

Abstract
Objective
To investigate the potential protective effects of amitriptyline and fluoxetine in a catecholamine cell model.
Methods
Cultured rat pheochromocytoma (PC12) cells were pretreated with amitriptyline or fluoxetine for 24 or 48 hours and were then subjected to neurotoxic insult (200 μmol/L hydrogen peroxide). Cell viability was determined by measurement of the reduction product of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT). The enzyme activity of superoxide dismutase (SOD) was determined by a commercial SOD assay kit.
Results
The decrease in cell viability induced by hydrogen peroxide was attenuated in PC12 cells pretreated with 100 !#!mu;mol/L amitriptyline for 24 hours or with 50 μmol/L amitriptyline or 50 μmol/L fluoxetine for 48 hours. Pretreatment with either amitriptyline or fluoxetine was associated with increased SOD activity in PC12 cells. Inhibition of SOD activity with diethyldithiocarbamic acid reduced the cytoprotective action of fluoxetine.
Conclusions
These data suggest that the neuroprotective actions of some antidepressants include the upregulation of SOD activity.
Medical subject headings: amitriptyline, fluoxetine, PC12 cells, cell viability, superoxide dismutase, oxidative stress
Abstract
Objectif
Étudier les effets protecteurs possibles de l'amitriptyline et de la fluoxétine dans un modèle cellulaire de catécholamines.
Méthodes
On a appliqué à des cellules cultivées de phéochromocytome (PC12) de rat un prétraitement de 24 ou de 48 heures à l'amitriptyline ou à la fluoxétine, puis on a induit une atteinte neurotoxique (200 μmol/L de peroxyde d'hydrogène). La viabilité des cellules a été établie par la mesure du produit de réduction du bromure de 3-[4,5-diméthylthiazol-2-yl]-2,5-diphényl-tétrazolium (MTT). L'activité enzymatique de la superoxide dismutase (SOD) a été établie au moyen d'une trousse du commerce pour le dosage de la SOD.
Résultats
Le prétraitement des cellules de PC12 au moyen de 100 μmol/L d'amitriptyline pendant 24 heures ou de 50 μmol/L d'amitriptyline ou de fluoxétine pendant 48 heures a atténué la diminution de la viabilité des cellules causée par le peroxyde d'hydrogène. Le prétraitement aussi bien à l'amitriptyline qu'à la fluoxétine a été associé avec une augmentation de l'activité enzymatique de la SOD dans les cellules de PC12. L'inhibition de l'activité de la SOD au moyen de l'acide diéthyldithiocarbamique a réduit l'action de cytoprotection de la fluoxétine.
Conclusions
Ces données semblent indiquer que l'action de neuroprotection de certains antidépresseurs comprend une régulation à la hausse de l'activité de la SOD.
Introduction
Neuroanatomic studies suggest that impairment of neuroplasticity and cellular resilience may be involved in the pathophysiology of major depressive disorder.1,2 Familial studies have highlighted the importance of genetic factors in the development of mood disorders, and it may be that depression occurs when neuronal systems lack the ability to exhibit adaptive plasticity in response to external stressors. Defects in these adaptive responses may be involved in the pathogenesis of depression, making vulnerable individuals more susceptible to mood disorders. Although a common mechanism of action of antidepressant therapies has eluded researchers for many years, recent evidence suggests that antidepressants and other mood stabilizers influence important signalling pathways that regulate neuroplasticity and cell survival. The clinical utility of antidepressant therapy may therefore be related to the downstream effects of these drugs, which are ultimately responsible for reversing dysfunctional adaptive responses and stimulating adaptive neuronal plasticity in patients with depression.3
