

Summary
Chimeric antigen receptor (CAR)-T therapy has shown superior efficacy against hematopoietic malignancies. However, many patients failed to achieve sustainable tumor control partially due to CAR-T cell exhaustion and limited persistence. In this study, by performing single-cell multi-omics data analysis on patient-derived CAR-T cells, we identify CD38 as a potential hallmark of exhausted CAR-T cells, which is positively correlated with exhaustion-related transcription factors and further confirmed with in vitro exhaustion models. Moreover, inhibiting CD38 activity reverses tonic signaling- or tumor antigen-induced exhaustion independent of single-chain variable fragment design or costimulatory domain, resulting in improved CAR-T cell cytotoxicity and antitumor response. Mechanistically, CD38 inhibition synergizes the downregulation of CD38-cADPR -Ca2+ signaling and activation of the CD38-NAD+-SIRT1 axis to suppress glycolysis. Collectively, our findings shed light on the role of CD38 in CAR-T cell exhaustion and suggest potential clinical applications of CD38 inhibition in enhancing the efficacy and persistence of CAR-T cell therapy.
Keywords: chimeric antigen receptor T, CAR-T, exhaustion, CD38, glycolysis, cADPR, SIRT1
Graphical abstract
Highlights
-
•
CD38 is identified as a potential hallmark of exhausted CAR-T cells
-
•
CD38 inhibition improves memory differentiation and counteracts CAR-T cell exhaustion
-
•
CD38 inhibition represses glycolysis via CD38-cADPR-Ca2+ and CD38-NAD+-SIRT1 axis
In this study, Huang et al. identify CD38 as a hallmark of exhausted CAR-T cells and demonstrate that inhibition of CD38 enzyme activity boosts CAR-T cell cytotoxic ability and antitumor efficacy by downregulation of CD38-cADPR-Ca2+ signaling and activation of the CD38-NAD+-SIRT1 axis to suppress glycolysis.
Introduction
The chimeric antigen receptor (CAR) consists of an antigen-recognition domain, the so-called single-chain variable fragment (scFv), and intracellular stimulatory domains (e.g., CD28 and 4-1BB) to rapidly expand, target antigens, and kill malignant cells in a major histocompatibility complex-independent manner.1 CAR-T cells have outperformed expectations for the treatment of B cell malignancies and provide the best example of the potential for synthetic biology to deliver novel therapeutics.2,3,4 Despite these successes, less than 50% of patients achieve sustainable disease control 1 year after treatment,5,6,7,8 and the majority result in inadequate T cell potency to eradicate solid tumor cells,9 which is mainly due to immunosuppression, T cell exhaustion, and senescence. T cell exhaustion hinders the efficacy of CAR-T cells driven by excessive CAR signaling triggered by a high antigen burden or constant signaling resulting from CAR receptor aggregation in an antigen-independent manner.10,11,12 This highlights exhaustion as a key barrier in the progress of cellular immunotherapy.
Several genes have been identified to be responsible for restraining cellular therapy through omics-based analysis of an in vitro exhaustion T cell model or patient-derived tumor-infiltrating lymphocytes, including the transcription factors IDH2,13 TOX,14,15 BATF,16 NR4A1,17 ID3, and SOX4.18 Other research based on CRISPR-Cas9 screening also demonstrated potential targets, such as MED12,19 TLE4, IKZF2,20 and EZH1,21 that regulate CAR-T durability and long-term cytotoxic function.
CD38, a single-chain transmembrane glycoprotein, was first recognized as a T cell activation marker that functions as an ectoenzyme possessing both ADP-ribosyl cyclase and hydrolase activities22 and exhibits NADase activity by balancing extra- and intracellular nicotinamide adenine dinucleotide (NAD+) levels.23,24 CD38 has a vital impact on T cell dysfunction in autoimmune diseases,25 tumors,26,27 and infectious diseases.28 Recent studies have used CD38 knockout as an induced pluripotent stem cell (iPSC)-derived NK gene-editing strategy, which exhibits enhanced metabolic fitness, together with elevated concentrations of glycolytic and antioxidant metabolites.29 However, its role in antitumor immunotherapy, especially CAR-T cell therapy, is not completely understood.
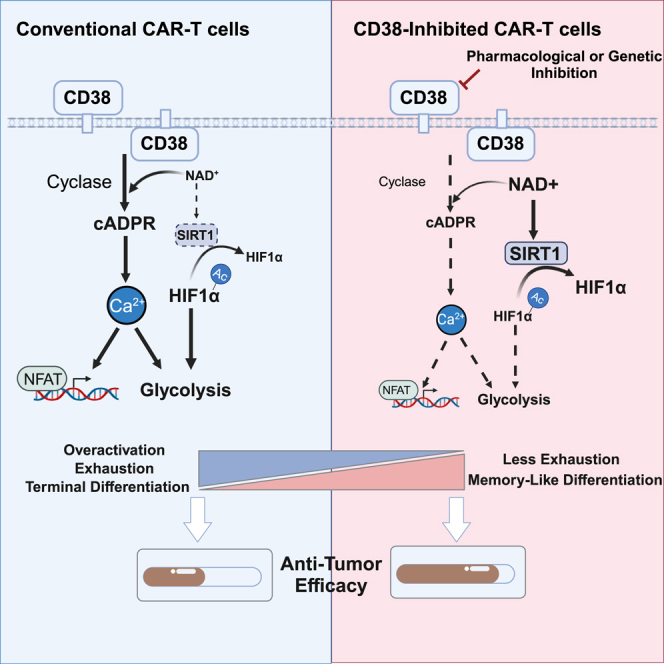
Here, we present evidence that CD38 is one of the hallmarks of exhausted CAR-T cells via our previous scATAC-seq (single-cell assay for transposase-accessible chromatin with high-throughput sequencing) data from patient-derived CAR-T cell populations and confirm our hypothesis with various in vitro CAR-T exhaustion models. Across multiple CAR-T cell models with different costimulatory domains, scFvs, and methods to drive exhaustion, we demonstrate that CD38 inhibition significantly counteracts CAR-T cell exhaustion and boosts the efficacy of CAR-T cells against hematological malignancies both in vitro and in vivo. Further research illustrates that perturbing CD38 enzyme activity rewires CAR-T cell glycolytic metabolism through CD38-cyclic ADP-ribose (cADPR)-Ca2+ signaling and the CD38-NAD+-SIRT1 axis.
Results
CD38 is identified as a potential hallmark of CAR-T cell exhaustion
To identify indicators and potential regulators of CAR-T cell exhaustion, we reanalyzed two sets of recently published single-cell sequencing data (Figure 1A). We first reanalyzed scATAC-seq data30 encompassing 10,929 CAR-T cells obtained from two patients at the expansion peak and later declining stages. CAR-T cells were divided into six subsets: central memory CD4+ T, effector memory CD4+ T, central memory CD8+ T, effector memory CD8+ T, effector CD8+ T, and exhausted CD8+ T cells. Among the top differentially expressed genes, CD38 was coordinately expressed with other well-defined transcription factors, including TOX, CTL4, BATF, and IRF4, as well as exhaustion markers (HAVCR2 and PDCD1) in exhausted CD8+ cells (Figures 1B and S1A), consistent with a previous report that CD38 was a marker of irreversible and not transitory exhausted T cells.31 To extend the scope of CAR-T types and clinical sample size, we further reanalyzed a recently published anti-CD19 CAR-T scRNA-seq dataset from 13 B cell malignancy patients on day 7 (D7) after CAR-T infusion.32 A total of ten distinct clusters were identified in the dataset (Figure 1C). A majority of the CD8+ CAR-T cells on D7 were composed of proliferating or effector subsets (cluster C0, C1, C2, C3, and C7), while a smaller population of CAR-T cells was characterized as the exhausted subset (C9). In accordance with our previous finding, exhausted CD8+ T cells were highly expressed with CD38 together with LAG3, TOX, and STAT2, as revealed by differential expression gene analysis (Figures S1B and S1D). Notably, CD38high CD8+ T cells manifested the highest transcriptomic signature score for T cell exhaustion33 in both in scRNA-seq and scATAC-seq datasets (Figure 1D). CD38 was significantly positively correlated with exhaustion (r = 0.459, p = 1.76e−8) in C9 cluster from scRNA-seq dataset (Figure S1C). These datasets indicate a positive correlation between CD38 expression and exhaustion features in CD8+ T cells.
Figure 1.

CD38 expression is positively correlated with CAR-T exhaustion
(A) Schematic depicting reanalysis of two recently published single-cell-level studies, including scATAC-seq data from two patient-derived CAR-T cells at the expansion peak stage (CAR-T-P) and the later declining stage (CAR-T-L) (upper panel) and scRNA-seq data from 13 B-ALL patient-derived CAR-T cells on day 7 after infusion (lower panel).
(B) In scATAC-seq data, top differentially activated transcription factors (TFs) for exhausted CD8 T subtypes. The color indicates Z score transformed TF deviation scores.
(C) In scRNA-seq data, UMAP shows the distribution of annotated cell subsets. Colors indicate cell subtypes.
(D) Boxplot of exhaustion score calculated by the mean expression of gene sets33 in CD38 high expression, low expression, and negative expression groups. Solid dots represent boxplot outliers.
(E) Schematic depicting two types of in vitro exhaustion model, tonic signaling-induced CD19-41BBz/GD2-CD28z CAR-T cell exhaustion and tumor antigen-stimulated CAR-T cell exhaustion.
(F) Flow cytometric analysis of CD38 expression in CD19-41BBz CAR-T cells from D4 to D14 after retroviral transduction. Mean fluorescence intensity is normalized to the control at each time point.
(G) Flow cytometric analysis of CD38 expression in CD19-41BBz CAR-T cells before and after NALM6 stimulation. Mean fluorescence intensity is normalized to the control at each time point.
(H and I) mRNA level of exhaustion-related or memory-like transcription factors in CD19-41BBz CAR-T cells in (H) D8, D13, and D18 after retroviral transduction or (I) before and after NALM6 stimulation (n = 3 biological replicates).
CAR-T cells manifest tonic signaling during in vitro manufacturing, leading to early exhaustion that limits their potency.10 In addition, exposure to tumor burdens induces exhaustion in the absence of tonic signaling.18 To fully investigate the contribution of CD38 in CAR-T cell exhaustion, two in vitro systems were employed to simulate tonic signaling-induced and tumor antigen-stimulated CAR-T exhaustion (Figure 1E). To confirm the tonic signal-induced exhaustion model, we detected tonic signaling index and exhaustion score34 during the CAR-T cell culture period from day 6 to day 18. Indeed, CD19-41BBz CAR-T exhibited an increasing level of tonic signaling and exhaustion (Figure S1E). By day 8, although the CD19-41BBz CAR construct resulted in lower levels of tonic signaling and exhaustion compared to the GD2-CD28z CAR construct, it still surpassed the control T cells transfected with an empty vector (Figure S1F). The tonic signaling index of CD19-41BBz CAR was three times higher than that of T cells, and the exhaustion score was nearly eight times higher than that of T cells. These results depicted that CD38 expression increased both forms of CAR-T exhaustion during long-term culture and after exposure to tumors in vitro (Figures 1F and 1G). Quantitative real-time PCR confirmed that there was a positive correlation between the expression of CD38 and exhaustion-related transcription factors, including TOX, PRDM1, BLIMP-1, TBX21, and ENTPD1, during the culture from day 8 to day 18, and a negative correlation with memory-associated factors or markers, including TCF7, LEF1 and IL-7R (Figure 1H). Similarly, after exposure to tumor antigens, an increase in CD38 levels was observed alongside elevated exhaustion transcription and decreased memory-like factors (Figure 1I). To confirm the universality of this phenomenon, we replicated the experiment in GD2-CD28z CAR-T cells, which are more prone to exhaustion, and obtained consistent results (Figures S1G–S1J). Taken together, these findings raise the possibility that CD38 is a key surface marker in CAR-T cell exhaustion, and it might be associated with T cell fate.
CD38 inhibition potentiates early memory differentiation and counteracts CAR-T cell exhaustion
To evaluate the function of CD38 in CAR-T cells, stimulated CD19-41BBz CAR-T cells were treated with three small-molecule inhibitors against CD38 at optimal concentrations, including non-competitive inhibitor compound 78C (a thiazoloquin(az)olin(on)),35 RBN013209 (a heterobicyclic amide),36 and luteolinidin (a flavanoid)37 (Figure 2A). None of the enzymatic inhibitors affected CD38 expression (Figure S2A). Since CAR-T cells exhibit a heterogeneous population and both clinical and preclinical evidence have shown that naive or memory-like subsets of CAR-T cells are critical for in vivo long-term persistence and superior antitumor capability,38,39 we assessed the distribution of different subtypes using CD62L and CD45RO as markers. Surprisingly, all three inhibitors enabled CAR-T cells to maintain the naive state (Tn; CD62L+ and CD45RO–) and central memory state (Tcm; CD62L+ and CD45RO+) (Figures 2B and 2C). CD38 inhibitors also endowed CAR-T cells with lower expression of the activation markers, CD25 and CD69 (Figure 2D), as well as the exhaustion-related inhibitory receptors, LAG-3, TIM-3, and PD-1 (Figure 2E), wherein compound 78C performed better than the other two inhibitors. CAR-T cells in the 78C-treated group had significantly lower co-expression of double- and triple-positive T cell inhibitory receptors than those in the control group (Figure 2F). Accordingly, compound 78C was selected as a representative small-molecule inhibitor of CD38. After drug washout, 78C-treated CAR-T cells sustained a better in vitro expansion than that of the control group (Figure 2G). Flow cytometry assessment further revealed that 78C significantly reduced apoptosis in CAR-T cells (Figures 2H and 2I). The enhanced expansion capacity and reduced apoptosis of CAR-T cells are likely attributable to increased memory potential.
