

Abstract
Mammalian embryogenesis commences with two pivotal and binary cell fate decisions that give rise to three essential lineages, the trophectoderm (TE), the epiblast (EPI) and the primitive endoderm (PrE). Although key signaling pathways and transcription factors that control these early embryonic decisions have been identified, the non-coding regulatory elements via which transcriptional regulators enact these fates remain understudied. Here, we have characterized, at a genome-wide scale, enhancer activity and 3D connectivity in embryo-derived stem cell lines that represent each of the early developmental fates. We observe extensive enhancer remodeling and fine-scale 3D chromatin rewiring among the three lineages, which strongly associate with transcriptional changes, although distinct groups of genes are irresponsive to topological changes. In each lineage, a high degree of connectivity, or “hubness”, positively correlates with levels of gene expression and enriches for cell-type specific and essential genes. Genes within 3D hubs also show a significantly stronger probability of coregulation across lineages, compared to genes in linear proximity or within the same contact domains. By incorporating 3D chromatin features, we build a predictive model for transcriptional regulation (3D-HiChAT), which outperforms models using only 1D promoter or proximal variables to predict levels and cell-type specificity of gene expression. Using 3D-HiChAT, we identify, in silico, candidate functional enhancers and hubs in each cell lineage, and with CRISPRi experiments we validate several enhancers that control gene expression in their respective lineages. Our study identifies 3D regulatory hubs associated with the earliest mammalian lineages and describes their relationship to gene expression and cell identity, providing a framework to comprehensively understand lineage-specific transcriptional behaviors.
INTRODUCTION
Mammalian development starts with two cell fate decisions giving rise to progenitors of embryonic and extraembryonic tissues required for embryogenesis1–4. First, cells of the totipotent morula segregate into either the inner cell mass (ICM), or trophectoderm (TE), a polarized epithelial cell layer that gives rise to trophoblast tissues of the placenta. While later, the ICM will generate the pluripotent epiblast (EPI) and the primitive endoderm (PrE) cells, which eventually form the embryo proper and the extraembryonic yolk sac tissue, respectively5. In vivo and in vitro studies have uncovered cellular and molecular hallmarks of these early embryonic decisions, including key signaling pathways (such as Notch, Wnt/B-catenin, Hippo etc.) and DNA-binding transcription factors (TF) that drive lineage specification and segregation6–8. However, little is known regarding the downstream non-coding DNA elements and regulatory networks that enforce these early embryonic fates.
Enhancers are essential regulatory elements that -together with TFs- regulate transcriptional activity of gene promoters often over large distances, establishing cell type-specific gene expression programs and hence cellular identities9,10. Chromatin profiling assays (ATAC-seq for chromatin accessibility, ChIP-seq for histone marks such as H3K27ac) have been useful for annotating hundreds of thousands of putative enhancers on a genome-wide scale in various tissues and cell lines11–15. However, these assays are limited in their accuracy to correctly assign enhancers to target genes, and to predict their relative regulatory impact on gene expression and cell identity, as shown by reporter assays16–18 and genetic or epigenetic engineering19,20. The emergence of 3D chromatin organization as an important regulatory layer of gene expression and cell identity21–27, highlights the necessity to study enhancer function in context of its own 3D neighborhood. This includes the specific long-range interactions of a given enhancer with one or more target genes, the insulating boundaries that may restrict enhancer function and the larger-scale compartmental organization28–35. Genome-wide Chromosome Conformation Capture (3C)-based chromatin assays, such as Hi-C36, Capture-C37,38, Micro-C39,40 or HiChIP41–45 in various cellular contexts have begun to map 3D enhancer-promoter interactions that are highly complex and largely cell-type specific. These 3D networks have significantly improved enhancer-promoter assignments and predictions of enhancer functionality compared to traditional approaches based on linear proximity10,46–48.
So far, 3D network analysis has not been utilized to study regulatory principles that govern early cell fate decisions. Applying genomics technologies in early embryogenesis in vivo is particularly challenging due to limited cell numbers in the mouse preimplantation blastocyst. Single cell technologies have begun to be used in this context, but they often suffer from poor genomic resolution49–54. In contrast, embryo-derived stem cell lines, known as Trophoblast Stem Cells (TSC), Embryonic Stem Cells (ESCs) and eXtraEmbryonic ENdoderm cells (XEN) have been valuable tools for studying mechanisms that govern the early embryonic lineages of TE, EPI and PrE derivatives, respectively55–59. Among them, mouse ESCs representing the naive EPI state have been extensively characterized by us and others using multiple -omics assays and functional screens26,27,60,61. However, only a few recent studies have started to interrogate the enhancer landscape and 3D chromatin organization of TSC and less for XEN cells62–69 whilst direct comparisons of the three lineages are still missing.
In this study, we used a multi-omics approach to comprehensively map the 1D and 3D putative regulatory landscapes in ESC, TSC and XEN cells as a means of identifying cis-regulatory elements and 3D networks that govern early embryonic lineages. Our integrative analysis revealed extensive enhancer remodeling and 3D rewiring between these closely related lineages with specific links to their transcriptional programs. Using a Random Forest machine learning approach with various 1D/3D features, we determined important 3D variables that improve prediction of transcriptional behaviors, such as levels and cell-type specificity of gene expression or gene coregulation. Our optimized 3D predictive model, coined 3D-HiChAT, was used to perform genome-wide in silico perturbations and predict putative enhancers with regulatory impact on one or more genes in each lineage. Finally, with a series of experimental perturbations in ESCs and XEN, we identified several functional enhancers and 3D hubs that control expression levels of one or more developmentally-relevant genes, including Tfcp2l1 and Klf2 in ESC and Mycn or Lmna in XEN cells70–72. In conclusion, our study provides a high-resolution 3D atlas of candidate regulatory interactions in early mouse embryonic lineages and reveals regulatory principles that determine the levels and cell-type specificity of gene expression.
RESULTS
Enhancer remodeling supports early embryonic lineage programs
To model and characterize the chromatin regulatory landscapes of the early developmental cell fates, we used three well-characterized TSC56, ESC73 and XEN cell lines56,57,74 (Fig. 1a). Independent characterization of each cell line by RNA-seq analysis and immunofluorescence (IF) validated the cell-type specific expression of key signature genes, including Cdx2, Eomes, Elf4 and Gata3 for TSCs, Nanog, Zfp42, Klf4 and Pou5f1 for ESCs and Gata4/6 and Sox17 for XEN (Fig. 1b and Extended Data Fig.1a). PCA integrating previously published RNA-seq datasets for TSC, ESC and XEN lines (Supplementary Table 1) further confirmed that our samples clustered together with their respective cell type and separated from the other lineages (Extended Data Fig. 1b).
Figure 1. Transcriptional changes and enhancer remodeling accompany early developmental decisions.
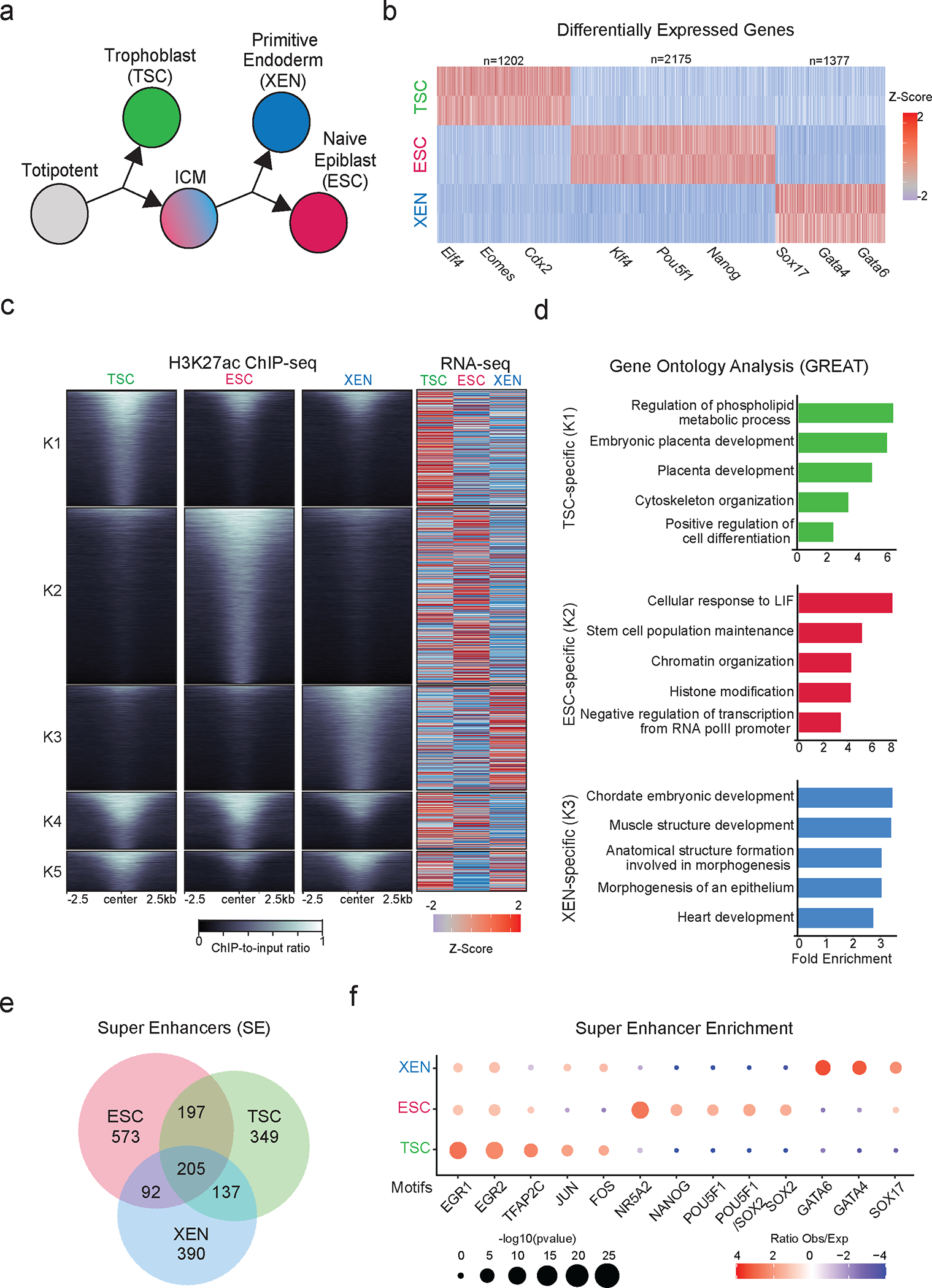
a. Schematic illustration depicting the cell lines used to model early developmental fate decisions.
b. Heatmap showing TSC, ESC and XEN signature genes, which are significantly upregulated in the respective cell line compared to the other two lineages (TPM>1, LogFC >2 and p-adjusted <0.01). Scale represents Z-score of normalized RNA-seq counts. RNA-seq was performed in two independent replicates for each sample. Examples of known regulators and markers of each lineage are highlighted on the bottom. (See Extended Data Fig.1.)
c. Tornado plot (left) illustrating H3K27ac ChIP-seq signal for TSC, ESC and XEN around different clusters of peaks (+/−2.5 kb), as defined by K-means clustering (K=5) using all H3K27ac peaks across cell lines. Scale bars denote normalized H3K27ac ChIP-seq signal over input. Heatmap (right) illustrates Z-score normalized RNA-seq levels of most proximal genes to each of the H3K27 peaks. (See Supplementary Table 2.)
d. Gene ontology analysis (using GREAT) of cell type specific enhancers as identified by K-means clustering shown in (1B). Significance was calculated using two-sided binomial test and “Region Fold Enrichment” is presented on the x-axis for selected significant (padj-value<0.05) biological processes shown in the graph. (See Supplementary Table 3.)
e. Venn-diagram showing overlap of Super Enhancers (SE) in TSC, ESC and XEN cell lines, called by the ROSE algorithm using H3K27ac peaks as input. (See Supplementary Table 2).
f. Relative enrichment of TF binding motifs found in cell-type specific SE. The enrichment plots depict selected significant motifs with −log10(p-value) higher in one cell type versus the other two. Size of dots indicates the p-value (two-sided Fisher’s exact test) while color indicates the ratio of observed versus expected frequency. (See Supplementary Table 3).
Note: all statistics are provided in Supplementary Table 9.
We next performed ChIP-seq for H3K27ac (a mark of putative active enhancers and promoters) and ATAC-seq to map the regulatory landscapes of TSC, ESC and XEN cells. PCA separated all three lineages based on either H3K27ac occupancy or chromatin accessibility (Extended Data Fig. 1b), suggesting genome-wide enhancer remodeling. K-means clustering of H3K27ac peaks across the three lineages revealed a large proportion of cell-type specific peaks (K1-K3) (Fig. 1c and Supplementary Table 2), predominantly located within distal intergenic and intronic regions (Extended Data Fig. 1c). Peaks shared among two or three lineages showed an overrepresentation of promoters (Extended Data Fig. 1c). Cell-type specific H3K27ac peaks were associated with elevated gene expression levels in the respective cell line (Fig. 1c). Gene ontology analysis using the GREAT tool75 for each of the cell-type specific peak clusters showed enrichment for lineage-specific processes and functions, such as placenta development for TSC, heart development for XEN, and LIF response for ESC (Fig. 1d and Supplementary Table 3). Using the ROSE algorithm, we also identified several hundreds of Super Enhancers (SE)76, the majority of which were unique for each lineage (Fig. 1e Supplementary Table 2), consistent with the suggested role of SEs in cell fate regulation69,76–78. Motif analysis of accessible sites within cell-type specific SE detected enrichment for known critical regulators of primitive endoderm (e.g GATA4/6 and SOX17) in XEN SE, naïve epiblast (e.g NANOG, POU5F1/SOX2, NR5A2) in ESC and trophoblast lineage (e.g TFAP2C and JUN/FOS) in TSC67,79–88 (Fig. 1f and Supplementary Table 3). These results document distinct transcriptional programs for each early developmental lineage, supported by the coordinated crosstalk of lineage-specific TFs and enhancer landscapes.
Multilayered 3D genomic reorganization in early embryonic lineages
To investigate potential large-scale 3D architectural changes among TSC, ESC, and XEN cells we initially performed in situ Hi-C (Supplementary Table 1). PCA analysis on the level of A/B compartments (100kb resolution) and TADs (40kb resolution) separated all three lineages (Extended Data Fig. 2a). A higher degree of similarity was observed between TSC and XEN, which are both extraembryonic lineages (Extended Data Fig. 2a). Pairwise comparisons of compartment scores showed that up to 33.5% of the genome (32.5% between ESC and XEN, 33.5% between ESC and TSC and 21.1% between TSC and XEN) underwent compartmentalization changes (e.g. A-to-B, B-to-A and A or B compartment strengthening with Delta c-score >0.2 or <−0.2), with ~500–2000 genomic windows switching from A-to-B or B-to-A (Fig. 2a). In agreement with previous studies in other cellular systems,89–91 compartmental reorganization in TSC, ESC and XEN cells associated with transcriptional and epigenetic changes. For example, A compartment strengthening, or B-to-A switches correlated with transcriptional upregulation and gain of H3K27ac signal, while B strengthening, and A-to-B shifts associated with gene downregulation and H3K27ac loss (Fig. 2b–c and Extended Data Fig. 2b). Although compartmental shifts occurred around several important developmental genes (see Sox2 and Foxa2 examples in Fig.2c), the majority (>80%) of cell type-specific genes and enhancers (K1/K2/K3) were not associated with compartmental changes (B-to-A). Large-scale topological changes can therefore only explain a fraction of the epigenetic and transcriptional reprogramming observed in these early developmental cell lineages. At 40kb resolution, despite few significant changes in insulation (<7%) between any pairwise comparison, we detected thousands (20,000–26,000) of genomic regions with significantly altered overall interactivity (within 0.5Mb window), especially when comparing ESCs with either of the extraembryonic lineages (Fig. 2d and Extended Data Fig. 2c). Gain or loss of interactivity associated with gain or loss of enhancer and transcriptional activity (Fig.2e and Extended Data Fig. 2d), respectively, documenting an extensive 3D chromatin reorganization occurring along with enhancer remodeling.
Figure 2. Hi-C and H3K27ac HiChIP reveals multilayered 3D genomic reorganization and complex networks of putative regulatory interactions in TSC, ESC and XEN.