One potential target of antidepressant regulation is the enzyme copper-zinc superoxide dismutase (SOD1, EC 1.15.1.1). This homodimeric metalloenzyme neutralizes the reactive oxygen species superoxide anion.4 Although SOD1 is found in all aerobic cells, it occurs in higher concentrations in the large pyramidal neurons of the hippocampus and neocortex of the mammalian brain.5,6 This endogenous antioxidant enzyme functions to reduce the oxidative stress of a cell and to prevent premature aging or neuronal death.7 Various neuroprotective factors have been shown to upregulate the transcription or enzyme activity of SOD1, including ginsenosides,8 neurotrophins9 and L-deprenyl.10 SOD1 is also upregulated by brain-derived neurotrophic factor (BDNF) and nerve growth factor.11 Furthermore, in vivo studies have established that upregulation of this enzyme is associated with neuroprotective capabilities in both ischemia9 and glutamate toxicity,12 whereas its downregulation induces apoptotic cell death of cultured neurons13 and PC12 cells.14 Glucocorticoids also cause downregulation of SOD1, in addition to their being implicated in the development of major depressive disorder.15 If episodes of clinical depression are accompanied by progressive hippocampal atrophy throughout the duration of the disease, antidepressant therapy or other forms of treatment that upregulate SOD1 gene expression may prevent worsening of affective symptoms that are either directly or indirectly related to hippocampal degeneration.16
The objectives of the present investigation involved testing the ability of the tricyclic antidepressant amitriptyline (a classic nonselective reuptake inhibitor) and of fluoxetine (a classic serotonin selective reuptake inhibitor) to protect PC12 cells from cell death when exposed to neurotoxic insult induced by hydrogen peroxide, as well as to assess the levels of SOD enzyme activity in these cell cultures.
Methods
Cell culture
The PC12 rat pheochromocytoma cell line was obtained from the American Type Culture Collection (Rockville, Md.) and cultured in RPMI 1640 medium (Media Laboratory, College of Veterinary Medicine, University of Saskatchewan, Saskatoon) supplemented with 0.03% glutamine, 5% fetal calf serum and 10% horse serum plus 100 IU/mL penicillin G sodium salt, as outlined in protocols provided by the supplier. Cells were grown in collagen-coated flasks at 37°C in 95% humidified air with 5% carbon dioxide. When the cells were 80% confluent, they were dislodged from the flask surface with a flow of medium from a pipette and then dispersed through a 22-gauge needle. The dispersed cells were plated onto collagen-coated 96-well plates at a density of 3 х 104 cells/well and were cultured under various combinations of times and drug regimens. The cells were cultured for either 24 or 48 hours in the presence of amitriptyline or fluoxetine and then cultured for an additional 4 hours in the presence of 200 μmol/L H2O2. Control cells were cultured with neither drug nor H2O2 treatment. All drugs were purchased from commercial sources.
Determination of cell viability
After incubation of the PC12 cells with the experimental reagents for the indicated time periods, cell viability was determined by colorimetric measurement of the reduction product of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT). The original medium was removed from the 96-well plates, and the cells were incubated for 4 hours at 37°C in the presence of RPMI 1640 medium with 1% fetal bovine serum containing 0.5 mg/mL MTT. A 100-μL aliquot of acid-isopropanol (0.04 mol/L hydrochloric acid) was then added to each well, and the plates were incubated at 37°C overnight to dissolve the formazan that had formed in the wells. Reduced MTT was measured by means of a kinetic microplate reader (Molecular Devices, Palo Alto, Calif.) at a wavelength of 570 nm. The protein contents of cell lysates were measured using a bicinchoninic acid protein assay reagent kit (Pierce Inc., Rockford, Ill.). The value for each treatment group was converted to the percentage of control.
Determination of SOD enzyme activity
The level of SOD enzyme activity in PC12 cells was measured using the SOD Assay Kit-WST (Dojindo Molecular Technologies, Inc., Gaithersburg, Md.) according to the protocol previously described by Wei et al.17 After incubation of the PC12 cells with the experimental reagents for the indicated time periods, the original medium was removed from the 96-well plates, and the PC12 cells were lysed with No-nidet P-40 lysis buffer (1% NP-40, 50 mmol/L Tris-HCl [pH 7.5], 0.05 mmol/L ethylenediamine tetra-acetate) for 20 minutes at 4°C. The lysates were centrifuged at 300g for 10 minutes, and 20 μL of this sample solution was used for determination of SOD enzyme activity according to the manufacturer's instructions. The value for each treatment group was converted to the percentage of control.
