

Summary
Immune checkpoint inhibitors, particularly PD-1/PD-L1 blockades, have been approved for unresectable hepatocellular carcinoma (HCC). However, high resistance rates still limit their efficacy, highlighting the urgent need to understand the underlying mechanisms and develop strategies for overcoming the resistance. In this study, we demonstrate that HCC with high MER proto-oncogene tyrosine kinase (MerTK) expression exhibits anti-PD-1/PD-L1 resistance in two syngeneic mouse models and in patients who received anti-PD-1/PD-L1 therapy. Mechanistically, MerTK renders HCC resistant to anti-PD-1/PD-L1 by limiting ferroptosis with the upregulation of SLC7A11 via the ERK/SP1 pathway and facilitating the development of an immunosuppressive tumor microenvironment (TME) with the recruitment of myeloid-derived suppressor cells (MDSCs). Sitravatinib, an inhibitor of MerTK, sensitizes resistant HCC to anti-PD-L1 therapy by promoting tumor ferroptosis and decreasing MDSC infiltration into the TME. In conclusion, we find that MerTK could serve as a predictive biomarker for patient stratification and as a promising target to overcome anti-PD-1/PD-L1 resistance in HCC.
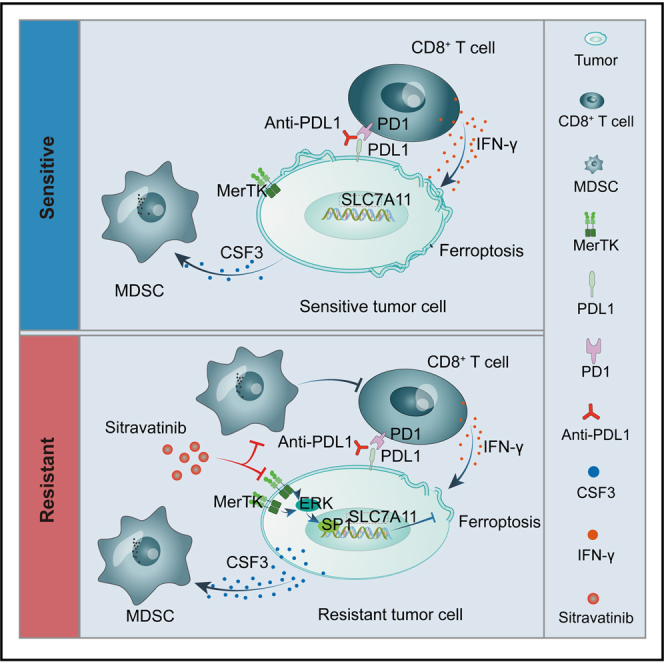
Graphical abstract
Highlights
-
•
MerTK renders anti-PD-1/PD-L1 therapy resistance in HCC
-
•
MerTK suppresses tumor cell ferroptosis and induces an immunosuppressive TME
-
•
Inhibition of MerTK increases the efficacy of PD-L1 antibody
-
•
Combination of sitravatinib and PD-L1 antibody has a synergistic antitumor effect
Dong et al. report that MerTK upregulates SLC7A11 expression to suppress tumor cell ferroptosis, favors a protumor TME by recruiting MDSCs in hepatocellular carcinoma (HCC), and drives anti-PD-L1 therapy resistance. Targeting MerTK increases ferroptosis and reduces MDSCs recruitment, which activates CD8+ T cells and sensitizes HCC to anti-PD-L1 blockade.
Introduction
Hepatocellular carcinoma (HCC) remains a global health challenge, with increasing incidence worldwide.1 Clinically, most HCC patients are diagnosed at advanced stages with limited treatment options. Recently, immunotherapies, such as the immune checkpoint inhibitors (ICIs) anti-CTLA4, anti-PD-1/PD-L1, their combination, or combinations with other drugs have achieved unparalleled success for patients with cancers.2,3 Moreover, ICIs such as nivolumab, pembrolizumab, and atezolizumab, among others, are currently approved by the US Food and Drug Administration (FDA) for the systemic treatment of HCC.4 However, because of frequent resistance to anti-PD-1/PD-L1 therapy, response rates are still far from satisfactory, as the overall benefit rate is observed only in approximately 15%–20% of HCC patients at present.2,3,5 Thus, it is urgent to identify biomarkers for optimal patient selection and to identify effective targets to overcome this resistance.
MER proto-oncogene tyrosine kinase (MerTK), a member of the TAM family (TYRO3, AXL, and MerTK) of receptor tyrosine kinases, is highly expressed in a variety of malignancies,6 including HCC (The Human Protein Atlas: proteinatlas.org). The homologous proteins growth arrest-specific protein 6 (Gas6) and protein S (Pros1) are both natural ligands for MerTK activation, which induce homodimerization and auto-phosphorylation of MerTK and activate downstream pathways, such as MEK/ERK, PI3K/AKT, and JAK/STAT, leading to tumor cell proliferation, anti-apoptosis, suppression of inflammation, and tumor growth.7,8,9 Recent studies have shown that MerTK plays a critical role in regulating HCC growth as a metabolic modulator by integrating aerobic glycolysis and oxidative phosphorylation.10 Furthermore, the receptor/ligand activity of MerTK has been documented in multiple immune cell types and potentiates antitumor immunity, which has implications for cancer immunotherapy.11,12,13
Ferroptosis, as a form of metabolically regulated cell death, has emerged to play an important role in cancer biology.14,15,16,17 In recent years, accumulating evidence suggests that the suppression of ferroptosis has been linked to resistance to conventional cancer therapies and that targeting ferroptosis might provide a therapeutic potential for cancers.18,19,20 Recently, it has been shown that the regulation of ferroptosis could reshape the tumor niche, leading to T-cell-mediated tumor eradication during immunotherapy.21,22 In HCC, ferroptosis has been demonstrated to confer drug resistance in systemic therapies such as targeted therapy and chemotherapy.23
In the present study, we reveal that MerTK is a major mechanism of resistance to PD-1/PD-L1 blockade, limiting tumor cell ferroptosis and causing an immunosuppressive tumor microenvironment (TME) by increasing myeloid-derived suppressor cell (MDSC) infiltration. MerTK blockade might be efficacious to counter this resistance and sensitize HCC to anti-PD-L1 therapy. Additionally, MerTK could serve as biomarkers for screening patients with HCC using sitravatinib plus anti-PD-L1 combination therapy.
Results
MerTK is identified to render HCC resistant to anti-PD-L1 treatment
To investigate the potential mechanisms involved in anti-PD-L1/PD-1 resistance in HCC, we used a syngeneic mouse tumor model to generate an in vivo resistance model (Figure 1A). Hepa1-6 subcutaneous tumor mouse models were established and treated with mouse anti-PD-L1 antibody along with serial passage and treatment (Figure S1A). As shown in Figure 1B, parental Hepa1-6 tumors were sensitive to anti-PD-L1 therapy, as evidenced by the tumor growth, which significantly decreased compared with tumors treated with IgG. After performing cycles of anti-PD-L1 antibody treatment along with serial passage, resistant Hepa1-6 (Res1-6) tumors were nonresponsive to anti-PD-L1 therapy, as shown by the similar tumor growth (Figure S1A) and survival rate (Figure 1C) compared with tumors treated with IgG. Res1-6 tumor xenografts were isolated and implanted into the liver to establish orthotopic implantation models and subsequently received anti-PD-L1 treatment. In line with the subcutaneous tumor mouse model, the Res1-6 tumors exhibited a poor response to the anti-PD-L1 treatment and increased lung metastases compared with the parental Hepa1-6 tumors (Figures S1B and S1C). Similarly, we used the same method to establish another hepatoma cell line HCA1-resistant mice model in vivo, and we obtained resistant Res-CA1 strains (Figure S1D). Similar to the Res1-6 tumor model, treatment with anti-PD-L1 antibody failed to yield an antitumor effect in the Res-CA1 groups, as indicated by the lack of difference in tumor growth, survival rate, and lung metastases between the anti-PD-L1 therapy and IgG control groups (Figures S1E–S1I). Collectively, these results indicate that we successfully established resistant mouse models to be considered as useful tools for elucidating the molecular mechanisms of anti-PD-1/PD-L1 resistance in HCC.
Figure 1.

MerTK mediates HCC resistant to anti-PD-L1 treatment
(A) Schematic illustrating the establishment of anti-PD-L1-resistant strains in vivo.
(B) Tumor growth curves of subcutaneous implantation models in Hepa1-6 and Res1-6 strains treated with anti-PD-L1 (aPD-L1) or IgG.
(C) Survival of orthotopic implantation models of Hepa1-6 and Res1-6 strains treated with aPD-L1 or IgG.
(D) Proteomic sequencing analysis was used to analyze the differently expressed proteins in Hepa1-6 and Res1-6. The top 20 differently expressed proteins are presented in a heatmap, including 10 upregulated and 10 downregulated proteins.
(E) Western blot of p-MerTK, MerTK, and β-actin in Hepa1-6, Res1-6, HCA-1, and Res-CA1 strains.
(F) IHC staining of p-MerTK and MerTK in Hepa1-6 and Res1-6 subcutaneous tumor tissues.
(G) Locations of sensitive and resistant tumors were exhibited by MRI, and AFP values before and after anti-PD-L1/PD-1 therapy between patient in sensitive group and patient in resistant group. IHC staining of MerTK expression in HCC tissues from sensitive and resistant patients received anti-PD-1/PD-L1 therapy.
(H and I) Subcutaneous xenograft mouse model of Res1-6, Res1-6-sh-MerTK, Hepa1-6, and Hepa1-6-OE-MerTK strains treated with anti-PD-L1 or IgG. When the tumor volume approximately reached 100 mm3 in size, tumor volume was measured every 3 days. After 25 days of treatment, mice were sacrificed. Shown are tumor appearance and tumor growth curves.
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Next, we screened for differential protein expression that may be involved in anti-PD-1/PD-L1 resistance using proteomic sequencing between Res1-6 and Hepa1-6 tumor cells (Figure S2A). Comparing the top 30 differentially expressed proteins, we identified that MerTK was highly overexpressed in the Res1-6 tumor cells (Figure 1D). Both western blot and immunohistochemistry (IHC) staining confirmed that the expression of p-MerTK and MerTK protein was significantly increased in the PD-L1-resistant HCC cells and tumor tissues from resistant mouse models (Figures 1E, 1F, and S2B). To further determine the correlation between MerTK and the anti-PD-1/PD-L1 therapeutic response, we analyzed the MerTK expression in HCC patients who received anti-PD-1/PD-L1 therapy and found that the expression level of MerTK was significantly higher in tumor tissues from HCC patients with resistance than that from HCC patients with sensitivity (Figure 1G). Clinically, the mRNA levels of MerTK in HCC tissues were highly expressed (Figure S2C) and significantly higher than that in paraneoplastic tissues (Figure S2D). The survival curves showed that a higher MerTK level was associated with the poor prognosis of HCC patients (Figure S2E).