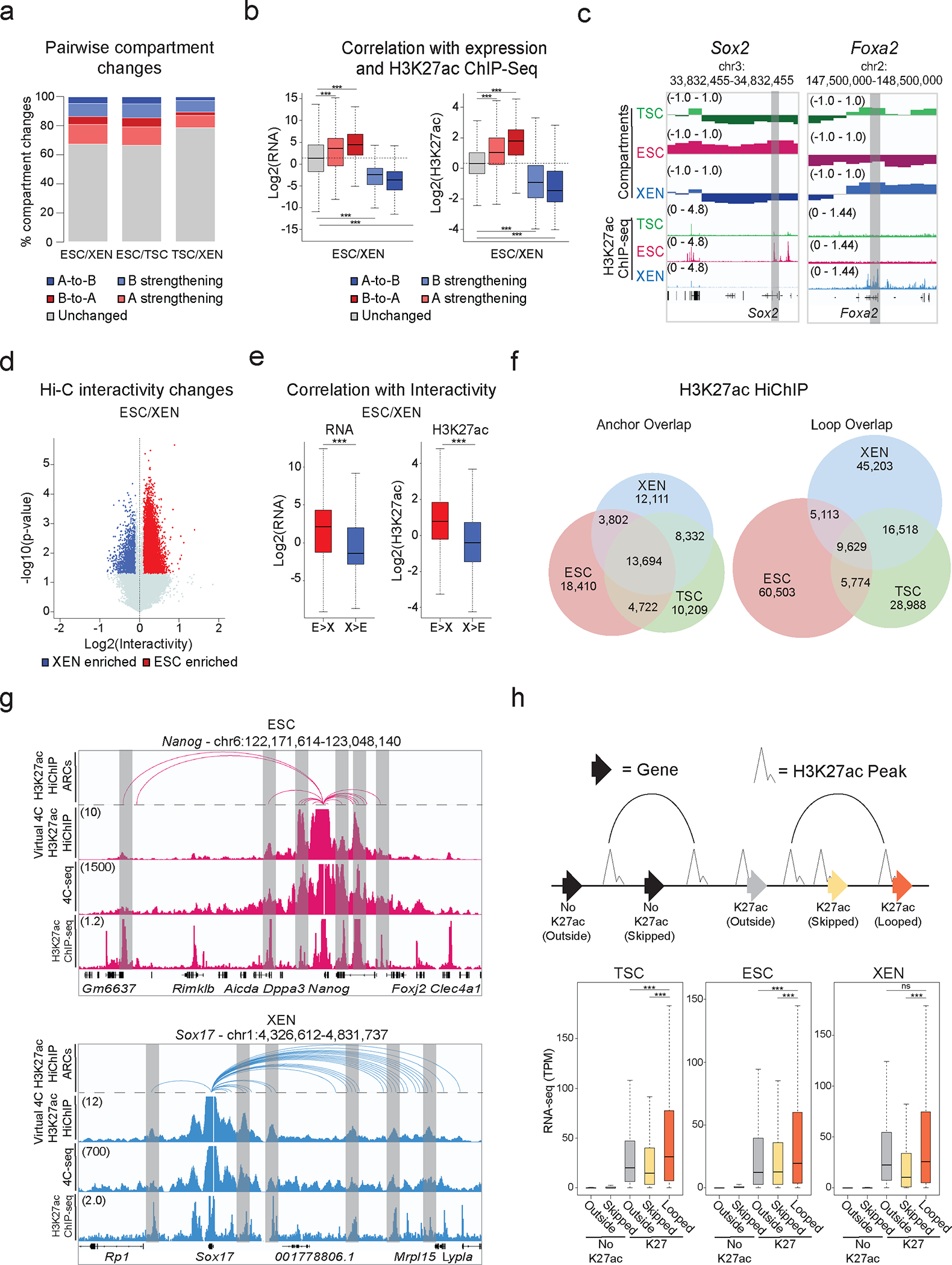
a. Stacked barplots showing A/B Hi-Ccompartment changes for pairwise comparisons. Compartment changes (100kb resolution) assigned based on A/B status and C-score difference between cell lines as: A-to-B shifts (dark blue), B-to-A shifts (dark red), A-strengthening (light red), B-strengthening (light blue) or unchanged (grey). See Methods for details.
b. Boxplots showing median expression changes (left, n=5230 genes) or H3K27ac ChIP-seq changes (right, n=12,225 peaks) between ESC and XEN cells for gene loci assigned to different groups described in (a). Asterisks indicate significance (p<0.001) by two-sided Wilcoxon rank test. (See Extended Data Fig. 2b).
c. Examples of A/B compartment switches around developmentally relevant genes, Sox2 (left-ESC) and Foxa2 (right-XEN).
d. Volcano plot showing differential Hi-C interactivity (40kb resolution) between ESC and XEN (n=2 independent replicates per sample). X-axis shows delta interactivity, y-axis shows −log10(p-value) calculated by two-sided Student’s t-test. Significant changes (p-value<0.05 and Diff>0.1 or <−0.1) are highlighted in blue (higher in XEN) or red (higher in ESC). (See Extended Data. Fig. 2c).
e. Boxplots showing changes in gene expression (left, n=645 genes) and H3K27ac ChIP-seq (right, n=11,174 peaks) between ESC and XEN at regions with delta interactivity, described in (d). (See Extended Data. Fig. 2d). Asterisks indicate significance (p<0.001) by two-sided Wilcoxon rank test.
f. Venn diagrams of shared and unique annotated anchors (left) and loops (right) in TSC (green), ESC (red) and XEN (blue) cells detected by H3K27ac HiChIP. Interactions identified by FitHiChIP 2.0 (5kb resolution).
g. IGV tracks showing concordance between H3K27ac HiChIP (Arcs on top, virtual 4C of normalized H3K27ac HiChIP signal in the middle) with independent in situ 4C-seq experiments around selected viewpoints (top = Nanog promoter, bottom = Sox17 promoter) and H3K27ac ChIP-seq tracks. Common interactions are highlighted in grey.
h. Schematic (top) defining gene categories by HiChIP loop status (looped, skipped, outside) and H3K27ac promoter presence (noK27ac vs K27ac). Boxplot (bottom) depicting median gene expression. n=26,742 genes across 2 independent RNA-seq samples per cell line. Asterisks indicate significant differences (p-val<0.001) by two-sided Wilcoxon rank test.
Note: all statistics are provided in Supplementary Table 9.
Encouraged by the 3D interactivity changes detected by Hi-C, we performed H3K27ac HiChIP43 to profile putative enhancer interactions in TSC, ESC and XEN cells at high genomic resolution (Supplementary Table 1). By applying FitHiChIP 2.092,93 at 5kb resolution with FDR<0.05 on all datasets, we called ~60,000–80,000 high-confidence pairwise interactions between ~35,000–40,000 anchors in each cell type (Fig. 2f), highlighting that many genomic regions engage in multiple chromatin contacts. Despite the large fraction of shared anchors among the 3 lineages, we observed poor overlap (12–16%) of chromatin interactions (“loops”) (Fig. 2f, right Venn diagram), in agreement with the regulatory rewiring indicated by Hi-C analysis. To independently validate the HiChIP loops, we confirmed their enrichment in recently published Micro-C data in mouse ESCs39 by aggregate plot analysis (Extended Data Fig. 2f). Moreover, we performed high-resolution in situ 4C-seq around enhancers and promoters of select cell-type specific genes (e.g., Sox17 for XEN and Nanog for ESC), and observed high concordance with the virtual 4C of HiChIP and the called HiChIP contacts in the respective cell type (Fig. 2g and Extended Data Fig. 2g).
HiChIP-detected interactions occurred over a large range of distances (10kb-2Mb) (Supplementary Table 4) with a similar size distribution among lineages (Extended Data Fig. 2e), often skipping multiple neighboring genes and enhancers, or crossing TAD boundaries (Supplementary Table 4). Genes whose promoters engaged in at least one HiChIP contact showed significantly higher expression levels compared to not-looped genes (whose promoters were skipped or entirely outside of loops) (Fig. 2h) in the respective cell type. Elevated expression of looped genes was also detected when we focused our comparison on looped and skipped genes with similar H3K27ac signal on their promoters (Extended Data Fig. 2h). These results support the notion that H3K27ac-HiChIP contacts represent active regulatory interactions in all three lineages that enhance transcriptional levels of engaged genes in a targeted manner.
3D “hubness” associates with gene expression and coregulation
The positive association between looping and gene expression suggests that engagement of promoters in multiple chromatin contacts should further enhance their transcriptional output. We therefore ranked promoters into quantiles based on their connectivity or “hubness” (number of distinct HiChIP-detected contacts per anchor) (Fig. 3a) and observed that higher hubness associated with progressively higher transcriptional levels (Fig. 3b) (Spearman correlation: TSC=0.35, ESC = 0.31, XEN=0.32). These observations were true across all cell lines, suggesting a potential additive regulatory impact of multiple connected anchors. Comparing the top 10% highly connected anchors (Q10) with the least connected ones (Q1) in each lineage, we found that genes in Q10 had not only significantly higher transcriptional levels (as shown in Fig. 3b), but also a strong preferential enrichment for gene ontology categories linked to either housekeeping processes or lineage-specific functions (Fig. 3c and Supplementary Table 3). In agreement, TSC, ESC or XEN signature genes (as defined in Fig. 1b) engaged in a significantly higher number of 3D interactions in the respective cell type (Fig. 3d). Loci encoding known master regulators were among the top connected genes in each cell type, including Klf4 in ESC (n=15 contacts) (Fig. 3e), Gata6 in XEN (n=27 contacts) and Cdx2 in TSC (n=26 contacts) (Extended Data Fig. 3a), suggesting multiple regulatory contacts contribute to robust and cell-type specific gene expression. Q10 anchors in ESC also showed a strong and preferential enrichment for genes identified as essential for ESC survival and proliferation by two independent CRISPR screen studies94,95 (Extended Data Fig. 3b). These results highlight that genes critical for survival or cell identity tend to establish multiple regulatory connections, which might act in a cooperative or redundant fashion to ensure tight regulation and robust expression.
Figure 3. Association of high 3D hubness with levels, cell-type specificity and coregulation of gene expression in early embryonic fates.
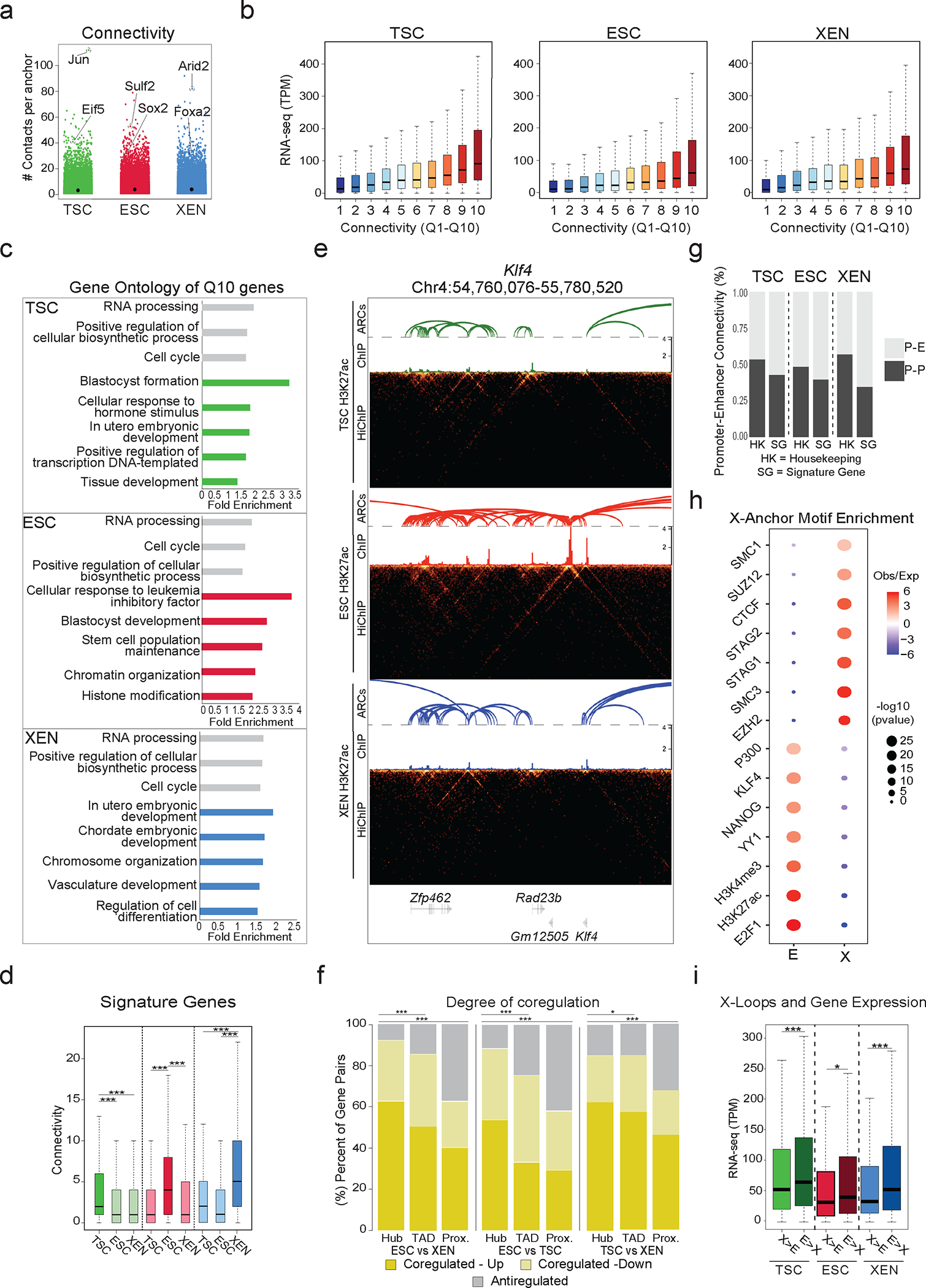
a. Plot showing connectivity range at 5kb HiChIP-detected anchors (hubness) in TSC, ESC and XEN cells. Highly connected example gene anchors are highlighted.
b. Boxplots showing median expression levels of genes with increasing HiChIP connectivity. All looped genes were binned into 10 quantiles by promoter connectivity (Q1 = least connected, Q10 = most connected). n=8870 (TSC), 9027 (ESC), 8848 (XEN) promoters across n=2 independent HiChIP and RNA-seq experiments.
c. Selected Gene ontology housekeeping (grey) or lineage-related (colored) terms enriched in multi-connected Q10 genes in ESC, XEN and TSC. (See Supplementary Table 3).
d. Boxplots showing distribution and median connectivity of signature genes in each cell type. Dark colors indicate signature genes origin (TSC n=892 (green), ESC n=1663 (red), XEN n=999 cells (blue). Asterisks indicate significant differences (p<.001) by two-sided Wilcoxon rank test.
e. HiGlass visualization of an ESC hub -Klf4 genomic locus- shown in TSC, ESC, and XEN with corresponding H3K27ac HiChIP-derived arcs and H3K27ac ChIP-seq signals.
f. Stacked barplots showing percentages of gene pairs coregulated (both upregulated or downregulated with log2 fold change>1 or <=1 and p.adj<−0.01) or anti-regulated (one upregulated, one downregulated). Gene pairs were from the same hub (shared anchor by HiChIP), the same TAD or nearest linear proximity. Statistics calculated by two-sided Fisher’s exact test (Supplementary Table 9).
g. Barplots showing percentages of Promoter-Enhancer (PE) or Promoter-Promoter (PP) pairs at housekeeping (HK) genes or signature genes (SG) in each cell type.
h. Relative enrichment of selected TF binding motifs in Enhancer (E) or X-linked anchors (X) in ESC calculated using LOLA. Dot size indicates p-value (two-sided Fisher’s exact test), color indicates ratio of observed versus expected. (See Supplementary Table 3).
i. Boxplots comparing gene expression for genes with different relative proportions of connected X and E anchors. The ratio of Enhancer vs X anchors is >2 (E>X hubs) or <0.5 (X>E hubs). n=1908 (TSC), 2668 (ESC) and 2410 (XEN) genes across n=2 independent HiChIP and RNA-seq experiments Asterisks indicate significant differences (* = p<0.05, *** = p<.001) by two-sided Wilcoxon rank test.
Note: all statistics are provided in Supplementary Table 9.
In addition to multiconnected promoters, we also identified highly interacting enhancers (or enhancer hubs) that form contacts with multiple genes. Such hubs could confer coordinated regulation of two or more genes during early cell fate decisions, as we and others have previously shown in other contexts42,96–98. To test this possibility, we focused on enhancers that interact with two or more differentially expressed genes in TSC, ESC or XEN, and examined if those gene pairs were expressed concordantly (Up-Up or Down-Down) or discordantly (Up-Down). A significantly higher proportion of coregulated genes occurred within hubs, when compared to gene pairs that were most proximal to one another or pairs within matched TADs (Fig. 3f). This highlights that 3D hubs harbor -and potentially actively control- coregulated genes. Integration of HiChIP interactions might therefore be superior to any other linear or 3D features (e.g., TAD organization) in predicting gene coregulation.
The positive correlation between connectivity and gene expression highlights the fact that H3K27ac HiChIP mostly detects putative active regulatory interactions. Indeed, the majority of HiChIP-detected interactions connected promoters (P: anchors contained one or more TSS) and/or putative enhancers (E: anchors with one or more H3K27ac peaks, none at a TSS) (Extended Data Fig. 3c). Lineage-specific genes predominantly formed interactions with enhancers over promoters (Fig. 3g), highlighting the importance of distal enhancers in cell-type specific gene regulation. In contrast, housekeeping genes had a higher proportion of P-P interactions in all lineages (Fig.3g), reminiscent of recently described 3D assemblies of housekeeping genes99. Thus, the type of contacts could also be informative for the levels or cell-type specificity of gene expression.
In addition to the P-P, P-E and E-E contacts, about ~25–30% of the called interactions involved one anchor without H3K27ac signal or TSS (X anchors). Overlap of accessible regions within X or E anchors in ESC with published ChIP-seq experiments (LOLA100) revealed a strong and preferential enrichment for CTCF and Cohesin binding at X anchors, as well as the Polycomb Repressive Complex (PRC), including EZH and SUZ12 (Fig. 3h and Supplementary Table 3). Therefore, X-anchored contacts might represent either structural or repressive loops. X-anchored loops also spanned significantly larger distances compared to E-E, E-P and P-P interactions (Extended Data Fig. 3d). In support to a potential repressive role, we noticed that multi-connected genes (n>3) with a higher proportion of X vs E anchors were associated with significantly lower expression levels compared to genes with higher proportion of E connections (Fig. 3i). This held true when focusing on hubs with similar total connectivity. For conserved interactions between lineages, we noticed that switches of the anchor status from X-to-E or from E-to-X associated with upregulation or downregulation of connected genes (Extended Data Fig. 3e). These results demonstrate that not all HiChIP-detected contacts associate with positive transcriptional regulation and suggest that categorization of interactions based on the features of the involved anchors enables a better understanding of the transcriptional fine-tuning around multi-connected gene loci.