Statistical analysis
All data are presented as mean (and standard error of the mean). Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Scheffé's post hoc comparison.
Results
Effects of antidepressants on viability of PC12 cells exposed to H2O2
Cell viability was reduced significantly after treatment for 24 hours with 200 μmol/L of fluoxetine or 400 μmol/L of amitriptyline (Fig. 1A) but was not affected by lower concentrations of these drugs. Further reduction in cell viability was observed after incubation for 48 hours (Fig. 1B). One-way ANOVA revealed that the viability of PC12 cells treated for 24 hours with 100 μmol/L amitriptyline and then exposed to 200 μmol/L H2O2 was significantly greater than the viability of PC12 cells exposed only to the H2O2 insult (F = 36.9, df = 3,28, p < 0.001; Fig. 2A). Statistical analysis also demonstrated that cells treated with 50 μmol/L amitriptyline or fluoxetine for 48 hours and subsequently exposed to H2O2 had significantly higher viability than untreated PC12 cells exposed to the same H2O2 insult (F = 84.2, df = 3,28, p < 0.001; Fig. 2B). When the cells were cotreated with 50 μmol/L fluoxetine and 1.0 mmol/L diethyldithiocarbamic acid (DETC, a metal-chelating agent that inhibits SOD1 activity) for 48 hours, the protective effect of fluoxetine was attenuated (Fig. 2C).
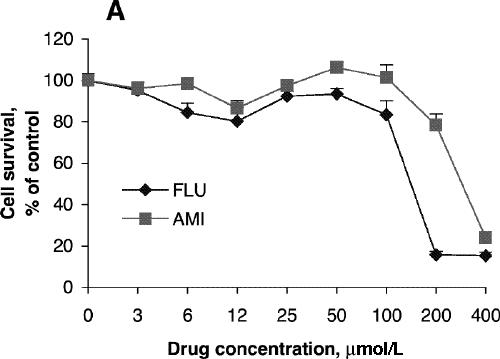
Fig. 1: Effects of antidepressants on 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) reduction and superoxide dismutase (SOD) activity of PC12 cells. Cells were treated with amitriptyline (AMI) or fluoxetine (FLU) for 24 hours (A, C) or 48 hours (B, D). MTT reduction assay (A, B) and test for SOD activity (C, D) were carried out according to the Methods. Data are presented as mean (and standard error of the mean), from 4 independent experiments for the MTT assay and 3 independent experiments for SOD activity.
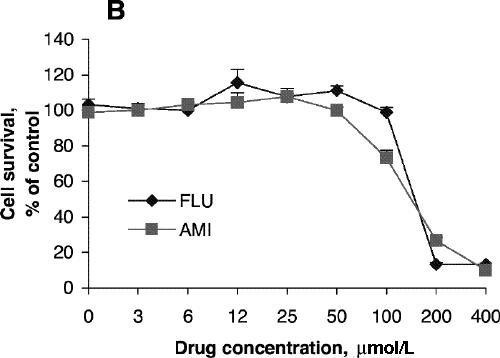
Figure 1. Continued.
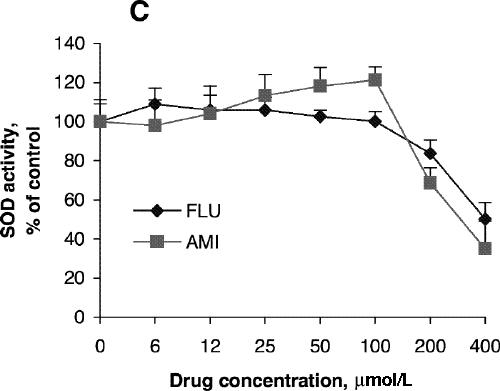
Figure 1. Continued.