Figure 2.

CD38 inhibition promotes central memory cell formation and counteracts CAR-T cell exhaustion to enhance antitumor efficacy
(A) Schematic depicting in vitro culture model. CAR-T cells were cocultured with NALM6 at the E:T ratio of 1:1 for 48 h, followed by CD38 inhibitor treatment (78C 10 μM, RBN013209 50 μM, luteolinidin 10 μM) for 72 h. Cell differentiation, activation, and exhaustion status were evaluated by flow cytometry.
(B) Flow cytometric analysis of CD62L and CD45RO in each group.
(C) Frequency of naive cells (CD62L+, CD45RO–), central memory cells (CD62L+, CD45RO+), effector memory cells (CD62L–, CD45RO+), and effector cells (CD62L–, CD45RO–) in control or inhibitor-treated CAR-T cells 12 days after T cell activation (n = 5 biological replicates). Two-tailed Student’s unpaired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, no significance. Tn, naive T; Tcm, central memory T; Tem, effector memory T; Teff, effector T. Statistical comparison is between each inhibitor-treated group with control.
(D) Frequency of CD25+ and CD69+ CAR-T cells in control or inhibitor-treated groups 12 days after T cell activation (n = 5 biological replicates).
(E) Frequency of LAG-3+, TIM-3+, and PD-1+ CAR-T cells in control or inhibitor-treated groups 12 days after T cell activation (n = 6, 3 biological replicates with two technical replicates for each donor).
(F) Frequency of inhibitory receptor co-expression (LAG-3, TIM-3, and PD-1) in control or 78C-treated groups (n = 3 biological replicates).
(G) Expansion kinetics of control and 78C-treated CD19-41BBz CAR-T cells during in vitro setting. Arrows indicate the time point of inhibitor treatment and drug washout (n = 3 technical replicates from one donor).
(H) Flow cytometric analysis of annexin V in each group.
(I) Frequency of apoptosis (annexin V+) in control or 78C-treated CAR-T cells 12 days after T cell activation (n = 6, 3 biological replicates with 2 technical replicates for each donor). Two-tailed Student’s unpaired t test.
(J) Specific lysis of NALM6-luciferase after coculture with control and 78C-treated CAR-T cells upon multiple rounds of tumor challenge. CAR-T cells were cocultured with NALM6-luci cells at the E:T ratio = 1:10 for every 24 h. Data are mean ± standard deviation (SD) of 3 technical replicates from one donor. Specific cytotoxicity is evaluated by (nontransduced T cell viability – CAR-T cell viability)/nontransduced T cell viability × 100%.
(K) Specific lysis of NALM6-luciferase after coculture with control and 78C-treated CD19-41BBz CAR-T cells for 72 h at low E:T ratio (n = 3 technical replicates from donor 1 and donor 2, respectively).
(L–O) Secretion of granzyme B, IL-2, IFNγ, and TNFα by control and 78C-treated CAR-T cells after the coculture of NALM6-luciferase for 72 h at E:T ratio of 1:1 and 1:128, respectively (n = 3 technical replicates).
To examine whether the effect of CD38 was restrained by tumor antigen stimulation-induced exhaustion, we employed 10 μM 78C on CAR-T cells without tumor exposure 9 days after T cell activation. Similarly, after 72 h of 78C treatment, we found an increased proportion of cells bearing naive and central memory phenotypes (Figures S2B and S2C). Lower levels of activation markers and inhibitory receptors were also detected using flow cytometry (Figures S2D–S2G). Furthermore, concordant with our findings in CD19-41BBz CAR-T cells, CD38-inhibited CD19-CD28z and GD2-CD28z CAR-T cells exhibited increased Tn and Tcm proportion (Figures S3A, S3B, S4A, and S4B), improved proliferation capability (Figures S3C and S4C), and decreased expression levels of activation (Figures S3D–S3F and S4D–S4F) and exhaustion markers (Figures S3G and S4G) compared to their control counterparts, confirming that the findings were not restricted to the costimulatory domain or scFv design.
To rule out potential off-target effects of the small-molecule CD38 inhibitors, we modified CD38-knockdown CAR-T cells using the transfection of a lentivirus-cooperating CD19-41BBz CAR construct and CD38-targeted short hairpin RNA (shRNA) (Figures S5A–S5C). Consistent with the aforementioned results described above, CD38 knockdown dramatically increased the proportion of CAR-T cells in the naive and central memory states (Figure S5D). In addition, the expression levels of exhaustion-related inhibitory receptors, including LAG-3, TIM-3, and PD-1, were significantly reduced upon CD38 knockdown (Figure S5E). Overall, these results demonstrate that CD38-inhibited CAR-T cells manifest diminished cell exhaustion and converted to a more memory-like state regardless of whether the CAR incorporated a CD28 or 41BB costimulatory domain or whether the CAR-T cells were stimulated by tumor antigens.
Perturbing CD38 enzymatic activity boosts CAR-T cell efficacy against malignancies
To evaluate the effector functions of CD38-inhibited CAR-T cells in antigen- or tonic signaling-induced exhaustion, we assessed cytokine levels using intracellular flow cytometry and an enzyme-linked immunosorbent assay (ELISA). CD38-inhibited CAR-T cells initially decreased effector cytokine production (IL-2, TNF-α, granzyme B, and IFNγ) in both CAR-T cell types, as shown in Figures S2H–S2S, indicating a less activated CAR-T cell state in vitro. To determine the impact of this “resting” state on CAR-T cell cytotoxicity upon tumor re-challenge, a luciferase-based cytolysis assay was performed under various conditions in CAR-T cells with different CAR structures (CD19-41BBz, CD19-CD28z, and GD2-CD28z). Remarkably, despite the impaired cytolytic capability upon repetitive tumor challenge, 78C-pretreated CAR-T cells displayed a significant improvement in cytotoxicity when subjected to exhaustion-inducing conditions (low E:T ratio at 1:10 and multiple rounds of tumor challenge) compared to that of the DMSO-pretreated group (Figure 2J). The effect of CD38 perturbation became more pronounced on anti-CD19 CAR-T cells as the E:T ratio decreased from 1:8 to 1:128 (Figures 2K and S3H) and on anti-GD2 CAR-T cells as the E:T ratio decreased from 1:1 to 1:4 (Figure S4H), consistent with the observed more significant increase of its cytokine secretion levels compared to that of the control group (Figures 2L–2O, S3I–S3L, and S4I–S4L). Consistently, CD38 knockdown also endowed CAR-T cells with superior cytotoxicity ability against tumor cells in vitro under exhaustion-induced conditions (multiple rounds of tumor challenge and low E:T ratio, Figures S5F and S5G) and higher cytotoxicity-related cytokine release (Figures S5H–S5K).
Based on the observation that CD38 inhibition sustained the proliferative ability, effector functions, and less exhausted and early memory phenotypes of CAR-T cells, we then tested whether CD38-inhibited CAR-T cells would confer enhanced in vivo antitumor efficacy. We employed a murine model wherein NALM6-GFP leukemia cells were inoculated into NOD-SCID-Il2rg−/− (NSG) mice and infused with 78C-treated CD19-41BBz or CD19-CD28z CAR-T cells 6 days post inoculation (Figures 3A and S3M). 78C-pretreated CAR-T cells successfully suppressed tumor growth in all treated mice in association with an overall prolongation of survival (Figures 3B–3E, S3O, and S3P). To characterize the CAR-T cell in vivo function and immunophenotype, mice were sacrificed 8 days after CAR-T cell inoculation. Notably, CAR-T cells pretreated with CD38 inhibitors for 72 h exhibited increased homeostatic expansion and persistence in both the bone marrow and spleen tissues (Figure 3F) and showed a significant increase in the ratio of CD8+:CD4+ cells (Figure S6D). The prominently expanded CAR-T cells were endowed with fewer exhaustion characteristics, represented by significantly diminished co-expression of multiple inhibitory receptor markers (double and triple positive) (Figures 3G and S6A–S6C). In addition, as observed in vitro, pretreatment with a CD38 inhibitor further reversed the terminated-differentiated stage (Figure 3H) and redirected CAR-T cell fate away from overactivation in vivo (Figures 3I and 3J). Interestingly, the differences in spleen tissue were more significant than those in bone marrow (Figure S6D). To investigate the in vivo effector function of CAR-T cells, we evaluated concentrations of serum cytokines concentrations. The 78C-treated group had elevated concentrations of IL-2 and IFNγ and comparable levels of granzyme B and TNF-α compared to those of the control group (Figure S6E), providing evidence that the memory-like state induced by CD38 inhibition did not compromise the cytotoxic activity of CAR-T cells in vivo. CD38-knockdown CD19-41BBz CAR-T cells also exhibited superior effector function in a NALM6-bearing NSG mice model (Figures S5L–S5P) and have higher granzyme B and IFNγ secretion in vivo (Figure S5O). Similarly, we observed superior tumor control and survival in a 143B osteosarcoma mice model treated with 78C-pretreated CAR-T cells expressing the GD2-CD28z receptor (Figures S4N–S4R).
Figure 3.

Perturbing CD38 boosts the efficacy of CAR-T cells against hematological malignanciesin vivo
(A) Schematic depicting in vivo experimental setup. NSG mice received 1 × 106 NALM6 cells on day −6 and 1.5 × 106 either nontransduced T cells (MOCK) or 4-1BB CD19-CAR-T cells on day 0. Bone marrow and spleen tissue were collected for fluorescence-activated cell sorting analysis on day 8.
(B) D0-D49 bioluminescence imaging (BLI) imaging of tumor clearance. n = 5 biological replicates for each group.
(C) The dorsal BLI signal is displayed for individual mice in each treatment group. n = 7 biological replicates pooled from two independent experiments.
(D) BLI imaging of tumor burden on D28 after CAR-T cell infusion.
(E) Kaplan-Meier survival plot for mice receiving mock T cells, control CAR-T cells, or CAR-T cells pretreated with CD38 inhibitors. n = 7 biological replicates pooled from two independent experiments (statistical analysis by Mantel-Cox test between control CAR-T and 78C-treated CAR-T group, ∗∗∗p = 0.0007).
(F) Absolute numbers of human T cells in the bone marrow (hindlimb) and spleen on day 8 after CAR-T cell injection. n = 3 or more mice per group.
(G) Frequency of inhibitory receptor co-expression (LAG-3, TIM-3, and PD-1) in bone marrow CAR-T cells in control (n = 3 biological replicates) or 78C-treated groups (n = 4 biological replicates).
(H) Frequency of effector CAR-T cell subset (CD62L–, CD45RO–) from bone marrow and spleen tissue in control (n = 3 biological replicates) or 78C-treated groups (n = 4 biological replicates).
(I) Frequency of CD25+ and CD69+ CAR-T cells from bone marrow and spleen tissue in control (n = 3 biological replicates) or 78C-treated groups (n = 4 biological replicates).
In summary, these data demonstrate that adoptive transfer of CD38-inhibited CAR-T cells induces more durable antitumor responses in the NSG mouse model. Enhanced therapeutic efficacy is largely associated with the phenotypic reprogramming of CAR-T cells toward a “resting” state, which is in line with the concept put forward by Weber et al. that transient cessation of receptor signaling, or “rest,” can enhance CAR-T cell efficacy by preventing or reversing exhaustion.40 In the presence of specific small molecules to reduce tonic CAR signaling, or drugs to switch off CAR expression temporarily, pre-exhausted CAR-T cells could rewire their cell fate to counteract exhaustion and redirect toward a memory-like state.
CD38 inhibition downregulates glycolysis metabolism in exhausted CAR-T cells
Next, we assessed the transcriptional differences between CD38-inhibited and control CAR-T cells. Bulk RNA-seq was performed on CD19-41BBz CAR-T cells after exposure to NALM6 cells for 48 h followed by a 72-h 78C or DMSO incubation period in vitro. Unbiased principal component analysis (PCA) displayed that CD38-inhibited CAR-T cells were transcriptionally distinct from control cells (Figure 4A). A total of 749 differentially expressed genes were identified between genotypes, illustrating the high degree of transcriptomic reprogramming induced by the CD38 inhibitor (Figure S7A). Consistent with the phenotypic and functional data, CD38-inhibited cells showed increased expression of early memory-related genes, including IL7R, TCF7, SELL, and KLF2. RNA sequencing also unleashed that transcripts associated with exhaustion (PDCD1, CD40LG, NR4A1, NR4A2, and NR4A3) and activation (GZMA, GZMB, and CD244) underwent rapid reversal (Figure 4B). Gene set enrichment analysis (GSEA) of our data with T cell exhaustion genes identified in the lymphocytic choriomeningitis virus mouse model41 revealed significant enrichment of terminally exhausted T cell populations in the DMSO group (Figure S7B). Consistent with the fact that CD38-inhibited CAR-T cells manifested lower cytokine levels, GSEA also revealed decreased enrichment of cytokine-related gene sets (Figure S7C). Gene set variation analysis (GSVA) further suggested that CD38-inhibited CD19-41BBz CAR-T cells were enriched in the gene expression involved in early memory and naive T cell differentiation (Figures 4C, S7E, and S7F) and exhibited decreased expression of key regulators of exhausted T cells, apoptosis, and hypoxia (Figures 4C and S7H). Representative exhaustion-related genes, including HAVCR2, NR4A2, CTLA4, and BATF, were downregulated in the 78C-treated group (Figure S7D).