In order to further detect whether MerTK is required to mediate anti-PD-1/PD-L1 resistance in HCC, we knocked down MerTK in Res1-6 tumor cells (Res1-6-shMerTK) (Figure S2F) and compared the responses to anti-PD-L1 therapy between Res1-6 and Res1-6-shMerTK cells in a mouse model (Figure S2G, treatment schedule). Remarkably, mice bearing Res1-6-shMerTK, but not Res1-6, tumors were sensitive to anti-PD-L1 treatment, as evidenced by the significant reduction in tumor growth (Figure 1H). Then, we generated a Hepa1-6 cell line with MerTK overexpression (Figure S2H) and compared the response to the PD-L1 antibody between the Hepa1-6 and Hepa1-6-OE-MerTK strains in a syngeneic mouse model (Figure S2I, treatment schedule). Consistently, PD-L1 therapy significantly decreased tumor growth in mice bearing Hepa1-6 tumors but not in those with Hepa1-6-OE-MerTK tumors (Figure 1I). These results indicate that MerTK renders HCC resistant to anti-PD-L1 treatment.
MerTK induces anti-PD-L1 resistance by suppressing ferroptosis
To further explore the potential mechanisms involved in anti-PD-L1 resistance, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the differential expression of proteins in the proteomic sequencing results revealed that the ferroptosis signaling pathway was remarkably enriched between Res1-6 and Hepa1-6 cells (Figure 2A). To test this possibility, we treated Res1-6 and Hepa1-6 cells with ferroptosis inducer erastin (5.0 μM) and ferroptosis inhibitor (Fer-1 or DFO), respectively. Using the fluorescent probe C11-BODIPY, we found that erastin treatment significantly decreased the lipid reactive oxygen species (ROS) levels in the Res1-6 cells compared with the Hepa1-6 cells and that the ferroptosis inhibitor Fer-1 or DFO eliminated these effects (Figure 2B). Consistent with these observations, SYTOX Green staining also demonstrated that the Res1-6 cells, but not Hepa1-6 cells, showed significant resistant to erastin-induced cell death. Importantly, erastin-induced cell death could be largely suppressed by the ferroptosis inhibitor Fer-1 or DFO (Figure 2C). Then, the Res1-6 and Hepa1-6 cells were labeled with red fluorescent dye and green fluorescent dye, respectively, and cocultured under erastin treatment conditions. We found that the cell survival ratio of the Res1-6 cells gradually increased and the ratio of the Hepa1-6 cells gradually decreased in a time-dependent manner (Figure S2J; Videos S1 and S2). These results demonstrate that ferroptosis could be involved in regulating anti-PD-L1 resistance and raises the possibility that MerTK-mediated anti-PD-L1 resistance could have been achieved by the suppression of tumor cell ferroptosis.
Figure 2.

MerTK promotes anti-PD-L1 resistance by suppressing ferroptosis in HCC
(A) All the differential genes between Hepa1-6 and Res1-6 cells were analyzed using KEGG pathway analysis using the functional gene sets in MSigDB (literature vs. databases containing signaling pathways).
(B) Fluorescence detection of lipid ROS by C11-BODIPY (left) and statistical analysis of relative lipid ROS fluorescence signal (right).
(C) Fluorescence detection of dead cells by SYTOX Green (left) and statistical analysis of percentage dead cells (right).
(D–F) Cell viability of Hepa1-6, Hepa1-6-OE-MerTK, Res1-6, and Res1-6-sh-MerTK strains treated with erastin (5.0 μM) in cocultured condition (left) and statistical analysis of cell survival rate in each time point (right).
(G–J) In subcutaneous xenograft mouse model, the statistical analysis of relative lipid ROS and MDA content in Hepa1-6, Hepa1-6-OE-MerTK, Res1-6, and Res1-6-sh-MerTK strains treated with anti-PD-L1 or IgG.
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗∗p < 0.01 and ∗∗∗p < 0.001.
We next investigated the potential function of MerTK in regulating ferroptosis of HCC. The Cancer Genome Atlas (TCGA) public database was used to determine the relationship between MerTK and ferroptosis-related genes in the mRNA expression levels. The results indicate that MerTK was positively correlated with ferroptosis suppressor genes (Slc7a11, Slc3a2, Nfe2l2, Gclc, Gclm, Gss, and Fth1) and negatively correlated with ferroptosis driver gene (Acsl4) (Figure S2K). The analysis of the ferroptosis sensitivity in the panel of HCC cell lines revealed that the HCC cell lines with high MerTK expression exhibited a lower half maximal inhibitory concentration (IC50) under erastin treatment condition (Figures S2L and S2M). Then, the Hepa1-6 and Hepa1-6-OE-MerTK cells were labeled using red and green fluorescent dye markers, respectively, and cocultured under erastin treatment conditions. We found that the cell survival ratio of the Hepa1-6-OE-MerTK cells gradually increased and the ratio of the Hepa1-6 cells gradually decreased in a time-dependent manner (Figures 2D and 2E; Video S3). Likewise, the Res1-6 and Res1-6-shMerTK cells were labeled with red and green fluorescent dye marker, respectively, and cocultured under erastin treatment conditions. The cell survival ratio of the Res1-6 cells exhibited a time-dependent increase, and the ratio of the Res1-6-shMerTK cells showed the opposite effect with a time-dependent decrease (Figures 2D and 2F; Video S4). We observed characteristic indicators related to ferroptosis, including lipid ROS and malonaldehyde (MDA) contents, in the mouse tumor models. The overexpression of MerTK significantly inhibited the lipid ROS and MDA contents in mice bearing Hepa1-6 tumors treated with PD-L1 (Figures 2G and 2H). Conversely, knockdown of MerTK significantly induced lipid ROS and MDA contents in the resistant tumor model with PD-L1 treatment (Figures 2I and 2J). Taken together, our results suggest that MerTK induced anti-PD-L1 resistance by protecting HCC cells from ferroptosis.
Suppression of SLC7A11-mediated ferroptosis is essential for anti-PD-L1 resistance by MerTK via the ERK/SP1 pathway
To investigate the underlying mechanisms involved in ferroptosis suppressed by MerTK in anti-PD-L1 resistance, we examined changes in the expression of genes related to ferroptosis in the above analysis of proteomic data. Notably, the volcano plot of the differentially expressed proteins indicated that the ferroptosis suppressor cystine/glutamate transporter (SLC7A11) was among the top genes identified in our proteomic analyses in the proteomic-level analysis (Figure 3A). Recently, accumulating studies have demonstrated that SLC7A11 is a key mediator in blocking ferroptosis.18,24,25 Then, we studied whether SLC7A11-mediated suppression of ferroptosis is sufficient to cause anti-PD-1/PD-L1 resistance induced by MerTK. To assess this possibility, we detected the expression of SLC7A11 in resistant tumor cells and tissues. Western blot analysis revealed that the SLC7A11 levels were significantly increased in anti-PD-L1-resistant tumor cells (Figure 3B), which was further confirmed in tissues from resistant tumor models with IHC staining (Figure 3C). We next analyzed the SLC7A11 expression in HCC patients who received anti-PD-1/PD-L1 therapy. The HCC patients who were resistant to anti-PD-L1/PD-1 therapy had higher levels of SLC7A11 expression compared with those whose tumors were controlled by anti-PD-L1/PD-1 therapy (Figure 3D). Interestingly, the Pearson correlation analysis showed a positive correlation between the expression of MerTK and SLC7A11 with IHC staining in anti-PD-L1/PD-1 therapy-resistant HCC patients (R = 0.91, p = 0.013) (Figure 3E).
Figure 3.

Suppression of SLC7A11 sensitizes anti-PD-L1 treatment caused by MerTK
(A) Volcano plot of differentially expressed proteins in protein mass spectrometry.
(B) Western blot of SLC7A11 expression in Hepa1-6, Res1-6, HCA-1, and Res-CA1.
(C) IHC staining of SLC7A11 in Hepa1-6, Res1-6, HCA-1, and Res-CA1 subcutaneous tumor tissues.
(D) IHC staining of SLC7A11 expression in HCC tissues from sensitive and resistant patients with anti-PD-1/PD-L1 therapy, and statistical analysis.
(E) The correlation between MerTK and SLC7A11 expression in tumor tissues from HCC patients received anti-PD-1/PD-L1 therapy, Pearson product-moment correlation coefficients and p values are shown.
(F and G) (F) Schematic illustrating the procedure of anti-PD-L1 or IgG treatment in Res1-6 and Res1-6-shSLC7A11 subcutaneous tumor model, and (G) the representative images of subcutaneous tumor in different groups.
(H) Statistical analysis of tumor growth curves.
(I and J) Western blot analysis of p-MerTK, MerTK, SLC7A11, and β-actin expression in Hepa1-6, Hepa1-6-OE-MerTK, Res1-6, and Res1-6-shMerTK subcutaneous tumor treated with anti-PD-L1 or IgG.
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗∗p < 0.01 and ∗∗∗p < 0.001.
Then Res1-6 and Res1-6-shSLC7A11 cells were labeled with red and green fluorescent dye marker, respectively, and cocultured under erastin treatment conditions. We found that erastin treatment showed time-dependent effects on the increased cell survival ratio of the Hepa1-6-OE-SLC7A11 cells and the decreased ratio of Hepa1-6 cells (Figures S3A–S3C). On the other hand, the Hepa1-6 and Hepa1-6-OE-SLC7A11 cells were labeled with red and green fluorescent dye marker, respectively, and cocultured under erastin treatment conditions. We found that the cell survival ratio of the Hepa1-6-OE-SLC7A11 cells gradually increased and the ratio of Hepa1-6 cells gradually decreased in a time-dependent manner (Figures S3A, S3B, and S3D). Consistent with these observations, the knockdown of SLC7A11 in Res1-6 significantly induced lipid ROS and MDA contents in the resistant muse model with PD-L1 treatment (Figures S3E and S3F). To further validate the correlation between the SLC7A11-mediated ferroptosis and anti-PD-L1 response in HCC, we compared the responses to anti-PD-L1 therapy between the Res1-6 and Res1-6-shSLC7A11 tumors in a resistant mouse model (Figure 3F). Compared with the Res1-6 tumors, mice bearing Res1-6-shSLC7A11 tumors showed more sensitivity to anti-PD-L1 therapy (Figures 3G and 3H). To demonstrate whether SLC7A11 is critical for anti-PD-L1 resistance induced by MerTK, we analyzed SLC7A11 expression in mouse tumor models bearing MerTK. Correspondingly, dramatic increases in SLC7A11 expression were demonstrated in subcutaneous xenografts of mouse models with MerTK overexpression (Figure 3I). On the other hand, SLC7A11 expression was downregulated in the tumor tissues of the resistant model when MerTK was knocked down (Figure 3J). We then analyzed the relationship between MerTK and SLC7A11 in the protein expression level in human HCC tissues using IHC on the TMA and found that MerTK expression was positively correlated with SLC7A11 expression in HCC (R = 0.31, p = 0.0035) (Figure S3G). Consistent with this, we observed a positive correlation between MerTK and SLC7A11 levels across a panel of HCC cell lines (Figure S3H). A survival analysis showed that patients with high MerTK and SLC7A11 expression had shorter overall survival (OS) (Figures S3I–S3K; Tables S2 and S3). Together, our analyses suggested that MerTK inhibits ferroptosis regulated by SLC7A11 in resistant HCC.