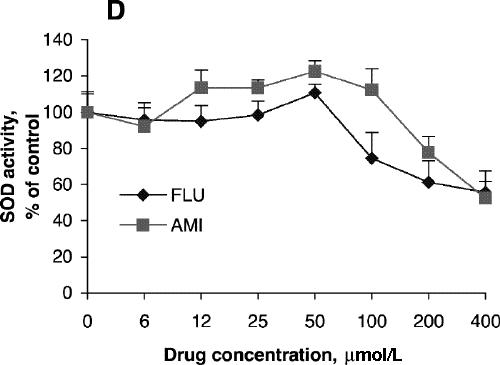
Figure 1. Continued.
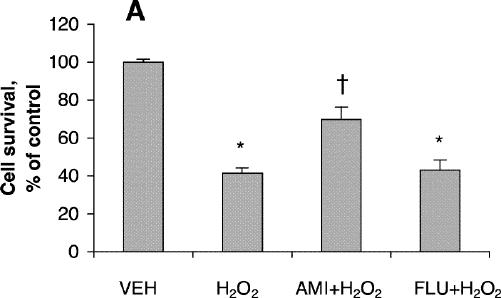
Fig. 2: Effects of antidepressants and diethyldithiocarbamic acid (DETC) on hydrogen peroxide-induced neurotoxicity in PC12 cells. Before being exposed to 200 μmol/L H2O2 for 4 hours, cells were pretreated with (A) 100 μmol/L amitriptyline (AMI) or 100 μmol/L fluoxetine (FLU) for 24 hours; (B) 50 μmol/L amitriptyline or 50 μmol/L fluoxetine for 48 hours; or (C) 50 μmol/L fluoxetine or 50 μmol/L fluoxetine plus 1.0 mmol/L DETC (D) for 48 hours. Data are presented as mean (and standard error of the mean) from 4 independent experiments. *p < 0.001 v. vehicle control (VEH); †p < 0.01 v. 200 μmol/L H2O2 treatment alone; ‡p < 0.001 v. 200 μmol/L H2O2 treatment alone.
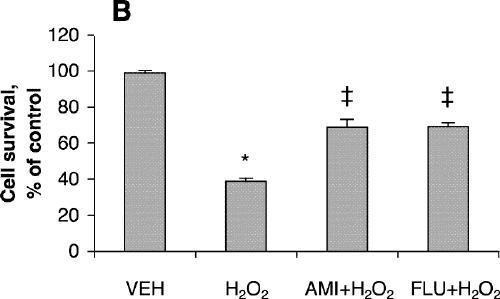
Figure 2. Continued.
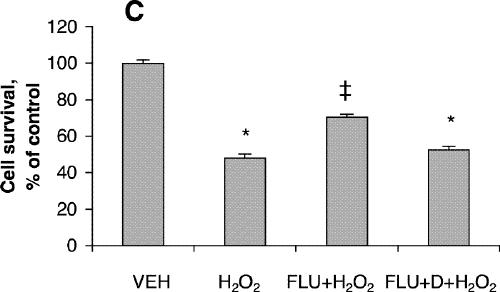
Figure 2. Continued.
Effects of antidepressants on SOD activity of PC12 cells
SOD activity increased with increasing concentrations of amitriptyline, reaching its highest level with incubation at 100 μmol/L for 24 hours (Fig. 1C), although cell viability was about the same as control at that concentration. No significant increase in SOD activity was observed after treatment with fluoxetine (Fig. 1C). SOD activity after treatment with amitriptyline or fluoxetine for 48 hours was highest at drug concentrations of 50 μmol/L (Fig. 1D), whereas cell viability was the same as control under these conditions. One-way ANOVA revealed that the SOD activity of PC12 cells treated with 100 μmol/L amitriptyline for 24 hours was significantly greater than SOD activity in control cells (Fig. 3A). No significant increase in SOD activity was observed in the cells treated with 100 μmol/L fluoxetine for 24 hours. Levels of SOD activity were significantly higher in PC12 cells treated with either 50 μmol/L amitriptyline or 50 μmol/L fluoxetine for 48 hours than in control cells (Fig. 3B). When the cells were treated with DETC, SOD activity decreased, and fluoxetine did not reverse the decrease caused by DETC (Fig. 3C).