Figure 4.

CD38 inhibition results in reduced glycolysis metabolism in CAR-T cells
(A) Principal component analysis of CAR-T cells in different groups.
(B) Volcano plot illustrating differential gene expression analysis in CD38-inhibited CAR-T compared to control CAR-T cells after coculture with NALM6 cells at 1:1 (E:T).
(C) Heatmap of selected pathways enriched in genes significantly upregulated or downregulated in 78C-treated CAR-T cells. A single sample enrichment score was calculated for each pathway, and the mean was taken per response group. A color gradient ranging from blue to red indicates the mean normalized enrichment score (ranging from −2 to +2) of pathways enriched in induced (red) or repressed (blue) genes.
(D–F) Metabolic rate as measured by Seahorse analysis of extracellular acidification rate (ECAR) of control (n = 5 technical replicates) or CD38-inhibited (n = 4 technical replicates) CAR-T cells after coculture with NALM6 cells.
(G) Heatmap of differentially expressed genes in canonical glycolysis pathway in comparison with the control group.
(H) mRNA level of glycolysis-related transcription factors in control or CD38-inhibited CAR-T cells after coculture with NALM6 cells. Data are mean ± SD of 3 technical replicates.
(I) Flow cytometric analysis of 2-NBDG uptake in each group.
(J) Median fluorescence intensity of 2-NBDG in control or 78C-treated CAR-T cells after coculture with NALM6 cells. Data are mean ± SD of 3 independent experiments from three different donors.
(K) Cellular lactate acid level in control or 78C-treated CAR-T cells after coculture with NALM6 cells (n = 4 technical replicates from two different donors).
(L) Flow cytometric analysis cytoplasmic ROS in control or 78C-treated CAR-T cells after coculture with NALM6 cells by CM-H2DCFDA staining (n = 3 biological replicates).
(M and N) Western blot analysis of PDH (M) and normalized PDH expression relative to TUBULIN in each group (N). NALM6-stimulated CAR-T cells were treated with DMSO/78C for 3 days. Quantitative analysis of western blot data obtained in n = 3 technical replicates is shown, normalized to tubulin.
GSVA showed that CD38-inhibited CD19-41BBz CAR-T cells exhibited decreased expression of key regulators of canonical glycolysis (Figure 4C), which is consistent with previous reports demonstrating that CD38 inhibition or knockout reprograms cell metabolism, especially glucose metabolism.29,42 Seahorse metabolic flux assay analysis further indicated that glycolysis and glycolytic capacity were decreased in 78C-treated CAR-T cells (Figures 4D–4F). Glycolytic enzymes, including ENO2, FOXK1, ALDOA, HK2, PFKM, PGAM1, PKM, and TPI, were significantly transcriptionally downregulated in 78C-treated CD19-41BBz CAR-T cells (Figures 4G and 4H). Consistently, lower glucose uptake (Figures 4I and 4J), lower intracellular lactate levels (Figure 4K), and intracellular reactive oxygen species (ROS) levels (Figure 4L) were detected in CD38-inhibited CD19-41BBz CAR-T cells than in the control group. The expression of pyruvate dehydrogenase (PDH), a cornerstone that links glycolysis to the citric acid cycle, as well as GLUT1 and GLUT3, the predominant glucose transporters in T cells, was also reduced in the 78C-treated group (Figures 4M, 4N, and S7I–S7L). Similarly, 78C-pretreated CAR-T cells expressing the CD19-CD28z or GD2-CD28z receptor also exhibited downregulated glycolytic enzymes transcriptionally compared to control groups (Figures S3R and S4S).
Moreover, 78C also improved the mitochondrial fitness of CAR-T cells. CD19-41BBz CAR-T cells treated with 78C exhibited lower levels of mitochondrial permeability transition pores and a lower mitochondrial membrane potential (Figures S8A–S8C), which aligns with the characteristics of long-persistent naive or central memory cells.43 To directly visualize the mitochondrial content, transmission electron micrographs of control and CD38-inhibited CD19-41BBz CAR-T cells were obtained after 12 days of culture (Figure S8D). 78C treatment increased the ratio of long to fragmented mitochondria (Figure S8F), but there was no difference in mitochondrial number (Figure S8E). However, despite increased mitochondrial fusion, Seahorse metabolic flux assay showed no significant differences in oxygen consumption rate in the 78C-exposed CD19-41BBz CAR-T cells (Figure S8G). Collectively, these results demonstrate that CD38 inhibition decreases glycolytic metabolism in CAR-T cells.
CD38 inhibition induced CD38-cADPR-Ca2+ signaling suppression in CAR-T cells
Subsequently, we investigated the mechanism by which CD38 inhibition downregulates glycolytic metabolism and represses CAR-T cell exhaustion. cADPR, ADP-ribose (ADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP) are three main products of the CD38 cyclase, hydrolase, and NAADP-synthase activity,44 which are vital cellular messengers activating Ca2+ flux in many other cell types.45,46,47,48 As demonstrated in a previous study, calcium signaling is hyperactivated during CAR-T cell exhaustion, and modulation of cytosolic calcium impedes exhaustion and reinvigorates T cell function by reducing nuclear factor of activated T-cells (NFAT) nuclear retention.49
We then investigated whether the CD38 inhibitor could reduce CAR-T cell exhaustion and improve therapeutic efficacy via second messengers and downstream Ca2+ signaling regulation (Figure 5A). We measured levels of intracellular second messengers in CAR-T cells after 72 h of 78C treatment. Upon 78C treatment, second messengers cADPR and ADPR were significantly reduced in CAR-T cells, while NAADP level remained comparable (Figures 5B and S9G). Consistently, Fluo-4 staining revealed a substantial decrease in cytosolic calcium concentrations in CAR-T cells following 78C treatment (Figure 5C). This was confirmed using Gene Ontology (GO) analysis of RNA-seq data, indicating the enrichment of 78C-induced downregulated genes in calcium-related pathways (Figures S9A–S9D). We then detected the expression of cytoplasmic and nuclear NFAT1 and found that 78C significantly blocked the soma-to-nucleus translocation of NFAT1 (Figures S9F and S9G), thereby inhibiting the transcription of exhaustion-related genes and glycolytic genes, which is concordant with a previous study.49
Figure 5.

CD38 inhibition induced CD38-cADPR-Ca2+signaling suppression in CAR-T cells
(A) Schematic of CD38-cADPR signaling and following activation of Ca2+ signaling.
(B) Intracellular cADPR and ADPR level in control or 78C-treated CAR-T cells after coculture with NALM6 cells (cADPR, n = 10, 5 biological replicates with 2 technical replicates for each donor. ADPR, n = 8, 4 biological replicates with 2 technical replicates for each donor).
(C) Median fluorescence intensity of Fluo-4 in CAR-T cells treated with DMSO, 78C, and 78C with cADPR, ADPR, or NAADP after coculture with NALM6 cells (n = 4 biological replicates).
(D) Flow cytometric analysis of CD62L and CD45RO in CAR-T cells treated with DMSO, 78C, and 78C with cADPR, ADPR, or NAADP.
(E) Frequency of CD62L+ CAR-T subset in each group (n = 4 biological replicates).
(F) Frequency of CD69+ CAR-T cells in CAR-T cells treated with DMSO, 78C, and 78C with cADPR, ADPR, or NAADP (n = 3 biological replicates).
(G) Frequency of LAG-3+, TIM-3+, and PD-1+ subsets in CAR-T cells treated with DMSO, 10 μM 78C, and 10 μM 78C with 30 μM cADPR, 500 μM ADPR, or 100 μM NAADP.
(H) mRNA level of glycolysis-related transcription factors in CAR-T cells treated with DMSO, 10 μM 78C, or 10 μM 78C with 5 μM cADPR (n = 3 biological replicates).
(I) Flow cytometric analysis of CD62L and CD45RO in CAR-T cells treated with DMSO or 10 μM 8-Br-cADPR.
(J) Frequency of CD62L+ CAR-T subset in each group (n = 3 donors).
(K) Frequency of CD25+ and CD69+ CAR-T cells in control or 8-Br-cADPR-treated CAR-T cells after coculture with NALM6 cells (n = 3 biological replicates).
(L) Frequency of LAG-3+, TIM-3+, and PD-1+ subsets in control or 8-Br-cADPR-treated CAR-T cells after coculture with NALM6 cells (n = 3 biological replicates).
(M) mRNA level of glycolysis-related transcription factors in CAR-T cells treated with DMSO or 8-Br-cADPR (n = 3 technical replicates).
To determine the possible downstream second messenger of CD38 involved in calcium signal regulation in CAR-T cells, rescue experiments were conducted. The supplementation of cADPR partially restored Ca2+ influx upon CD38 inhibition (Figure 5C), indicating a potential association between cADPR and the reduced calcium flux caused by 78C. Consistently, incubating CAR-T cells with cADPR after 78C treatment reversed the phenotypic features to a large extent, leading to a decrease in CD62L expression (Figures 5D and 5E), an increase in activation-related marker expression (Figure 5F), and an increase in exhaustion-related inhibitory receptor expression (Figure 5G). The cADPR supplementation also re-upregulated the transcripts of glycolytic enzyme genes (ALDOA, ALDOC, HK2, TPI, etc.), indicating a reversal in glycolytic metabolism (Figure 5H). We then used 8-Br-cADPR, a competitive inhibitor of cADPR, to confirm the involvement of cADPR in the regulation of exhaustion-related phenotypes in CAR-T cells. Similarly, 8-Br-cADPR treatment exhibited the same trend as CD38 inhibition on CAR-T cell differentiation, exhaustion, activation, and glycolytic metabolism (Figures 5I–5M), confirming that the suppression of CD38-cADPR-Ca2+ signaling may be one potential mechanism underlying the superior functionality of CAR-T cells upon CD38 inhibition.
ADPR is the primary product of NAD+ hydrolase. We observed a reduction in intracellular ADPR levels following 78C treatment, suggesting a potential inhibition of CD38 hydrolase activity. The supplementation of ADPR also had a trend toward increasing intracellular calcium levels, although this difference was not statistically significant (Figure 5C). As for NAADP, we did not observe a significant restoration of Ca2+ flux following NAADP supplementation (Figures 5C and S9G). Regarding phenotypic impact, neither ADPR nor NAADP supplementation exerted significant rescue of differentiation state in CAR-T cells. ADPR supplementation also didn’t result in any reverse in exhaustion-related inhibitory receptor expression in CAR-T cells (Figure 5G). Although NAADP partially rescued the expression of exhaustion-related inhibitory markers (LAG-3 and TIM-3) in CAR-T cells (Figure 5G), the NAADP antagonist, Ned-19, failed to reverse any phenotypic effect of CD38 inhibition on CAR-T cells (Figures S9H–S9J).
SARM1 is another enzyme capable of metabolizing NAD+ into secondary adenosinergic products, which exhibit higher cyclase activity and efficiency.50 Two SARM1 inhibitors, dehydronitrosonisoldipine51 and DSRM-3716,52 both significantly promoted memory-like differentiation (Figure S9K), albeit not as effectively as CD38 inhibitors. No significant reduction was observed in the exhaustion-related inhibitor receptors upon SARM1 inhibitor treatment (Figure S9L). These results may be attributed to the low SARM1 gene expression in CAR-T cells, especially in the exhausted cell subset. Indeed, the expression of SARM1 at the transcriptional level was over 10-fold lower than CD38, which was confirmed both by our bulk RNA-seq data and reanalyzed scRNA-seq data (Figures S9M and S9N).
Together, the repression of CD38-Ca2+ signaling represents a potential mechanism contributing to the enhanced functionality of CD38-inhibited CAR-T cells, which is primarily dependent on the second messenger cADPR. Our results also demonstrated the potential roles of ADPR and possibly NAADP in the functional mechanism of CAR-T cells, which require further studies to establish the kinetic preponderance of each CD38-dependent second messenger on CAR-T cell activity.
CD38-NAD+-SIRT1 signaling is enhanced upon CD38 inhibition in CAR-T cells
As supplementation of second messengers partially reversed the impact of the CD38 inhibitor on CAR-T cells, we subsequently endeavored to propose additional underlying mechanisms that collaborate with CD38-cADPR-Ca2+ signaling. Considering from the perspective of substrate, CD38 is one of the main NAD+-degrading enzymes in mammalian tissues and plays a key role in NAD-related signaling. The CD38-NAD+-SIRT1 axis is well-defined in CD8+ and CD4+ T cell cytotoxicity and antitumor efficacy.25,27 Hence, we hypothesized that the observed effect of the CD38 inhibitor on CAR-T cell exhaustion and cell state was attributable to CD38-NAD+-SIRT1 signaling (Figure 6A). The total NAD+ level and ratio of NAD+ to NADH were significantly increased in the 78C-treated group (Figures 6B and 6C). SIRT1, an NAD+-dependent protein deacetylase, acts as an epigenetic modulator of key transcription factors that regulate immune cell functions.53 Augmented NAD+ levels led to increased SIRT1 expression (Figures 6D and 6E) and 4-fold higher SIRT1 deacetylase activity in 78C-pretreated CAR-T cells (Figure 6F). Activating of SIRT1 activity by small-molecule SRT210454 phenocopied the effect of CD38 inhibition in CAR-T cells, which increases memory-like differentiation and decreases the expression of exhaustion-related receptors without compromising their apoptotic susceptibility (Figures S10A–S10C).