Distinct classes of genes vary in sensitivity to 3D rewiring
Our HiChIP results document extensive fine-scale 3D reorganization during early embryonic decisions, which we independently validated for select loci by 4C-seq (Fig. 4a). To determine to what degree 3D rewiring associates with transcriptional changes, we generated an atlas of all promoter-centric contacts across the three lineages and plotted differential HiChIP connectivity vs differential RNA-seq levels between any pair of early embryonic cell types (Fig. 4b, Extended Data Fig. 4a). In every pairwise comparison, we observed a concordance of expression changes with 3D connectivity remodeling (R=0.422 for ESC/XEN, 0.318 for ESC/TSC and 0.367 for TSC/XEN), which was stronger than the correlation between transcriptional and compartmental changes (R= 0.214 for ESC/XEN, 0.098 for ESC/TSC and 0.126 for TSC/XEN). This means that gain or loss of specific HiChIP contacts at the promoter correlates with gene up- or down-regulation, respectively (3D-concordant). However, not all genes behaved the same way. We also identified gene loci that experienced significant changes in 3D connectivity without any transcriptional changes (termed “3D-insensitive”) (Fig. 4b. Extended Data Fig. 4A and Supplementary Table 5). Gene ontology analysis for the 3D-concordant gene set showed a strong enrichment for lineage-related processes (pluripotency-associated signaling (ESC), tube morphogenesis (XEN) and placenta development (TSC)) (Fig. 4c–d, Extended Data Fig. 4b–e and Supplementary Table 3). In contrast, 3D-insensitive genes strongly enriched for housekeeping functions, such as RNA processing, metabolism and cell cycle (Fig. 4c–d and Extended Data Fig. 4b–e). Unlike 3D-concordant genes, 3D-insensitive genes showed constitutively high expression levels and stronger promoter H3K27ac and ATAC-seq signals across all cell types (Fig. 4e and Extended Data Fig. 4f). This analysis suggests that housekeeping and lineage-specific genes have differential sensitivity or dependence on 3D connectivity changes in early embryonic lineages.
Figure 4. Association of 3D rewiring with cell-type specific gene expression.
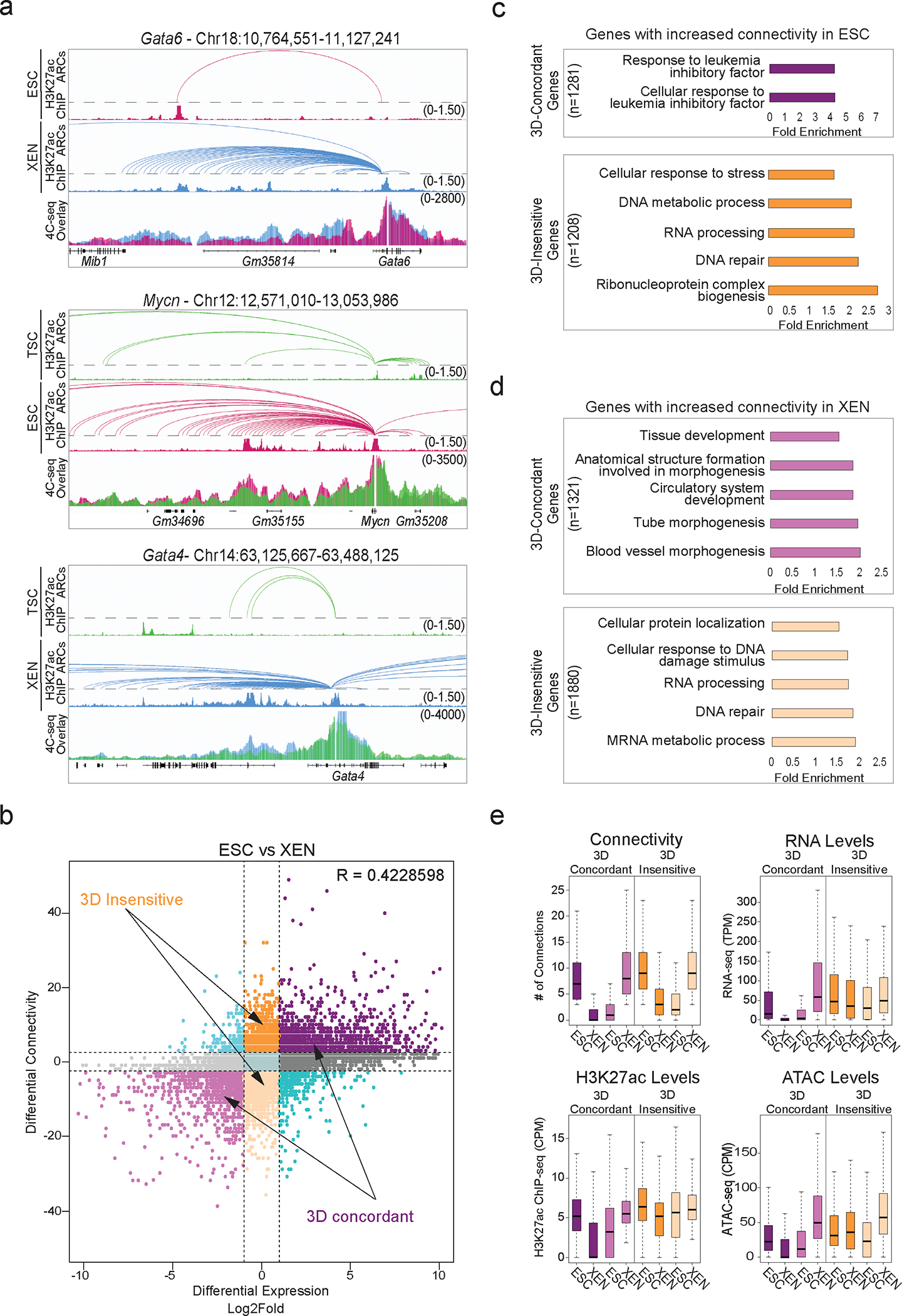
a. Examples of 3D rewiring at developmental genes in TSC, ESC and XEN detected by H3K27ac HiChIP (arcs on top with respective H3K27ac ChIP-seq tracks), validated by 4C-seq (merged tracks at the bottom). Averaged 4C-seq signals from n=3 independent replicates.
b. Correlation between differential HiChIP connectivity/hubness and differential gene expression in ESC vs XEN cells. R represents Spearman correlation. Two prominent groups are highlighted: 3D-insensitive genes with differential connectivity >3 but no transcriptional changes (log2FC<1 or >−1) and 3D-concordant genes with connectivity and expression changes (log2FC >1 or <−1) positively correlating (Supplementary Table 5).
c. Gene ontology depicting select biological processes significantly enriched in 3D concordant (purple) and 3D insensitive (orange) groups in ESC cells as defined in (b). (See Supplementary Table 3).
d. Same as (c), but for XEN cells. (See Supplementary Table 3).
e. Comparison of connectivity, gene expression (TPM), H3K27ac and ATAC CPM levels at promoters of 3D-concordant (n=2,235) and 3D-insensitive genes (n = 2581) in ESC and XEN cells. Two-sided Wilcoxon rank sum test was used for all comparisons (Supplementary Table 9).
Note: all statistics are provided in Supplementary Table 9.
3D features improve predictive modeling of gene expression
So far, our analyses established strong links between 3D connectivity and transcriptional regulation, with notable exceptions. Therefore, we sought to systematically investigate which 3D features were most important for predicting transcriptional output. To this end, we built an optimized Random Forest machine-learning model, coined 3D-HiChAT, utilizing 1D-information extracted from ATAC-seq and H3K27ac ChIP-seq datasets and 3D-information from HiChIP (Fig.5a). Specifically, we generated a list of ten 1D, 3D or composite variables originating either from gene promoters (5kb anchor containing the TSS) or interacting anchors-enhancers (Supplementary Table 6). By applying recursive feature selection to eliminate features with low importance, we nominated eight predictive features (Extended Data Fig. 5a), that individually showed variable correlation with gene expression (ranging from 0.17–0.58) (Extended Data Fig. 5b). We also constructed models that utilize only 1D-information from ChIP-seq and ATAC-seq either from the promoter region (“Promoter-centric model”) or from the extended linear neighborhood (“Linear proximity models” n=25 ranging from 10kb to 2Mb distance from promoter) to compare against 3D-HiChAT (Fig. 5a). Random Forest classification or regression methodology was used for each model to predict the top 10% or bottom 10% expressed genes (classification) or absolute gene transcription levels (correlation) in each cell type. We performed Leave One Chromosome Out (LOCO) methodology to train our data in TSC for all chromosomes but mitochondrial (chrM) and chromosome Y (chrY) (n=20, chr1–19 & chrX) prior to testing on the rest of the chromosomes and cell lines.
Figure 5. Predictive modeling using 3D chromatin features outperforms promoter- or 1D-based models for gene expression levels or cell-type specificity.
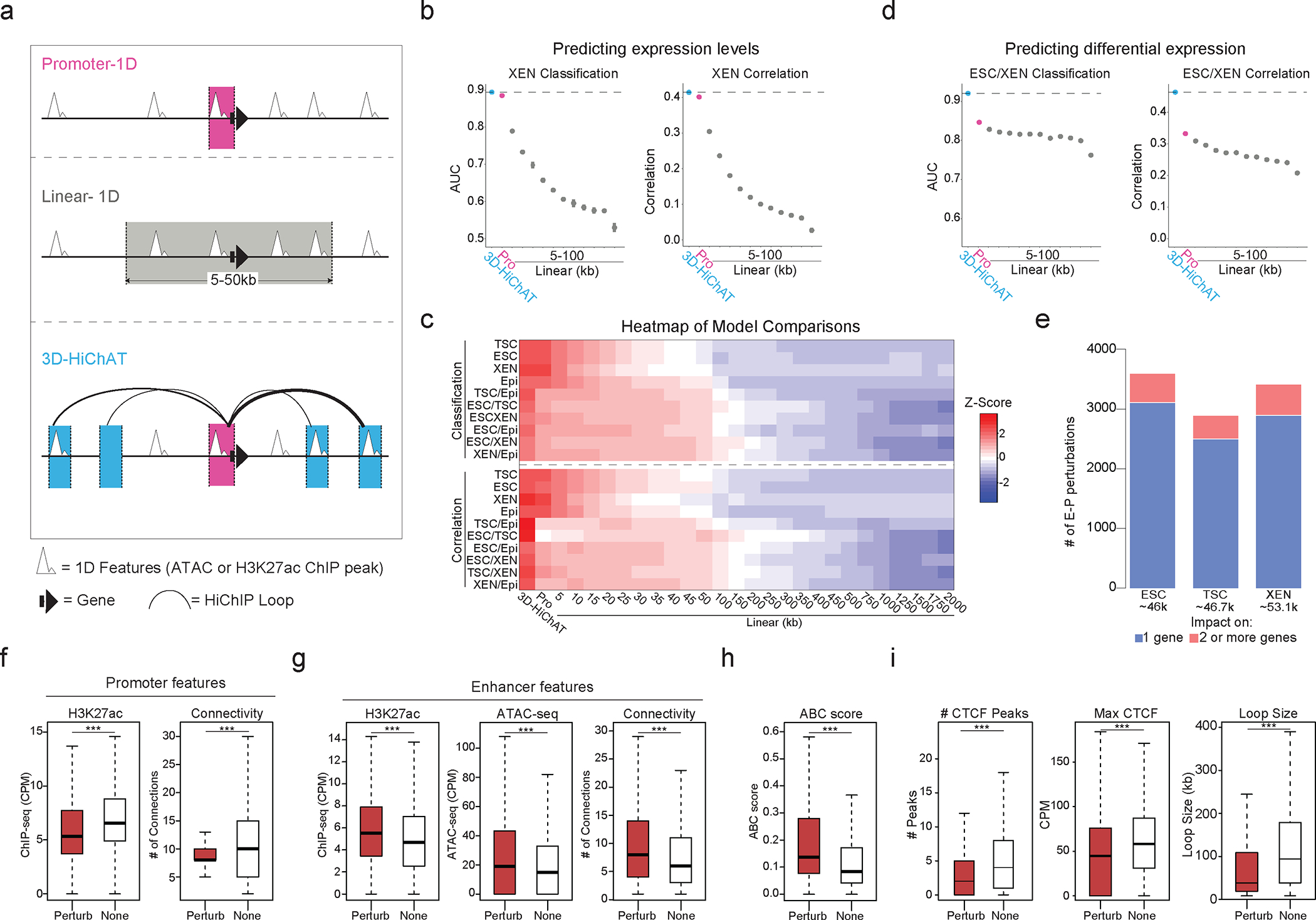
a. Schematic illustration of 1D or 3D variables used for modeling gene expression. (See Supplementary Table 6).
b. Area Under Curve (AUC) scores and Spearman Correlation for classifying gene expression (top 10% high vs low, left graph) and predicting absolute levels (right graph) in XEN using 3D-HiChAT, Promoter and Linear models. Dots represents average scores from the LOCO training approach, error bars show standard deviation. (See Extended Data Figure 5 and Supplementary Table 6).
c. Top: Heatmap of z-scored normalized AUC values across tested models for classification of gene expression or differential gene expression (top 10% high or low) in each cell line. Bottom: Heatmap of z-scored normalized Spearman correlation values across all models for prediction of gene expression levels or differential expression in each lineage. (See Supplementary Table 6).
d. AUC scores and Spearman Correlation generated for classifying differential expression (top 10% up or downregulated, left) and predicting expression (right) between XEN and ESCs using 3D-HiChAT, Promoter and Linear models. Dots represents average scores from the LOCO training approach, error bars show standard deviation. (See Extended Data Figure 5 and Supplementary Table 6).
e. Barplots showing numbers of E-P perturbations predicted to reduce one (blue) or more (pink) target gene expression using 3D-HiChAT. (See Extended Data Fig. 5f).
f. Boxplots showing median H3K27ac signals (left) or Connectivity (right) at promoter anchors of either perturbed (Perturb, n=4231) or unaffected E-P pairs (None, n= 4231) in ESC as described in (e). Asterisks indicate significance pval<0.001 by Two-sided Wilcoxon rank test.
(g-h). Boxplots showing median H3K27ac signal, ATAC-seq signal, Connectivity (g) and ABC score (h) at enhancer anchors of either perturbed (Perturb n=4231) or unaffected E-P pairs (None n=4231) in ESC as described in (e). Asterisks indicate significance pval<0.001 by two-sided Wilcoxon rank test.
i. Boxplots showing median numbers and max intensities of intervening CTCF peaks and genomic distance between perturbed (Perturb n=4231) or unaffected E-P pairs (None n=4231) in ESC as described in (e). Asterisks indicate significance pval<0.001 by two-sided Wilcoxon rank test.
Note: all statistics are provided in Supplementary Table 9.
When classifying gene expression (high vs low) in each cell type, we noticed that 3D-HiChAT consistently outperformed (Area Under Curve, AUC 0.89–0.93) the promoter-centric model (AUC ranging from 0.88–0.92), albeit by a small margin. Linear proximity models showed drastically lower accuracy when included information from distal regions (Fig. 5b and Extended Data Fig. 5c). Therefore, although the epigenetic features of gene promoters are largely sufficient to explain transcriptional output, incorporating 3D features specifically from distal interacting elements rather than from the extended linear neighborhood can improve our understanding of gene expression. The same conclusions were reached when we applied Random Forest regression analysis for predicting absolute transcriptional levels (instead of classification to high or low expressing genes) with 3D-HiChAT outperforming both promoter and linear 1D models (Spearman Correlation coefficient 0.42–0.49 for 3D vs 0.40–0.46 for Promoter-centric models) (Fig. 5b and Extended Data Fig.5c). 3D-HiChAT showed similar performance and accuracy across different cell lines and species using published HiChIP, ATAC-seq and RNA-seq datasets42, suggesting that it is stable and generalizable (Extended Data Fig 5d).
Next, we used similar methodology (see Methods for details) to test and compare the ability of our models to predict differential gene expression among the three embryonic lineages. To avoid using the same cell lines both for training and testing, which could result in overfitting, we generated RNA-seq, ATAC-seq, H3K27ac ChIP-seq and HiChIP from a fourth embryonic cell type, mouse Epiblast Stem Cells (EpiSCs)57, using same methods and QC standards. The models were trained using the LOCO approach on TSC versus EpiSC data prior to testing in all other pairwise lineage comparisons using the same eight predictive features shown in Extended Data Fig. 5a. Both classification and regression analysis demonstrated a clear superiority of 3D-HiChAT over promoter-centric or Linear proximity models in predicting differential gene expression (Fig.5c–d and Extended Data Fig. 5e). Promoter-based models showed poor overall predictability, highlighting that promoter features are insufficient to explain/predict cell-type specific gene expression. (Fig.5c–d and Extended Data Fig. 5e). These results highlight the importance of distal regulatory elements in cell-type specific gene expression and demonstrate that HiChIP features can enable accurate prediction of context-specific transcriptional output.