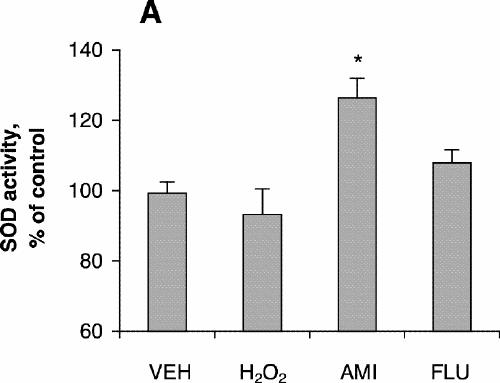
Fig. 3: Effects of antidepressants and diethyldithiocarbamic acid (DETC) on superoxide dismutase (SOD) activity of PC12 cells. PC12 cells were treated with (A) vehicle (VEH), 200 μmol/L hydrogen peroxide for 4 hours, 100 μmol/L amitriptyline (AMI) for 24 hours or 100 μmol/L fluoxetine (FLU) for 24 hours; (B) vehicle, 200 μmol/L H2O2 for 4 hours, 50 μmol/L amitriptyline for 48 hours or 50 μmol/L fluoxetine for 48 hours; (C) vehicle, 50 μmol/L fluoxetine for 48 hours, 1.0 mmol/L DETC for 48 hours, or 50 μmol/L fluoxetine plus 1.0 mmol/L DETC for 48 hours. Data are presented as mean (and standard error of the mean) from 4 independent experiments. *p < 0.05 v. vehicle control.
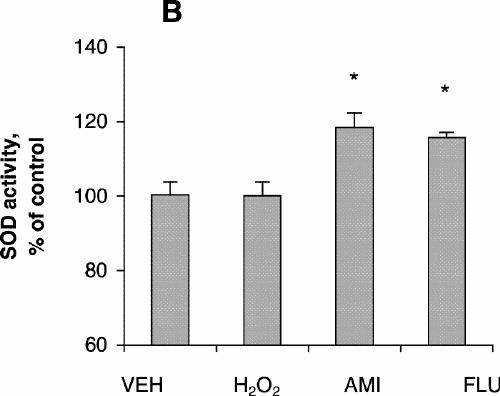
Figure 3. Continued.
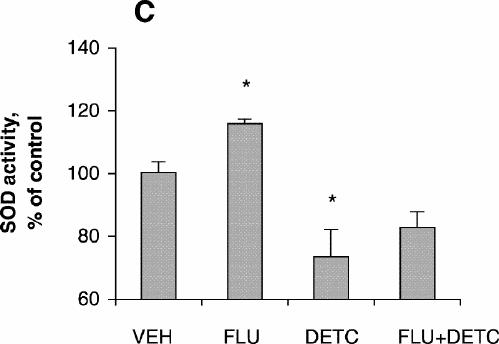
Figure 3. Continued.
Discussion
Endogenous H2O2 that is formed as a natural byproduct of enzymatic oxidase action or as a product of the dismutation of superoxide anion catalyzed by SOD1 contributes to the background level of cellular oxidative stress. It is generally accepted that the antioxidant enzymes catalase and glutathione peroxidase can protect cells from the effects of basal H2O2 production.18 However, the administration of exogenous H2O2 can elevate oxidative stress beyond the protective capacities of endogenous antioxidant defence systems, resulting in apoptosis or necrosis of cultured neurons19 and PC12 cells.17 There is good evidence from in vitro studies to suggest that astrocytes respond to oxidative insults by upregulating protective molecules, including SOD1.20 Other researchers have observed an increase in SOD1 in activated astrocytes following addition of the cytotoxic agent quinolinic acid.21 Upregulation of SOD1 and similar antioxidative enzymes may protect these cells from apoptosis or necrosis upon exposure to a range of neurotoxicants.