As previous studies have reported that there are several classical downstream signaling pathways of MerTK,7,26 we questioned which downstream signaling pathway was activated and mediated upregulation of SLC7A11 in resistant tumors. Western blot analysis demonstrated that the ERK/SP1 pathway was remarkably altered.27 Both MerTK knock down and ERK inhibitor-HY-126288 treatment in the Res1-6 tumor cells significantly decreased the expression of p-ERK, p-SP1, and SLC7A11 (Figure 4A). On the other hand, overexpression of MerTK in Hepa1-6 cells significantly increased SLC7A11 protein expression, and ERK inhibitor-HY-126288 treatment decreased SLC7A11 protein expression in Hepa1-6-OE-MerTK cells (Figure 4B). The fluorescent probe C11-BODIPY assay revealed that Hepa1-6-OE-MerTK cells had decreased lipid ROS levels induced by erastin, while the ERK inhibitor treatment in Hepa1-6-OE-MerTK cells increased lipid ROS levels (Figures 4C and 4D). Compared with Res1-6 cells, the Res1-6-sh-MerTK cells had increased lipid ROS levels induced by erastin, and ERK inhibitor treatment in Res1-6 cells increased lipid ROS levels (Figures 4C and 4E). We further found that overexpression of MerTK in Hepa1-6 cells significantly decreased cell death induced by erastin, and ERK inhibitor treatment in Hepa1-6-OE-MerTK cells increased cell death induced by erastin (Figures 4F and 4G). Compared with Res1-6 cells, Res1-6-sh-MerTK cells increased cell death induced by erastin and ERK inhibitor treatment in Res1-6 cells also increased cell death (Figures 4F and 4H). These observations indicated that MerTK suppresses SLC7A11-mediated ferroptosis for anti-PD-L1 resistance via the ERK/SP1 pathway.
Figure 4.

MerTK regulates HCC tumor cell ferroptosis via the ERK/SP1 pathway
(A) Western blot analysis of p-MerTK, MerTK, p-ERK, ERK, p-SP1, SP1, SLC7A11, and β-actin expression in Res1-6, Res1-6-shMerTK, and Res1-6 treated with ERK1/2 inhibitor (HY-126288).
(B) Western blot analysis of p-MerTK, MerTK, p-ERK, ERK, p-SP1, SP1, SLC7A11, and β-actin expression in Hepa1-6, Hepa1-6-OE-MerTK, and Hepa1-6-OE-MerTK treated with ERK1/2 inhibitor.
(C–E) (C) Fluorescence detection of lipid ROS by C11-BODIPY and (D) statistical analysis of relative lipid ROS fluorescence signal in Hepa1-6 tumor cells and (E) in Res1-6 tumor cells.
(F–H) (F) Fluorescence detection of dead cells by SYTOX Green, (G) statistical analysis of percentage dead cells in Hepa1-6 tumor cells and (H) in Res1-6 tumor cells.
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗∗p < 0.01 and ∗∗∗p < 0.001.
MerTK induces anti-PD-L1 resistance by favoring a protumor TME with recruitment of MDSCs
There is emerging evidence for the importance of the TME in the immunotherapy response in cancer. To better understand the mechanistic role of MerTK in anti-PD-1/PD-L1 resistance, we looked further into the immune characterization of the resistant HCCs. Compared with Hepa1-6 tumors, we found a significant reduction in CD8+ T cells and IFNγ+CD8+ T cells in Res1-6 tumors, whereas the percentage of MDSCs significantly increased using flow cytometry (Figures S4A and S4B). To detect the specific alteration of the MDSCs in vivo, mouse MDSCs were further classified into gMDSCs (CD45+, CD11b+, Gr1+, Ly6C−, and Ly6G+) and mMDSCs (CD45+, CD11b+, Gr1+, Ly6C+, and Ly6G−) (Figures S4C). Compared with Hepa1-6 tumors, we found a significant increase in the percentage of MDSCs, and gMDSCs in Res1-6 tumors, and the mMDSCs also increased but did not show a significant difference (Figures S4D). Then, multiplex immunofluorescence (mfIHC) was used to visualize the MerTK and SLC7A11 expressions and immune subsets in the HCC patients who received anti-PD-1/PD-L1 therapy and found elevated expression levels of MerTK and SLC7A11, as well as an increased infiltration of MDSCs (CD11b+CD15+/CD11b+CD14+), gMDSCs (CD11b+CD15+CD14−), and mMDSCs (CD11b+CD15−CD14+) in the tumor tissues of resistant HCC patients compared with that in HCC patients with sensitivity (Figures S4E–S4G). Correspondingly, MerTK expression was positively correlated with SLC7A11 expression in anti-PD-1/PD-L1-resistant HCC patients (R = 0.821, p = 0.034) (Figures S4E and S4H). In addition, the mfIHC results also showed a positive correlation between the expression level of MerTK and the proportion of total MDSCs (R = 0.857, p = 0.024) (Figures S4E and S4I), and gMDSCs (R = 0.786, p = 0.048) (Figures S4E and S4J) in anti-PD-1/PD-L1-resistant HCC patients. The correlation between the expression level of MerTK and the proportion of mMDSCs did not show significantly correlation in anti-PD-1/PD-L1-resistant HCC patients (Figures S4E and S4K). Next, to explore the functions of MerTK in the TME, we evaluated the correlation between MerTK expression and the antitumor activity of cytotoxic T cells in TCGA’s database and found that cytotoxic T cells mediated prolonged overall survival in patients with low expression of MerTK but not in patients with high MerTK expression (Figure 5A), suggesting that MerTK reduces the antitumor effects of cytotoxic T cells. In addition, TCGA’s database also showed a negative correlation between the expression level of MerTK and the enrichment of cytotoxic CD8+ T cells (Figure 5B). Then, mfIHC was further used to visualize the MerTK expression and immune subsets in human HCC tissue microarray and found that patients with high MerTK expression had significantly lower infiltration of CD8+ T cells (Figures 5C and 5D) and higher infiltration of MDSCs (CD11b+, CD15+/CD14+) (Figures 5C and 5E). Specifically, the group with high MerTK expression had significantly higher infiltration of gMDSCs (CD11b+, CD15+, and CD14−) (Figures 5C and 5F). The enrichment of mMDSCs (CD11b+, CD15−, and CD14+) did not show a significantly difference in the HCC tumor tissue between the groups with high and low MerTK expression (Figures 5C and 5G). The correlation between the expression level of MerTK and the proportion of MDSCs show significantly correlation in HCC patients (Figure 5H). We further evaluated the immunological profiles in mouse models bearing stable expression of MerTK. Compared with Hepa1-6 tumors, overexpression of MerTK resulted in a significant reduction of CD8+ T cells and IFNγ+CD8+ T cells and an increase in the percentage of MDSCs (Figures 5I and S5A). On the contrary, knockdown of MerTK in Res1-6 significantly increased the population of CD8+ T cells and IFNγ+CD8+ T cells, and decrease the levels of MDSCs (Figures 5J and S5B). The, we further determine the specific MDSCs populations affected by MerTK alterations in vivo, and found that overexpression of MerTK resulted in a significant increase in the percentage of MDSCs and gMDSCs, and the percentage of mMDSCs also increase but did not show significant difference (Figures S5C and S5D). On the other hand, knockdown of MerTK in Res1-6 significantly decreased the percentage of MDSCs and gMDSCs, and the percentage of mMDSCs also decreased (Figures S5E and S5F). To explore the possibility of antitumor immune mediators underlying MDSCs accumulation in anti-PD-L1 resistance induced by MerTK, we evaluated the chemokine profiles using qRT-PCR and found that the levels of CSF3 significantly increased in resistant tumors (Figure S5G). Furthermore, overexpression of MerTK resulted in a significant increase in CSF3 in vivo (Figure S5H). Similar changes in CSF3 expression were observed in Hepa1-6-OE-MerTK culture medium with an ELISA assay (Figure S5I). To further explore whether MerTK affects the chemotactic migration of MDSCs in resistant HCC, we established an in vitro coculture system using a Transwell migration assay of MDSCs. Hepa1-6-OE-MerTK tumor cells significantly increased the migration of MDSCs. Similarly, adding CSF3 chemokine to the Hepa1-6 tumor cells’ culture medium significantly increased MDSCs migration. Both the neutralizing antibody of CSF3 and G-CSFR (CSF3 receptor) significantly decreased MDSCs migration induced by lower chamber Hepa1-6-OE-MerTK tumor cells (Figures S5J and S5K). We further assessed the interplay between CSF3 and MDSCs recruitment in vivo using the anti-CSF3 antibody. Compared with Hepa1-6-OE-MerTK tumor, anti-CSF3 antibody treatment decreased tumor weight and tumor growth in vivo (Figures S6A–S6C). Flow cytometric analysis revealed that anti-CSF3 treatment significantly reduced MDSC infiltration (Figures S6D and S6E).
Figure 5.

MerTK induces anti-PD-L1 resistance by favoring a protumor microenvironment
(A) Relationship between overall survival and CTL levels in HCC patients with low and high MerTK gene copy numbers.
(B) The correlations between the mRNA expression levels of MerTK and cytotoxic CD8+ T cells.
(C) The representative image of HCC tissue stained with MerTK (red), CD8 (gold), CD11b (purple), CD15 (green), and CD14 (pink).
