs because lineage genes tended to have higher logfold change values in general, but both cutoffs filtered down to ∼5% of the total DEGs and therefore have comparable biological significance. To avoid overlap between our gene sets, genes that appeared in more than one set were assigned to the one for which they had the highest logfold change. To determine CASCADE scores in bulk transcriptomic data, the expression of high-E, low-E, high-L, and low-L genes were calculated for each sample in the bulk cohorts. The final ecological score was calculated as the high-E score minus the low-E score, normalized to the range of 0–1. The same procedure was performed on the high-L and low-L scores to calculate the lineage score. Finally, tumor score was calculated as the product of ecological score and lineage score. These metrics are all in the range of 0–1, but none are assumed to have a uniform distribution or any natural mean. We therefore used a median value for all subsequent metric cutoffs and survival analyses in bulk datasets. We didn’t apply CibersortX to deconvolute the bulk transcriptome data due to a large number of cellular subtypes derived from the single-cell data in this study and the close relation between subtypes of the same major cell type.
Survival analysis
We used a Cox proportional hazards model via the survival package (version 3.2–13) to perform survival analysis in this study. For the analysis of lineage-ecological quadrants in the CLARITY retrospective cohort, we observed a small number of samples in some of the classes. To reduce bias due to the small sample size, we applied Firth’s penalized maximum likelihood bias reduction method for Cox regression using the coxphf package (version 1.13.1).
Mapping to published HCC signatures
Samples from the prospective NCI-CLARITY cohort were mapped to classes reported in published studies of HCC subsets (CLARITY, TCGA, Hoshida, Yamashita, Lee) using the NearestTemplatePrediction module (version 6) from GenePattern following the method by Candia et al.33 Class assignments were calculated using the default parameters of cosine distance and 1000 resamplings. The visualization was created using circlize (version 0.4.15).
Quantification and statistical analysis
Statistical analysis was performed using R (version 3.4.3) and GraphPad Prism (version 9). Wilcoxon rank-sum test and Student’s t-test were used in this study. log rank test and log rank test for trend were used in survival analysis.
Acknowledgments
We thank Melody Lee for generating the graphical abstract; Eytan Ruppin and Kun Wang for advice on data analysis; and the patients, families, and nurses for contribution to this study. This work was supported by grants (ZIA BC 012079, ZIA BC 012083, ZIA BC 010877, ZIA BC 010876, ZIA BC 010313, and ZIA BC 011870) from the intramural research program of the Center for Cancer Research, National Cancer Institute of the United States.
Author contributions
L.M. and X.W.W. developed the study concept. T.F.G. directed the clinical study. L.M., X.W.W., and M.R. directed the experimental design and interpreted the data. M.R. performed computational analysis. L.W., M.F., S.B., A.J.C., M.L., B.T., M.K., A.B., C.M., C.X., J.M.H., and T.F.G. conducted experiments and additional data analysis. M.R., X.W.W., and L.M. wrote the manuscript with help from L.W. All authors read, edited, and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 26, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101394.
Contributor Information
Xin Wei Wang, Email: xw3u@nih.gov.
Lichun Ma, Email: lichun.ma@nih.gov.