We next used 3D-HiChAT to predict the relative regulatory impact of putative enhancers on multiconnected (n>2) genes in each cell line by performing genome-wide in silico perturbations. Specifically, we predicted the degree of expression changes (% of perturbation) for each target gene after systematically removing each connected anchor/enhancer and recalculating all variables. E-P pairs were ranked based on their perturbation scores (%) in each cell line separately and cut-offs (for high-confidence perturbation) were determined at the points where the slope of the tangent along the curve exceeded the value of one (Extended Data Fig. 5f). While we observed perturbations in both directions (positive and negative perturbation), we focused on perturbations that caused gene downregulation, suggesting a putative enhancer function. This strategy identified ~4,300 out of the 46,000 interrogated E-P pairs that passed the cut-off (<−9.91%) in ESCs, ~3,400 out of 46,700 E-P pairs in TSC (< −12.55%) and ~4,200 out of 53,100 in XEN (<−11.20%) (Fig. 5e and Extended Data Fig. 5f).
To understand which features confer susceptibility or resistance to expression changes upon in silico perturbation, we directly compared the predicted functional enhancer-promoter pairs (Perturb) with an equal number of non-perturbed pairs (None). Genes within the perturbed group were characterized by significantly lower ChIP-seq signal at their promoters as well as lower overall promoter connectivity compared to non-affected genes (Fig. 5f). This suggests that genes with high promoter activity, and/or high hubness are less responsive to individual perturbations or to connectivity changes, aligning with our observations about the “3D-insensitive” genes (Fig. 4e). On the other hand, anchors predicted to perturb gene expression -compared to the non-perturbing ones- had significantly stronger H3K27ac signal and contact probabilities (Fig. 5g), in agreement with the recently published Activity-By-Contact (ABC) model46. Although, 3D-HiChAT predictions showed a good correlation with ABC scores, (R=−0.40795) (Extended Data Fig. 5g, Fig. 5h), we observed several enhancers with high 3D-HiChAT scores but low ABC. These enhancers were at higher distances (median = 50kb / mean=90.75 kb) compared to the ones with high ABC (median = 15kb / mean = 20.47 kb), suggesting that our model might be able to capture more distal functional enhancers (Extended Data Fig. 5h). Additional comparison between the Perturb or None groups showed that predicted impactful enhancers were significantly closer to their target genes and crossed significantly fewer and weaker CTCF binding sites (Fig. 5i). This is consistent with the notion that functional enhancers reside within the same insulated neighborhood or TAD with their target genes30,34,101,102, although we predicted a small fraction (589/42331=13.92%) of impactful enhancers that crossed TAD boundaries.
Finally, we observed that predicted impactful enhancers were characterized by significantly higher hubness (Fig.5g), supporting the notion that enhancer 3D connectivity indicates stronger regulatory impact and reflects centrality in regulatory networks. This finding could suggest that multiconnected enhancers have regulatory impact on multiple genes, and operate as 3D regulatory hubs. In total, 3D-HiChAT identified 484 putative enhancer hubs in ESC (controlling 1108 genes), 392 hubs in TSC (controlling 904 genes) and 523 hubs in XEN (controlling 1317 genes) whose deletions predicted downregulation of two and up to eight different genes (Supplementary Table 6) (Fig.5e).
3D-HiChAT model reveals functional enhancers and hubs in early embryonic lineages
Our results suggest that the 3D-HiChAT model could enable discovery of core enhancers dictating early embryoniccell fates. To experimentally test this, we focused on a complex locus in ESCs that spans ~1.3Mb and harbors, two important genes for maintenance or acquisition of pluripotency, Tfcp2l1 and Gli2103–108. According to our HiChIP data, both genes reside in the same A compartment in ESCs and form connections with a total of 17 proximal and distal putative enhancers; each with variable perturbation scores based on 3D-HiChAT (Fig. 6a–b and Extended Data Fig. 6a). We experimentally tested two shared putative enhancers, Enh3 and Enh14, with the expectation that Enh3 will only affect Tfcp2l1 while Enh14 will affect both genes. We transduced an ESC line stably expressing dCas9-BFP-KRAB (CRISPRi) with guide RNAs targeting each of the shared enhancers or the gene promoters (Extended Data Fig. 6b). After antibiotic selection, RT-qPCR was used to determine impact on gene expression compared to an empty vector control (n≥3 independent experiments per gRNA). In agreement with our predictions, CRISPRi silencing of Enh3 caused significant downregulation of Tfcp2l1 only (Extended Data Fig. 6b), while silencing of Enh14 significantly reduced the expression of both Tfcp2l1 and Gli2 (Fig.6c–e). The concordant downregulation of both enhancer-connected genes validates its function as a 3D regulatory hub. CRISPRi-mediated silencing of Enh14 had no significant impact on other connected genes, consistent with lower predicted perturbation scores on these genes.
Figure 6. Experimental validation of predicted enhancers in ESC and XEN.
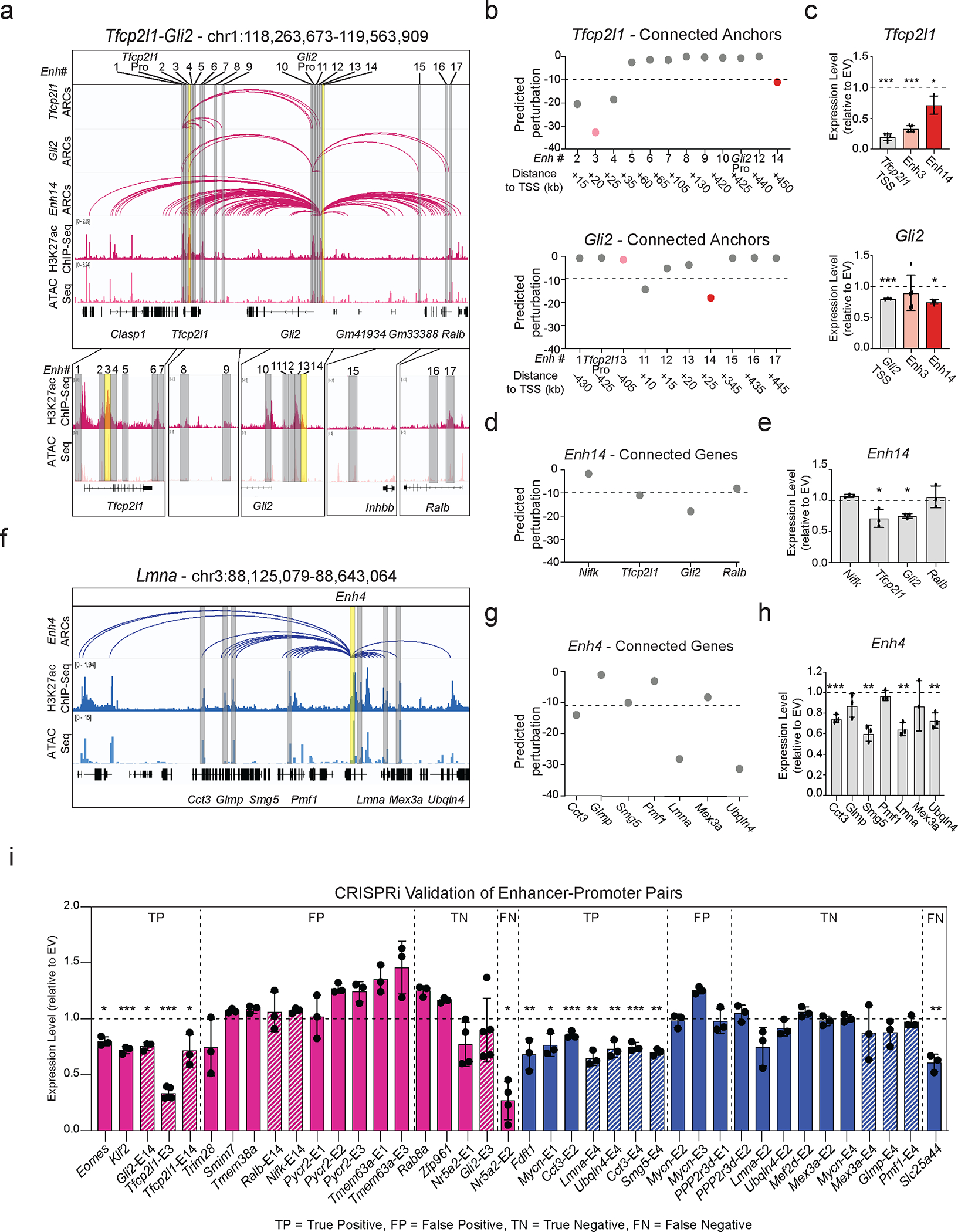
a. IGV tracks of the Tfcp2l1-Gli2 locus depicting putative regulatory elements. HiChIP arcs from both promoters and Enh14 are shown with H3K27ac ChIP-Seq and ATAC-seq.
b. Predicted 3D-HiChAT perturbation scores for Tfcp2l1, Gli2 promoter connected putative enhancers. Dotted line indicates cut-off (<−9.9) for impactful hits in ESC (see Extended Data Fig.5f and Methods).
c. Relative mRNA levels of Tfcp2l/Gli2 upon CRISPRi-targeting compared to empty vector (EV). Dots indicate biological replicates (n= 3 for Enh14 and n=5 for, independent experiments). Error bars indicate mean ± SD. Statistical performed by one-tailed unpaired student t-test. Asterisks indicate significance < 0.05.
d. 3D-HiChAT perturbation scores for Enh14 connected genes. Dotted line indicates cut-off (<−9.9) for impactful hits in ESC (see Extended Data Fig.5f and Methods).
e. Relative mRNA levels of genes upon Enh14 CRISPRi perturbation in ESCs compared to empty vector (EV). Dots indicate biological replicates (n = 3 independent experiments). Error bars indicate mean ± SD. Statistics performed by one-tailed unpaired student t-test. Asterisks indicate significance < 0.05.
f. IGV tracks of Enh4 interacting with 7 genes (yellow) in XEN. HiChIP arcs originating from Enh4, H3K27ac ChIP-Seq and ATAC-seq are shown.
g. Predicted perturbation scores for Enh4 hub connected genes. Dotted line indicates cut-off (<−11.20) chosen for impactful hits in XEN (see Extended Data Fig.5f and Methods).
h. Relative mRNA levels of genes upon Enh4 CRISPRi in XEN compared to empty vector (EV). Dots indicate biological replicates (n= 3 independent experiments). Error bars indicate mean ± SD. Statistics performed by one-tailed unpaired student t-test. Asterisks indicate significance < 0.05.
i. Barplots summarizing expression changes upon CRISPRi perturbations of 40 enhancer-promoter pairs in ESC (Pink) and XEN (blue), with positive (cut-off <−9 for ESC, <−11.2 in XEN) or negative predictions, by 3D-HiChAT. Bars represent RT-qPCR values relative to Empty Vector (EV) with housekeeping normalization (Hprt for ESC, Gapdh for XEN). Shaded bars indicate data from above (c), (e) or (h). Dots indicate biological replicates (n=3 independent experiments). Error bars represent mean ± SD. Statistical analysis was performed by one-tailed unpaired student t-test. Asterisks indicate significance < 0.05.
Note: all statistics and source PCR data are provided in Supplementary Table 9.
By establishing a similar CRISPRi system in XEN cells (Extended Data Fig. 6c) we also validated an enhancer hub (Enh4) connected to 7 genes across a 520kb region (Fig.6f) with different predicted impact on each gene (Fig. 6g). CRISPRi-mediated targeting of this hub led to significantly downregulated levels of Lmna, Cct3, Smg5 and Ubqln4, while other connected genes (Glmp, Pmf1 and Mex3a) remained unaffected (Fig. 6h), in agreement with our model predictions.
To build robustness in our models predictions, we extended our experimental perturbations to a total of 40 enhancer-promoter pairs in ESC (n=20, pink) or XEN (n=20, blue), with moderate connectivity (between 2–12 connections) and variable 3D-HiChAT perturbation scores (ranging from −0.02 to −46.8) (Fig. 6i and Supplementary Table 6). We identified 12 true positive hits (including enhancers around developmental genes Klf2, Eomes and Mycn) and 13 true negative hits. By ranking E-P pairs based on their 3D-HiChAT perturbation scores and classifying genes as perturbed or not based on CRISPRi results, we calculated an overall accuracy of 0.71 (Extended Data Fig. 6d). Although this is potentially an underestimation due to variable efficiencies of gRNAs, it calls for additional improvements and metrics to improve prediction accuracy. Of note, more than half of our validated enhancers had very low ABC scores (<0.2), (Extended Data Fig. 6e) partly reflecting their higher distance to their target genes, suggesting that our model might be more suitable in predicting distal functional enhancers.
Together, these results demonstrate the ability of 3D-HiChAT to predict complex regulatory relationships, including enhancer hierarchies around multiconnected genes and enhancer-promoter specificity of multiconnected enhancers. Future, systematic combinatorial perturbations could further dissect the regulatory logic around multiconnected loci. Given the stable performance of the model across different cell types and species (see Extended Data Fig.5d), 3D-HiChAT could be applied in different biological systems to nominate candidate functional enhancers or help interpretation of disease-associated structural variants.
DISCUSSION
Cell-type specific transcriptional programs are controlled by the activity of transcription factors and their target enhancers109–112. Studying the mechanisms of enhancer activity and specificity is essential for understanding and modulating the mechanisms that dictate cell fate decisions. In this study, we applied H3K27ac HiChIP and other genomics technologies to capture the remodeling of enhancer landscapes and 3D interactomes in the first embryonic lineages and establish associations with transcriptional behavior and cell identity. Our results generated detailed 3D networks of enhancer-promoter connections in mouse TSCs, ESCs and XEN cells and provided a resource of predicted functional enhancers for each lineage and proof-of-concept validations. Our integrative analysis and predictive model revealed -potentially universal- insights into the functional interplay between 3D connectivity and transcription.
Physical proximity -but not necessarily physical contact- is considered the most likely mechanism for functional communication between genes and distal regulatory elements101 and an important feature for assigning enhancers to their cognate target genes113. In agreement with previous studies in various cellular contexts42,98,114,115, we show a strong positive correlation between 3D connectivity -or “hubness”-and gene expression across lineages, but also important exceptions which reflect the intricate nature of transcriptional regulation in the context of complex and dynamic 3D networks. Specifically, we uncovered distinct principles and 1D/3D features that influence (i) the relative susceptibility of multi-connected genes to topological changes or enhancer perturbations and (ii) the relative regulatory impact of individual enhancers on one or more target genes. For example, we observed a strong concordance between transcriptional and topological changes around lineage-specific genes, suggesting that the de novo establishment (or strengthening) of long-range interactions with distal enhancers is critical for robust and context-specific activation of these genes. Meanwhile, housekeeping genes appeared insensitive to 3D rewiring, suggesting that their high expression levels are likely driven from their promoters, which are saturated or irresponsive to additional regulatory input. This result aligns with recent high-throughput reporter assays that interrogated enhancer-promoter compatibility and found a reduced responsiveness of housekeeping promoters to distal enhancers116. Moreover, our in silico and experimental perturbations showed that highly connected genes -both housekeeping and developmental- tend to be less susceptible to individual enhancer deletions, suggesting functional redundancy among enhancers and phenotypic robustness in line with previous studies in different cellular contexts117,118
Several computational models have been developed to predict putative functional enhancers in various cellular contexts either based on 1D features (e.g. chromatin accessibility, histone marks, TF/co-factor binding, nascent transcription etc.)119–124 and/or 3D features, such as CTCF binding, insulation33,125 or contact probability with target genes46,126,127. These predictions become particularly challenging in the context of highly interacting hubs where multiple genes and putative regulatory elements come in spatial proximity (albeit not necessarily all at the same time and allele) making it hard to dissect which of these interactions have positive, negative or neutral regulatory impact. 3D-HiChAT predictions and functional validations show that consideration of 3D features extracted from 3D enhancer-promoter networks enables better predictions of (i) transcriptional behaviors, such as levels and cell-type specificity of gene expression or probability of gene co-regulation and (ii) of complex regulatory relationships, including enhancer hierarchies or redundancies and enhancer-promoter specificities. We were able to identify and validate several “dominant” enhancers around multiconnected developmental genes, as well as functional enhancer hubs, responsible for the coordinated regulation of more than two genes in ESC or XEN. Not all connected genes respond to the same enhancer and not all putative enhancers contributed to the regulation of their interacting genes. In agreement with previous studies, 3D-HiChAT showed that the relative contact frequency between enhancers and promoters and their putative activity/accessibility (as indicated by H3K27ac ChIP-seq and ATAC-seq) are important predictors of their regulatory relationships. However, our model also took into consideration the secondary interactions of each enhancer and showed that high degree of enhancer hubness is predictive of stronger regulatory impact upon perturbation, and potentially on multiple connected/coregulated genes. These findings nominate 3D hubness as an important predictive feature of regulatory centrality and suggest that mapping of 3D hubs could help dissect regulatory hierarchies and predict core modules (both critical genes and enhancers) that instruct cell-type-specific transcriptional programs.
Collectively, our study shows that 3D-HiChAT is a stable model, generalizes to different cell-types and species, performs better than 1D-based models and enables prediction of complex regulatory relationships around multiconnected genes and enhancers. However, generation and utilization of ultra-resolution (sub-kb) 3D genomics datasets and consideration of additional variables, such as binding of CTCF or lineage-specific transcription factors or enhancer-associated co-factors, could further improve model performance. Moreover, systematic high-throughput functional screens of putative positive and negative regulatory elements (e.g. X anchors) during dynamic cell fate transitions, will enable a deeper understanding of the regulatory relationships (hierarchies, redundancies, synergies or competitions) and inform development of better modeling approaches for prediction of core regulatory enhancers and hubs.
In conclusion, our study systematically mapped the dynamic 3D enhancer chromatin networks within the first embryonic (EPI) and extraembryonic (TE and PrE) cell fates and nominated candidate core enhancers for future high-throughput functional perturbations in vitro or in vivo. Our integrative analysis and 3D-HiChAT predictive model revealed conserved principles of transcriptional regulation through long-range interactions, providing a framework for understanding and modulating lineage-specific transcriptional behaviors.