(D–G) (D) The percentage statistical analysis of CD8+ T cells (E) MDSCs, (F) gMDSCs, and (G) mMDSCs in tumor tissues.
(H) The correlation analysis between the expression of MerTK and the enrichment of MDSCs.
(I and J) T-distributed stochastic neighbor embedding (t-SNE) plot of tumor-infiltrating leukocytes overlaid with color-coded clusters and the frequency of clusters of the indicated immune cell subsets, including CD3+ T cells, CD8+ T cells, IFNγ+CD8+ T cells, CD4+ T cells, CD11b+ cells, and MDSCs in Hepa1-6, Hepa1-6-OE-MerTK, Res1-6, and Res1-6-shMerTK subcutaneous tumor model treated with anti-PD-L1 or IgG (left) and the statistical analysis (right).
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To detect whether MDSCs are the main immune mechanism in mediating resistance, we isolated MDSCs from MerTK overexpression tumor and evaluated CD8+ T cell killing efficiency using a T cell/MDSC coculture assay. The results show that CD8+ T cell cytotoxic efficiency was inhibited by MDSCs in the coculture system (Figures S6F). With the increase in the ratio of MDSCs, the cell viability of Hepa1-6-OE-MerTK tumor cells significantly increased (Figure S6G). The concentration of IFN-γ and granzyme B secreted from CD8+ T cells was effectively inhibited by MDSCs in the coculture assay (Figures S6H and S6I). The assay of carboxyfluorescein succinimidyl ester (CFSE)-labeled CD8+ T cells demonstrated that MDSCs, derived from tumors overexpressing MerTK, could effectively inhibit the proliferation of CD8+ T cells in vitro (Figure S6J). Then, we assessed the therapeutic efficacy of anti-Gr-1, which could eliminate MDSCs, in combination with anti-PD-L1 in Hepa1-6-OE-MerTK cells in the mouse model. Compared with monotherapy, the combination therapy significantly decreased tumor weight and tumor growth in the subcutaneous Hepa1-6-OE-MerTK tumors (Figures S6K–S6M). Collectively, these data suggest that MerTK induces anti-PD-L1 resistance by favoring a protumor TME through the recruitment of MDSCs and a significant reduction of CD8+ T cells and IFNγ+CD8+ T cells.
Inhibition of MerTK increases the efficacy of PD-L1 antibody
Given the ability of MerTK to manipulate ferroptosis and favor an immunosuppressive TME, we sought to determine whether targeting MerTK could increase ferroptosis and thereby enhance HCC response to anti-PD-L1 blockade. We treated Res1-6 cells with different kinds of MerTK inhibitors, including UNC5293, UNC2541, UNC1267, and sitravatinib in vitro and found that MerTK inhibitors could effectively inhibit the viability of Res1-6 cells. Among these drugs, sitravatinib displayed a much lower IC50 in inhibiting Res1-6 cells compared with the others including UNC5293, UNC2541, and UNC1267 (Figure 6A). PI staining assay demonstrated that sitravatinib promoted erastin-induced ferroptosis of Res1-6 cells in vitro (Figure 6B). Using the fluorescent probe C11-BODIPY, we observed that sitravatinib increased lipid ROS levels induced by erastin in Res1-6 cells (Figure 6C). Compared with erastin treatment alone, we further confirmed the significant increase in death of Res1-6 cells when treatment with erastin and sitravatinib was combined, using SYTOX Green staining assay (Figure 6D). These results demonstrate that targeting MerTK increases the ferroptosis of Res1-6 cells induced by erastin.
Figure 6.

Inhibition of MerTK promotes ferroptosis and increases the efficacy of PD-L1 antibody in resistant HCC
(A) Cell viability (percentage) analysis of Res1-6 cells following MerTK inhibitors (sitravatinib, UNC5293, UNC2541, and UNC1267) at different concentrations (0, 0.1, 0.5, 1.0, 2.0, 4.0, 8.0, 16.0, and 32.0 μM).
(B) Cell death detection by propidium iodide (PI) staining (left) and statistical analysis (right).
(C) Fluorescence detection of lipid ROS by C11-BODIPY (left) and statistical analysis of relative lipid ROS fluorescence signal (right). (D) Fluorescence detection of dead cells by SYTOX Green (left) and statistical analysis of percentage dead cells (right).
(E) The representative images of subcutaneous tumor in Res1-6 strains were treated with IgG, sitravatinib, anti-PD-L1 or their combination (left) and the statistical analysis of tumor growth curves (right).
(F) The representative images of orthotopic tumor in Res1-6 strains were treated with IgG, sitravatinib, anti-PD-L1 or their combination (left) and the statistical analysis of survival curves (right).
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
The potential role of MerTK in triggering resistance provided us with a strong rationale for further targeting MerTK to sensitize resistant HCC to anti-PD-L1 therapy. To this end, we further investigated sitravatinib therapy in the anti-PD-L1-resistant tumor model and treated mice bearing Res1-6 tumors with sitravatinib, anti-PD-L1 antibody, and their combination. Compared with monotherapy or the control, the combination therapy significantly decreased the tumor weight and tumor growth in the subcutaneous Res1-6 tumors (Figures 6E, S7A, and S7B). To further validate the efficacy of the combination treatment, we established an orthotopic Res1-6 mouse model. We observed significant tumor regression, reduced number of lung metastases and prolonged mouse survival for the combination compared with that of control or single agent alone (Figures 6F and S7C). We performed the same combinatory treatment in the subcutaneous Res-HCA-1 tumors and found that the combination treatment of sitravatinib plus PD-L1 antibody inhibited tumor growth more effectively than monotherapy or control (Figures S7D–S7F). Similar results were further confirmed in the orthotopic HCA-1 mouse model, in which the combination treatment resulted in significantly higher antitumor activity and longer mouse survival than the control or any single agent (Figures S7G–S7K). In addition, no significant changes in the biochemical indicators of the liver and kidney functions were observed (Figures S7L–S7O). Collectively, these results demonstrate that targeting MerTK in combination with anti-PD-L1 has the potential to overcome anti-PD-1/PD-L1 resistance.
Sitravatinib plus PD-L1 antibody activates ferroptosis and reduces MDSC infiltration in HCC microenvironment
To further understand the antitumor response to the combination of sitravatinib and anti-PD-L1, we evaluated the immunological profiles in the Res1-6 subcutaneous tumors. Compared with control group, flow cytometric analysis of the Res1-6 tumors revealed a significant reduction in MDSCs in mice treated with sitravatinib or in combination with anti-PD-L1. Moreover, the infiltrating of CD8+ T cells, IFNγ+CD8+ T cells, and the secretion of IFN-γ increased in the combination treatment group compared with sitravatinib or anti-PD-L1 alone (Figures 7A, 7B, and S7P). In addition, the expression of CSF3 and MDSC infiltration decreased in sitravatinib treatment groups (Figures 7A, 7B, S7Q, and S7R). Western blot showed that sitravatinib could effectively inhibit MerTK activity and downregulate SLC7A11 expression, while their combination demonstrated a synergistic effect on p-MerTK and SLC7A11 levels in Res1-6 tumors (Figure 7C). Consistent with the western blot findings, IHC scores for p-MerTK and SLC7A11 in the combinational treatment group were significantly lower than those of the other groups (Figure 7D). In addition, we also carried out IHC staining of Ki-67 in the Res1-6 subcutaneous tumors. The results indicate that resistant tumors treated with their combination expressed lower Ki-67 compared with those treated with a single agent alone (Figure 7D). Then, to investigate the regulation of ferroptosis after combined therapy, we evaluated biomarkers indicative of ferroptosis, such as lipid ROS and MDA contents, in resistant tumors with different treatments. Compared with each agent alone, the combination of sitravatinib and anti-PD-L1 significantly increased the levels of lipid ROS and MDA contents in both in Res1-6 tumors (Figures 7E and 7F) and Res-HCA-1 tumors (Figures 7G and 7H).
Figure 7.

Sitravatinib combined with PD-L1 antibody increases ferroptosis and reduces MDSC infiltration in HCC microenvironment
(A and B) (A) T-SNE plot of tumor-infiltrating leukocytes overlaid with color-coded clusters and the frequency of clusters of the indicated immune cell subsets, including CD3+ T cells, CD8+ T cells, IFNγ+CD8+ T cells, CD4+ T cells, CD11b+ cells, and MDSCs in Res1-6 strains treated with IgG, sitravatinib, anti-PD-L1, or their combination and (B) the statistical analysis.
(C) Western blot analysis of p-MerTK, MerTK, SLC7A11, and β-actin expression in different groups.
(D) The representative imagines of IHC staining of p-MerTK, MerTK, and Ki-67 from subcutaneous tumors treated with IgG, sitravatinib, anti-PD-L1 or their combination. Scale bar: 100 μm.
(E–H) In subcutaneous xenograft mouse model, the statistical analysis of relative lipid ROS and MDA content in Res1-6 and Res-CA1 strains treated with IgG, sitravatinib, anti-PD-L1, or their combination.
All results are shown as mean ± SEM (n = 5). One- or two-way ANOVA was used to analyze the data; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
In recent years, ICI-based therapies (PD-1/PD-L1 blockade) have achieved unprecedented success for the treatment of advanced HCC patients. However, only a fraction of HCC patients derive long-term benefit, and a large number of responders acquire resistance after initial responses,2,3,5 highlighting the urgent need to understand the underlying mechanisms of resistance. Our results from the resistant mouse models demonstrate that MerTK induces anti-PD-1/PD-L1 resistance by suppressing ferroptosis and favoring a protumor TME as a potential mechanism by which HCC escapes from PD-1/PD-L1 blockade. Moreover, the analysis of clinical HCC data also supports a positive association between MerTK and SLC7A11. Notably, our findings indicate that blocking MerTK increases ferroptosis and enhances the efficacy of immune-checkpoint-based therapies in HCC, which supports that MerTK inhibitor may be used as an adjuvant for immune-checkpoint-based treatment of HCC.
Individual tumors induce distinct pathways of immune checkpoint reprogramming to escape immune surveillance. Previous work reported that resistance to PD-1/PD-L1 blockade therapy can be caused by both tumor-intrinsic and tumor-extrinsic factors.28 Here, we established in vivo resistant mouse models to investigate the potential mechanisms involved in anti-PD-L1/PD-1 resistance in HCC. Through an analysis of the proteomics in models with PD-L1 resistance, our study demonstrates that the HCC cells developed resistance to PD-L1 through the upregulation of MerTK. Previous studies have found that MerTK is required for tumor proliferation and metastasis.7 In addition, the MerTK signaling pathway plays an essential role in contributing to an immunosuppressive TME via the regulation of macrophage, MDSC, T cell, and natural killer (NK) cell functions.29 Recently, Liu et al.10 found that MerTK ablation increased ROS production and promoted a switch from a glycolytic metabolism to oxidative phosphorylation in HCC. Our data, obtained in humans and mice, demonstrate that MerTK is critical for immunotherapy resistance, suggesting that MerTK could be considered a possible predictive biomarker for patient selection and a promising therapeutic target to overcome anti-PD-1/PD-L1 resistance in HCC.