Figure 6.

CD38-NAD+-SIRT1 signaling is enhanced upon CD38 inhibition in CAR-T cells
(A) Schematic of CD38-NAD-SIRT1-HIF1a signaling.
(B and C) Total NAD+ level and the ratio of NAD+ to NADH in control or 78C-treated CAR-T cells after coculture with NALM6 cells (n = 4 biological replicates).
(D and E) Western blot analysis of SIRT1 (D) and normalized SIRT1 expression relative to ACTIN in each group (E). CAR-T cells were treated with DMSO/78C for 3 days after coculture with NALM6 cells. Quantitative analysis of western blot data obtained in n = 5 experiments from three donors is shown, normalized to β-actin.
(F) SIRT1 activity in control or 78C-treated CAR-T cells after coculture with NALM6 cells (n = 3 biological replicates).
(G) mRNA level of HIF1A transcription factor in control or 78C-treated CAR-T cells after coculture with NALM6 cells (n = 3 biological replicates).
(H and I) Cell lysates from control or 78C-treated CAR-T cells were subjected to immunoprecipitation with a HIF1A antibody followed by western blot analysis with lysine acetylation or HIF1A antibodies. Data are presented as the means ± SD of 6 technical replicates from three different donors.
(J) Frequency of naive cells, central memory cells, effector memory cells, and effector cells in CAR-T cells treated with DMSO, 10 μM 78C, or 10 μM 78C plus 100 μM IOX2 (n = 3 biological replicates).
(K) Frequency of CD69+ CAR-T cells in each group (n = 3 biological replicates).
(L) Frequency of LAG-3+, TIM-3+, and PD-1+ CAR-T cells in each group (n = 3 biological replicates).
(M) mRNA level of glycolysis-related transcription factors in each group of CAR-T cells (n = 3 technical replicates).
The deacetylase activity of SIRT1 modulates the acetylation/deacetylation status of various transcription factors involved in the regulation of key cellular responses.55 Emerging evidence indicates that SIRT1 directly deacetylates and inactivates HIF1A,56 directing cell differentiation and metabolic reprogramming.57,58 Therefore, we investigated whether CD38 inhibition affected the acetylation level of HIF1A by performing an intracellular staining assay on the control and 78C-treated CAR-T cells (Figure 6G). CD38 inhibition decreased global expression of HIF1A (Figure 6G). Immunoprecipitated HIF1A from CD38-inhibited CAR-T cells displayed lower levels of acetylation compared to that of control CAR-T cells (Figures 6H and 6I). To confirm the role of HIF1A in CAR-T phenotypic reprogramming, one of the PHD2 inhibitors, IOX2, was used to promote HIF1A stabilization and increase its expression. The changes in CAR-T cell phenotype by 78C, including increased Tn and Tcm subset frequencies (Figure 6J), decreased expression of the activation receptor, CD69 (Figure 6K), and exhausted inhibitory receptors LAG-3, TIM-3, and PD-1 (Figure 6L) were partially restored by IOX2. IOX2 also reversed 78C-mediated reduction in glycolysis by re-evaluating the transcripts of glycolytic enzyme genes (ALDOA, ALDOC, HK2, TPI, etc.) (Figure 6M). In summary, these data indicate that the increased NAD+ levels in CD38-inhibited CAR-T cells activate SIRT1 expression and deacetylase activity, which in turn decreases the acetylation status of HIF1A to inactivate it and repress glycolysis.
As small-molecule CD38 inhibitors are not approved for human applications, therapeutic anti-CD38 antibodies are currently being evaluated in large-scale clinical trials.59 To investigate their potential impact on CAR-T cells, we tested daratumumab and isatuximab in vitro. However, both antibodies did not significantly influence CAR-T cell differentiation, activation, and exhaustion state or have a pronounced impact on cytotoxicity ability (Figures S10D–S10G). Treatment with the monoclonal antibodies led to only a slight increase in intracellular NAD+ levels in CAR-T cells, which was significantly lower compared to the effect observed with CD38 small-molecule inhibitors (Figure S10H).
Discussion
The remarkable success of adoptive T cell therapy is accompanied by a plausible obstacle, namely, relapse. Given the therapeutic inadequacy of CAR-T cells, attributable to exhaustion and short persistence, pivotal genes associated with CAR-T cell exhaustion have emerged as a new research trend. In the current study, we identified CD38 as a membrane hallmark of CAR-T cell exhaustion and extended this finding by demonstrating that inhibition of CD38 enzyme activity or CD38 knockdown reversed tonic signaling- or tumor antigen-induced CAR-T cell exhaustion and terminal differentiation state independent of the CAR construct. Such phenotypic improvement boosts CAR-T cell cytotoxic ability and, therefore, contributes to antitumor response enhancement in vivo.
The function of CD38 in T cells has been widely delineated.60 Recent studies have demonstrated that elevated levels of CD38 reduce CD8+ T cell cytotoxicity and increase susceptibility to infections in patients with systemic lupus erythematosus.25,61 In the context of chronic viral infections, CD38 plays a cell-intrinsic role in virus-specific exhausted T cells through its interplay with NAD+ or metabolic regulation.62,63,64 However, its role in CD8+ T cell antitumor immunotherapy is not completely understood, and different studies have reported controversial conclusions. Chatterjee et al. reported that Th1/Th17 T cells featuring diminished expression of CD38 displayed heightened glutamine metabolism and mitochondrial dynamics, which greatly enhanced their ability to restrain tumor growth.27 Depleting dysfunctional PD-1+CD38hi CD8+ cells relieved the anti-PD-1 therapeutic resistance in a mouse tumor model.65 Nonetheless, another study demonstrated that CD38 knockout could not rescue terminal-exhausted differentiation or increase the antitumor efficacy of CD8+ T cells in vivo.66 The incongruity among various investigations is possibly attributable to T cell heterogeneity, different tumor models, and experimental conditions. Consistent with the majority of these studies, we observed enhanced mitochondrial fitness, attenuated T cell cytotoxicity, and enhanced therapeutic efficacy upon CD38 inhibition in CAR-T cells.
We further investigated the regulatory mechanisms that drive CAR-T cell dysfunction. Using RNA-seq analysis, we found that CD38 inhibition resulted in glycolysis signaling suppression (Figure 4). Repressing the activity of glycolysis67 or its key metabolic regulators, such as PI3K,68,69 AKT,70 and mTOR,71 during the in vitro setting of antitumor T or CAR-T cells results in a less-exhausted and memory-like profile, yielding improved tumor clearance. Several factors are known to regulate glycolysis. Specifically, inhibition of intracellular Ca2+ signaling and its downstream NFAT reprograms intracellular glucose metabolism of T cells72 to alleviate the excessive activation of CAR-T cells and impede exhaustion,49 which is confirmed by our current study. CD38 is an NAD-glycohydrolase responsible for the formation of extracellular ADPR and intracellular cADPR from NAD substrate, as well as for the formation of NAADP from nicotinamide adenine dinucleotide phosphate (NADP) and nicotinic acid as substrates, respectively. ADPR, cADPR, and NAADP are all vital cellular messengers activating Ca2+ flux.45,46,47,73 We discovered that cADPR and ADPR, rather than NAADP, exhibited a noticeable decrease upon CD38 inhibition. 78C is reported to be a potent inhibitor of the hydrolase activity of CD38, and that it is 10-fold less potent against the cyclase activity. Since our mass spectrometry results revealed that both cADPR and ADPR were significantly reduced after 78C application, we assume that although there may be differences in degree, 10 μM of 78C exhibits inhibitory effects on both the hydrolase and cyclase activities of CD38. However, though ADPR has the potential to regulate intracellular Ca2+ levels, it didn’t exhibit any reversal effect on 78C-treated CAR-T cells like cADPR. We acknowledge the need for further detailed investigations into the pharmacokinetic properties of 78C in CAR-T cells. In addition, there might be other indirect downstream second messengers, apart from cADPR, involved in the regulation of CAR-T cell functionality. Among these, cGAMP might emerge as a highly promising second messenger involved in T cell function74 and T cell-mediated antitumor immunity.75 Further experiments are warranted to validate this hypothesis. Furthermore, other NAD+-consuming enzymes with cyclase activity including SARM1 and PARP1 are also worthy of consideration to regulate CAR-T cell function.
In addition to the CD38-cADPR-Ca2+ pathway, we found that CD38 inhibition positively regulates CD38-NAD+-SIRT1 signaling. CD38 is one of the main NAD-consuming enzymes in mammalian cells, and NAD+ plays a vital role in T cell metabolism27,76 and immune function.25 A CD38-related decline in NAD+ has been reported to be tightly associated with T cell function.61,77 Wang et al. reported that NAD+ precursor supplementation with CAR-T cells strongly enhanced the tumor-killing function in vivo.78 In addition, NAD+ is an essential cofactor for a series of key regulatory enzymes,79 including the SIRT family (HDACIII), PARP, SARM, etc. SIRT1 deacetylates histones80 and other key proteins, such as FOXO1,81 PGC1α, and EZH2,82,83 for signal modulation. SIRT1 has been reported to directly deacetylate and inactivate HIF1A, a key transcription factor that orchestrates aerobic glycolysis by regulating glycolytic enzyme expression.56 The SIRT1-HIF1A axis in immune cells directs cell differentiation and metabolic reprogramming.57,58,84 In our study, we found that CD38 inhibition significantly increased NAD+ levels and upregulated SIRT1 expression and activity. HIF1A expression was downregulated, and its acetylation level significantly decreased after CD38 inhibition, which was consistent with repressed glycolytic metabolism. We applied the pyruvate dehydrogenase inhibitor, IOX2, to stabilize HIF1A, after which a series of CAR-T cell phenotypes were rescued, thus demonstrating the important role of CD38/SIRT1-HIF1A in the functional impact of CAR-T cells.
As a widely used CD38 monoclonal antibody, daratumumab is reported to partially influence the cyclase enzymatic function of CD38, whereas isatuximab does so with greater potency in tumor cells.85 However, no beneficial effect was seen on CAR-T cells after either CD38 antibody treatment. Although daratumumab is reported to deplete CD38+ immune regulatory cells and promote T cell expansion,60 there is currently no literature reporting a direct effect of CD38 monoclonal antibodies on CD8 T cell function, which may be attributed to the design of CD38 monoclonal antibodies. On the other hand, enzymatic potency of CD38 monoclonal antibody may be cell type specific. CD38 monoclonal antibodies are large protein molecules that cannot directly enter cells like small molecules; thus they may not directly influence intracellular NAD+ in CAR-T cells to regulate second messenger metabolism.
