

Abstract
Atrial fibrillation (AF) is the most common arrhythmia in adults, with a prevalence increasing with age. Current clinical management of AF is focused on tertiary prevention (i.e., treating the symptoms and sequelae) rather than addressing the underlying molecular pathophysiology. Robust animal models of AF, particularly those that do not require supraphysiologic stimuli to induce AF (i.e., showing spontaneous AF), enable studies that can uncover the underlying mechanisms of AF. Several mouse models of AF have been described to exhibit spontaneous AF, but pathophysiologic drivers of AF differ among models. Here, we describe relevant AF mechanisms and provide an overview of large and small animal models of AF. We then provide an in-depth review of the spontaneous mouse models of AF, highlighting the relevant AF mechanisms for each model.
Introduction
AF affects 1–2% of adults > 20 years old in the United States as well as an estimated 46.3 million individuals worldwide (Benjamin et al. 2019), with 70% of individuals with AF being older than 65 years old (Feinberg et al. 1995). Data from the Framingham Heart Study have demonstrated a three-fold increase in AF prevalence over the last 50 years (Schnabel et al. 2015). AF is a progressive arrhythmia disorder (Wijffels et al. 1995), with AF risk increasing with age. Patients typically progress from paroxysmal AF, which spontaneously terminates within seven days, to persistent AF to long-standing persistent AF, which lasts more than 12 months. Treatment of acute AF involves rate control with beta blockers, non-dihydropyridine calcium channel blockers, or digoxin for patients with decompensated heart failure (HF). Patients who fail rate control are treated with rhythm control via pharmacologic, electrical, or interventional cardioversion. Anticoagulation is given to patients as guided by the CHA2DS2-VASc score. In addition to having adverse side effects, these therapies are limited in that they only address the symptoms and sequalae of AF, not the underlying cause. Moreover, while anticoagulation decreases risk of stroke up to 60% (Piccini and Fonarow 2016), patients still have a residual stroke risk along with greater risk of HF, cognitive impairment, and mortality. Thus, better treatments targeting the underlying pathobiology of AF are needed for the primary and secondary prevention of AF. Robust animal models are critical in understanding such molecular mechanisms as well as uncovering novel therapeutic targets.
Atrial fibrillation mechanisms
In this review, key aspects of AF mechanisms are described with a focus on those recapitulated in animal models. The two major mechanisms contributing to the formation of AF are reentry and ectopic electrical activity (Fig. 1) (Heijman et al. 2016). Reentry-prone substrates are formed because of electrical and structural remodeling. Structural remodeling is characterized by left atrial (LA) enlargement and fibrosis, which creates heterogenous conduction and alters cardiomyocyte excitability (Fig. 1). Electrical remodeling is characterized by abbreviated refractoriness and slowed conduction (Nattel and Dobrev 2016). Ectopic electrical activity, which provides the actual trigger to initiate arrhythmic episodes, occurs in the form of abnormal automaticity, early after-depolarizations (EADs), or delayed after-depolarization (DADs), all of which can trigger an ectopic action potential (Fig. 1) (Wit and Boyden 2007).
Fig. 1.
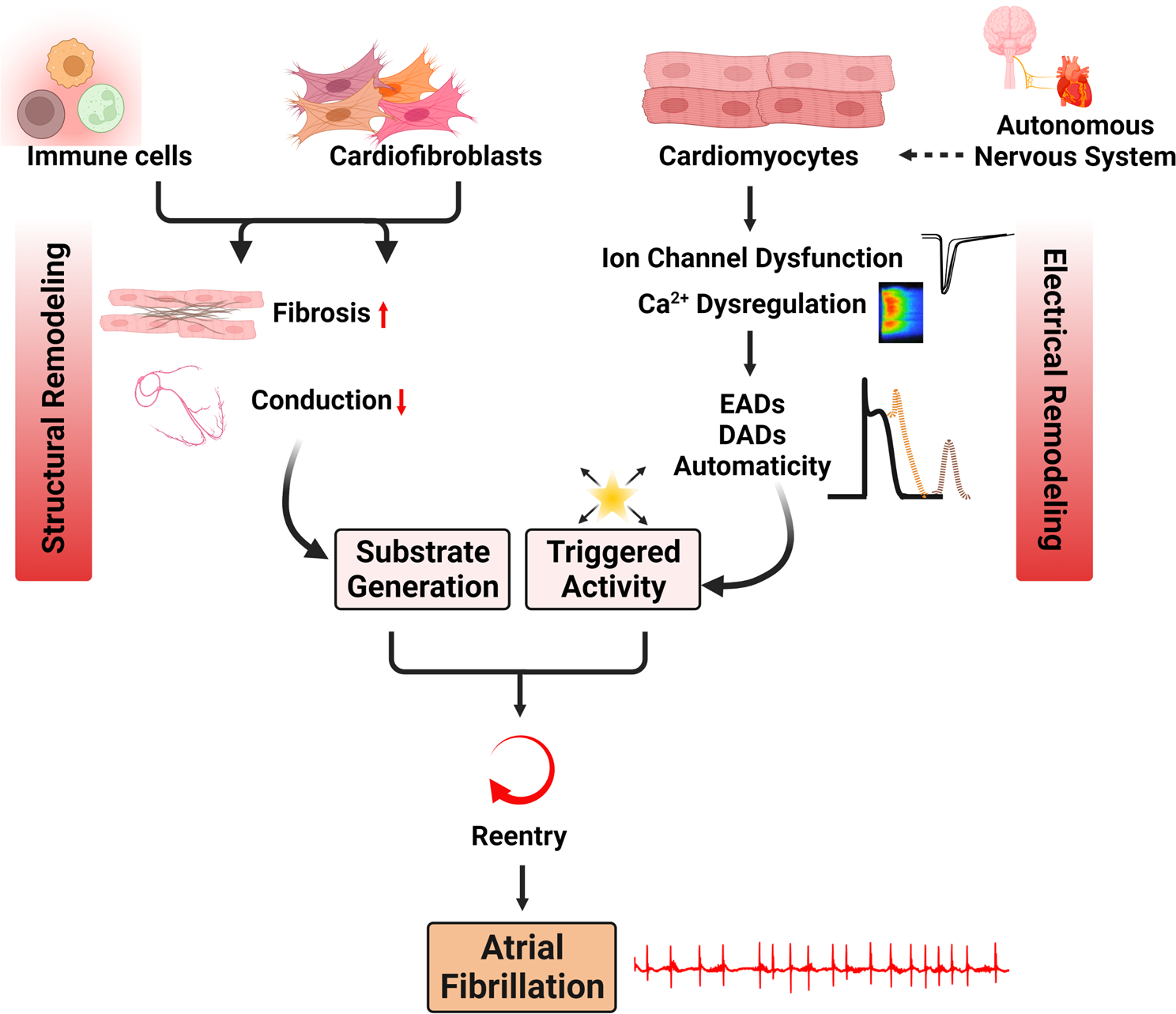
Major AF mechanisms present in mouse models of spontaneous AF. Altered substrate and ectopic electrical activity are the two major mechanisms. Altered substrate is characterized by fibrosis and slowed conduction. Ectopy is characterized by EADs, DADs, and automaticity, which arise due to ion channel dysfunction and calcium dysregulation. AF atrial fibrillation, Ca2+ calcium, DADs delayed afterdepolarizations, EADs early afterdepolarizations