Supplemental information
References
- 1.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Andor N., Graham T.A., Jansen M., Xia L.C., Aktipis C.A., Petritsch C., Ji H.P., Maley C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016;22:105–113. doi: 10.1038/nm.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polyak K., Haviv I., Campbell I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Merlo L.M.F., Pepper J.W., Reid B.J., Maley C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 5.Huang D.Q., Singal A.G., Kono Y., Tan D.J.H., El-Serag H.B., Loomba R. Changing global epidemiology of liver cancer from 2010 to 2019: NASH is the fastest growing cause of liver cancer. Cell Metab. 2022;34:969–977.e2. doi: 10.1016/j.cmet.2022.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegel R.L., Miller K.D., Wagle N.S., Jemal A. Cancer statistics, 2023. CA. Cancer J. Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 7.Villanueva A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 8.Wang X.W., Thorrgeirsson S.S. The Biological and clinical challenge of liver cancer heterogeneity. Hepatic Oncology. 2014;1:5. doi: 10.2217/hep.14.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 10.Yau T., Kang Y.K., Kim T.Y., El-Khoueiry A.B., Santoro A., Sangro B., Melero I., Kudo M., Hou M.M., Matilla A., et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020;6 doi: 10.1001/jamaoncol.2020.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monge C., Pehrsson E.C., Xie C., Duffy A.G., Mabry D., Wood B.J., Kleiner D.E., Steinberg S.M., Figg W.D., Redd B., et al. A Phase II Study of Pembrolizumab in Combination with Capecitabine and Oxaliplatin with Molecular Profiling in Patients with Advanced Biliary Tract Carcinoma. Oncol. 2022;27:e273–e285. doi: 10.1093/oncolo/oyab073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maley C.C., Aktipis A., Graham T.A., Sottoriva A., Boddy A.M., Janiszewska M., Silva A.S., Gerlinger M., Yuan Y., Pienta K.J., et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer. 2017;17:605–619. doi: 10.1038/nrc.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suvà M.L., Tirosh I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell. 2019;75:7–12. doi: 10.1016/j.molcel.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Lim B., Lin Y., Navin N. Advancing cancer research and medicine with single-cell genomics. Cancer Cell. 2020;37:456–470. doi: 10.1016/j.ccell.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawson D.A., Kessenbrock K., Davis R.T., Pervolarakis N., Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018;20:1349–1360. doi: 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science (New York, N.Y.) 2021;374:abe6474. doi: 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 17.Lavie D., Ben-Shmuel A., Erez N., Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer. 2022;3:793–807. doi: 10.1038/s43018-022-00411-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma R.-Y., Black A., Qian B.-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546–563. doi: 10.1016/j.it.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Kurachi M. Springer; 2019. CD8+ T Cell Exhaustion; pp. 327–337. [DOI] [PubMed] [Google Scholar]
- 20.Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greten T.F., Schwabe R., Bardeesy N., Ma L., Goyal L., Kelley R.K., Wang X.W. Immunology and immunotherapy of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023;20:349–365. doi: 10.1038/s41575-022-00741-4. [DOI] [PubMed] [Google Scholar]
- 22.Marjanovic N.D., Hofree M., Chan J.E., Canner D., Wu K., Trakala M., Hartmann G.G., Smith O.C., Kim J.Y., Evans K.V., et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell. 2020;38:229–246.e13. doi: 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma L., Hernandez M.O., Zhao Y., Mehta M., Tran B., Kelly M., Rae Z., Hernandez J.M., Davis J.L., Martin S.P., et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell. 2019;36:418–430.e6. doi: 10.1016/j.ccell.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma L., Wang L., Khatib S.A., Chang C.W., Heinrich S., Dominguez D.A., Forgues M., Candia J., Hernandez M.O., Kelly M., et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2021;75:1397–1408. doi: 10.1016/j.jhep.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma L., Heinrich S., Wang L., Keggenhoff F.L., Khatib S., Forgues M., Kelly M., Hewitt S.M., Saif A., Hernandez J.M., et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat. Commun. 2022;13:7533. doi: 10.1038/s41467-022-35291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taavitsainen S., Engedal N., Cao S., Handle F., Erickson A., Prekovic S., Wetterskog D., Tolonen T., Vuorinen E.M., Kiviaho A., et al. Single-cell ATAC and RNA sequencing reveal pre-existing and persistent subpopulations of cells associated with relapse of prostate cancer. bioRxiv. 2021 doi: 10.1101/2021.02.09.430114. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Black J.R.M., McGranahan N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer. 2021;21:379–392. doi: 10.1038/s41568-021-00336-2. [DOI] [PubMed] [Google Scholar]