PC12 cells have been widely employed to investigate the mechanisms involved in neurotoxicity, neuroprotection and neuronal repair,22,23 and previous research has implicated SOD1 in the action of antidepressants on PC12 cells.16 Whereas a decrease in SOD1 corresponds with an increase in apoptotic cell death,13,14 an increase in endogenous SOD1 activity has been shown to protect neurons from apoptosis induced by staurosporine.24 However, the effects of SOD1 may become deleterious when this enzyme is overexpressed and the resulting H2O2 overwhelms catalase or glutathione peroxidase detoxification mechanisms.25 Some researchers have even suggested that overexpression of SOD1 leading to excessive H2O2 production may be partly responsible for the brain injury observed after perinatal hypoxia–ischemia.26
The results of the investigation reported here indicate that some antidepressants increase SOD1 activity in PC12 cells. Our finding that fluoxetine could not reverse the inhibition of SOD1 activity caused by DETC (i.e., DETC attenuates the protective effect of this drug) supports the hypothesis that increased SOD1 activity may be involved in the mechanisms by which fluoxetine and amitriptyline provide cytoprotective action to PC12 cells. However, the underlying mechanisms by which these drugs increase SOD1 activity are still unknown.
Although the SOD1 activity was approximately 10%–20% higher in cells treated with subcytotoxic concentrations of fluoxetine or amitriptyline than in control cells, there were no differences in cell viability between the 2 groups. Moreover, no obvious signs of cell death were observed in the treated cells. Other researchers have similarly reported that subcytotoxic concentrations of these antidepressants increase levels of reduced glutathione in C6 cells, but that higher concentrations of fluoxetine and amitriptyline decrease glutathione levels.27 In our experiment, treating the PC12 cells with antidepressants increased the levels of SOD1 activity, which ultimately led to the production of more endogenous H2O2. Because the viability of cells treated with subcytotoxic concentrations of antidepressants was essentially identical with that of control cells, we propose that catalase or glutathione peroxidase or both were upregulated to participate in the cellular defence against H2O2 produced by the activation of SOD1. Thus, when the challenge H2O2 was added, the defence system had already been induced. This hypothesis is consistent with the fact that the viability of cells pretreated with antidepressants and subsequently subjected to exogenous H2O2 was significantly greater than the viability of untreated cells subjected to the same neurotoxic insult. Our results suggest that subcytotoxic concentrations of antidepressants may provide protection to PC12 cells exposed to H2O2 by increasing levels of the antioxidative enzyme SOD1; future studies should attempt to determine whether antidepressants increase production of catalase or glutathione peroxidase. Further work will also be necessary to elucidate the mechanisms by which different doses and exposure times initiate reactive responses and whether these doses approach or fall within the therapeutic range for achieving remission from depression in humans. A major limitation of an in vitro model is the impossibility of accurately comparing concentrations that produce changes in the culture dish with those to which the endogenous cells may be exposed in the central nervous system. At present there are no relevant data, so caution must be exercised in extrapolation of results from a culture system to an in vivo situation.
Although major depressive disorder is currently considered a heterogeneous disease, both stress28 and dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis29 have been implicated in the origin and exacerbation of this disease.30 Stress and glucocorticoid injections have also been shown to initiate dendritic atrophy in the hippocampus31,32,33 and to reduce cellular resilience, making neurons in this region more vulnerable to injury from other insults.34 However, the findings that repeated administration of selective norepinephrine or serotonin reuptake inhibitors increase the rate of hippocampal cell proliferation35 and that long-term, but not short-term, administration of antidepressants enhances neurogenesis36,37 provide an explanation as to why these drugs may help reverse or prevent the symptoms of depression in most patients. Given that glucocorticoids have been shown to decrease BDNF messenger RNA,38 one of the key mediators in the therapeutic response of antidepressant medications, further research should be directed at investigating the feasibility of upregulation of SOD1 as a protective measure against glucocorticoid toxicity and an important mechanism whereby antidepressants could protect against further cell death or atrophy caused by HPA dysregulation. In those patients whose depression is caused, or accompanied, by stress or glucocorticoid-induced neurotoxicity, continued efforts aimed at maintaining or increasing levels of neuroprotective enzymes such as SOD1 may prove extremely beneficial. In short, increased levels of SOD1 activity may account, at least in part, for the observed clinical effects of certain antidepressant drugs used in the treatment of depression.