METHODS
Cell culture
Detailed culture conditions of all stem cell lines used in this manuscript (including feeder dependent ESC, feeder independent ESC, Trophoblast and XEN cell lines) are provided in the supplementary information.
Lentiviral production and infection
293T cells were transfected with overexpression constructs along with the packaging vectors VSV-g, Tat, Rev and Gag-pol using PEI reagent (PEI MAX, Polyscience, 24765–2). The supernatant was collected after 48 and 72 h, and the virus was concentrated using polyethylglycol (Sigma, P4338). Cells were infected in medium containing 5 μg ml−1 polybrene (Millipore, TR-1003-G), followed by centrifugation at 1300g for 90 min at 32°C.
CRISPRi
XEN cells were infected with lentiviruses harboring the pHR–SFFV–dCas9–BFP–KRAB vector (Addgene, cat. no. 46911), while ESC v6.5 cells were infected with a modified version of the plasmid in which the SFFV promoter was replaced with an EF1a promoter 42. Cells expressing BFP were selected by 3 consecutive rounds of FACS sorting (enriching only for the high expressing cells each time). The resulting, ESC stably expressing the dCas9–BFP-KRAB cells, were then infected with a lentivirus harboring the pLKO5.GRNA.EFS.PAC vector (Addgene, cat. no. 57825) containing either a single or 2 gRNAs targeting the region of interest. Due to the Purmocyin resistance the XEN-dCas9-BFP-KRAB cells were infected with a modified version of the pLKO5.GRNA.EFS.PAC vector (Addgene, cat. no. 57825) replacing puromycin with blasticidin resistance. Cells were selected with puromycin (LifeTech, K210015) or blasiticidin for 4 days and subsequently collected for RT–qPCR analysis. All used guide RNAs and RT–qPCR primers for each target enhancer and gene are described in Supplementary Table 7.
H3K27ac ChIP-seq
ChIP-seq was performed as previously described42, with a few modifications. 10 million cells were used per replicate for TSC, ESC and XEN and in vitro derived EpiSC cells. Initially cells were crosslinked in 1% formaldehyde at RT for 10 minutes and quenched with 125mM glycine for 5 mins at RT. As a normalization control, 5 million formaldehyde-fixed Drosophila nuclei were added to each sample. Cell pellets were washed twice in 1xPBS and resuspended in 300ul lysis buffer (10mM Tris pH8, 1mM EDTA, 0.5% SDS) for at least 15 minutes. Next, chromatin was sonicated in a Pico bioruptor device for 10 cycles with the length of the intervals being 30sec on/off, in order to produce 300–800 bp chromatin fragments. Sonicated chromatin was then spun down for 15 minutes at 4°C at 22,000g and 10μl of the sheared soluble chromatin solution was used in order to check the shearing efficiency and the rest was kept at 4°C. 5% of each sample was kept as an input while the rest of the supernatants were diluted 5 times with dilution buffer (0.01% SDS, 1.1% triton,1.2mM EDTA,16.7mM Tris pH8, 167mM NaCl) and incubated with 3μg H3K27ac antibody (ab4729) O/N under agitation at 4°C. Next day, protein G-Dynabeads were pre-washed 3 times in ice cold 0,01% Tween-20/1xPBS, pre-blocked for 30 minutes at 4°C with 1% BSA/1xPBS and finally added to each sample (30ul Dynabeads per sample) and incubated for 3.5 hours at 4°C in order to bind the specific chromatin-antibody complexes. Upon IP, beads were washed twice in low salt buffer (0.1% SDS,1% triton, 2mM EDTA, 150mM NaCl, 20mM Tris pH8), twice in high salt buffer (0.1% SDS,1% triton, 2mM EDTA, 500mM NaCl, 20mM Tris pH8), twice in LiCl buffer (0.25M LiCl, 1% NP40, 1% deoxycholic acid, 1mM EDTA, 10mM Tris pH8) and once in TE buffer. DNA was then eluted from the beads by incubating with 150ul elution buffer (1% SDS, 100mM NaHCO3) for 30 minutes at 65°C (vortexing every 10min). Input and bound fractions of supernatants were reversed overnight at 65°C with 20mg/ml proteinase K. Next day samples were treated with 100mg/ml RNase and DNA was purified using a ZYMO Kit (D4014) following manufacturer’s instructions. Finally, 25ng of immunoprecipitated material and input were used for ChIP-seq library preparation using the KAPA Hyper prep kit (KK8502) according to manufacturer’s instructions. Libraries were sequenced on an Illumina NextSeq2000 platform on SR100 mode. ChIP-seq data have been deposited in the Short Read Archive (SRA) under the accession codes GSE212992.
ATAC-seq
ATAC-seq was carried out as previously described with minor modifications128. For each cell line 2 replicates were performed and analyzed. Briefly, a total of 50,000 cells were washed with 50 μL of cold 1xPBS and then nuclei were isolated in 50 μL lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.2% (v/v) IGEPAL CA-630). Nuclei were then centrifuged for 10min at 800g at 4°C, followed by the addition of 50 μL transposition reaction mix (25 μL TD buffer, 2.5 μL Tn5 transposase and 22.5 μL ddH2O) using reagents from the Nextera DNA library Preparation Kit (Illumina #FC-121–103). Samples were then incubated at 37°C for 30min. DNA was isolated using a ZYMO Kit (D4014). ATAC-seq libraries were prepared using NEBNext High-Fidelity 2X PCR Master Mix (NEB, #M0541), a uniquely barcoded primer per sample, and a universal primer. Samples were first subjected to 5 cycles of initial amplification. To determine the suitable number of cycles required for the second round of PCR (to minimize PCR bias) the library was assessed by quantitative PCR128. Briefly, a 5 μL aliquot of the initial amplification sample was used for 20 cycles of qPCR. Linear Rn versus cycle was plotted to determine cycle number corresponding to 1/3 of maximum fluorescent intensity. For each sample, the remaining 45 μL of initial tagmented PCR product was further amplified for 5 more cycles using Nextera primers. Samples were subject to a dual size selection (0.55x–1.5x) using SPRIselect beads (Beckman Coulter, B23317). Fragment distribution of libraries was assessed with an Agilent Bioanalyzer and finally, the ATAC libraries were sequenced on an Illumina Hi-Seq (2500) platform for 50bp paired-end reads.
In situ Hi-C
The protocol was performed as previously described42 with minor modifications. Hi-C was performed starting with 2 million cells per replicate and using the Arima-Hi-C kit (Arima, A510008) according to manufacturer’s instructions. Approximately 500ng of DNA was used for each Hi-C sample to prepare libraries using the KAPA Hyper Prep Kit (KAPA, KK8502) and performing 5 cycles of amplification. Libraries were sequenced using the Illumina Nextseq 2000 in PE50 mode.
In situ 4C-seq
The protocol was performed as previously described with minor modifications129. Briefly, 10 million cultured ESC, TSC and XEN cells were fixed with 12 ml of 1% formaldehyde (Thermo Scientific, 28908) in 10% FBS for 10 min at room temperature (RT) (tumbling). Quenching of the cross-linking was performed with the addition of 1.8 ml of freshly prepared ice-cold 1 M glycine (Sigma-Aldrich #500046). Tubes were transferred directly on ice and centrifuged for 5 min 500g at 4°C. Cells were washed with 1xPBS and centrifuged for 5 min 500g at 4°C, and pellets were frozen in liquid nitrogen and stored at −80°C. Next, cells were then vigorously resuspended in 1 ml of fresh ice-cold lysis buffer (10 mM tris (pH 8), 10 mM NaCl, 0.2% NP-40, and 1 tablet of complete protease inhibitor (Roche, 04693159001)], transferred to 9 ml of prechilled lysis buffer, and incubated for 20 min on ice. Following centrifugation at 500g for 5 min at 4°C, the pellet was resuspended in 50uL of 0.5% SDS and incubated for 10 min at 65°C. SDS was quenched with 145uL ddH2O and 25uL of 10% Triton X-100 for 15 mins at 37°C. At this point, 5 μl of the sample was taken as the “undigested control”. Next, 25ul of CutSmart buffer (NEB, B7204S) was added with 10μl DpnII enzyme (NEB, R0543M) and the samples were incubated overnight at 37°C under agitation (750rpm). Upon first digestion, 5μl of the sample was taken as the “digested control” while the efficiency of chromatin digestion was verified after DNA extraction from 5 l of undigested and digested controls and loading in a 1.5% agarose gel. After verification of chromatin digestion (smear between 0.2 and 2 kb), DpnII was deactivated by 20 min incubation at 62°C (under agitation 750 rpm). Ligation of DNA ends between the cross-linked DNA fragments was performed by diluting the samples in 669 μL ddH2O and adding 120 μL T4 ligation buffer (NEB, B0202), 60 μL 10mM ATP (NEB, P0756S), 120 μL 10% Triton X-100, 6 μL 20mg/ml BSA and 5 μL 400U/μl T4 DNA Ligase (NEB, M0202) overnight at 16°C (tumbling) followed by 30min at RT. 10μl of the ligated sample was tested as “ligated control,” on a 1.5% agarose gel. The samples were then treated with proteinase K and reverse crosslinked overnight at 65°C. Following RNase treatment, phenol/chloroform extraction and DNA precipitation, the pellets were dissolved in 100 μL of 10mM Tris pH 8 and incubated for 1 hour at 37°C. Efficiency of extraction and purification were verified on a 1.5% agarose gel. For the second digestion 20 μL of 10x buffer B (Fermentas), 10 μL Csp6I (Fermentas, ER0211), 80 μL ddH2O were added to the DpnII-ligated 3C template and samples were incubated overnight at 37°C under agitation (750rpm). Csp6I was inactivated at 65°C for 20 min, and DNA fragmentation was tested on 1.5% agarose gel. A second ligation was performed by adding 300 μL T4 ligation buffer, 150 μL 10mM ATP, 5μL T4 DNA Ligase, and ddH2O to 3mL and incubating overnight at 16°C. After 30 min of incubation at RT, samples were PCI-extracted, ethanol-precipitated, resuspended in 200 μl of sterile water, and purified using the Qiaquick PCR Purification Kit (Qiagen). DNA concentration of each digested sample was calculated using the Qubit brDNA HS assay kit (Invitrogen). For library preparation, primers were designed either around the enhancer or the promoter of lineage specific genes. Library preparation was then performed using the inverse PCR strategy. Briefly, 4×200 ng of 4C-template DNA was used to PCR amplify the libraries using the Roche Expand long template PCR system (Roche, 11681842001) with the following PCR conditions: 94 °C for 2 min, 16 cycles: 94 °C for 10 seconds; [primer specific] °C for 1 min; 68 °C for 3 min, followed by a final step of 68 °C for 5 min. Amplified material was pooled, and primers were removed using SPRIselect beads (Beckman Coulter, B23317). A second round of PCR with the following conditions: (94 °C for 2 min, 20 cycles: 94 °C for 10 seconds; 60 °C for 1 min; 68 °C for 3 min and 68 °C for 5 min) was performed using the initial PCR library as a template, with overlapping primers to add the P5/P7 sequencing primers and indexes. The samples quantity and purity were determined using a NanoDrop spectrophotometer while the 4C PCR library efficiency and the absence of primer dimers were reconfirmed by Agilent Bioanalyzer. For each cell line 3 replicates were performed, and the libraries were sequenced on a HiSeq4000 in SE150 mode. All the 4C-seq primer sequences are provided in Supplementary Table 7.
H3K27ac HiChIP
ESC cells were processed for each HiChIP replicate using the Abcam H3K27ac antibody (ab4729) and following the HiChIP protocol as previously described42. TSC, XEN and EpiSC cells were used for each HiChIP replicate using the Arima-HiC+ kit (Arima, A101020) and the H3K27ac antibody (active motif H3K27ac 91193) according to manufacturer’s instructions with few modifications. The efficiencies of H3K27ac antibodies were tested by ChIP-seq, and both antibodies resulted in similar distribution and number of peaks. In order to improve the sonication efficiency, a modified lysis buffer was used containing 10mM Tris pH8, 1mM EDTA and 0.5% SDS. Prior to over-night incubation with the antibody the sample was diluted in a buffer to bring it back the original composition of the Arima R1 buffer (10mM Tris pH8, 140mM NaCl, 1mM EDTA, 1% triton, 0.1% SDS, 0.1% sodium deoxycholate). 5ng of immunoprecipitated DNA material was used to make libraries using the Swift Biosciences Accel-NGS 2S Plus DNA Library Kit (Cat #21024) according to manufacturer’s instructions and performing between 8–14 cycles of amplification for all samples. Final libraries were sequenced using the Illumina Nextseq 2000 in PE50 mode.
Modeling
Random Forest methodology was used for classification of gene expression levels and gene expression level prediction. A set of 28 variables that contain information from 1D (H3K27ac, ATAC-seq) and 3D (HiChIP) experiments were calculated for all hubs in our 4 cell types (Supplementary Table 6). After eliminating features with high correlation among them from 1D, 3D and combined 3D we ended up with 10 features. Recursive feature elimination (rfe function in “caret” library in R) was used for feature selection which led to the use of 8 out of the 10 features both in classification and regression Random Forest models.
Classification of hubbed genes based on their expression levels was achieved by separating looped genes into 10 equally sized groups (Q1 to 10). Cross validation was performed with “leave one chromosome out” method (L.O.C.O.) where we train our data in all chromosomes but one which we use for testing. This process is repeated until we leave every chromosome out of the training test for chromosomes 1–19 and chrX. AUC and correlation scores are calculated in each round of LOCO (n=20) and average AUC and correlation is calculated for all of our models tested (promoter, linear 2D and 3D). TSC promoter hubs for Q1 and Q10 were used for training, with ntree=1000 and mtry=floor (sqrt(# Variables) in TSC and tested classification of Q10 and Q1 gene groups in ESC, XEN and EPISC. In order to evaluate the models, we calculated average AUC score for each model in all cell lines. None of the model showed over-fitting since both training and testing sample showed similar accuracy. The same methodology was used to identify differential expression. For each cell type pair (n=6) we merged looped genes and calculated the difference for all of our 8 variables. We selected TSC/EPISC pair as our initial dataset which was split into training and test dataset as before with LOCO by selecting the Q1 and Q10 promoter hubs based on fold change. Random Forest was applied as before and average AUC scores were calculated for the rest cell type pairs (n=5, ESC/XEN, ESC/EPISC, TSC/ESC, TSC/XEN, XEN/EPISC).
Gene expression prediction was achieved with Random Forest regression model and ntree=1000 and mtry=floor(#Variables/3). Again, TSC was used for training and testing for all hubbed genes. The same steps were followed when we performed RF to predict fold changes between cell type pairs. Evaluation of RF model was performed with average spearman rank correlation coefficient.
To estimate the effect of each enhancer in our cell lines we performed in silico perturbation of each hub by removing one enhancer at a time in ESC, XEN and TSC. All 8 variables (hub metrics) were recalculated after each enhancer removal and gene expression levels were estimated based on the new hub metrics. In-silico perturbation was estimated as the percentage of change between Predicted and In-silico predicted gene expression levels for each of the genes and were separated into two groups based on their gene expression changes (Perturbed vs None).
Hi-ChAT score calculation
HiChAT score is calculated for each promoter anchor taking into account accessibility, enhancer and loop strength similar to ABC score46. For each gene only their interacting-looped enhancers within a 4Mb regions were used. ATAC signal was used for estimating accessibility of the enhancer identified by H3K27ac. For each promoter hub HiChAT was calculated with the following formula:
where is the number of connected enhancer anchors for a given promoter. HiChAT calculation provides an ABC-like score46 for all promoters by aggregating the Activity by contact signal of all connected enhancers. Two HiChAT scores (1 & 2) were generated by calculating the combined ATAC/H3K27ac signal at the enhancer and accessible regions respectively and tested in our gene expression predicting models.
Statistical methods and plots
All bioinformatic analysis on the genomic datasets generated in this study (ChIP-seq, ChIP-exo, ATAC-seq, RNA-seq, 4C-seq, Hi-C and HiChIP) are reported in the supplementary file.
Extended Data
Extended Data Fig. 1. Related to Figure 1.