- 28.Chaisaingmongkol J., Budhu A., Dang H., Rabibhadana S., Pupacdi B., Kwon S.M., Forgues M., Pomyen Y., Bhudhisawasdi V., Lertprasertsuke N., et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell. 2017;32:57–70.e3. doi: 10.1016/j.ccell.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roessler S., Long E.L., Budhu A., Chen Y., Zhao X., Ji J., Walker R., Jia H.L., Ye Q.H., Qin L.X., et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012;142:957–966.e12. doi: 10.1053/j.gastro.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barkley D., Moncada R., Pour M., Liberman D.A., Dryg I., Werba G., Wang W., Baron M., Rao A., Xia B., et al. Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat. Genet. 2022;54:1192–1201. doi: 10.1038/s41588-022-01141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S., et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611–1624.e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budhu A., Pehrsson E.C., He A., Goyal L., Kelley R.K., Dang H., Xie C., Monge C., Tandon M., Ma L., et al. Tumor biology and immune infiltration define primary liver cancer subsets linked to overall survival after immunotherapy. Cell Rep. Med. 2023;4:101052. doi: 10.1016/j.xcrm.2023.101052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Candia J., Bayarsaikhan E., Tandon M., Budhu A., Forgues M., Tovuu L.O., Tudev U., Lack J., Chao A., Chinburen J., Wang X.W. The genomic landscape of Mongolian hepatocellular carcinoma. Nat. Commun. 2020;11:4383. doi: 10.1038/s41467-020-18186-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao T., Shooshtari P., Haeryfar S.M.M. Leveraging Public Single-Cell and Bulk Transcriptomic Datasets to Delineate MAIT Cell Roles and Phenotypic Characteristics in Human Malignancies. Front. Immunol. 2020;11:1691. doi: 10.3389/fimmu.2020.01691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruf B., Bruhns M., Babaei S., Kedei N., Ma L., Revsine M., Benmebarek M.-R., Ma C., Heinrich B., Subramanyam V., et al. CSF1R+PD-L1+ Tumor-associated macrophages trigger MAIT Cell dysfunction at the HCC Invasive Margin. Cell. 2023;186:3686–3705.e32. doi: 10.1016/j.cell.2023.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q., et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 37.Nowell P.C. The Clonal Evolution of Tumor Cell Populations: Acquired genetic lability permits stepwise selection of variant sublines and underlies tumor progression. Science (New York, N.Y.) 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 38.Rozenblatt-Rosen O., Regev A., Oberdoerffer P., Nawy T., Hupalowska A., Rood J.E., Ashenberg O., Cerami E., Coffey R.J., Demir E., et al. The human tumor atlas network: charting tumor transitions across space and time at single-cell resolution. Cell. 2020;181:236–249. doi: 10.1016/j.cell.2020.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata T., Aburatani H. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014;11:340–349. doi: 10.1038/nrgastro.2014.6. [DOI] [PubMed] [Google Scholar]
- 40.Ding X., He M., Chan A.W., Song Q.X., Sze S.C., Chen H., Man M.K., Man K., Chan S.L., Lai P.B., et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology. 2019;157:1630–1645.e6. doi: 10.1053/j.gastro.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Affo S., Yu L.-X., Schwabe R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017;12:153–186. doi: 10.1146/annurev-pathol-052016-100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pathria P., Louis T.L., Varner J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40:310–327. doi: 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Godfrey D.I., Koay H.F., McCluskey J., Gherardin N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019;20:1110–1128. doi: 10.1038/s41590-019-0444-8. [DOI] [PubMed] [Google Scholar]
- 44.Pfister D., Núñez N.G., Pinyol R., Govaere O., Pinter M., Szydlowska M., Gupta R., Qiu M., Deczkowska A., Weiner A., et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450–456. doi: 10.1038/s41586-021-03362-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roessler S., Jia H.L., Budhu A., Forgues M., Ye Q.H., Lee J.S., Thorgeirsson S.S., Sun Z., Tang Z.Y., Qin L.X., Wang X.W. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70:10202–10212. doi: 10.1158/0008-5472.CAN-10-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The scRNA-seq data are available through the Gene Expression Omnibus (accession number GSE229772). The data can also be visualized at scAtlasLC (https://scatlaslc.ccr.cancer.gov).
-
•
Original code of CASCADE is available at https://github.com/MaLab621/CASCADE (https://doi.org/10.5281/zenodo.10424656). Any additional code is available upon reasonable request.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.