Previous studies have shown that tumor cells that escape from ferroptosis promote anti-PD-1/PD-L1 therapy resistance.22,24 On the other hand, immune cell ferroptosis in the TME such as neutrophil ferroptosis increases the release of oxygenated lipids to create an immunosuppressive TME and limits the activity of tumor-infiltrating CD8+ T cells.30 In this study, we found that MerTK induces anti-PD-L1 resistance by suppressing tumor cell ferroptosis through the upregulation of the ferroptosis inhibitor SLC7A11 via the ERK/SP1 pathway. Our findings revealed a direct link between MerTK and ferroptosis networks in resistant HCC, and broaden our understanding of the regulatory mechanism of immunotherapy resistance on the basis of PD-1/PD-L1 blockade. Because of the multiple biological functions of the MerTK signaling pathway,7,26 the finding that MerTK is a regulator of ferroptosis in HCC with acquired resistance to PD-1/PD-L1 blockade implies that ferroptosis contributes to many kinds of physiological and pathological states.
In the context of ICI-based therapies, a comprehensive analysis of the TME needs to be implemented. Here, we uncovered that anti-PD-L1 therapy-resistant HCC exhibited increased MDSC infiltration via CSF3 induced by MerTK. MDSCs are universally acknowledged as the cornerstone of an immunosuppressive TME.31 Our study reveals that the TME undergoes adaptive reprogramming with the recruitment of MDSCs under the continued pressure of PD-L1/PD-1 blockade, which provides an insight into how MerTK regulates an immunosuppressive TME in HCC with acquired resistance to PD-1/PD-L1 inhibitors.
There is great interest in developing rational combination therapy strategies by overcoming immune resistance and further enhancing antitumor efficacy. Combining ferroptosis induction with MDSCs blockade renders primary tumors and metastases in the liver sensitive to immune checkpoint blockade.32 Here, we demonstrated that the MerTK axis affects the efficacy of immunotherapy by regulating ferroptosis, suggesting that targeting MerTK is an attractive strategy to enhance the efficacy of anti-PD-1 antibody in HCC. Using preclinical animal models, we found that sitravatinib, a tyrosine kinase inhibitor (TKI) that targets MerTK, could overcome immune checkpoint blockade resistance by inducing ferroptosis and the infiltration of CD8+ T cells and IFNγ+CD8+ T cells, and reducing MDSCs recruitment in the TME. Sitravatinib, an oral TKI that potently targets the TAM (TYRO3, AXL, and MERTK) receptors, vascular endothelial growth factor receptor (VEGFR) family, c-Kit, c-MET, FLT3, and the platelet-derived GFR (PDGFR) family.33,34 Du W. et al. found that sitravatinib could effectively inhibit MerTK activation and possessed potent antitumor activity via targeting the TME, resulting in innate and adaptive immune cell changes that augment immune checkpoint blockade.9 As with all preclinical model systems and human clinical and pathological correlations, our results suggested a potential translational strategy to overcome ICI resistance mediated by MerTK in HCC therapy. Interestingly, the combination of sitravatinib and ICIs is being assessed in a phase I trial in HCC (NCT03941873), which further substantiates the hypotheses of our work. Furthermore, our study showed that level of MerTK expression was positively correlated with the efficacy of the anti-PD-L1 therapy in two syngeneic mouse models and in patients who had received anti-PD-1/PD-L1 therapy, suggesting that MerTK could be used for clinical application as a predictor and target of the response to cancer immunotherapy.
In conclusion, the present study demonstrated that MerTK upregulates SLC7A11 expression to suppress tumor cell ferroptosis, favors a protumor TME by recruiting MDSCs in the HCC microenvironment, and, consequently, drives anti-PD-L1 therapy resistance. Targeting MerTK is an effective strategy to increase the therapeutic efficacy of PD-L1 antibody. The combination of MerTK inhibitor, sitravatinib, and ICIs can efficiently increase tumor cell ferroptosis, reduce MDSCs recruitment, and promote CD8+ T cell activation in PD-L1-resistant TME.
Limitations of the study
Our study has a clear translational outlook, and it is important to know that sitravatinib is an inhibitor of many other receptor tyrosine kinases, such as Axl, KIT, FLT3, VEGFR3, VEGFR2, and VEGFR1. Some of them are targeted by therapies, such as bevacizumab, which has been successfully combined with other treatments, or other approved TKIs. Nevertheless, our goal was to discover the potential underlying mechanisms of anti-PD-1/PD-L1 therapy resistance and find strategies for overcoming this resistance. We believe that the combination strategy of sitravatinib and immune checkpoint blockade to treat HCC may demonstrate a long-term benefit and therapeutic value for HCC patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse PD-L1 antibody | Bio X Cell | Cat#BE0361; RRID:AB_2927503 |
| Anti-mouse SLC7A11 antibody | Proteintech | Cat#26864-1-AP; RRID:AB_2880661 |
| Anti-mouse MerTK antibody | SAB Signalway Antibody | Cat#32851 |
| Anti-mouse p-MerTK antibody | SAB Signalway Antibody | Cat#13320 |
| Anti-mouse ki67 antibody | Abcam | Cat# ab92742; RRID:AB_10562976 |
| Anti-mouse CD3 antibody | Abcam | Cat#ab16669; RRID:AB_443425 |
| Anti-mouse CD8 antibody | Abcam | Cat#ab237709; RRID:AB_2892677 |
| Anti-mouse CD11b antibody | Abcam | Cat#ab52478; RRID:AB_868788 |
| Anti-mouse Gr1 antibody | Abcam | Cat#ab25377; RRID:AB_470492 |
| Anti-mouse ERK 1/2 antibody | Cell Signaling Technology | Cat#9102; RRID:AB_330744 |
| Anti-mouse p-ERK1/2 (T202/Y204) antibody | Cell Signaling Technology | Cat#4370; RRID:AB_2315112 |
| Anti-mouse SP1 antibody | Proteintech | Cat#21962-1-AP; RRID:AB_10898171 |
| Anti-mouse p-SP1 (T453) antibody | Abcam | Cat#ab59257; RRID:AB_946335 |
| Anti-human CD15 antibody | Abcam | Cat#ab135377 |
| Anti-human CD14 antibody | Abcam | Cat#ab133335; RRID:AB_2889158 |
| Anti-human CD11b antibody | Abcam | Cat#ab133357; RRID:AB_2650514 |
| Anti-mouse β-actin antibody | Proteintech | Cat#60008-1-Ig; RRID:AB_2289225 |
| Anti-mouse IgG antibody | Bio X Cell | Cat#BE0089; RRID:AB_1107769 |
| Anti-human HRP secondary antibody | Yuanxibio | Cat#A10011-60 |
| BUV395 anti-mouse CD45 antibody | BD Biosciences | Cat#564279; RRID:AB_2651134 |
| PE/cy7 anti-mouse CD3 antibody | BioLegend | Cat# 100219; RRID:AB_1732068 |
| BV/785 anti-mouse CD4 antibody | BioLegend | Cat#100552; RRID:AB_2563053 |
| BV/510 anti-mouse CD8 antibody | BioLegend | Cat#100752; RRID:AB_2563057 |
| Bv421 anti-mouse IFN-γ antibody | BioLegend | Cat#505829; RRID:AB_10897937 |
| PE anti-mouse CD11b antibody | BioLegend | Cat#101208; RRID:AB_312791 |
| FITC anti-mouse Ly6G antibody | BioLegend | Cat#127606; RRID:AB_1236494 |
| Bv421 anti-mouse Ly6C antibody | BioLegend | Cat# 128031; RRID: AB_2562177 |
| Percp/cy5.5 anti-mouse Gr1 antibody | BioLegend | Cat#108428; RRID:AB_893558 |
| APC anti-mouse F480 antibody | BioLegend | Cat#123115; RRID:AB_893493 |
| Bacterial and virus strains | ||
| Escherichia coli | Tsingke | Cat# Tsingke-001 |
| Biological samples | ||
| Human PAAD tissue microarrays | Huashan Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sitravatinib | MCE | Cat#HY-16961 |
| SYBR | TaKaRa | Cat#RR067A |
| Red blood cell lysis buffer | Tiangen | Cat#RT122-01 |
| CFSE | Invitrogen | Cat# 65-0850-84 |
| BODIPY 581/591 C11 | Invitrogen | Cat# D3861 |
| Pkh67 Green Fluorescent Cell Linker Kit | Sigma-Aldrich | Cat# PKH67GL |
| Propidium iodide | Invitrogen | Cat#P1304MP |
| SYTOX Green | Invitrogen | Cat#S7020 |
| Critical commercial assays | ||
| IFN-γ ELISA Kit | R&D Systems | Cat# MIF00 |
| CSF3 ELISA Kit | R&D Systems | Cat# MCS00 |
| Tumor Dissociation Kit | Miltenyi Biotec | Cat#130-096-730 |
| Cell Counting Kit-8 | Dojindo | Cat#CK04-11 |
| MDSCs Isolation Kit | Miltenyi Biotec | Cat#130-094-538 |
| Zombie NIRTM Fixable Viability Kit | BioLegend | Cat#423105 |
| Critical commercial assays | ||
| Lipid Peroxidation MDA Assay Kit | Beyotime | Cat#S0131S |
| Deposited data | ||
| Raw and analyzed expression profiling by array | https://www.ncbi.nlm.nih.gov/geo/ | GEO: GSE76427 |
| Raw and analyzed expression profiling by array | https://www.ncbi.nlm.nih.gov/geo/ | GEO: GSE36376 |
| Proteomics data | This paper; iProX Data | iProX: IPX0007790000 |
| Experimental models: Cell lines | ||
| Human: 293T | Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai China | Cat# SCSP-502 |
| Human: HepG2 | Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai China | Cat# SCSP-510 |
| Human: Hep3B | Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai China) | Cat# SCSP-5045 |
| Human: Huh7 | Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai China) | Cat# SCSP-526 |
| Human: MHCC97-H | Liver Cancer Institute, University of Fudan University, China | N/A |
| Human: HCC-LM3 | Liver Cancer Institute, University of Fudan University, China | N/A |
| Mouse: Hepa1-6 | Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai China | Cat# SCSP-512 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Beijing Vital River Laboratory | Cat#219 |
| Mouse: C3H | Beijing Vital River Laboratory | Cat#212 |
| Oligonucleotides | ||
| shRNA target sequences for SLC7A11/MerTK, see Table S1 | This paper | N/A |
| SLC7A11/MerTK cDNA clone, see Table S1 | This paper | N/A |
| PCR primers for β-actin/SLC7A11/MerTK/Csf1/Csf2/Csf3/Cxcl1/Cxcl2, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pCDH-CMV-MCS-EF1-puro | Ke Lei Biological Technology | Cat# kl-zl-0287-01 |
| psPAX | Ke Lei Biological Technology | Cat#12260-20ul |
| pMD2.G | Ke Lei Biological Technology | Cat#12259-20ul |
| pLKO.1-Puro | Ke Lei Biological Technology | Cat#kl-zl-0961-01 |
| pCMV-Gag-pol | Ke Lei Biological Technology | Cat# kl-zl-0178-01 |
| VSVG | Ke Lei Biological Technology | Cat# kl-zl-0955-01 |
| Software and algorithms | ||
| Prism 9.0.0 | GraphPad Software | https://graphpad.com/scientific software/prism/ |
| RStudio (v1.4.1106) | RStudio | https://www.rstudio.com/ |
| TIMER2.0 | Dana Farber Cancer Institute | http://timer.cistrome.org/ |
| FlowJo v10.7.2 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| Fiji | ImageJ | https://imagej.net/software/fiji/ |
| SlideViewer | 3DHISTECH | https://www.3dhistech.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Qiongzhu Dong (qzhdong@fudan.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data needed to evaluate the conclusions in the paper are present in the paper or the supplemental information.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Datasets, patients and follow-up
The RNA-sequencing-based gene expression data and clinical data of 370 HCC tumor samples were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/). Data from GSE76427 and GSE36376 were retrieved from the public GEO database (http://www.ncbi.nlm.nih.gov/geo). The “R” software was used to normalize the original RNA-sequencing data to transcript per million reads (TPM).