Limitations of the study
The present study has several limitations. First, the small clinical sample size was used for the analytical methods, and the limited scope of CAR-T types evaluated necessitates an expansion of the clinical sample size to confirm the correlation between CD38 expression and cell exhaustion during the CAR-T production and in vivo expansion phases. As for the in vitro model, tonic signaling is achieved by long-term culture and was confirmed by terminated T cell differentiation, overactivation, and exhaustion10 in our system. Although relatively mild, CD19-41BBz CAR-T cells still exhibit tonic signaling.86,87,88 However, recent research has reported that not all tonic signals are harmful to CAR-T activity, and CD19-41BBz CAR-T may require CAR clustering and tumor-independent activation for its persistence and effective function.34,86 Since CD19− exhaustion-inducing tonic signaling in 41BBz CAR-T cells is still under investigation, GD2-CD28z CAR is the better model for studying tonic signaling-induced exhaustion currently. Second, given that none of the CD38 inhibitors are FDA approved and can only be applied in vitro during CAR-T cell cultivation, the effects of small-molecule inhibitors on tumors and the host’s immune system in vivo warrant further investigation. Third, the current strategy for screening small molecules targeting CD38 is relatively imprecise. In the future, it is essential to propose a rational framework for identifying the optimal CD38 inhibitor. Two points that are worth considering include the type of enzymatic activity that small molecules primarily target (hydrolytic or cyclizing activity) and the enzymatic inhibition characteristics of small molecules (competitive or non-competitive). Fourth, it is imperative to acknowledge the inherent complexity of cellular regulatory networks in vivo. In addition to tumor antigen stimulation, the tumor microenvironment is influenced by various factors including metabolites and other soluble small molecules such as adenosine, indoleamine 2,3-dioxygenase (IDO), and extracellular vesicles.89 More general exhaustion models should be considered in further study. Finally, though we have proposed two mechanisms underlying cell exhaustion alleviation and CAR-T cell efficacy enhancement, delving deeper into the interplay between these signals is still worth investigating.90,91 Additional studies are required to further validate the two mechanisms. In summary, this study identified CD38 as a critical regulator of CAR-T cell effector differentiation and exhaustion. The pharmacological inhibition of CD38 enzyme activity or CD38 knockdown induces broad functional enhancements in CAR-T cells, including enhanced CAR-T cell-specific cytotoxicity and superior therapeutic efficacy. Our study also provides a deeper understanding of how CD38 mediates CAR-T cell exhaustion through two potential mechanisms.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human CD3-PE-Cy7 | BioLegend | Cat#300420, RRID: AB_439781 |
| APC/Cyanine7 Mouse IgG1, κ isotype Ctrl | Biolegend | Cat#400128; RRID: AB_2892538; RRID |
| APC Mouse IgG1, κ isotype Ctrl | Biolegend | Cat#400120; RRID: AB_2888687 |
| PE/Cyanine7 Mouse IgG1, κ isotype Ctrl | Biolegend | Cat#400126; RRID: AB_2861433 |
| Brilliant Violet 605™ Mouse IgG1, κ Isotype Ctrl | Biolegend | Cat#400162; RRID: AB_11125373 |
| FITC Mouse IgG1, κ Isotype Ctrl | Biolegend | Cat#400108; RRID: AB_326429 |
| Brilliant Violet 421™ Mouse IgG1, κ Isotype Ctrl | Biolegend | Cat#400158; RRID: AB_11150232 |
| PE Mouse IgG1, κ isotype Ctrl | Biolegend | Cat#400112; RRID: AB_3076354 |
| anti-human CD69-FITC | BioLegend | Cat#310904; RRID: AB_314839 |
| anti-human PD1-APC | BioLegend | Cat#329908; RRID: AB_940475 |
| anti-human TIM-3-PE | BioLegend | Cat#345006; RRID:AB_2116576 |
| anti-human LAG-3-PE-Cy7 | BioLegend | Cat#369208; RRID: AB_2629834 |
| anti-human CD62L-PE | BioLegend | Cat#304806; RRID: AB_314465 |
| anti-human CD45RO-APC | BioLegend | Cat#304210; RRID: AB_314426 |
| anti-human IL-2-APC | BioLegend | Cat#500310; RRID: AB_315096 |
| anti-human TNFα-PE | BioLegend | Cat#502909; RRID: AB_315261 |
| anti-human Granzyme B-PE | BioLegend | Cat#372208; RRID: AB_2687031 |
| anti-human IFNγ-APC | BioLegend | Cat#502512; RRID: AB_315237 |
| anti-human Annexin-V-APC | BioLegend | Cat#640920; RRID: AB_2941659 |
| anti-human PD1-BV421 | BD Biosciences | Cat#564323; RRID: AB_10900818 |
| anti-human GLUT1 mAb | Abcam | Cat#ab115730; RRID: AB_10903230 |
| anti-human GLUT3 mAb | Abcam | Cat#ab191071; RRID: AB_2736916 |
| anti-human CD38 mAb | Abcam | Cat#ab108403; RRID: 10890803 |
| Chemicals, peptides, and recombinant proteins | ||
| 78C | Selleck | Cat#S8960 |
| RBN013209 | MCE | Cat#HY-144987 |
| Luteolinidin Chloride | Targetmol | Cat#TN1895 |
| 8-Bromo-cADP-Ribose (8-Br-cADPR) | SANTA | Cat#sc-201514A |
| cADP-Ribose (cADPR) | Sigma | Cat#C7344 |
| IOX2 | Selleck | Cat#S2919 |
| Adenosine 5′-diphosphoribose sodium (ADPR sodium) | MCE | Cat#HY-100973A |
| Nicotinic acid adenine dinucleotide phosphate sodium salt (NAADP sodium) | sigma | Cat#N5655 |
| Dehydronitrosonisoldipine | MCE | Cat#HY-Z0816 |
| DSRM-3716 | MCE | Cat#HY-W021879 |
| Ned19 | Targetmol | Cat#T12205 |
| Critical commercial assays | ||
| Cell Counting Kit-8 (CCK-8) | DOJINDO | Cat#CK04 |
| MitoProbe™ DiIC1(5) Assay Kit | Invitrogen | Cat#M34151 |
| Fluo-4 a.m. ester | Thermo Fisher | Cat#F14201 |
| RNA Isolation Kit | Qiagen | Cat#931636 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat#23225 |
| Cytoplasmic Extraction Reagents Kit | Thermo Scientific | Cat#78833 |
| ELISA kit | MLUTI SCIENCES | Cat#E2620 |
| Seahorse XF Glycolytic Stress Assay Kit | Agilent | Cat#103020-100 |
| Seahorse XF Mito Stress Assay Kit | Agilent | Cat#103015-100 |
| NAD+/NADH ratio Assay Kit | Beyotime | Cat#S0175 |
| Universal Sirt1 Activity Assay Kit | Abcam | Cat#ab156065 |
| by Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) ELISA Kit | MyBioSource | Cat#MBS2000162 |
| Deposited data | ||
| Bulk RNA-seq data in this paper | Genome Sequence Archive in China National Genomics Data Center | GSA:HRA004078 |
| scRNA-seq data | Gene Expression Omnibus | GEO: GSE197268 |
| scATAC-seq data | China National GeneBank DataBase (CNGBdb) | CNP0002442 |
| Experimental models: Cell lines | ||
| 293T cell line | ATCC | Cat#CRL-1573 |
| Nalm6 cell line | ATCC | Cat#CRL-3273 |
| 143B cell line | ATCC | Cat#CRL-8303 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD-SCID-Il2rg−/− (NSG) | Shanghai Model Biological Center | N/A |
| Oligonucleotides | ||
| Primers | See Table S1 for primers and RNA sequences. | N/A |
| Recombinant DNA | ||
| Anti-CD19 scFv (FMC63) | Auther W. Hohman lab | N/A |
| Anti-GD2 scFv (14g2a-E101K) | Carl June lab | N/A |
| pLV-shRNA-CAR | this manuscript | N/A |
| Software and algorithms | ||
| Signac 1.10.0 | https://doi.org/10.1038/s41592-021-01282-5 | https://stuartlab.org/signac/ |
| FlowJo 10.0 | FlowJo, LLC | https://www.flowjo.com/ |
| Graphpad Prism version 10.0 | GraphPad software | https://www.graphpad.com |
| ChIPseeker R package | https://doi.org/10.1002/cpz1.585 | https://bioconductor.org/packages/ChIPseeker/ |
| R version 4.2.0 | R Core Team 2017 | https://www.r-project.org |
| Seraut | Rahul Satija Lab | https://github.com/satijalab/seurat |
| GO | GO Consortium | http://geneontology.org/ |
| GSEA | Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| Clusterprofiler | Guangchuang Yu lab | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| HISAT2 version 2.0.4 | https://doi.org/10.1038/nmeth.3317 | http://www.ccb.jhu.edu/software/hisat/ |
| StringTie version 1.3.4days | https://doi.org/10.1038/nbt.3122 | https://github.com/gpertea/stringtie |
| edgeR | https://doi.org/10.1093/bioinformatics/btp616 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, He Huang (huanghe@zju.edu.cn).
Materials availability
This study did not generate new materials.
Data and code availability
-
(1)
Single-cell RNA-seq data from the previous study (Haradhvalal et al. 2022, Nat Med) are publicly available at Gene Expression Omnibus with GEO accession GSE197268.
-
(2)
Single-cell ATAC-seq data from the previous study (Jiang et al., 2022, Leukemia) are publicly available at CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0002442 (db.cngb.org/search/project/CNP0002442).
-
(3)
Bulk RNA-seq data have been deposited in Genome Sequence Archive in National Genomics Data Center, GSA: HRA004078, publicly accessible at https://ngdc.cncb.ac.cn/gsa.
-
(4)
All the original code is available at Zenodo (https://doi.org/10.5281/zenodo.8403894).
-
(5)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines and Cell culture
The 293T cell line (ATCC, CRL-1573), NALM6 cell line (ATCC, CRL-3273), and 143B cell line (ATCC, CRL-8303) were purchased from ATCC. Cell lines were stably transfected with GFP and firefly luciferase when required for certain experiments. NALM6 and luciferase-GFP-NALM6 cell lines were cultured in RPMI-1640 (Corning) supplemented with 10% fetal bovine serum (FBS, Corning). 293T cells were cultured in DMEM (Corning) containing 10% FBS (Corning). Prior to in vivo experiments, cells were periodically tested for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza) and determined to be negative. Small molecule drugs including compound 78C (10μM, Selleck, S8960), RBN013209 (50μM, MCE, HY-144987), Luteolinidin Chloride (10μM, Targetmol, TN1895), 8-Bromo-cADP-Ribose (8-Br-cADPR) (8uM, SANTA, sc-201514A), cADP-Ribose (cADPR) (30μM, Sigma, C7344), IOX2 (100μM, Selleck, S2919), Adenosine 5′-diphosphoribose sodium (ADPR sodium) (500μM, MCE, HY-100973A), Nicotinic acid adenine dinucleotide phosphate sodium salt (NAADP sodium) (100μM, sigma, N5655), Dehydronitrosonisoldipine (3uM, MCE, HY-Z0816), DSRM-3716 (30μM, MCE, HY-W021879) and Ned19 (20μM, 50uM, Targetmol, T12205).
Mice and in vivo studies
NSG mice were purchased from the Shanghai Model Biological Center. Briefly, 6- to 8-week-old female mice were randomly assigned to each treatment group and no mice were excluded prior to CAR-T cell treatment. For leukemic model, 1 × 106 Luciferase-GFP-NALM6 cells were injected into each group via tail vein and 1–1.5 × 106 CAR-T cells were inoculated intravenously 5 days later. After CAR-T cell injection, the tumor burden progression was measured weekly by bioluminescence imaging (BLI) using the IVIS imaging system (IVIS Luminu III, Perkin-Elmer). Living Image software (Perkin-Elmer) was utilized to analyze the data. Mice were humanely euthanized when they showed signs of morbidity and/or hindlimb paralysis. 8 days after CAR-T cells injection, spleen and bone marrow tissues were collected and analyzed by flow cytometry. As for osteosarcoma solid tumor model, NSG mice received 1 × 106 143B cells via right hind leg intramuscular injection on day −5. On Day 0, 1 × 107 nontransduced T cells (NT) or CAR-T cells were incubated via tail vein. Tumor area was assessed every 2-5 days to monitor tumor burden using caliper.
Method details
Study design
Our objective of this research was to determine the functional role of CD38 inhibitions in CART cells exhaustion and therapeutic efficacy. The peripheral blood samples were acquired from two patients with multiple myeloma treated with anti-BCMA CAR-T cells in one clinical trial study, which was approved by The First Affiliated Hospital of Zhejiang University, China.30 The number of biological replicates (referring to T cells from different healthy donors), type of statistical methods used, and p values are reported in the figure legends. in vitro experiments were carried out at least three times with no data points excluded from analyses. As to in vivo experiments, NSG mice were treated with CART cells from two healthy donors in two independent experiments. Investigators were not blinded to group allocation during experiments and assessment of outcomes.
CAR structure design and lentivirus production
CD19-41BB/28ζ and GD2-28ζ CAR were constructed of a single-chain variable fragment (scFv) from Clone FMC6392 and 14g2a-E101K11 respectively, followed by CD8 hinge, the 4-1BB/CD28 costimulatory domain and the CD3ζ intracellular region. The ScFv was inserted in EF-1a-P2A-mCherry vector (Figures S1K and S1L). CD19-41BB/28ζ and GD2-28ζ CAR lentiviral supernatant were produced by 293T cell line transfection as us previously described.93 Briefly, we co-transfected 7.5 μg CAR plasmids, and 5.625 μg psPAX2, 1.875 μg pMD2.G enveloping plasmids into 293T cells using PEI (Polysciences) at the confluence of 60% in 10-cm plates. We harvested the virus supernatant at 48h and 72h and concentrated the lentivirus supernatant by ultracentrifugation (Beckman) at 25000rpm for 2 h. The concentrated virus was stocked at −80°C for future use.
Primary human T cell isolation and CAR-T cell production
We obtained healthy volunteer’s peripheral blood mononuclear cells (PBMC) from the Institute of Hematology Zhejiang University (Hangzhou, China) with informed consent. Primary human T cells were isolated and stimulated using anti-CD3/CD28 beads (Life Technologies) at a bead-to-cell ratio of 3:1. 2 × 106 T cells were transfected with condensed virus supernatant at the MOI (multiplicity of infection) of 2 in RPMI 1640 with 10% FBS (corning) and 200 IU mL−1 IL-2 (Peptech). We added 6ug polybrene to increase infection efficiency and detected the infection efficiency of CAR after 72 h.
Cell proliferation assays
Cell Counting Kit-8 (CCK-8; DOJINDO, CK04) is used for cell proliferation assay. Briefly, flat-bottomed 96-well plates were inoculated in triplicate with 2000 cells/100 μL. Dye solution was added to each well and incubated at 37°C for 3–4 h. The absorbance was then measured at 450 nm using a microplate reader.
Flow cytometry
Cells were harvested and stained according to related protocols. All phenotypic FACs results of CAR-T cells are based on a gating strategy for mCherry/GFP-positive CAR-T cells. Positive cell populations were determined using isotype antibody (IGg1 k, Clone MOPC-21; Biolegend). The FACs data were analyzed by CytoFLEX (Beckman) and data were analyzed using FlowJo software. Beckman Moflo Astrios EQ (Beckman) was used for flow sorting.
Intracellular cytokines staining
After CAR-T cell culture or coculture with target cells at different effector-to-target (E:T) ratio, 1× monensin (eBioscience, # 00-4505-51) was added in culture medium for 6 h to allow for intracellular cytokines enrichment. Intracellular proteins were fixed, permeabilized and stained for 40min using a Foxp3/Transcription Factor Staining Buffer Set (eBioscience, #00-5523-00) according to the instructions.
Glucose uptake
Glucose uptake ability was assessed with uptake of the fluorescent analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol4-yl)amino]-2-deoxy-D-glucose (2-NBDG, Invitrogen, N13195). After cells were collected and suspended in RPMI 1640, 2-NBDG was introduced for 20–30 min at a concentration. Each tube was then removed from the incubator and washed twice with PBS. The mean fluorescence intensity of 2-NBDG uptake was determined by flow cytometry.
Cellular ROS level measurement
To measure cellular ROS levels, cells were loaded with a chloromethyl derivative of H2DCFDA 2′,7′-Dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Invitrogen, C6827) (2 μM) at 37 C for 15 min. ROS (H2O2) levels in the gated CAR-T cell population were quantified using flow cytometry.