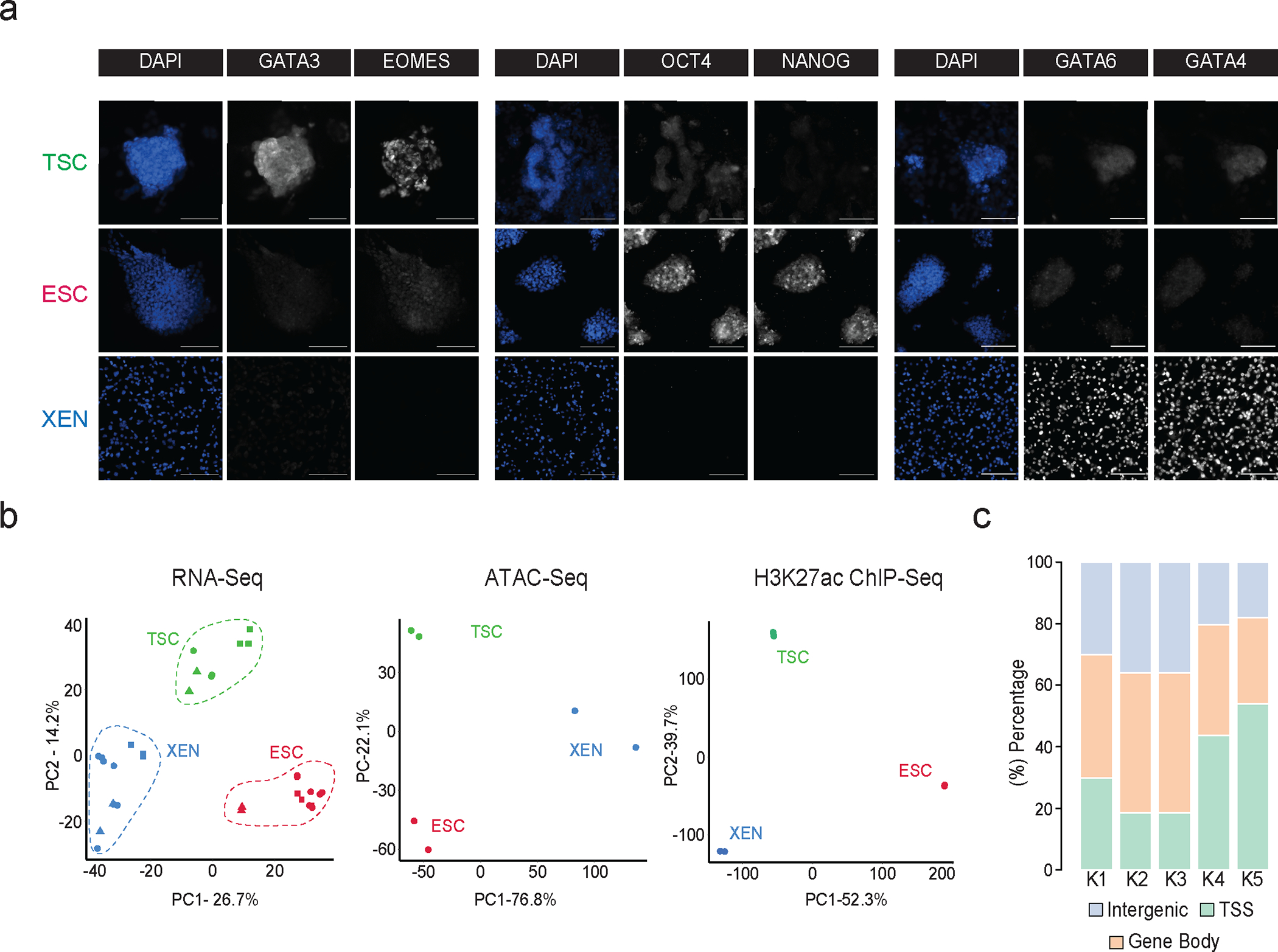
a. Representative single (xy) stack epifluorescence images of immunofluorescence experiments showing expression of key lineage markers (greyscale) in TSC, ESC and XEN cells. Cells were counterstained with DAPI (blue) for DNA content. n=3 independent experiments. Scale bar 100μm.
b. Principal component analysis (PCA) of all TSC, ESC and XEN replicates based on their RNA-seq, ATAC-seq and H3K27ac ChIP-seq profiles. PCA plots were designed based on the top10% of most variable genes or peaks in all three cell lines. In each plot, circles indicate the experimental data presented in this study, while squares and triangles correspond to publicly available RNA-seq data (Supplementary Table 8) or independent -unpublished- studies from our lab, respectively.
c. Stacked barplot showing the distribution of H3K27 occupancy among intergenic regions, gene bodies or TSS (promoter +/− 1.5kb) for each K-Mean cluster as identified in Fig.1c.
Note: all statistics are provided in Supplementary Table 9.
Extended Data Fig. 2. Related to Figure.
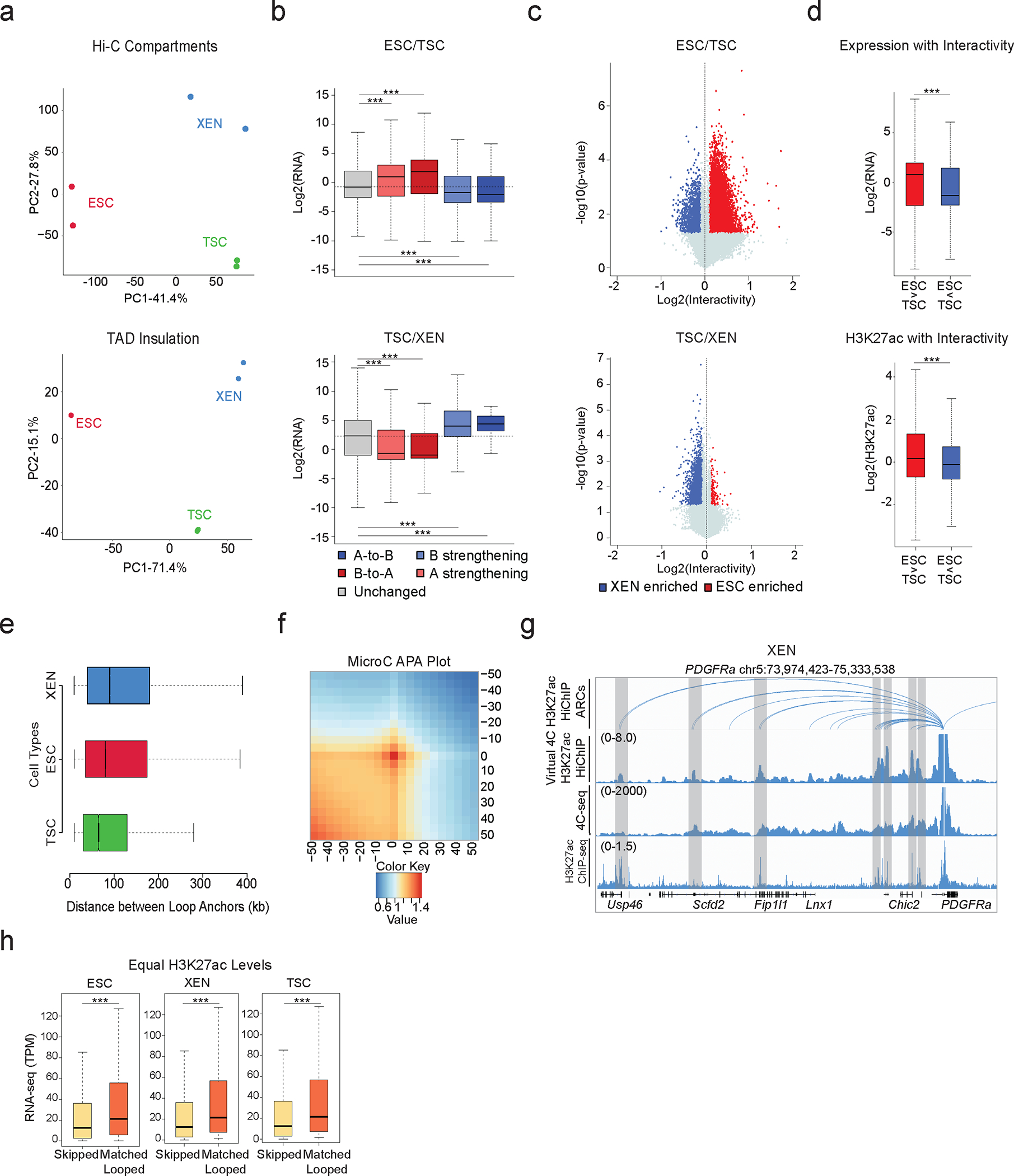
a. Principal Component Analysis (PCA) plot of all lineages and replicates based on their compartment scores at 100kb resolution (top) and on their TAD insulation levels at 40kb resolution (bottom).
b. Boxplots showing median expression changes between ESC and TSC (n = 4,327 genes), and TSC and XEN (n = 4156 genes) cells of genes in unaltered compartments (grey box and dashed line) or compartments with shifts as described in Fig 2b. Asterisks indicate significance (p<0.001) by two-sided Wilcoxon rank test.
c. Volcano plot showing differential Hi-C interactivity at 40kb resolution between ESC – TSC and TSC - XEN. X-axis shows delta interactivity while y-axis shows −log10(p-value) calculated by two-sided Student’s t-test. Significant changes (p-value<0.05 and Diff>0.1 or <−0.1) are noted with blue and red color.
d. Boxplots showing gene expression (n=2352 genes) and enhancer strength (n=15,982 peaks) changes between ESC-TSC regions with connectivity changes as described in Fig. 2c. Asterisks indicate significance (p<.001) by two-sided Student’s t-test.
e. Boxplot comparing the sizes of HiChIP-detected loops in the three cell lineages across n=2 independent Hi-C samples.
f. Aggregate peak analysis (APA) showing the aggregate signal of MicroC data in ESC 39 centered around ESC HiChIP interacting regions identified by FitHiChIP2.0 at 5 kb resolution. (See Supplementary Table 4 and Methods).
g. IGV tracks aligning H3K27ac HiChIP results (arcs on top and virtual 4C representation in the middle) with 4C-seq normalized signals around PDGFRA promoter in XEN along with corresponding H3K27ac ChIP-seq occupancy.
h. Boxplot showing the median expression levels of a curated list of skipped and looped genes in ESC, XEN and TSC across n=2 independent HiChIP and RNA-seq samples. Selected genes have similar ranges of H3K27ac signal at promoters. Asterisks indicate significance (p-value<0.05), as calculated by two-sided Wilcoxon rank sum test. (See Supplementary Table 4).
Note: all statistics are provided in Supplementary Table 9.
Extended Data Fig. 3. Related to Figure 3.
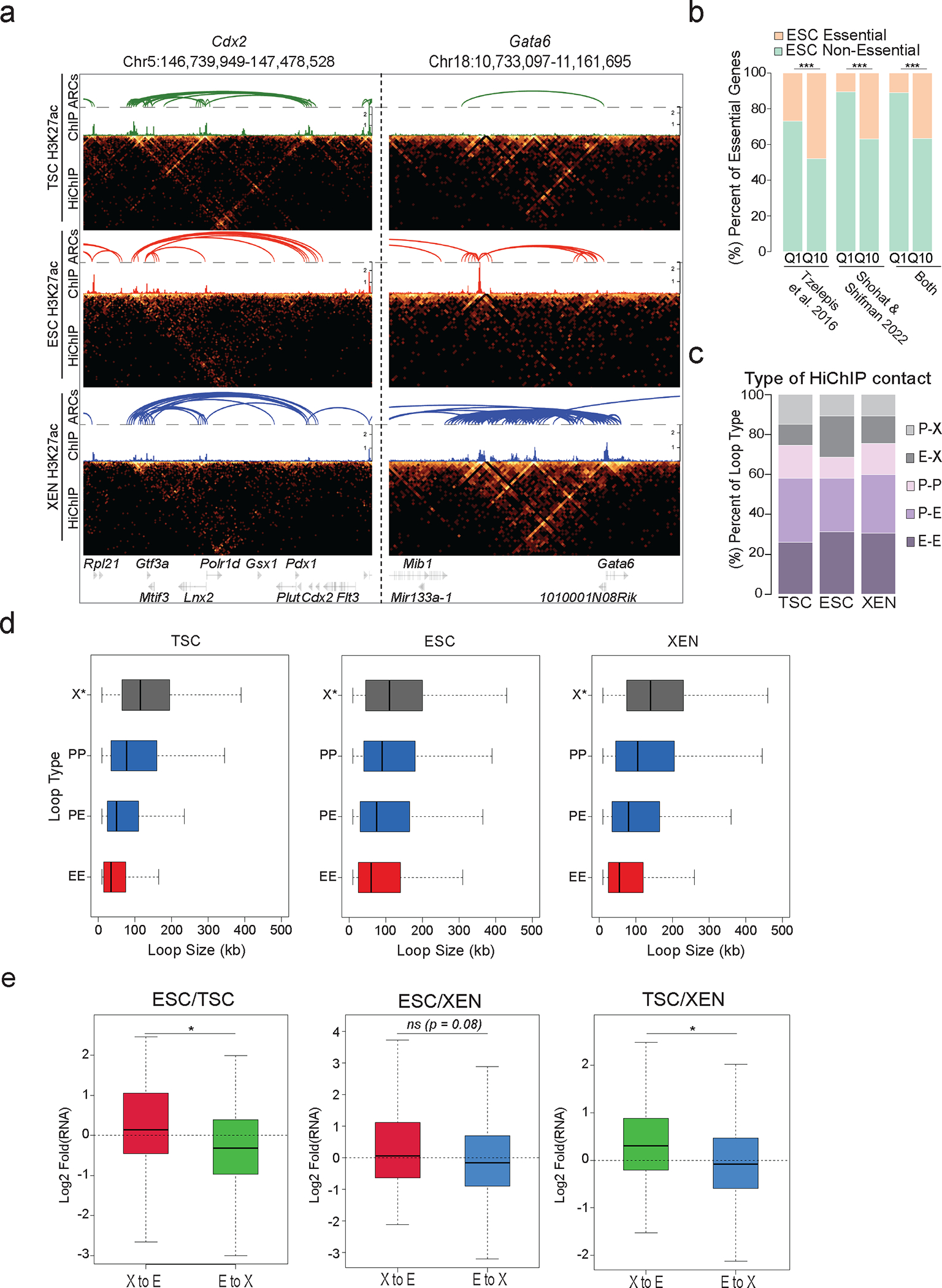
a. HiGlass visualization of H3K27ac HiChIP results around a TSC related hub (Cdx2) and a XEN-related hub (Gata6) in TSC, ESC, and XEN along with the corresponding H3K27ac HiChIP derived arcs and H3K27ac ChIP-seq signals. Interacting scores are presented in 5kb resolution.
b. Barplot showing the percentages of essential genes -as identified in two recent studies 94,95- within the least (Q1) versus most (Q10) connected hubs. The preferential enrichment of essential genes in Q10 is significant (p-value<0.001, two-sided Fisher’s exact test).
c. Stacked barplots showing the proportions of different HiChIP loop subtypes in TSC, ESC and XEN cells. Loops were separated into 5 chromatin interaction categories based on the presence of regulatory elements, such as promoter/TSS (P) or putative enhancer (E, H3K27ac peak). X- anchors were defined as anchors that do not contain any TSS nor an H3K27ac peak.
d. Boxplot showing the size distribution of X loops (X-E and X-P) compared to E-E, E-P and P-P loops in all cell lines. (n=60,909 (TSC), 81,679 (ESC), 77,124 (TSC) loops across n=2 independent HiChIP experiments)
e. Boxplots showing expression changes between any two cell types around multiconnected genes (n>=5 in both cell types of interest), when at least one of their conserved anchors switches chromatin states: either from X-to-E (enhancer gain) or from E-to-X (enhancer loss) Asterisks indicate significance < 0.05 by two-sided Wilcoxon rank-sum test. (See also Supplementary Table 9).
Note: all statistics are provided in Supplementary Table 9.
Extended data Fig. 4. Related to Figure 4.
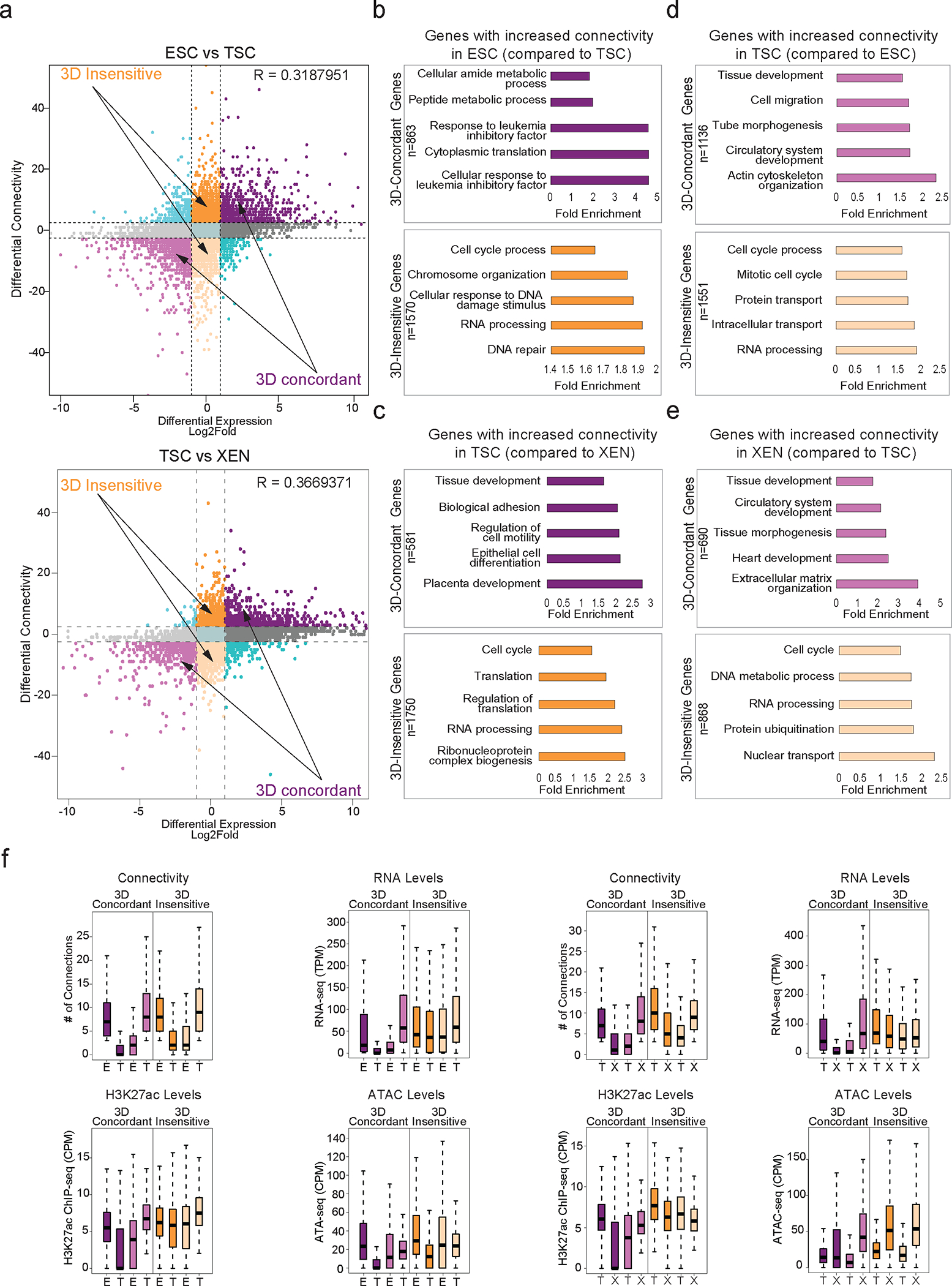
a. Correlation between differential HiChIP connectivity/hubness and differential gene expression in connectivity and differential gene expression in ESC and TSC cells (top) and TSC and XEN cells (bottom). R represents Spearman correlation identifies distinct groups of genes. We focus on the two most prominent groups: 3D-insensitive genes, defined as genes with differential connectivity >3 but no transcriptional changes (log2FC<1 or >−1) and 3D-concordant genes for which connectivity and expression changes (log2FC >1 or <−1) positively correlate (Supplementary Table 5).
b-e. Gene ontology analysis depicting the most significant biological processes enriched in the 3D concordant and 3D-insensitive genes in each pairwise comparison (ESC vs TSC and TSC vs XEN) as defined in (a). All genes in A compartments were used as background. For further details see also Supplementary Table 3.
f. Comparison of connectivity, gene expression levels as well as H3K27ac and ATAC CPM levels between ESC and TSC cells (left) and TSC and XEN cells (right) at promoters of 3D-concordant (n=1818 for ESC/TSC and n=1108 for TSC/XEN) and 3D-insensitive genes (n=2637 for ESC/TSC, n=2230 for TSC/XEN) as defined in (a). Insensitive genes show higher levels of connectivity, H3K27ac, ATAC and expression in both cell types. Two-sided Wilcoxon rank sum test was used for all comparisons with p > 0.05 indicating significance. (Supplementary Table 9).