Acknowledgments
We thank Dr. A.V. Juorio for his valuable discussions. This work was supported by the Canadian Psychiatric Research Foundation and the Saskatchewan Health Research Foundation.
Footnotes
Contributors: The study was conceived by Drs. Kolla and Wei, and all authors participated in the experimental design. Drs. Kolla and Wei were responsible for data acquisition. All authors participated in data interpretation and drafting the article, and gave final approval for the version to be published.
Competing interests: None declared.
Correspondence to: Dr. Xin-Min Li, Neuropsychiatry Research Unit, Medical Research Building, University of Saskatchewan, 103 Wiggins Rd., Saskatoon SK S7N 5E4; fax 306 966-8830; xin-min.li@usask.ca
Submitted Jan. 15, 2004; Revised June 18, 2004; Accepted Aug. 30, 2004
References
- 1.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry 1997;54:597-606. [DOI] [PubMed]
- 2.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry 1999;46:1181-91. [DOI] [PubMed]
- 3.Vaidya VA, Duman RS. Depression — emerging insights from neurobiology. Br Med Bull 2001;57:61-79. [DOI] [PubMed]
- 4.Hartz JW, Deutsch HF. Subunit structure of human superoxide dismutase. J Biol Chem 1972;247:7043-50. [PubMed]
- 5.Delacourte A, Defossez A, Ceballos I, Nicole A, Sinet PM. Preferential localization of copper zinc superoxide dismutase in the vulnerable cortical neurons in Alzheimer's disease. Neurosci Lett 1988;92:247-53. [DOI] [PubMed]
- 6.Ceballos I, Javoy-Agid F, Delacourte A, Defossez A, Lafon M, Hirsch E, et al. Neuronal localization of copper-zinc superoxide dismutase protein and mRNA within the human hippocampus from control and Alzheimer's disease brains. Free Radic Res Commun 1991;12-13 Pt 2:571-80. [DOI] [PubMed]
- 7.Greenlund LJ, Deckwerth TL, Johnson EM Jr. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron 1995;14:303-15. [DOI] [PubMed]
- 8.Kim YH, Park KH, Rho HM. Transcriptional activation of the Cu,Zn-superoxide dismutase gene through the AP2 site by ginsenoside Rb2 extracted from a medicinal plant, Panax ginseng. J Biol Chem 1996;271:24539-43. [DOI] [PubMed]
- 9.Wengenack TM, Curran GL, Poduslo JF. Postischemic, systematic administration of polyamine-modified superoxide dismutase reduces hippocampal CA1 neurodegeneration in rat global ischemia. Brain Res 1997;754:46-54. [DOI] [PubMed]
- 10.Li XM, Juorio AV, Qi J, Boulton AA. L-Deprenyl potentiates NGF-induced changes in superoxide dismutase mRNA in PC12 cells. J Neurosci Res 1998;53:235-8. [DOI] [PubMed]
- 11.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 1995;65:1740-51. [DOI] [PubMed]
- 12.Furukawa K, Estus S, Fu W, Mark RJ, Mattson MP. Neuroprotective action of cycloheximide involves induction of bcl-2 and antioxidant pathways. J Cell Biol 1997;136:1137-49. [DOI] [PMC free article] [PubMed]
- 13.Rothstein JD, Bristol LA, Hosler B, Brown RH, Kuncl RW. Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc Natl Acad Sci U S A 1994;91:4155-9. [DOI] [PMC free article] [PubMed]
- 14.Troy CM, Shelanski ML. Down-regulation of copper/zinc superoxide dismutase causes apoptotic cell death in PC12 neuronal cells. Proc Natl Acad Sci U S A 1994;91:6384-7. [DOI] [PMC free article] [PubMed]
- 15.Cvijic G, Radojicic R, Djordjevic J, Davidovic V. The effect of glucocorticoids on the activity of monoamine oxidase, copper-zinc superoxide dismutase and catalase in the rat hypothalamus. Funct Neurol 1995;10:175-81. [PubMed]