Mitochondrial permeability transition pore assay
Mitochondrial permeability transition pore (MPTP) activity was analyzed through calcein-AM and CoCl2. In brief, 1x10E6 CAR-T cells from different experimental groups were collected and washed with PBS and incubated with calcein-AM in 1mL detection buffer. CoCl2 was then added and incubated for 30 min at 37 C to quench the cytosolic calcein. Cells were finally washed twice with PBS, and the intracellular calcein fluorescence intensity was measured by flow cytometry.
Mitochondrial membrane potential assay
We assess the mitochondrial membrane potential using MitoProbe DiIC1(5) Assay Kit (Invitrogen, M34151). The mitochondrial membrane potential was measured using DiIC(5) staining according to the manufacturer’s instructions. CCCP treatment was used as a positive control for depolarization.
FACS analysis of cytosolic calcium
Cytosolic Calcium was analyzed using Fluo-4 a.m. ester (Thermo Fisher F14201) according to the product manuals. Briefly, CAR-T cells were collected and stained with indicators for 15–60 min at 20°C–37°C. After washing with indicator-free medium, cells were further incubated for another 30 min to allow complete de-esterification of intracellular AM esters. The mean fluorescence intensity of bound Fluo-4 was determined using CytoFLEX (Beckman).
FACS analysis of CAR expression
Mouse-derived CAR expression was analyzed using Goat Anti-Mouse IgG, F(ab')2 fragment primary antibody (Jackson,#115-066-006) and streptavidin-PE secondary antibody (BD, 554061). Briefly, CAR-T cells were collected and stained with primary antibody for 20 min at room temperature. After washing with cold PBS, cells were further incubated with streptavidin-PE for another 20 min. The positive subset was determined using CytoFLEX (Beckman).
Quantitative real-time PCR
We extracted mRNA from different experiment cell groups using an RNA Isolation Kit (Qiagen) according to the instruction manual. cDNA reverse transcription was performed with Super Script First-Strand Synthesis System (Life Technologies). All reactions were performed on an Applied Biosystems Step One Plus real-time PCR machine with TaqMan Fast Universal PCR Master Mix (Applied Biosystems).
Western Blot
Whole-cell lysates were collected by lysing 5 × 106 in 100ul RIPA buffer (Beyotime) with the addition of protease inhibitors cocktail. Proteins were examined by Western blot analysis using the Pierce BCA Protein Assay Kit (Thermo Scientific, 23225). Cytoplasmic and nuclear proteins were generated using NE-PER Nuclear and Cytoplasmic Extraction Reagents kits (Thermo Scientific, 78833).
Luciferase-based cytolysis assay and Elisa assay
Luciferase-based cytotoxicity assays were used to measure the cytolytic ability of CAR-T cells. Approximately 2 × 104 target luciferase-expressing NALM6 cells were co-incubated with CAR-T cells for 24h or 72 h at different E:T ratios from 1:1 to 1:128 using black-walled 96-well plates. Triplicate wells were plated for each condition. Mixed cells were harvested by centrifugation and added to a Bright-Glo Luciferase Assay system (Promega, E2620) for 2 min to allow complete cell lysis, and cell viability was measured using a luminometer (SpectraMax iD5, Molecular Devices).
After incubation with NALM6 cells, the supernatant of each well was collected by centrifugation and analyzed using a human ELISA kit (MLUTI SCIENCES, China, IL-2 #EK101HS, IFN-γ #EK180HS, TNF-α #EK182HS, Granzyme B #E-EL-H1617c). ELISAs were performed using purified antiIL-2, anti-IFN-γ, anti-TNF-α, and anti-Granzyme B mAbs as capture Abs; the corresponding biotinylated anti-IL-2, anti-IFN-γ, anti-TNF-α, and anti-Granzyme B mAbs; HRP-conjugated streptavidin (Sigma Aldrich); and tetramethylbenzidine microwell peroxidase substrate and stop solution (Kirkegaard and Perry Laboratories) according to the manufacturer’s instructions.
Multiple rounds of tumor cell challenge
2 × 103 CAR-T cells were co-cultured with NALM6-luci at the E: T ratio of 1:10 in clear 96-well plates. Triplicate wells were plated for each condition. After co-culture for 24 h, the remaining CAR-T cells were added with fresh tumor cells at a 1:10 E:T ratio for 24 h. By the analogy, the experiment was performed for 3 rounds in which CAR-T cells almost lost cytolysis ability. The percentage of specific lysis was evaluated using luciferase-based cytolysis assay after every round co-culture.
Transmission electron microscope analysis
Each sample was incubated in 2% paraformaldehyde (PFA, EMS), 2.5% glutaraldehyde (GA, EMS) in 0.1 M Sodium Cacodylate (EMS) buffer, pH 7.4 at RT for 30 min. Then 100 μL of cell suspension was embedded by centrifugation (5 min at 1230 g at 30°C) in 100 μL 2% Low Melting Agarose (LMA, Gibco BRL) in 0.1 M cacodylate buffer. Then warm the sample in 2% osmium tetroxide (OsO4, EMS), 1.5% potassium ferricyanide (EMS) in 0.1 M cacodylate buffer for 60 min at RT followed by incubation in 1% thiophenylhydrazine (TCH, EMS) for 20 min. After wash of TCH, incubate for a second time in OsO4 (2% OsO4 in H2O) for 30 min at RT and wash. The samples were incubated overnight at 4°C in uranyl acetate substitute (UA, EMS) in H2O (1:3). Remove UAR the next day and add Walton’s lead solution at 60°C for 30 min. Dehydrate the sample with EtOH (70%, 90% and 2 × 100%) for 5 min respectively, followed by propylene oxide (Aldrich) for 2 × 10 min. Subsequently, they were infiltrated with resin (Spurr, EMS) and incubated in 50% propylene oxide resin for 2 h, followed by at least 3 changes of fresh 100% resin. For FIB-SEM imaging, the embedded cells were mounted on an aluminum SEM stub (12 mm diameter) and the samples were coated with ∼8 nm Platinum (Quorum Q150T ES). We performed FIB-SEM imaging using a Zeiss Crossbeam 540 system and Atlas 5 software. The focused ion beam (FIB) was set to remove a 5 nm fraction by advancing gallium ions on the surface. Imaging was performed at 1.5 kV using an ESB (backscattered electron) detector.
mRNA library construction and sequencing
Total mRNA was extracted from CAR-T cells in each group using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Then Poly (A) RNA was purified using Dynabeads Oligo (dT)25–61005 (Thermo Fisher) through two rounds of purification. Next the poly(A) RNA was fragmented into pieces with the Magnesium RNA Fragmentation Module (NEB, e6150). The cleaved RNA fragments were reverse-transcribed and next used to synthesize U-labeled second-stranded DNAs. An A-base was subsequently added to the blunt ends of each strand. Each adapter contained a T-base overhang to ligate the adapter to the A-tailed fragmented DNA. Single- or dual-index adapters were ligated to the fragments. After the heat-labile UDG enzyme (NEB, m0280) treatment of the U-labeled second-stranded DNAs, the ligated products were amplified by PCR. The theaverageinsert size of the final cDNA library was 300 ± 50 bp. Finally, the 2 × 150 bp paired-end sequencing (PE150) was performed on an Illumina Novaseq 6000 (LC-Bio Technology Co., Ltd.)
Sequence Alignment and gene expression analysis
Sequenced reads were aligned to the human reference genome (GRCh38) using the HISAT2 software (version 2.0.4).94 StringTie (v 1.3.4days. Linux_x86_64)95 was used to estimate the expression levels of all transcripts. The edgeR package (v 3.26.8) in R (v 4.2.0) was used to perform PCA and differential expression analysis. The DEGs with absolute log2(fold change) > 1.0 and p value <0.05, were considered as significant. GO annotation, GSEA and GSVA were carried out using the clusterProfiler package (v 3.12.0)96 with default settings. The gene signatures used were consistent with previous research38 as follows: T cell differentiation,97,98 T cell exhaustion,99 T cell activation100 and hypoxia.101 As for metabolic gene profiles, ‘CP:REACTOME: Reactome gene sets’ and ‘GO biological process sets’ were also selected: http://www.broadinstitute.org/gsea/msigdb/.
scATAC-seq data and scRNA-seq data analysis
The method of scATAC-seq analysis is the same as that we described before.30 The Seurat function FindAllMarkers was used to identify the cell-type specific chromatin accessible regions (peaks) with thresholds: logfc >0.5, minimum cells per group = 50, adjusted p value <0.05. Peak sets were annotated to the nearest genes by R package ChIPseeker102 function annotatePeak (promoter region defined as TSS ±3 kb).
The method of scRNA-seq analysis is the same as that we described before.103 Anchors were identified using FindIntegrationAnchors with 1–20 dimensions and 3,000 anchor features. Downstream analysis after integration included data feature scaling (ScaleData), principal-component analysis (PCA; RunPCA), and SNN (shared nearest neighbor) graph building (FindNeighbors). The Function FindAllMarkers was used to identify the cell-type-specific expressed genes.
Genomic track profile
The chromatin accessibility of particular genomic regions was depicted using the functional tool CoveragePlot. DNase-seq data and ChIP-seq data of histone modifications (H3K27ac and H3K4me1) were acquired from the ENCODE database for human primary T cells, with the identifiers ENCFF930DNG, ENCFF234VLS, ENCFF608TNF.104 The obtained DNase-seq and ChIP-seq data were then visualized utilizing the WashU Epigenome Browser and were integrated with the visual results of scATAC-seq using genomic coordinates.
Metabolic state analysis in CAR-T cells
Seahorse XF assays
CAR-T cells were resuspended in Seahorse XF Assay Medium (Agilent Technologies) and seeded at 6000 cells per well in a 96-well plate. The cell culture microplate (101085-004, Agilent) was precoated with Cell-Tak (354240, Corning) for 20 min at room temperature. The XF 96 sensor cartridge was hydrated with 200 μL of calibration buffer per well in a 37°C non-CO2 incubator overnight. The extracellular acidification rate (ECAR) was measured in real time in an XFe96 analyzer using a Seahorse XF Glycolytic Stress Assay Kit (103020-100, Agilent). ECAR measurements were performed under basal conditions followed by the sequential addition of 1.5 mM glucose, 1.5 mM oligomycin, and 50 microM 2-deoxy-D-glucose (2-DG). The oxygen consumption rate (OCR) was measured in real time in an XFe96 analyzer using a Seahorse XF Mito Stress Assay Kit (103015-100, Agilent). OCR measurements were performed under basal conditions followed by the sequential addition of 1.5 mM Oligomycin (OL), 0.5 mM FCCP, and 0.5 mM Rotenone and Antimycin A (ROT/AAz).
LC-MS/MS analysis of cADPR, ADPR and lactate
For sample preparation, CAR-T cells were treated with 10uM 78C for 72h and were harvested and washed with cold normal saline. The cell pellet was subjected to 20 cycles (30 s on and 30 s off for every cycle) of ultrasonic lysis with a mixture of water and acetonitrile at a ratio of 1:3. Subsequently, the sample was centrifuged at 12,000g for 30 min at 4°C, and the supernatant was collected for further analysis.
A Waters acuity UPLC I-class system coupling with Xevo TQ-S micro triple quadrupole mass spectrometer (Waters Corporation, USA) was used for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. Analytes (5 μL) were injected and separated on a Waters HSS T3 column (150 mm × 2.1 mm id, 1.8 μm), and the column temperature was maintained at 30°C. The mobile phase was set at a flow rate of 0.2 mL/min, and the gradient was programmed as follows: 0–1.0 min hold 1% A, 1.0–3.0 min to 5% A, 3.0–11.0 min to 90% A, 11.0–12.0 min to 1% A, and hold 1% A for 3 min afterward. In positive mode, the mobile phase A and B were acetonitrile and pure water with 0.1% formic acid, respectively. For negative mode, the mobile phase A was pure water with 0.1% formic acid 10 mM ammonium acetate, and the mobile phase B was acetonitrile. MS analysis was carried out in multiple-reaction monitoring (MRM) mode, and an external standard method was used for quantification. ADPR and cADPR were analyzed in positive ion mode, while the lactic acid was performed in negative mode. MRM transitions were monitored at 560.1 > 348, 666 > 649, 89 > 43, respectively, for ADPR, cADPR and lactic acid.
ELISA assay for NAADP level
Intracellular NAADP level was evaluated by Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) ELISA Kit (MyBioSource, MBS2000162). Briefly, 50μL standard or samples were added to each well. Then add 50μL prepared Detection Reagent A immediately. After shaking and mixing, incubate the plate for 1 h at 37°C and aspirate and wash 3 times. Next, add 100μL prepared Detection Reagent B. Incubate 30 min at 37°C and then aspirate and wash 5 times; Finally, add 90μL Substrate Solution and Incubate 10–20 min at 37°C; Add 50μL Stop Solution and read at 450 nm immediately.
Total NAD+ level and NAD+/NADH ratio measurement
Total NAD+ level and NAD+/NADH ratio were measured using the NAD+/NADH ratio Assay Kit (S0175, Beyotime), according to the manufacturer’s instructions.