Note: all statistics are provided in Supplementary Table 9.
Extended data Fig. 5. Related to Figure 5.
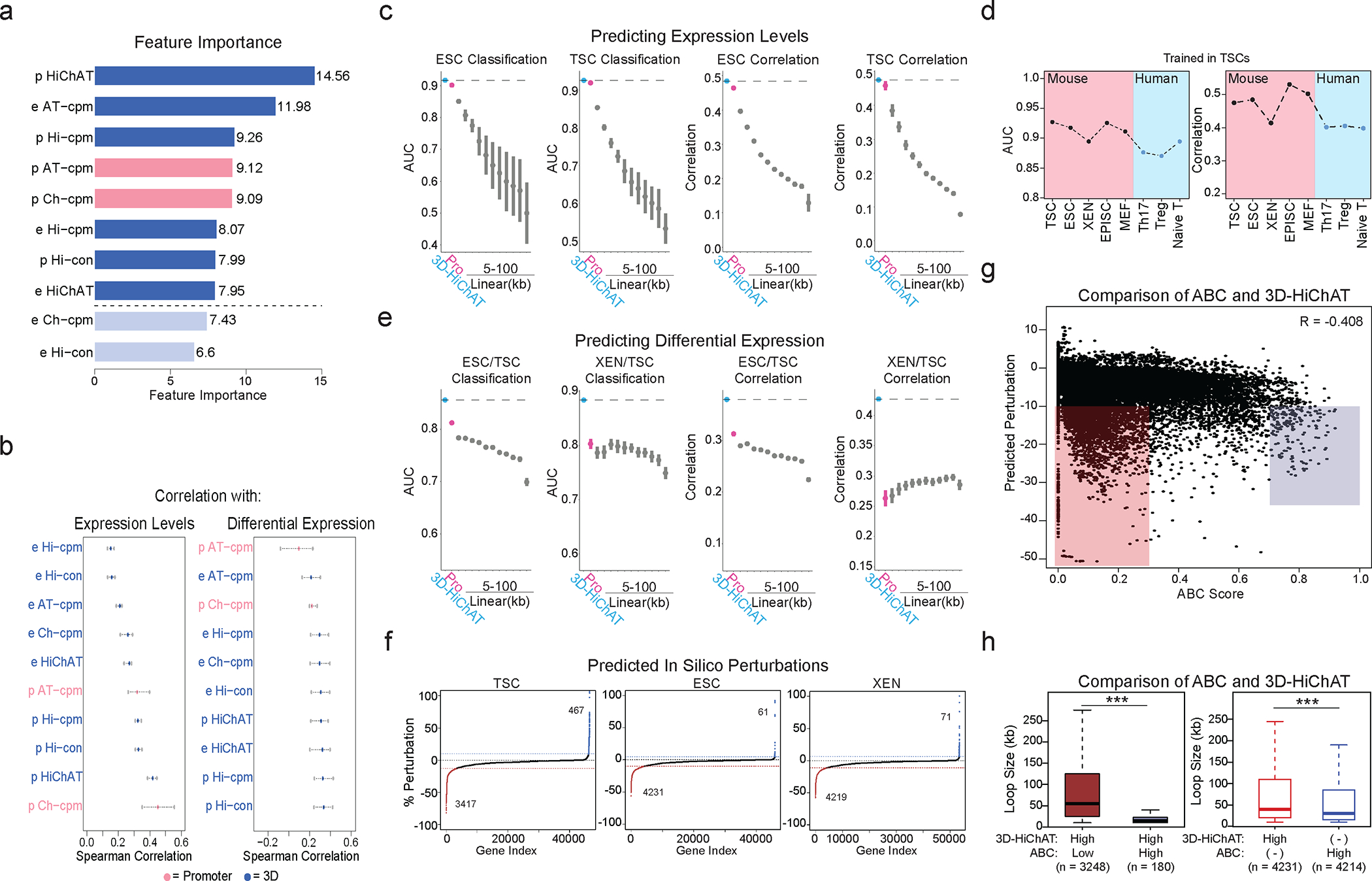
a. Barplot of feature importance showing 10 1D (pink) and 3D (blue) features, ranked from high to low. Light blue indicates features not selected. (See Supplementary Table 6).
b. Spearman correlation values for each variable considered for our 3D model with gene expression (left) and differential expression (right). Dots represent minimum, mean and maximum correlation scores. (See Supplementary Table 6).
c. Area Under Curve (AUC) scores and Spearman Correlation for classifying gene expression (top 10% high vs low, left graph) and predicting levels (right graph) in ESC or TSC cells using 3D-HiChAT, Promoter and Linear models. Dots represent average scores from LOCO training approach (n=20). Error bars show standard deviation. (See Extended Data Figure 5, Supplementary Table 6).
d. Plots showing AUC and Spearman correlation for classifying gene expression (top 10% high vs low, left graph) and predicting levels (right graph) using 3D-HiChAT in various lineages including mouse lineages and published human data: Naïve T cells, T-Helper 17 Cells (Th17), and T regulatory cells (Tregs)130,131.
e. AUC scores and Spearman Correlation generated for classifying differential expression (top 10% up or downregulated, left) and predicting expression changes (right) between XEN and ESCs using 3D-HiChAT, Promoter and Linear models. Dots represent average scores from LOCO training approach (n=20). Error bars show standard deviation. (See Supplementary Table 6).
f. Ranked perturbation scores (%) predicted by in silico perturbations of ~46K E-P pairs in ESC, ~46.7K in TSC and ~53.1K in XEN using 3D-HiChAT. Dotted horizontal lines indicate selected cut-offs for impactful perturbations.
g. Scatterplot comparing predicted perturbation scores from 3D-HiChAT with respective ABC scores. R Spearman correlation values are shown on the top.
h. Boxplots showing that enhancers with high 3D-HiChAT-predicted perturbation scores and low ABC scores (red) are more distal to their target genes (loop size) than those with high scores in both models (blue) (left plot n=3,428 enhancers). Enhancers/anchors with high 3D-HiChAT scores are more distal to the ones with high ABC scores (>0.7) (right plot n=8,445 enhancers). Asterisks indicates significance calculated by two-sided Wilcoxon rank-sum test, p-val<0.001.
Note: all statistics are provided in Supplementary Table 9.
Extended Data Fig. 6. Related to Figure 6.
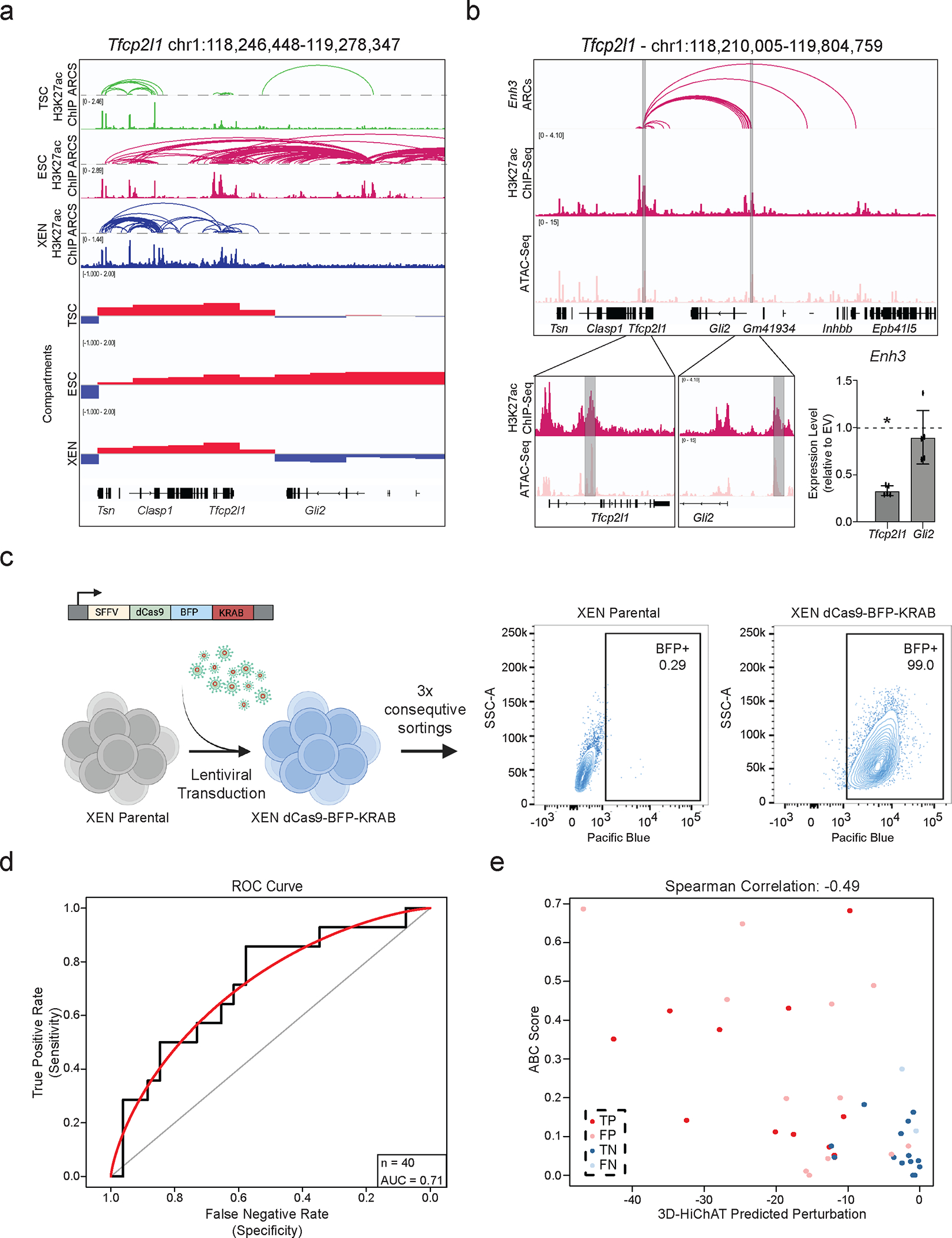
a. Visualization of the Tfcp2l1 Locus showing H3K27ac HiChIP arcs, H3K27ac ChIP and Compartment c-scores called by Hi-C for TSC, ESC, and XEN. Notably, a group of putative enhancers upstream of Gli2 are uniquely expressed and only in an A compartment in ESCs.
b. IGV tracks of the Tfcp2l1-Gli2 locus showing the two enhancers chosen for functional validation, Enh3 and Enh14. H3K27ac HiChIP derived arcs originating from both enhancers are shown as well. RT-qPCR showing relative expression levels of Tfcp2l and Gli2 upon CRISPRi perturbation of Enh3 compared to control cells infected with empty vector (EV). Dots indicate independent experiments (n=3). Error bars represent mean ± SD. Asterisks indicate significance, with p-value <0.05, as calculated using unpaired one-tailed t-test.
c. Schematic showing experimental strategy for generating a stable XEN line expressing dCas-BFP-KRAB (CRISPRi). Representative FACs plot from n>10 independent experiments.
d. AUC curve (red) showing a value of 0.71 when comparing our precited perturbation scores to our experimental validations presented in Fig.6i for n=40 different E-P pairs.
e. Scatter plot comparing the predicted perturbation scores and the ABC scores for each of the 40 experimentally tested E-P pairs. Spearman Correlation value of −0.49. Different colors indicate different groups reflecting the concordance or discordance between predictions and experimental validations as shown in Fig.6i. TP: true positive, TN: true negative, FP: false positive, FN: false negative.
Note: all statistics are provided in Supplementary Table 9.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to all member from the Apostolou and Stadtfeld groups for critical reading of the manuscript and input. We also thank Julian Pulecio and Danwei Huangfu for advice on the functional experiments and Christina Leslie for advice on the modeling. This work was partly supported by a HiChIP research grant from Arima Genomics. DM was supported by the T32 HD060600. EA is a recipient of the Mark Foundation Emerging Leader Award and supported by the NIH (1R01GM138635, 1U01DK128852, RM1GM139738) and the Tri-Institutional Stem Cell Initiative of the Starr Foundation.
Footnotes
Conflict of interest statement
The authors declare that the above study was conducted in the absence of any commercial, financial, or personal relationships that could have appeared to influence the work reported in this article. All authors have approved the submitted version.
Code availability
Custom R scripts used for data analysis in this study have been developed in our lab and are available upon request.
Data availability
All genomic datasets generated in this study (ChIP-seq, ATAC-seq, RNA-seq, 4C-seq, Hi-C and HiChIP) have been uploaded in the Gene Expression Omnibus (GEO) under GSE213645 accession number. Source data are provided with this paper.