Ten patients who had curative resection for HCC at authors’ institutes from September 2019 to October 2021 were enrolled in this study. All patients who had surgical resection were diagnosed with HCC and agreed with written informed consent in accordance with ethical approval by the Ethics Committee of Huashan Hospital, Fudan University (Shanghai, China). All patients had complete clinical cases and follow-up data. During the follow-up period, the routine blood tests, liver and kidney function and tumor markers were re-examined every month. Magnetic resonance imaging (MRI) scan or computed tomography (CT) was monitored every 6 months.
Cell cultures and animals
HEK 293T cells, human HCC cell lines (HepG2, Hep3B, and Huh7) and mouse HCC cell line (Hepa1-6) were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai China). Human HCC cell lines (MHCC97-H, HCC-LM3), and mouse HCC cell line (HCA-1) were obtained from the Liver Cancer Institute at Fudan University (Shanghai, China) as previously described.35,36 These cell lines were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) (Hyclone) supplemented with 10% fetal bovine serum (FBS) (Gibco). All cell lines were routinely maintained at 37.0°C in a humidified incubator with 5% CO2.
Male C57BL/6 mice (8-weeks-old, 25-27g) and C3H mice (8-weeks-old, 25-30g) were purchased from Shanghai Slac Laboratory Animal Co. and maintained on standard rodent chow and water ad libitum. Prior to any surgical procedures, all mice were given intraperitoneal (i.p.) injection of Pentobarbital Sodium (40 mg/kg; Sigma Aldrich, St.Louis, MO, USA) and conducted in the SPF laboratory. All procedures involving animals were approved by The Animal Care and Use Committee of Fudan University (Shanghai, China).
Animal studies and establishment of resistance models
To establish the subcutaneous HCC model, Hepa1-6 cells or HCA-1 cells (2 × 106 cells in 100 μL of phosphate buffer saline [PBS]) were injected into the right flanks of mice. Mice bearing tumors about 50 mm3 in volume were divided in 2 groups. For treatment with antibody, 200 μg mouse anti-PD-L1 antibody (BE0361, Bio X Cell) or IgG control (BE0089, Bio X Cell) was injected intraperitoneally (i.p.) twice a week. For the in vivo anti-PD-L1 resistant model, Hepa1-6 cells or HCA-1 cells were inoculated into the right flanks of mice. Anti-PD-L1 antibody was administered to the mice 6 times at 3-day intervals. Tumors were harvested and dissociated for culturing. The treatment cycle was repeated 3 more times to obtain Hepa1-6 anti-PD-L1 antibody resistant cells (Res1-6 cells) or HCA1 anti-PD-L1 antibody resistant cells (Res-CA1 cells). For the orthotopic implantation HCC model, subcutaneous tumors were collected and minced into 1 mm3 sections, and then inoculated into the left liver lobes of mice to establish orthotopic implantation model. To evaluate tumor metastasis in vivo, lungs were collected and fixed in 4% formalin when the mice were sacrificed. Lung metastatic nodules were calculated by hematoxylin and eosin (HE) staining on consecutive sections. Survival analysis was performed using GraphPad Prism 8 (GraphPad Software).
For mouse HCC cells including Res1-6-sh-control, Res1-6-shMerTK, Hepa1-6-OE-control, and Hepa1-6-OE-MerTK cells (2 × 106 cells in 100 μL of PBS) were injected into the right flank of C57BL/6 mice (5 mice/group). All tumors were collected, and the tumor weight was measured. For the sitravatinib and anti-PD-L1 antibody combination treatment study, mice were treated daily with vehicle control (90% corn oil and 10% DMSO) or sitravatinib (20 mg/kg, HY-16961, MCE) once per day for 6 days a week by oral administration, and 200 μg mouse anti-PD-L1 antibody (BE0361, Bio X Cell) or IgG control (BE0089, Bio X Cell) (i.p.) twice a week. Tumor volume was measured using the formula: 1/2 × (length × width2), that the length is the longest diameter of the tumor and width is the shortest diameter.
Method details
Enzyme-linked Immunosorbent assays (ELISAs)
The cell culture medium or tumor tissue homogenate were obtained before operation and separated by centrifugation (10,000 g for 10 min) at 4°C. Double-blinded quantitative detection of IFN-γ and CSF3 were performed using the mouse IFN-γ ELISA kit (R&D Systems, USA) and CSF3 ELISA kit (R&D Systems, USA) according to the manufacturer’s instructions. ELISA plates (MaxiSorp, Nunc) were coated over night at 4°C with 4 μg/mL goat anti-mouse IgM (KPL) in PBS pH 7.4. After washing the plates two times with Blocking Buffer (PBS pH 7.4, 0.25% BSA, 0.05% Tween 20), plates were blocked with Blocking Buffer for 2 h at RT. Plates were washed two times with PBS pH 7.4, stabilized and dried using a commercial plate stabilizer (Liquid PlateSealer, Candor) according to the manufacturer’s recommendations. The anti-IFN-γ and anti-CSF3 antibodies in medium were diluted 1:100 in Dilution Buffer (PBS pH 7.4, 0.05% Proclin 300, 0.01% phenol red). Per well, 50 μL of cell culture medium was applied and the plates were incubated for 60 min at RT (23°C). After washing the plates three times with Wash Buffer (100 mM Tris/HCl pH 7.4, 150 mM NaCl, 0.05% Tween 20, 0.005% ProClin 300, 300 μL/well), 50 μL HRP-labeled CCHFV-NP (final dilution 1:50,000 in Conjugate Dilution Buffer (PBS pH 7.4, 1% BSA, 0.5% FCS, 1% Nonidet P40, 0.1% ProClin 300) was added and the plates were incubated for 60 min at RT (23°C). Plates were washed again three times with Wash Buffer (300 μL/well), and 100 μL SureBlue Reserve TMB Microwell Peroxidase Substrate (KPL) was added per well. Plates were incubated for 20 min at RT (23°C) and the reaction was stopped by the addition of 100 μL 1N sulfuric acid (Merck) per well. The HRP reaction product was quantified by measuring optical density at 450 nm and 620 nm on a Spectrostar Nano ELISA reader (BMG Labtech).
TMT proteomics analysis and bioinformatic analysis
Hepa1-6 and Res1-6 tumor cells were harvested and sonicated three times on ice using a high intensity ultrasonic processor (Scientz) in lysis buffer (8 M urea, 5 μM TSA and 10 mM NAM, and 1% Protease Inhibitor Cocktail). Then, 1:50 trypsin-to-protein mass ratio trypsin digestion was performed overnight with the protein solution, 1:100 trypsin-to-protein mass ratio digestion was performed for 4 h. Tryptic peptides were firstly dissolved in 0.5 M TEAB. Each peptide channel was labeled with its respective TMT reagent (based on manufacturer’s protocol, Thermo Fisher Scientific, #90068). Measurement conditions of liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed on a Q Exactive mass spectrometer (Thermo Scientific) that was coupled to Easy nLC (Proxeon Biosystems, now Thermo Fisher Scientific) for 90 min. The mass spectrometer was operated in positive ion mode. MS data was acquired using a data-dependent top10 method dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. Automatic gain control (AGC) target was set to 3e6, and maximum inject time to 10 ms. Dynamic exclusion duration was 40.0 s. Survey scans were acquired at a resolution of 70,000 at m/z 200 and resolution for HCD spectra was set to 17,500 at m/z 200, and isolation width was 2 m/z. Normalized collision energy (NCE) was 30 and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled. The MS raw data for each sample were searched using the MASCOT engine (Matrix Science, London, UK; version 2.2) embedded into Proteome Discoverer (PD) 1.4 software (Thermo Scientific) for identification and quantitation analysis.
Functional analysis was performed with Encyclopedia of Genes and Genomes (KEGG) database (http://kobas.cbi.pku.edu.cn/), and Gene Ontology (GO) annotation (UniProt-GOA database: http://www.ebi.ac.uk/GOA/). Fisher’s exact test was used to test differentially modified proteins with a p value less than 0.05. The significance of pathway enrichment was calculated by Fisher exact test and corrected by BH algorithm, multiple tests with a corrected p value <0.05 are considered significant.
Migration and invasion assays
The migration and invasion ability of HCC cells were determined by using 24-well transwell chambers, with upper and lower culture compartments separated by polycarbonate membranes with 8 μm pores (Costar) coated with or without Matrigel (Corning). The bottom chamber was filled with DMEM containing 20% FBS as a chemoattractant. The upper chamber, 5 × 104 cells, for migration/invasion, were seeded into and maintained at 37°C in a humidified incubator containing 5% CO2 with serum-free medium. Cells that migrated to the underside of the membrane were stained with crystal violet, imaged, and counted with light microscope (×100, Leica). All the in vitro assays were conducted three times with three repetitions.