SIRT1 activity assay
Sirt1 activity (Universal Sirt1 Activity Assay Kit, Abcam) is determined as per the manufacturer’s protocol. Briefly, SIRT Assay Buffer, SIRT Substrate, HDAC Inhibitor, and SIRT Co-factor were added to 10ug nuclear extracts from each group. The total volume was 50 μL/well. Each well is incubated at 37°C for 60–90 min. Add Capture Antibody to each well for 60 min and wash twice. Detection Antibody is then added for 30min and washed with wash buffer. Developer Solution and Stop Solution is next added and the absorbance is read on a microplate reader within 2–10 min at 450 nm.
Generation of CD38 knockdown CAR-T cells
The anti-CD19 CAR scFv described above was inserted in pSico-EF-1a-IRES-EGFP vector. The scramble and gene target shRNA were inserted in the multiple cloning sites co-expressed with the CAR expression vectors. The shRNA sequences included scramble shRNA: GCGCGATAGCGCTAATAAT, CD38 shRNA-1: 5’- GCATACCTTTATTGTGATCTA-3’, CD38 shRNA-2: 5’- CCAAAGTGTATGGGATGCTTT-3’. Plasmid-expressed firefly luciferase was constructed from pSico-EF-1a-IRESmCherry and pHIV-Luc-ZsGreen (Addgen).
Quantification and statistical analysis
Data are shown as mean ± standard deviation (SD). All in vitro experiments consisted of three or more biological or technical replicates per experimental group and are demonstrated as individual data points. As for in vivo studies, data consisted of more than five mice per group. No data were excluded from the statistical analysis. For comparisons between two groups, a two-tailed unpaired Student’s t-test was used. For comparisons between three or more groups, one-way ANOVA and Dunnett’s post hoc test were used as indicated. For in vivo experiments, overall survival was described by Kaplan-Meier curves, and log rank tests were used to compare differences in survival between groups. p-values <0.05 were considered the threshold of significance for all analyses. p-values were calculated for ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. Analyses were performed by Prism 8 (GraphPad) software.
Acknowledgments
We are grateful to Wenzhao Zhou from Liangzhu Laboratory who provided assistance with liquid chromatography-tandem mass spectrometry analysis. We are grateful to Xin Wei from Life Sciences Institute, Zhejiang University, who assisted in the data visualization. We are grateful to Xinghui Song, Yingying Huang, and Jiajia Wang from the Core Facilities, Zhejiang University School of Medicine, for the technical support.
Funding was provided by the National Key Research and Development Program of China (2022YFA1103500), National Natural Science Foundation of China (82130003, 82270234, 82270235, 82222003,92268117, and 82161138028), and Provincial Key R&D Program Project of Zhejiang (2021C03010 and Z24H080001).
Author contributions
Y.H., M.S., X.T., and X.S. contributed equally to this work. Y.H. and M.S. designed the study and wrote the manuscript. X.T. and X.S. analyzed and interpreted the data. L.W., B.C., and L.L. performed the experiments and analyzed and interpreted the data. X.W. and K.L. constructed the plasmid and performed the GD2-CD28z CAR lentivirus package. P.J. and Y.F. analyzed the scATAC-seq and RNA-seq data, respectively. Y.L.H. and Z.Z. assisted in construction of CD38 knockdown CAR-T cells. J.C. and M.Z. performed flow cytometric analysis. H.H., P.Q., and Y.X.H. conceived of and supervised the study. All authors read, edited, and agreed to the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: February 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101400.
Contributor Information
Yongxian Hu, Email: 1313016@zju.edu.cn.
Pengxu Qian, Email: axu@zju.edu.cn.
He Huang, Email: huanghe@zju.edu.cn.
Supplemental information
References
- 1.Sadelain M., Brentjens R., Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu Y., Wu Z., Luo Y., Shi J., Yu J., Pu C., Liang Z., Wei G., Cui Q., Sun J., et al. Potent Anti-leukemia Activities of Chimeric Antigen Receptor-Modified T Cells against CD19 in Chinese Patients with Relapsed/Refractory Acute Lymphocytic Leukemia. Clin. Cancer Res. 2017;23:3297–3306. doi: 10.1158/1078-0432.CCR-16-1799. [DOI] [PubMed] [Google Scholar]
- 3.Munshi N.C., Anderson L.D., Jr., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 4.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 5.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K., Bleakley M., Brown C., Mgebroff S., Kelly-Spratt K.S., et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N., et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Lancet O. CAR T-cell therapy for solid tumours. Lancet Oncol. 2021;22:893. doi: 10.1016/S1470-2045(21)00353-3. [DOI] [PubMed] [Google Scholar]
- 10.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynn R.C., Weber E.W., Sotillo E., Gennert D., Xu P., Good Z., Anbunathan H., Lattin J., Jones R., Tieu V., et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y., Si X., Shao M., Teng X., Xiao G., Huang H. Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells. J. Hematol. Oncol. 2022;15:38. doi: 10.1186/s13045-022-01255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Si X., Shao M., Teng X., Huang Y., Meng Y., Wu L., Wei J., Liu L., Gu T., Song J., et al. Mitochondrial isocitrate dehydrogenase impedes CAR T cell function by restraining antioxidant metabolism and histone acetylation. Cell Metab. 2024;36:176–192.e10. doi: 10.1016/j.cmet.2023.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Alfei F., Kanev K., Hofmann M., Wu M., Ghoneim H.E., Roelli P., Utzschneider D.T., von Hoesslin M., Cullen J.G., Fan Y., et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265–269. doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 15.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X., Zhang C., Qiao M., Cheng C., Tang N., Lu S., Sun W., Xu B., Cao Y., Wei X., et al. Depletion of BATF in CAR-T cells enhances antitumor activity by inducing resistance against exhaustion and formation of central memory cells. Cancer Cell. 2022;40:1407–1422.e7. doi: 10.1016/j.ccell.2022.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Liu X., Wang Y., Lu H., Li J., Yan X., Xiao M., Hao J., Alekseev A., Khong H., Chen T., et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567:525–529. doi: 10.1038/s41586-019-0979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Good C.R., Aznar M.A., Kuramitsu S., Samareh P., Agarwal S., Donahue G., Ishiyama K., Wellhausen N., Rennels A.K., Ma Y., et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell. 2021;184:6081–6100.e26. doi: 10.1016/j.cell.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freitas K.A., Belk J.A., Sotillo E., Quinn P.J., Ramello M.C., Malipatlolla M., Daniel B., Sandor K., Klysz D., Bjelajac J., et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science. 2022;378 doi: 10.1126/science.abn5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D., Prager B.C., Gimple R.C., Aguilar B., Alizadeh D., Tang H., Lv D., Starr R., Brito A., Wu Q., et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021;11:1192–1211. doi: 10.1158/2159-8290.CD-20-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jing R., Scarfo I., Najia M.A., Lummertz da Rocha E., Han A., Sanborn M., Bingham T., Kubaczka C., Jha D.K., Falchetti M., et al. EZH1 repression generates mature iPSC-derived CAR T cells with enhanced antitumor activity. Cell Stem Cell. 2022;29:1181–1196.e6. doi: 10.1016/j.stem.2022.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malavasi F., Deaglio S., Funaro A., Ferrero E., Horenstein A.L., Ortolan E., Vaisitti T., Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 23.Aksoy P., White T.A., Thompson M., Chini E.N., Chini E.N. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 2006;345:1386–1392. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 24.Chini C.C.S., Peclat T.R., Warner G.M., Kashyap S., Espindola-Netto J.M., de Oliveira G.C., Gomez L.S., Hogan K.A., Tarragó M.G., Puranik A.S., et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020;2:1284–1304. doi: 10.1038/s42255-020-00298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katsuyama E., Suarez-Fueyo A., Bradley S.J., Mizui M., Marin A.V., Mulki L., Krishfield S., Malavasi F., Yoon J., Sui S.J.H., et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep. 2020;30:112–123.e4. doi: 10.1016/j.celrep.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Arena G., Musto P., Cascavilla N., Dell'Olio M., Di Renzo N., Perla G., Savino L., Carotenuto M. CD38 expression correlates with adverse biological features and predicts poor clinical outcome in B-cell chronic lymphocytic leukemia. Leuk. Lymphoma. 2001;42:109–114. doi: 10.3109/10428190109097682. [DOI] [PubMed] [Google Scholar]
- 27.Chatterjee S., Daenthanasanmak A., Chakraborty P., Wyatt M.W., Dhar P., Selvam S.P., Fu J., Zhang J., Nguyen H., Kang I., et al. CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018;27:85–100.e8. doi: 10.1016/j.cmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hua S., Lécuroux C., Sáez-Cirión A., Pancino G., Girault I., Versmisse P., Boufassa F., Taulera O., Sinet M., Lambotte O., Venet A. Potential role for HIV-specific CD38-/HLA-DR+ CD8+ T cells in viral suppression and cytotoxicity in HIV controllers. PLoS One. 2014;9 doi: 10.1371/journal.pone.0101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woan K.V., Kim H., Bjordahl R., Davis Z.B., Gaidarova S., Goulding J., Hancock B., Mahmood S., Abujarour R., Wang H., et al. Harnessing features of adaptive NK cells to generate iPSC-derived NK cells for enhanced immunotherapy. Cell Stem Cell. 2021;28:2062–2075.e5. doi: 10.1016/j.stem.2021.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang P., Zhang Z., Hu Y., Liang Z., Han Y., Li X., Zeng X., Zhang H., Zhu M., Dong J., et al. Single-cell ATAC-seq maps the comprehensive and dynamic chromatin accessibility landscape of CAR-T cell dysfunction. Leukemia. 2022;36:2656–2668. doi: 10.1038/s41375-022-01676-0. [DOI] [PubMed] [Google Scholar]
- 31.Philip M., Fairchild L., Sun L., Horste E.L., Camara S., Shakiba M., Scott A.C., Viale A., Lauer P., Merghoub T., et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haradhvala N.J., Leick M.B., Maurer K., Gohil S.H., Larson R.C., Yao N., Gallagher K.M.E., Katsis K., Frigault M.J., Southard J., et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 2022;28:1848–1859. doi: 10.1038/s41591-022-01959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R., et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 34.Chen J., Qiu S., Li W., Wang K., Zhang Y., Yang H., Liu B., Li G., Li L., Chen M., et al. Tuning charge density of chimeric antigen receptor optimizes tonic signaling and CAR-T cell fitness. Cell Res. 2023;33:341–354. doi: 10.1038/s41422-023-00789-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarragó M.G., Chini C.C.S., Kanamori K.S., Warner G.M., Caride A., de Oliveira G.C., Rud M., Samani A., Hein K.Z., Huang R., et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018;27:1081–1095.e10. doi: 10.1016/j.cmet.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shambharkar P., Blackwell D.J., Vasbinder M.M., Schenkel L.B., Kunii K., Lemera J.L., Kuplast-Barr K.G., Ren Y., Bamberg E., Church W.D., et al. Abstract 1344: Small molecule inhibitor of CD38 modulates its intra- and extracellular functions leading to antitumor activity. Cancer Res. 2021;81:1344. [Google Scholar]
- 37.Boslett J., Hemann C., Zhao Y.J., Lee H.C., Zweier J.L. Luteolinidin Protects the Postischemic Heart through CD38 Inhibition with Preservation of NAD(P)(H) J. Pharmacol. Exp. Therapeut. 2017;361:99–108. doi: 10.1124/jpet.116.239459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O'Connor R.S., Hwang W.T., et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung I.Y., Narayan V., McDonald S., Rech A.J., Bartoszek R., Hong G., Davis M.M., Xu J., Boesteanu A.C., Barber-Rotenberg J.S., et al. BLIMP1 and NR4A3 transcription factors reciprocally regulate antitumor CAR T cell stemness and exhaustion. Sci. Transl. Med. 2022;14:eabn7336. doi: 10.1126/scitranslmed.abn7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber E.W., Parker K.R., Sotillo E., Lynn R.C., Anbunathan H., Lattin J., Good Z., Belk J.A., Daniel B., Klysz D., et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372 doi: 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beltra J.C., Manne S., Abdel-Hakeem M.S., Kurachi M., Giles J.R., Chen Z., Casella V., Ngiow S.F., Khan O., Huang Y.J., et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity. 2020;52:825–841.e8. doi: 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kar A., Mehrotra S., Chatterjee S. CD38: T Cell Immuno-Metabolic Modulator. Cells. 2020;9 doi: 10.3390/cells9071716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sukumar M., Liu J., Mehta G.U., Patel S.J., Roychoudhuri R., Crompton J.G., Klebanoff C.A., Ji Y., Li P., Yu Z., et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016;23:63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horenstein A.L., Faini A.C., Malavasi F. CD38 in the age of COVID-19: a medical perspective. Physiol. Rev. 2021;101:1457–1486. doi: 10.1152/physrev.00046.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calcraft P.J., Ruas M., Pan Z., Cheng X., Arredouani A., Hao X., Tang J., Rietdorf K., Teboul L., Chuang K.T., et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y., Winkler P.A., Sun W., Lü W., Du J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature. 2018;562:145–149. doi: 10.1038/s41586-018-0558-4. [DOI] [PubMed] [Google Scholar]
- 47.Lee H.C., Deng Q.W., Zhao Y.J. The calcium signaling enzyme CD38 - a paradigm for membrane topology defining distinct protein functions. Cell Calcium. 2022;101 doi: 10.1016/j.ceca.2021.102514. [DOI] [PubMed] [Google Scholar]