REFERENCES
- 1.Alberio R Regulation of Cell Fate Decisions in Early Mammalian Embryos. Annual Review of Animal Biosciences (2020). doi: 10.1146/annurev-animal-021419-083841 [DOI] [PubMed] [Google Scholar]
- 2.Bardot ES & Hadjantonakis AK Mouse gastrulation: Coordination of tissue patterning, specification and diversification of cell fate. Mech. Dev. (2020). doi: 10.1016/j.mod.2020.103617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossant J Making the Mouse Blastocyst: Past, Present, and Future. in Current Topics in Developmental Biology (2016). doi: 10.1016/bs.ctdb.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 4.Rossant J & Tam PPL Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development (2009). doi: 10.1242/dev.017178 [DOI] [PubMed] [Google Scholar]
- 5.Grabarek JB et al. Differential plasticity of epiblast and primitive endoderm precursors within the ICM of the early mouse embryo. Development (2012). doi: 10.1242/dev.067702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui W & Mager J Transcriptional Regulation and Genes Involved in First Lineage Specification During Preimplantation Development. in Advances in Anatomy Embryology and Cell Biology (2018). doi: 10.1007/978-3-319-63187-5_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frum T & Ralston A Cell signaling and transcription factors regulating cell fate during formation of the mouse blastocyst. Trends in Genetics (2015). doi: 10.1016/j.tig.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muñoz-Descalzo S, Hadjantonakis AK & Arias AM Wnt/ß-catenin signalling and the dynamics of fate decisions in early mouse embryos and embryonic stem (ES) cells. Seminars in Cell and Developmental Biology (2015). doi: 10.1016/j.semcdb.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim B & Levine MS Enhancer-promoter communication: hubs or loops? Current Opinion in Genetics and Development (2021). doi: 10.1016/j.gde.2020.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoenfelder S & Fraser P Long-range enhancer–promoter contacts in gene expression control. Nature Reviews Genetics (2019). doi: 10.1038/s41576-019-0128-0 [DOI] [PubMed] [Google Scholar]
- 11.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A. (2010). doi: 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J et al. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature (2018). doi: 10.1038/s41586-018-0080-8 [DOI] [PubMed] [Google Scholar]
- 13.Birney E et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature (2007). doi: 10.1038/nature05874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature (2015). doi: 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yue F et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature (2014). doi: 10.1038/nature13992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnold CD et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science (80−. ). (2013). doi: 10.1126/science.1232542 [DOI] [PubMed] [Google Scholar]
- 17.Babbitt CC, Markstein M & Gray JM Recent advances in functional assays of transcriptional enhancers. Genomics (2015). doi: 10.1016/j.ygeno.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murtha M et al. FIREWACh: High-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat. Methods (2014). doi: 10.1038/nmeth.2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barakat TS et al. Functional Dissection of the Enhancer Repertoire in Human Embryonic Stem Cells. Cell Stem Cell (2018). doi: 10.1016/j.stem.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopes R, Korkmaz G & Agami R Applying CRISPR-Cas9 tools to identify and characterize transcriptional enhancers. Nature Reviews Molecular Cell Biology (2016). doi: 10.1038/nrm.2016.79 [DOI] [PubMed] [Google Scholar]
- 21.Apostolou E et al. Genome-wide chromatin interactions of the nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell (2013). doi: 10.1016/j.stem.2013.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beagan JA et al. Local genome topology can exhibit an incompletely rewired 3D-folding state during somatic cell reprogramming. Cell Stem Cell (2016). doi: 10.1016/j.stem.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dekker J et al. The 4D nucleome project. Nature (2017). doi: 10.1038/nature23884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denholtz M et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell (2013). doi: 10.1016/j.stem.2013.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature (2012). doi: 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Giammartino DC & Apostolou E The Chromatin Signature of Pluripotency: Establishment and Maintenance. Current Stem Cell Reports (2016). doi: 10.1007/s40778-016-0055-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorkin DU, Leung D & Ren B The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell (2014). doi: 10.1016/j.stem.2014.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allahyar A et al. Enhancer hubs and loop collisions identified from single-allele topologies. Nat. Genet. (2018). doi: 10.1038/s41588-018-0161-5 [DOI] [PubMed] [Google Scholar]
- 29.Beagrie RA et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature (2017). doi: 10.1038/nature21411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dowen JM et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell (2014). doi: 10.1016/j.cell.2014.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hnisz D, Day DS & Young RA Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell (2016). doi: 10.1016/j.cell.2016.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang T et al. Identification of multi-loci hubs from 4C-seq demonstrates the functional importance of simultaneous interactions. Nucleic Acids Res. (2016). doi: 10.1093/nar/gkw568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell (2012). doi: 10.1016/j.cell.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun F et al. Promoter-Enhancer Communication Occurs Primarily within Insulated Neighborhoods. Mol. Cell (2019). doi: 10.1016/j.molcel.2018.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao L, Berman BP & Farnham PJ Demystifying the secret mission of enhancers: Linking distal regulatory elements to target genes. Crit. Rev. Biochem. Mol. Biol. (2015). doi: 10.3109/10409238.2015.1087961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-. ). (2009). doi: 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Downes DJ et al. High-resolution targeted 3C interrogation of cis-regulatory element organization at genome-wide scale. Nat. Commun. (2021). doi: 10.1038/s41467-020-20809-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hughes JR et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. (2014). doi: 10.1038/ng.2871 [DOI] [PubMed] [Google Scholar]
- 39.Hsieh THS et al. Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol. Cell (2020). doi: 10.1016/j.molcel.2020.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krietenstein N et al. Ultrastructural Details of Mammalian Chromosome Architecture. Mol. Cell (2020). doi: 10.1016/j.molcel.2020.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crispatzu G et al. The chromatin, topological and regulatory properties of pluripotency-associated poised enhancers are conserved in vivo. Nat. Commun. (2021). doi: 10.1038/s41467-021-24641-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Giammartino DC et al. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat. Cell Biol. (2019). doi: 10.1038/s41556-019-0390-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mumbach MR et al. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods (2016). doi: 10.1038/nmeth.3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez RN, Chowdhary K, Leon J, Mathis D & Benoist C FoxP3 associates with enhancer-promoter loops to regulate Treg-specific gene expression. Sci. Immunol. (2022). doi: 10.1126/sciimmunol.abj9836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee R et al. CTCF-mediated chromatin looping provides a topological framework for the formation of phase-separated transcriptional condensates. Nucleic Acids Res. (2022). doi: 10.1093/nar/gkab1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fulco CP et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nature Genetics (2019). doi: 10.1038/s41588-019-0538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galouzis CC & Furlong EEM Regulating specificity in enhancer–promoter communication. Current Opinion in Cell Biology (2022). doi: 10.1016/j.ceb.2022.01.010 [DOI] [PubMed] [Google Scholar]
- 48.Shlyueva D, Stampfel G & Stark A Transcriptional enhancers: From properties to genome-wide predictions. Nature Reviews Genetics (2014). doi: 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
- 49.Collombet S et al. Parental-to-embryo switch of chromosome organization in early embryogenesis. Nature (2020). doi: 10.1038/s41586-020-2125-z [DOI] [PubMed] [Google Scholar]
- 50.Glaser LV et al. Assessing genome-wide dynamic changes in enhancer activity during early mESC differentiation by FAIRE-STARR-seq. Nucleic Acids Res. (2021). doi: 10.1093/nar/gkab1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo F et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. (2017). doi: 10.1038/cr.2017.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mittnenzweig M et al. A single-embryo, single-cell time-resolved model for mouse gastrulation. Cell (2021). doi: 10.1016/j.cell.2021.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pijuan-Sala B et al. Single-cell chromatin accessibility maps reveal regulatory programs driving early mouse organogenesis. Nat. Cell Biol. (2020). doi: 10.1038/s41556-020-0489-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nowotschin S et al. The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature (2019). doi: 10.1038/s41586-019-1127-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Latos PA & Hemberger M From the stem of the placental tree: Trophoblast stem cells and their progeny. Development (Cambridge) (2016). doi: 10.1242/dev.133462 [DOI] [PubMed] [Google Scholar]
- 56.Tanaka S, Kunath T, Hadjantonakis AK, Nagy A & Rossant J Promotion to trophoblast stem cell proliferation by FGF4. Science (80-. ). (1998). doi: 10.1126/science.282.5396.2072 [DOI] [PubMed] [Google Scholar]
- 57.Tesar PJ et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature (2007). doi: 10.1038/nature05972 [DOI] [PubMed] [Google Scholar]
- 58.Martin GR Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U. S. A. (1981). doi: 10.1073/pnas.78.12.7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evans MJ & Kaufman MH Establishment in culture of pluripotential cells from mouse embryos. Nature (1981). doi: 10.1038/292154a0 [DOI] [PubMed] [Google Scholar]
- 60.Li QV, Rosen BP & Huangfu D Decoding pluripotency: Genetic screens to interrogate the acquisition, maintenance, and exit of pluripotency. Wiley Interdiscip. Rev. Syst. Biol. Med. (2020). doi: 10.1002/wsbm.1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pelham-Webb B, Murphy D & Apostolou E Dynamic 3D Chromatin Reorganization during Establishment and Maintenance of Pluripotency. Stem Cell Reports (2020). doi: 10.1016/j.stemcr.2020.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loof G et al. 3D genome topologies distinguish pluripotent epiblast and primitive endoderm cells in the mouse blastocyst. bioRxiv (2022). [Google Scholar]
- 63.Schoenfelder S et al. Divergent wiring of repressive and active chromatin interactions between mouse embryonic and trophoblast lineages. Nat. Commun. (2018). doi: 10.1038/s41467-018-06666-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee BK et al. Super-enhancer-guided mapping of regulatory networks controlling mouse trophoblast stem cells. Nat. Commun. (2019). doi: 10.1038/s41467-019-12720-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thompson JJ et al. Rapid redistribution and extensive binding of NANOG and GATA6 at shared regulatory elements underlie specification of divergent cell fates. bioRxiv (2021). [Google Scholar]
- 66.Tomikawa J et al. Exploring trophoblast-specific Tead4 enhancers through chromatin conformation capture assays followed by functional screening. Nucleic Acids Res. (2020). doi: 10.1093/nar/gkz1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wamaitha SE et al. Gata6 potently initiates reprograming of pluripotent and differentiated cells to extraembryonic endoderm stem cells. Genes Dev. (2015). doi: 10.1101/gad.257071.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y et al. Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat. Genet. (2018). doi: 10.1038/s41588-017-0003-x [DOI] [PubMed] [Google Scholar]
- 69.Jia R et al. Super Enhancer Profiles Identify Key Cell Identity Genes During Differentiation From Embryonic Stem Cells to Trophoblast Stem Cells Super Enhencers in Trophoblast Differentiation. Front. Genet. (2021). doi: 10.3389/fgene.2021.762529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo G & Smith A A genome-wide screen in EpiSCs identifies Nr5a nuclear receptors as potent inducers of ground state pluripotency. Development (2010). doi: 10.1242/dev.052753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Festuccia N, Owens N, Chervova A, Dubois A & Navarro P The combined action of Esrrb and Nr5a2 is essential for murine naïve pluripotency. Dev. (2021). doi: 10.1242/DEV.199604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heng JCD et al. The Nuclear Receptor Nr5a2 Can Replace Oct4 in the Reprogramming of Murine Somatic Cells to Pluripotent Cells. Cell Stem Cell (2010). doi: 10.1016/j.stem.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 73.Rideout WM et al. Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat. Genet. (2000). doi: 10.1038/72753 [DOI] [PubMed] [Google Scholar]
- 74.Kunath T et al. Imprinted X-inactivation in extra-embryonic endoderm cell lines from mouse blastocysts. Development (2005). doi: 10.1242/dev.01715 [DOI] [PubMed] [Google Scholar]
- 75.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. (2010). doi: 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell (2013). doi: 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou HY et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. (2014). doi: 10.1101/gad.248526.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hnisz D et al. Transcriptional super-enhancers connected to cell identity and disease. Cell (2014). [Google Scholar]
- 79.Artus J, Piliszek A & Hadjantonakis AK The primitive endoderm lineage of the mouse blastocyst: Sequential transcription factor activation and regulation of differentiation by Sox17. Dev. Biol. (2011). doi: 10.1016/j.ydbio.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McDonald ACH, Biechele S, Rossant J & Stanford WL Sox17-mediated XEN cell conversion identifies dynamic networks controlling cell-fate decisions in embryo-derived stem cells. Cell Rep. (2014). doi: 10.1016/j.celrep.2014.09.026 [DOI] [PubMed] [Google Scholar]
- 81.Ling KW et al. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J. Exp. Med. (2004). doi: 10.1084/jem.20031556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo B et al. Expression, regulation and function of Egr1 during implantation and decidualization in mice. Cell Cycle (2014). doi: 10.4161/15384101.2014.943581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takahashi K & Yamanaka S Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell (2006). doi: 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 84.Renaud SJ, Kubota K, Rumi MAK & Soares MJ The FOS transcription factor family differentially controls trophoblast migration and invasion. J. Biol. Chem. (2014). doi: 10.1074/jbc.M113.523746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Knöfler M, Vasicek R & Schreiber M Key regulatory transcription factors involved in placental trophoblast development - A review. Placenta (2001). doi: 10.1053/plac.2001.0648 [DOI] [PubMed] [Google Scholar]
- 86.Benchetrit H et al. Direct Induction of the Three Pre-implantation Blastocyst Cell Types from Fibroblasts. Cell Stem Cell (2019). doi: 10.1016/j.stem.2019.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujikura J et al. Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev. (2002). doi: 10.1101/gad.968802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kubaczka C et al. Direct Induction of Trophoblast Stem Cells from Murine Fibroblasts. Cell Stem Cell (2015). doi: 10.1016/j.stem.2015.08.005 [DOI] [PubMed] [Google Scholar]
- 89.Fraser J et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature (2015). doi: 10.1038/nature14222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hu G et al. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity (2018). doi: 10.1016/j.immuni.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhattacharyya S, Chandra V, Vijayanand P & Ay F Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. (2019). doi: 10.1038/s41467-019-11950-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang L, Hill MC, Ellinor PT & Li M Bacon: a comprehensive computational benchmarking framework for evaluating targeted chromatin conformation capture-specific methodologies. Genome Biol. (2022). doi: 10.1186/s13059-021-02597-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shohat S & Shifman S Genes essential for embryonic stem cells are associated with neurodevelopmental disorders. Genome Res. (2019). doi: 10.1101/gr.250019.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tzelepis K et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. (2016). doi: 10.1016/j.celrep.2016.09.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krijger PHL & De Laat W Regulation of disease-associated gene expression in the 3D genome. Nature Reviews Molecular Cell Biology (2016). doi: 10.1038/nrm.2016.138 [DOI] [PubMed] [Google Scholar]
- 97.Miguel-Escalada I et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat. Genet. (2019). doi: 10.1038/s41588-019-0457-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Madsen JGS et al. Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells. Nat. Genet. (2020). doi: 10.1038/s41588-020-0709-z [DOI] [PubMed] [Google Scholar]
- 99.Dejosez M et al. Regulatory architecture of housekeeping genes is driven by promoter assemblies. CellReports 42, 112505 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sheffield NC & Bock C LOLA: Enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics (2016). doi: 10.1093/bioinformatics/btv612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zuin J et al. Nonlinear control of transcription through enhancer–promoter interactions. Nature 604, 571–577 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Luo R et al. Dynamic network-guided CRISPRi screen reveals CTCF loop-constrained nonlinear enhancer-gene regulatory activity in cell state transitions. bioRxiv (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang X et al. the transcription factor TFCP2L1 induces expression of distinct target genes and promotes self-renewal of mouse and human embryonic stem cells. J. Biol. Chem. (2019). doi: 10.1074/jbc.RA118.006341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ye S, Li P, Tong C & Ying QL Embryonic stem cell self-renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. (2013). doi: 10.1038/emboj.2013.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sun H et al. Tfcp2l1 safeguards the maintenance of human embryonic stem cell self-renewal. J. Cell. Physiol. (2018). doi: 10.1002/jcp.26483 [DOI] [PubMed] [Google Scholar]
- 106.Qiu D et al. Klf2 and Tfcp2l1, Two Wnt/β-Catenin Targets, Act Synergistically to Induce and Maintain Naive Pluripotency. Stem Cell Reports (2015). doi: 10.1016/j.stemcr.2015.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Papathanasiou M et al. Identification of a dynamic gene regulatory network required for pluripotency factor-induced reprogramming of mouse fibroblasts and hepatocytes. EMBO J. (2021). doi: 10.15252/embj.2019102236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li Y et al. Gene expression profiling reveals the heterogeneous transcriptional activity of Oct3/4 and its possible interaction with Gli2 in mouse embryonic stem cells. Genomics (2013). doi: 10.1016/j.ygeno.2013.09.004 [DOI] [PubMed] [Google Scholar]
- 109.Higgs DR Enhancer–promoter interactions and transcription. Nat. Genet. (2020). doi: 10.1038/s41588-020-0620-7 [DOI] [PubMed] [Google Scholar]
- 110.Spitz F & Furlong EEM Transcription factors: From enhancer binding to developmental control. Nature Reviews Genetics (2012). doi: 10.1038/nrg3207 [DOI] [PubMed] [Google Scholar]
- 111.Philips T & Hoopes L Transcription Factors and Transcriptional Control in Eukaryotic Cells. Nat. Educ. (2008). [Google Scholar]
- 112.Panigrahi A & O’Malley BW Mechanisms of enhancer action: the known and the unknown. Genome Biology (2021). doi: 10.1186/s13059-021-02322-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li J & Pertsinidis A New insights into promoter-enhancer communication mechanisms revealed by dynamic single-molecule imaging. Biochemical Society Transactions (2021). doi: 10.1042/BST20200963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schmitt AD et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. (2016). doi: 10.1016/j.celrep.2016.10.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Di Giammartino DC, Polyzos A & Apostolou E Transcription factors: building hubs in the 3D space. Cell Cycle (2020). doi: 10.1080/15384101.2020.1805238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bergman DT et al. Compatibility rules of human enhancer and promoter sequences. Nature 607, (Springer US, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Osterwalder M et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature (2018). doi: 10.1038/nature25461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kvon EZ, Waymack R, Gad M & Wunderlich Z Enhancer redundancy in development and disease. Nature Reviews Genetics (2021). doi: 10.1038/s41576-020-00311-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Beer MA, Shigaki D & Huangfu D Enhancer Predictions and Genome-Wide Regulatory Circuits. Annual Review of Genomics and Human Genetics (2020). doi: 10.1146/annurev-genom-121719-010946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tobias IC et al. Transcriptional enhancers: From prediction to functional assessment on a genome-wide scale. Genome (2021). doi: 10.1139/gen-2020-0104 [DOI] [PubMed] [Google Scholar]
- 121.Ernst J & Kellis M ChromHMM: Automating chromatin-state discovery and characterization. Nature Methods (2012). doi: 10.1038/nmeth.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tippens ND et al. Transcription imparts architecture, function and logic to enhancer units. Nat. Genet. (2020). doi: 10.1038/s41588-020-0686-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cao Q et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. (2017). doi: 10.1038/ng.3950 [DOI] [PubMed] [Google Scholar]
- 124.Whalen S, Truty RM & Pollard KS Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. (2016). doi: 10.1038/ng.3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Luo R et al. Dynamic network-guided CRISPRi screen reveals CTCF loop-constrained nonlinear enhancer-gene regulatory activity in cell state transitions. bioRxiv 2023.03.07.531569 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Karbalayghareh A, Sahin M & Leslie CS Chromatin interaction-aware gene regulatory modeling with graph attention networks. Genome Res. (2022). doi: 10.1101/gr.275870.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bigness J, Loinaz X, Patel S, Larschan E & Singh R Integrating Long-Range Regulatory Interactions to Predict Gene Expression Using Graph Convolutional Networks. J. Comput. Biol. 29, 409–424 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. (2015). doi: 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Krijger PHL, Geeven G, Bianchi V, Hilvering CRE & de Laat W 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods (2020). doi: 10.1016/j.ymeth.2019.07.014 [DOI] [PubMed] [Google Scholar]
- 130.M.R. M. et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rubin AJ et al. Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell (2019). doi: 10.1016/j.cell.2018.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic datasets generated in this study (ChIP-seq, ATAC-seq, RNA-seq, 4C-seq, Hi-C and HiChIP) have been uploaded in the Gene Expression Omnibus (GEO) under GSE213645 accession number. Source data are provided with this paper.