- 16.Li XM, Chlan-Fourney J, Juorio AV, Bennett VL, Shrikhande S, Bowen RC. Antidepressants up-regulate messenger RNA levels of the neuroprotective enzyme superoxide dismutase (SOD1). J Psychiatry Neurosci 2000;25:43-7. [PMC free article] [PubMed]
- 17.Wei Z, Bai O, Richardson JS, Mousseau DD, Li XM. Olanzapine protects PC12 cells from oxidative stress induced by hydrogen peroxide. J Neurosci Res 2003;73:364-8. [DOI] [PubMed]
- 18.Calderón FH, Bonnefort A, Muñoz FJ, Fernández V, Videla LA, Inestrosa NC. PC12 and neuro 2a cells have different susceptibilities to acetylcholinesterase-amyloid complexes, amyloid25-35 fragment, glutamate, and hydrogen peroxide. J Neurosci Res 1999;56:620-31. [DOI] [PubMed]
- 19.Whittemore ER, Loo DT, Watt JA, Cotman CW. A detailed analysis of hydrogen peroxide-induced cell death in primary neuronal culture. Neuroscience 1995;67:921-32. [DOI] [PubMed]
- 20.Pentreath VW, Slamon ND. Astrocyte phenotype and prevention against oxidative damage in neurotoxicity. Hum Exp Toxicol 2000;19:641-9. [DOI] [PubMed]
- 21.Noack H, Lindenau J, Rothe F, Asayama K, Wolf G. Differential expression of superoxide dismutase isoforms in neuronal and glial compartments in the course of excitotoxically mediated neurodegeneration: relation to oxidative and nitrergic stress. Glia 1998;23:285-97. [DOI] [PubMed]
- 22.Greene LA, Tischler A. PC12 pheochromocytoma cultures in neurobiological research. Adv Cell Neurobiol 1982;3:373-414.
- 23.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol 1997;10:299-305. [DOI] [PubMed]
- 24.Patel M. Inhibition of neuronal apoptosis by a metalloporphyrin superoxide dismutase mimic. J Neurochem 1998;71:1068-74. [DOI] [PubMed]
- 25.Lindenau J, Noack H, Possel H, Asayama K, Wolf G. Cellular distribution of superoxide dismutases in the rat CNS. Glia 2000;29:25-34. [DOI] [PubMed]
- 26.Dittelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. Brain injury after perinatal hypoxia–ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res 1996;39:204-8. [DOI] [PubMed]
- 27.Slamon ND, Pentreath VW. Antioxidant defense against antidepressants in C6 and 1321N1 cells. Chem Biol Interact 2000;127:181-99. [DOI] [PubMed]
- 28.Lindefors N, Brodin E, Metsis M. Spatiotemporal selective effects on brain-derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shock. Neuroscience 1995; 65:661-70. [DOI] [PubMed]
- 29.Plotsky PM, Owens MJ, Nemeroff CB. Psychoneuroendocrinology of depression: hypothalamic-pituitary-adrenal axis. Psychiatr Clin North Am 1998;21:293-307. [DOI] [PubMed]
- 30.Gold PV, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry 2002;7:254-75. [DOI] [PubMed]
- 31.Magarinos AM, McEwen BS, Flugge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci 1996;16:3534-40. [DOI] [PMC free article] [PubMed]
- 32.Sapolsky RM, Uno H, Rebert CS, Finsh CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci 1990;10:2897-902. [DOI] [PMC free article] [PubMed]
- 33.Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res 1990;531:225-31. [DOI] [PubMed]
- 34.Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 2000;57:925-35. [DOI] [PubMed]
- 35.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult hippocampus. J Neurosci 2000;20:9104-10. [DOI] [PMC free article] [PubMed]
- 36.D'Sa C, Duman RS. Antidepressants and neuroplasticity. Bipolar Disord 2002;4:183-94. [DOI] [PubMed]
- 37.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9. [DOI] [PubMed]
- 38.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 1995;15:1768-77. [DOI] [PMC free article] [PubMed]