- 48.Carty M., Kearney J., Shanahan K.A., Hams E., Sugisawa R., Connolly D., Doran C.G., Muñoz-Wolf N., Gürtler C., Fitzgerald K.A., et al. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity. 2019;50:1412–1424.e6. doi: 10.1016/j.immuni.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Shao M., Teng X., Guo X., Zhang H., Huang Y., Cui J., Si X., Ding L., Wang X., Li X., et al. Inhibition of Calcium Signaling Prevents Exhaustion and Enhances Anti-Leukemia Efficacy of CAR-T Cells via SOCE-Calcineurin-NFAT and Glycolysis Pathways. Adv. Sci. 2022;9 doi: 10.1002/advs.202103508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Z.Y., Xie X.J., Li W.H., Liu J., Chen Z., Zhang B., Li T., Li S.L., Lu J.G., Zhang L., et al. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience. 2019;15:452–466. doi: 10.1016/j.isci.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li W.H., Huang K., Cai Y., Wang Q.W., Zhu W.J., Hou Y.N., Wang S., Cao S., Zhao Z.Y., Xie X.J., et al. Permeant fluorescent probes visualize the activation of SARM1 and uncover an anti-neurodegenerative drug candidate. Elife. 2021;10 doi: 10.7554/eLife.67381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes R.O., Bosanac T., Mao X., Engber T.M., DiAntonio A., Milbrandt J., Devraj R., Krauss R. Small Molecule SARM1 Inhibitors Recapitulate the SARM1(-/-) Phenotype and Allow Recovery of a Metastable Pool of Axons Fated to Degenerate. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2020.108588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang T., Berrocal J.G., Frizzell K.M., Gamble M.J., DuMond M.E., Krishnakumar R., Yang T., Sauve A.A., Kraus W.L. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 2009;284:20408–20417. doi: 10.1074/jbc.M109.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W., Zhou X., Hou W., Chen E., Ye C., Chen M., Lu Q., Yu X., Li W. Reversing the imbalance in bone homeostasis via sustained release of SIRT-1 agonist to promote bone healing under osteoporotic condition. Bioact. Mater. 2023;19:429–443. doi: 10.1016/j.bioactmat.2022.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang T., Kraus W.L. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta. 2010;1804:1666–1675. doi: 10.1016/j.bbapap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim J.H., Lee Y.M., Chun Y.S., Chen J., Kim J.E., Park J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell. 2010;38:864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 57.Liu G., Bi Y., Shen B., Yang H., Zhang Y., Wang X., Liu H., Lu Y., Liao J., Chen X., Chu Y. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014;74:727–737. doi: 10.1158/0008-5472.CAN-13-2584. [DOI] [PubMed] [Google Scholar]
- 58.Liu G., Bi Y., Xue L., Zhang Y., Yang H., Chen X., Lu Y., Zhang Z., Liu H., Wang X., et al. Dendritic cell SIRT1-HIF1alpha axis programs the differentiation of CD4+ T cells through IL-12 and TGF-beta1. Proc. Natl. Acad. Sci. USA. 2015;112:E957–E965. doi: 10.1073/pnas.1420419112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Donk N.W.C.J., Richardson P.G., Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. 2018;131:13–29. doi: 10.1182/blood-2017-06-740944. [DOI] [PubMed] [Google Scholar]
- 60.Krejcik J., Casneuf T., Nijhof I.S., Verbist B., Bald J., Plesner T., Syed K., Liu K., van de Donk N.W.C.J., Weiss B.M., et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128:384–394. doi: 10.1182/blood-2015-12-687749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen P.M., Katsuyama E., Satyam A., Li H., Rubio J., Jung S., Andrzejewski S., Becherer J.D., Tsokos M.G., Abdi R., Tsokos G.C. CD38 reduces mitochondrial fitness and cytotoxic T cell response against viral infection in lupus patients by suppressing mitophagy. Sci. Adv. 2022;8:eabo4271. doi: 10.1126/sciadv.abo4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoffmann M., Pantazis N., Martin G.E., Hickling S., Hurst J., Meyerowitz J., Willberg C.B., Robinson N., Brown H., Fisher M., et al. Exhaustion of Activated CD8 T Cells Predicts Disease Progression in Primary HIV-1 Infection. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Montali I., Ceccatelli Berti C., Morselli M., Acerbi G., Barili V., Pedrazzi G., Montanini B., Boni C., Alfieri A., Pesci M., et al. Deregulated intracellular pathways define novel molecular targets for HBV-specific CD8 T cell reconstitution in chronic hepatitis B. J. Hepatol. 2023;79:50–60. doi: 10.1016/j.jhep.2023.02.035. [DOI] [PubMed] [Google Scholar]
- 64.DeRogatis J.M., Neubert E.N., Viramontes K.M., Henriquez M.L., Nicholas D.A., Tinoco R. Cell-Intrinsic CD38 Expression Sustains Exhausted CD8(+) T Cells by Regulating Their Survival and Metabolism during Chronic Viral Infection. J. Virol. 2023;97 doi: 10.1128/jvi.00225-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verma V., Shrimali R.K., Ahmad S., Dai W., Wang H., Lu S., Nandre R., Gaur P., Lopez J., Sade-Feldman M., et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat. Immunol. 2019;20:1231–1243. doi: 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma K., Sun L., Shen M., Zhang X., Xiao Z., Wang J., Liu X., Jiang K., Xiao-Feng Qin F., Guo F., et al. Functional assessment of the cell-autonomous role of NADase CD38 in regulating CD8(+) T cell exhaustion. iScience. 2022;25 doi: 10.1016/j.isci.2022.104347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sukumar M., Liu J., Ji Y., Subramanian M., Crompton J.G., Yu Z., Roychoudhuri R., Palmer D.C., Muranski P., Karoly E.D., et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng W., O'Hear C.E., Alli R., Basham J.H., Abdelsamed H.A., Palmer L.E., Jones L.L., Youngblood B., Geiger T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32:1157–1167. doi: 10.1038/s41375-017-0008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Funk C.R., Wang S., Chen K.Z., Waller A., Sharma A., Edgar C.L., Gupta V.A., Chandrakasan S., Zoine J.T., Fedanov A., et al. PI3Kdelta/gamma inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood. 2022;139:523–537. doi: 10.1182/blood.2021011597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Urak R., Walter M., Lim L., Wong C.W., Budde L.E., Thomas S., Forman S.J., Wang X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer. 2017;5:26. doi: 10.1186/s40425-017-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Araki K., Turner A.P., Shaffer V.O., Gangappa S., Keller S.A., Bachmann M.F., Larsen C.P., Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaeth M., Maus M., Klein-Hessling S., Freinkman E., Yang J., Eckstein M., Cameron S., Turvey S.E., Serfling E., Berberich-Siebelt F., et al. Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity. 2017;47:664–679.e6. doi: 10.1016/j.immuni.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee H.C., Zhao Y.J. Resolving the topological enigma in Ca(2+) signaling by cyclic ADP-ribose and NAADP. J. Biol. Chem. 2019;294:19831–19843. doi: 10.1074/jbc.REV119.009635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Horenstein A.L., Bracci C., Morandi F., Malavasi F. CD38 in Adenosinergic Pathways and Metabolic Re-programming in Human Multiple Myeloma Cells: In-tandem Insights From Basic Science to Therapy. Front. Immunol. 2019;10:760. doi: 10.3389/fimmu.2019.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li W., Lu L., Lu J., Wang X., Yang C., Jin J., Wu L., Hong X., Li F., Cao D., et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aay9013. [DOI] [PubMed] [Google Scholar]
- 76.Chini E.N. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr. Pharmaceut. Des. 2009;15:57–63. doi: 10.2174/138161209787185788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Camacho-Pereira J., Tarragó M.G., Chini C.C.S., Nin V., Escande C., Warner G.M., Puranik A.S., Schoon R.A., Reid J.M., Galina A., Chini E.N. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23:1127–1139. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Y., Wang F., Wang L., Qiu S., Yao Y., Yan C., Xiong X., Chen X., Ji Q., Cao J., et al. NAD(+) supplement potentiates tumor-killing function by rescuing defective TUB-mediated NAMPT transcription in tumor-infiltrated T cells. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109516. [DOI] [PubMed] [Google Scholar]
- 79.Covarrubias A.J., Perrone R., Grozio A., Verdin E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021;22:119–141. doi: 10.1038/s41580-020-00313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kong S., Kim S.J., Sandal B., Lee S.M., Gao B., Zhang D.D., Fang D. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J. Biol. Chem. 2011;286:16967–16975. doi: 10.1074/jbc.M111.218206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doan A.E., Mueller K.P., Chen A., Rouin G., Daniel B., Lattin J., Chen Y., Mozarsky B., Markovska M., Arias-Umana J., et al. 247 FOXO1 is a master regulator of CAR T memory programming. Journal for ImmunoTherapy of Cancer. 2023;11:A286. [Google Scholar]
- 82.Cantó C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C., Elliott P.J., Puigserver P., Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F., Messadeq N., Milne J., Lambert P., Elliott P., et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y., Bi Y., Chen X., Li C., Li Y., Zhang Z., Wang J., Lu Y., Yu Q., Su H., et al. Histone Deacetylase SIRT1 Negatively Regulates the Differentiation of Interleukin-9-Producing CD4(+) T Cells. Immunity. 2016;44:1337–1349. doi: 10.1016/j.immuni.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 85.Martin T.G., Corzo K., Chiron M., Velde H.V., Abbadessa G., Campana F., Solanki M., Meng R., Lee H., Wiederschain D., et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells. 2019;8 doi: 10.3390/cells8121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh N., Frey N.V., Engels B., Barrett D.M., Shestova O., Ravikumar P., Cummins K.D., Lee Y.G., Pajarillo R., Chun I., et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021;27:842–850. doi: 10.1038/s41591-021-01326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gomes-Silva D., Mukherjee M., Srinivasan M., Krenciute G., Dakhova O., Zheng Y., Cabral J.M.S., Rooney C.M., Orange J.S., Brenner M.K., Mamonkin M. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017;21:17–26. doi: 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gomes da Silva D., Mukherjee M., Madhuwanti S., Dakhova O., Liu H., Grilley B., Gee A., Neelapu S.S., Rooney C., Heslop H., et al. Direct Comparison of in Vivo Fate of Second and Third-Generation Cd19-Specific Chimeric Antigen Receptor (Car)-T Cells in Patients with B Cell Non-Hodgkin Lymphoma (B-Nhl): Reversal of Toxicity from Tonic Signaling. Cytotherapy. 2017;19:S9. [Google Scholar]
- 89.Zhu X., Li Q., Zhu X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.1034257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu H., Chen I., Shimoda L.A., Park Y., Zhang C., Tran L., Zhang H., Semenza G.L. Chemotherapy-Induced Ca(2+) Release Stimulates Breast Cancer Stem Cell Enrichment. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2020.108605. [DOI] [PubMed] [Google Scholar]
- 91.Azimi I. The interplay between HIF-1 and calcium signalling in cancer. Int. J. Biochem. Cell Biol. 2018;97:73–77. doi: 10.1016/j.biocel.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 92.Nicholson I.C., Lenton K.A., Little D.J., Decorso T., Lee F.T., Scott A.M., Zola H., Hohmann A.W. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol. 1997;34:1157–1165. doi: 10.1016/s0161-5890(97)00144-2. [DOI] [PubMed] [Google Scholar]
- 93.Zhang H., Hu Y., Shao M., Teng X., Jiang P., Wang X., Wang H., Cui J., Yu J., Liang Z., et al. Dasatinib enhances anti-leukemia efficacy of chimeric antigen receptor T cells by inhibiting cell differentiation and exhaustion. J. Hematol. Oncol. 2021;14:113. doi: 10.1186/s13045-021-01117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luckey C.J., Bhattacharya D., Goldrath A.W., Weissman I.L., Benoist C., Mathis D. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 2006;103:3304–3309. doi: 10.1073/pnas.0511137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C., et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McKinney E.F., Lee J.C., Jayne D.R.W., Lyons P.A., Smith K.G.C. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. doi: 10.1038/nature14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bangs S.C., Baban D., Cattan H.J., Li C.K.F., McMichael A.J., Xu X.N. Human CD4+ memory T cells are preferential targets for bystander activation and apoptosis. J. Immunol. 2009;182:1962–1971. doi: 10.4049/jimmunol.0802596. [DOI] [PubMed] [Google Scholar]
- 101.Winter S.C., Buffa F.M., Silva P., Miller C., Valentine H.R., Turley H., Shah K.A., Cox G.J., Corbridge R.J., Homer J.J., et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007;67:3441–3449. doi: 10.1158/0008-5472.CAN-06-3322. [DOI] [PubMed] [Google Scholar]
- 102.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 103.Li X., Guo X., Zhu Y., Wei G., Zhang Y., Li X., Xu H., Cui J., Wu W., He J., et al. Single-Cell Transcriptomic Analysis Reveals BCMA CAR-T Cell Dynamics in a Patient with Refractory Primary Plasma Cell Leukemia. Mol. Ther. 2021;29:645–657. doi: 10.1016/j.ymthe.2020.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
(1)
Single-cell RNA-seq data from the previous study (Haradhvalal et al. 2022, Nat Med) are publicly available at Gene Expression Omnibus with GEO accession GSE197268.
-
(2)
Single-cell ATAC-seq data from the previous study (Jiang et al., 2022, Leukemia) are publicly available at CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0002442 (db.cngb.org/search/project/CNP0002442).
-
(3)
Bulk RNA-seq data have been deposited in Genome Sequence Archive in National Genomics Data Center, GSA: HRA004078, publicly accessible at https://ngdc.cncb.ac.cn/gsa.
-
(4)
All the original code is available at Zenodo (https://doi.org/10.5281/zenodo.8403894).
-
(5)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.