Plasmid and cell transfections
To establish stable MerTK or SLC7A11 overexpression cell lines, MerTK cDNA (Mouse: NP_032613.1) or SLC7A11 cDNA (Mouse: NP_036120.1) was cloned into pCDH-CMV-MCS-EF1-puro vector. The recombinant vector was co-transfected into HEK293T cells with two other packaging plasmids psPAX2 and pMD2.G using Lipofectamine 3000 (Invitrogen, MA, USA). For the stable MerTK or SLC7A11 knockdown cell lines, short hairpin RNA (shRNA) sequences against the MerTK (mouse) sequence or SLC7A11 (mouse) sequence were cloned into the pLKO.1 puro retroviral vector. The recombinant vectors were co-transfected into HEK293T cells with GAG-POL and VSVG plasmid using Lipofectamine 3000. All the primers are listed in Table S1.
RNA isolation and real-time quantitative reverse transcription PCR (qRT-PCR)
RNA isolation and cDNA obtaining were performed as described previously.35,37 For qRT-PCR analysis, cDNA was amplified using SYBR Green Real-time PCR Master Mix (Takara, Japan). qRT-PCR reactions were performed in triplicates as the following conditions: 95°C/20s, 40 cycles of 95°C/60s and 60°C/20s using the ABI PRISM 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and repeated at least three times. Relative mRNA levels were analyzed by the -ΔΔCt method using β-actin as the endogenous control and presented as 2ˆ(−ΔCt). All the primers are listed in Table S1.
Flow cytometry
The single cells were isolated from mouse subcutaneous xenograft tumor by a semi-automated combined mechanical/enzymatic process using the gentleMACS Dissociator and the Tumor Dissociation Kit (mouse, Miltenyi Biotec, Germany). Red blood cells were lysed using red blood cell lysis buffer (Tiangen, China). Cell suspensions were blocked with mouse FcR blocking reagent (Miltenyi Biotec, USA) and then stained with antibodies against mouse CD45 (BD Biosciences, #564279), CD3 (BioLegend, #100219), CD4 (BioLegend, #100552), CD8 (BioLegend, #100752), IFN-γ (BioLegend, #505829), CD11b (BioLegend, #101208), Gr-1 (BioLegend, #108428), F480 (BioLegend, #123115) and matched isotype controls, depending on the experiment. For IFN-γ staining, single cells were restimulated with 1 μL/mL Cell Activation Cocktail (with Brefeldin) in 5% CO2 before intracellular staining at 37°C for 5h. These antibodies were detected by: BUV395 anti-mouse CD45 antibody, PE/cy7 anti-mouse CD3 antibody, BV/785 anti-mouse CD4 antibody, BV/510 anti-mouse CD8 antibody, Bv421 anti-mouse IFN-γ antibody, PE anti-mouse CD11b antibody, Percp/cy5.5 anti-mouse Gr1 antibody, APC anti-mouse F480 antibody. Samples were run on a BD FACSVerse (BD Biosciences, USA) and analyzed using FlowJo software (BD Biosciences, USA).
Immunohistochemistry (IHC) and tissue microarrays (TMA) of HCC tissues
IHC staining was performed as described previously.37 Briefly, following deparaffinization, rehydrating and antigen retrieval, primary antibodies were applied to slides, incubated at 4°C overnight, followed by incubation with secondary antibody (Dako Denmark A/S, Glostrup, Denmark) at 37°C for 30 min. SLC7A11 antibody (Proteintech, #26864-1-AP), MerTK antibody (SAB Signalway Antibody, #32851), p-MerTK antibody (SAB Signalway Antibody, #13320), Ki67 antibody (Abcam, #ab92742), CD3 antibody (Abcam, #ab16669), CD8 antibody (Abcam, #ab237709), CD11b antibody (Abcam, #ab52478), Gr1 antibody (Abcam, #ab25377) were used as primary antibodies. Staining was carried out with DAB (3,3′-diaminobenzidine) and counterstaining was performed with hematoxylin.
We applied IHC staining analysis on a cohort of 98 HCC patients. Clinical samples from patients were obtained after acquiring their consent in accordance with the protocol approved by the Ethics Boards of Huashan Hospital, Fudan University (Shanghai, China). Formalin-fixed and paraffin-embedded tissues were used to construct TMA as previously described.37 Briefly, two four-micron core biopsies from the donor blocks were taken and transferred to the recipient paraffin block at predefined array positions and constructed 98 cases of TMA blocks in this study. Scoring for MerTK and SLC7A11 staining was conducted using percentage score × staining intensity score. The percentage of positive-staining cells: 0%–5% scored 0, 6–25% scored 1, 26–50% scored 2, 51–75% scored 3 and more than 75% scored 4; staining intensity: no staining scored 0, weakly staining scored 1, moderately staining scored 2 and strongly staining scored 3.
Multiplex immunofluorescence (mfIHC)
The steps of multiplex immunofluorescence staining are as follows. Briefly, immunofluorescence (IF) staining was performed on 4 μm-thick formalin-fixed, paraffin-embedded whole-tissue sections using standard primary antibodies, followed by the TSA 7-color kit (WiSee Bio, #D110071-50T) and subsequent DAPI staining. For example, the deparaffinized slides were incubated with the primary antibody for 30 min, followed by a 10-min treatment with an anti-rabbit/mouse horseradish peroxidase (HRP) secondary antibody (Yuanxibio, #A10011-60). The procedure for immunofluorescence labeling was executed meticulously, employing TSA 620 and adhering strictly to the manufacturer’s guidelines. Slides were washed in TBST buffer and then transferred to a pre-warmed citrate solution (98°C) for 15 min. The slides were then allowed to cool to room temperature in the same solution. The slides were rinsed with Tris buffer in between each stage. This procedure was replicated for each antibody/fluorescent dye. Thereafter, two drops of DAPI were applied to each slide, followed by a rinse in distilled water and manual cover-slipping. The slides were dried in the air, mounted with anti-fade mounting media, and photographed using the Aperio Versa 8 tissue imaging equipment (Leica). Indica Halo software was used to analyze the images. The antibodies used in this study were MerTK (Preoteintech, #27900-1-AP), SLC7A11 (Preoteintech, #26864-1-AP), CD8 (Abcam, #ab237709), CD15 (Abcam, #ab135377), CD14 (Abcam, #ab133335), and CD11b (Abcam, # ab133357).
Cell proliferation assays
The cell proliferation assay was carried out by using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Cancer cells were seeded into 96-well plates at a density of 3 × 103/100μL cells per well (n = 5 for each time point) and incubated for 12, 24, 36, 48, 60 and 72 h. Then the medium was replaced with 10 μL CCK-8 solution and 90 μL complete medium. The absorbance at 450 nm was measured after incubation for 1 h at 37°C in 5% CO2.
MDSCs isolation and coculture assays
MDSCs were isolated from tumor tissue collected from tumor-bearing mice using a mouse MDSCs Isolation Kit (Miltenyi Biotec, America) according to the manufacturer’s instruction. For coculture assays, prior to the experiment, the plates (6-well) were pre-coated with 1mL of PBS containing 5 μg/mL anti-CD3 antibody and incubated overnight at 4°C. The extracted CD8+ T lymphocytes were counted, centrifuged, and the supernatant was discarded. The T lymphocytes were then resuspended in 2 mL of RPIM 1640 complete culture medium containing 5 μg/mL CFSE and incubated for 20 min at 37°C. The incubation was terminated with 10 mL of complete culture medium, followed by centrifugation and discarding of the supernatant. Each well was then seeded with 1 × 106 CFSE-labeled CD8+ T lymphocytes. In the wells containing T lymphocytes with activators, 0.5 × 106 MDSCs, 2 × 106 MDSCs, and 5 × 106 MDSCs were added respectively. The culture medium was adjusted to 2.5 mL, and 5 μg/mL anti-CD28 was added. The plates were then incubated at 37°C with 5% CO2 for four days. After four days, the cells in the culture plate were collected and analyzed for CFSE level.
MDSCs chemotaxis assay
HCC cells were firstly added to the bottom layer of a 12-well 5 μm transwell chamber at the concentration of 104 cells/well, cultured in DMEM containing 10% fetal bovine serum (FBS) at 37.0°C in a humidified incubator with 5% CO2 for 6 h. The MDSCs were pre-labeled with CSFE fluorescence and added to the upper layer of transwell chamber at the concentration of 105 cells/well, cultured at 37.0°C in a humidified incubator with 5% CO2 for another 3 h. The fluorescence positive MDSCs were photographed with a fluorescence microscope and analyzed with ImageJ software. The experiment was repeated 5 times independently.
Western blot
Western blot was carried out as described before.35 Total protein was extracted by RIPA buffer containing protease cocktail inhibitor. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% skim milk in TBST (BD Pharmingen), PVDF membranes were incubated with the primary antibody overnight at 4°C and then with the secondary antibody for 1 h at room temperature. The following antibodies were used: MerTK antibody, p-MerTK antibody, ERK antibody, p-ERK antibody, SP1 antibody, p-SP1 antibody, SLC7A11 antibody, and β-actin antibody. Protein bands were detected by image acquisition using ImageQuantTM LAS 4000 (GE Healthcare Life Sciences).
BODIPY-C11 staining
Hepa1-6 and Res1-6 cells (1 × 105 cells/well) were seeded in 6-well plates and treated with erastin, and the cells were then stained with DMEM culture containing 5 μM BODIPY 581/591 C11 and incubated for 30 min at 37°C in a cell culture incubator. Cells were washed twice using PBS and photographed with a fluorescence microscope and analyzed with ImageJ software. For tumor cells in vivo, the single cells were isolated from mouse subcutaneous xenograft tumor by a semi-automated combined mechanical/enzymatic process using the gentleMACS Dissociator and the Tumor Dissociation Kit (mouse, Miltenyi Biotec, Germany), and the cells were then stained with 5 μM BODIPY 581/591 C11. Cells were washed twice using PBS and analyzed immediately by flow cytometer, and the signals from both nonoxidized C11 (phycoerythrin [PE] channel) and oxidized C11 (FITC channel) were monitored. The experiment was repeated 5 times independently and shown as relative lipid ROS.
Cell survival ability and cell death assays
To examine cell viability of Hepa1-6 and Res1-6 strains under erastin treatment coculture condition, Hepa1-6 cells were labeled with the green fluorescent dye PKH67 (Sigma-Aldrich, St. Louis, MO) and Res1-6 cells were labeled with the red fluorescent dye PKH26 (Sigma-Aldrich, St. Louis, MO), respectively. The fluorescence positive cells were photographed with a fluorescence microscope and analyzed with ImageJ software (the National Institutes of Health, Bethesda, MD, USA). Cell death was detected by staining for SYTOX Green (Invitrogen) or propidium iodide (Invitrogen) followed by fluorescence microscope or flow cytometry. All experiments were repeated 5 times independently.
Malonaldehyde (MDA) assay
MDA content in tumor cells was measured using the Lipid Peroxidation MDA Assay Kit (S0131S, beyotime). The single cells were isolated from mouse subcutaneous xenograft tumor by a semi-automated combined mechanical/enzymatic process using the gentleMACS Dissociator and the Tumor Dissociation Kit (mouse, Miltenyi Biotec, Germany) and homogenized in MDA lysis buffer, and the cell lysate was sonicated and centrifuged to remove insoluble material. The supernatant (200 μL) was mixed with 600 μL TBA solution and incubated at 95°C for 1 h. The MDA-TBA complex concentration was measured on a microplate reader with relative fluorescence units at Ex/Em = 532/553 nm or OD = 532 nm and was normalized to the protein concentration.
Quantification and statistical analysis
Statistical analyses were performed using the Statistical Package for GraphPad Prism 8.0 software. The Student’s t test and one- or two-way ANOVA analysis were used for comparison between groups. For all tests, significance was determined with a 95% confidence interval (ns, p > 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Acknowledgments
The authors acknowledge the members of the Huashan Hospital Central Laboratory, especially Xiao Xu for excellent technical assistance. This work was supported by the National Key Research and Development Program of China (grant 2023YFC2413200/2023YFC2413201), the National Natural Science Foundation of China (grant 82373017), the China Postdoctoral Science Foundation (grant 2023M734043), and a grant provided by the National Research Center for Translational Medicine at Shanghai, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine (Shanghai, China) (NRCTM[SH]-2023-03).
Author contributions
Data analysis, Q.D. and S.W.; visualization, validation, writing, and original draft preparation, S.W., L. Zhu, Z.Z., T.L., X.S., D.X., L. Zhang, N.L., and X.L.; project administration, Q.D. and L.Q.; funding acquisition: Q.D. and L.L.; clinical data collection, S.W., L.L., and Y.G.; experiment assistance, Y.F., H.W., T.Z., X.W., and Y.Z.
Declaration of interests
The authors declare no competing interests.
Published: February 20, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101415.
Contributor Information
Lu Lu, Email: lulu@huashan.org.cn.
Lunxiu Qin, Email: qinlx@fudan.edu.cn.
Qiongzhu Dong, Email: qzhdong@fudan.edu.cn.
Supplemental information
References
- 1.Llovet J.M., Kelley R.K., Villanueva A., Singal A.G., Pikarsky E., Roayaie S., Lencioni R., Koike K., Zucman-Rossi J., Finn R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021;7:6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 2.El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., Kim T.Y., Choo S.P., Trojan J., Welling T.H., Rd, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu A.X., Finn R.S., Edeline J., Cattan S., Ogasawara S., Palmer D., Verslype C., Zagonel V., Fartoux L., Vogel A., et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–952. doi: 10.1016/S1470-2045(18)30351-6. [DOI] [PubMed] [Google Scholar]
- 4.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 5.Llovet J.M., Castet F., Heikenwalder M., Maini M.K., Mazzaferro V., Pinato D.J., Pikarsky E., Zhu A.X., Finn R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022;19:151–172. doi: 10.1038/s41571-021-00573-2. [DOI] [PubMed] [Google Scholar]
- 6.Graham D.K., DeRyckere D., Davies K.D., Earp H.S. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer. 2014;14:769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 7.Huelse J.M., Fridlyand D.M., Earp S., DeRyckere D., Graham D.K. MERTK in cancer therapy: Targeting the receptor tyrosine kinase in tumor cells and the immune system. Pharmacol. Ther. 2020;213 doi: 10.1016/j.pharmthera.2020.107577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Y., Zhang Y., Leung J.Y., Fan C., Popov K.I., Su S., Qian J., Wang X., Holtzhausen A., Ubil E., et al. MERTK mediated novel site Akt phosphorylation alleviates SAV1 suppression. Nat. Commun. 2019;10:1515. doi: 10.1038/s41467-019-09233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du W., Huang H., Sorrelle N., Brekken R.A. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI insight. 2018;3 doi: 10.1172/jci.insight.124184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y., Lan L., Li Y., Lu J., He L., Deng Y., Fei M., Lu J.W., Shangguan F., Lu J.P., et al. N-glycosylation stabilizes MerTK and promotes hepatocellular carcinoma tumor growth. Redox Biol. 2022;54 doi: 10.1016/j.redox.2022.102366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao X., Jiang G., Zhang S., Hu S., Fan Y., Li G., Yu H., He S. LncRNA SNHG16 contributes to osteosarcoma progression by acting as a ceRNA of miR-1285-3p. BMC Cancer. 2021;21:355. doi: 10.1186/s12885-021-07933-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nyakas M., Fleten K.G., Haugen M.H., Engedal N., Sveen C., Farstad I.N., Flørenes V.A., Prasmickaite L., Mælandsmo G.M., Seip K. AXL inhibition improves BRAF-targeted treatment in melanoma. Sci. Rep. 2022;12:5076. doi: 10.1186/s12885-021-07933-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng J., Zhou M., Liao T., Kuang W., Xia H., Yin Z., Tan Q., Li Y., Song S., Zhou E., Jin Y. Targeting Cancer Cell Ferroptosis to Reverse Immune Checkpoint Inhibitor Therapy Resistance. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.818453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stockwell B.R. A powerful cell-protection system prevents cell death by ferroptosis. Nature. 2019;575:597–598. doi: 10.1038/d41586-019-03145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascón S., Hatzios S.K., Kagan V.E., et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang M., Luo H., Yi X., Wei X., Jiang D.S. The epigenetic regulatory mechanisms of ferroptosis and its implications for biological processes and diseases. MedComm. 2023;4:e267. doi: 10.1002/mco2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang X., Green M.D., Wang W., Yu J., Choi J.E., Jiang L., Liao P., Zhou J., Zhang Q., Dow A., et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9:1673–1685. doi: 10.1158/2159-8290.CD-19-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C., Liu X., Jin S., Chen Y., Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol. Cancer. 2022;21:47. doi: 10.1186/s12943-022-01530-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang C., Zhang X., Yang M., Dong X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019;31 doi: 10.1002/adma.201904197. [DOI] [PubMed] [Google Scholar]
- 21.Friedmann Angeli J.P., Krysko D.V., Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer. 2019;19:405–414. doi: 10.1038/s41568-019-0149-1. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Z., Lim S.O., Yan M., Hsu J.L., Yao J., Wei Y., Chang S.S., Yamaguchi H., Lee H.H., Ke B., et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Invest. 2021;131 doi: 10.1172/JCI139434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L., Min J., Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Targeted Ther. 2022;7:378. doi: 10.1038/s41392-022-01229-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W., Green M., Choi J.E., Gijón M., Kennedy P.D., Johnson J.K., Liao P., Lang X., Kryczek I., Sell A., et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koppula P., Zhuang L., Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620. doi: 10.1007/s13238-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du W., Zhu J., Zeng Y., Liu T., Zhang Y., Cai T., Fu Y., Zhang W., Zhang R., Liu Z., Huang J.A. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2021;28:1284–1300. doi: 10.1038/s41418-020-00651-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Q., Liu M., Huang W., Chen X., Zhang B., Zhang T., Wang Y., Liu D., Xie M., Ji X., et al. IL-1β-Induced Elevation of Solute Carrier Family 7 Member 11 Promotes Hepatocellular Carcinoma Metastasis Through Up-regulating Programmed Death Ligand 1 and Colony-Stimulating Factor 1. Hepatology. 2021;74:3174–3193. doi: 10.1002/hep.32062. [DOI] [PubMed] [Google Scholar]
- 28.Pitt J.M., Vétizou M., Daillère R., Roberti M.P., Yamazaki T., Routy B., Lepage P., Boneca I.G., Chamaillard M., Kroemer G., Zitvogel L. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y., Fei M., Zhang G., Liang W.C., Lin W., Wu Y., Piskol R., Ridgway J., McNamara E., Huang H., et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity. 2020;52:357–373.e9. doi: 10.1016/j.immuni.2020.01.014. [DOI] [PubMed] [Google Scholar]
- 30.Kim R., Hashimoto A., Markosyan N., Tyurin V.A., Tyurina Y.Y., Kar G., Fu S., Sehgal M., Garcia-Gerique L., Kossenkov A., et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338–346. doi: 10.1038/s41586-022-05443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veglia F., Perego M., Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conche C., Finkelmeier F., Pešić M., Nicolas A.M., Böttger T.W., Kennel K.B., Denk D., Ceteci F., Mohs K., Engel E., et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774–1782. doi: 10.1136/gutjnl-2022-327909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patwardhan P.P., Ivy K.S., Musi E., de Stanchina E., Schwartz G.K. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget. 2016;7:4093–4109. doi: 10.18632/oncotarget.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minchom A., Popat S. Sitravatinib and Acquired Immune Checkpoint Inhibitor Resistance: A Gem for the Future? J. Thorac. Oncol. 2023;18:830–833. doi: 10.1016/j.jtho.2023.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Zhu Y., Yang J., Xu D., Gao X.M., Zhang Z., Hsu J.L., Li C.W., Lim S.O., Sheng Y.Y., Zhang Y., et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68:1653–1666. doi: 10.1136/gutjnl-2019-318419. [DOI] [PubMed] [Google Scholar]
- 36.Qin Z., Xiang C., Zhong F., Liu Y., Dong Q., Li K., Shi W., Ding C., Qin L., He F. Transketolase (TKT) activity and nuclear localization promote hepatocellular carcinoma in a metabolic and a non-metabolic manner. J. Exp. Clin. Cancer Res. 2019;38:154. doi: 10.1186/s13046-019-1131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li T.E., Wang S., Shen X.T., Zhang Z., Chen M., Wang H., Zhu Y., Xu D., Hu B.Y., Wei R., et al. PKM2 Drives Hepatocellular Carcinoma Progression by Inducing Immunosuppressive Microenvironment. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.589997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data needed to evaluate the conclusions in the paper are present in the paper or the supplemental information.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.