Abnormal automaticity often arises in ectopic pacemaker cells and is due to upregulation of the hyperpolarization-activated cyclic nucleotide-gated (HCN) funny current (Tse 2016). EADs occur when the refractory period is prolonged due to premature reactivation of the sarcolemmal voltage-gated L-type calcium channel (LTCC) prior to full repolarization. In some cases, EADs occur due to premature reactivation of fast sodium currents (Sato et al. 2017). However, DADs are thought to be the predominant AF trigger and arise in states of abbreviated refractoriness such as secondary to spontaneous ryanodine receptor (RyR2)-mediated sarcoplasmic reticulum (SR) diastolic calcium leak (Wit and Boyden 2007). Spontaneous diastolic calcium release events can activate the sodium/calcium exchanger (NCX)—which is upregulated in patients with persistent AF (Voigt et al. 2012)—to pump out one calcium ion in exchange for three sodium ions, resulting in a net depolarizing current that can trigger an action potential (Voigt et al. 2012). Repeated focal ectopic firing can also sustain AF in the absence of an AF-prone substrate or initiate an AF-sustaining reentrant circuit.
One important mechanistic driver of reentry and ectopic electrical activity is oxidative stress. Oxidative stress has been shown to result in atrial electrical remodeling, characterized by increased SR calcium leak (Joseph et al. 2016), conduction slowing via alterations in gap junction expression (Gemel et al. 2017; Smyth et al. 2010), and decreased inward sodium current density (Liu et al. 2012). Oxidative stress also causes atrial structural remodeling through several pathways such as the receptor for advanced glycation endproducts (RAGE) axis (Kato et al. 2008), transforming growth factor (TGF) β1 expression (Mira et al. 2013), and the Ras homolog gene family member A (RhoA) GTPase/ROCK pathway (Chen et al. 2014; Sah et al. 1999). Oxidative stress is a key determinant of AF susceptibility and contributes to spontaneous AF in many of the mouse models discussed below.
Overview of animal models of atrial fibrillation
Large animal models
Large animals of AF include goats, dogs, sheep, pigs, and horses. As discussed (Frydrychowski et al. 2020), advantages of large animal models include the possibility of detailed experimental procedures, in-depth determination of cardiac function, high availability of tissue samples (Odening et al. 2021) and anatomical-physiological similarity to human hearts. However, these models often incur higher experimental costs and longer study periods. Moreover, spontaneous AF is uncommon except in dogs (Buchanan 1965) and horses (Hesselkilde et al. 2019). In large animal models, AF is often induced via atrial structural remodeling or rate-induced electrical remodeling such as atrial burst pacing to shorten ERP (Nishida et al. 2010). AF in goats (Dosdall et al. 2013), sheep (Anne et al. 2007), and dogs (Cardin et al. 2007) is induced via atrial tachypacing (ATP) and reflects the progressive nature of human AF as maintenance of AF progressively shortens the atrial ERP and increases AF duration. Sheep enable the study of ischemia-induced AF due to the presence of a unique coronary branch supplying the posterior LA (Avula et al. 2018). Spontaneous AF in dogs is associated with structural heart disease (e.g., myxomatous mitral valve disease (Jung et al. 2016)) and carries mechanistic similarities to humans as canine pulmonary veins have a shorter action potential duration (APD) compared to the surrounding LA (Ehrlich et al. 2003). While similarity to humans in terms of size and EP properties as well as ability for genetic manipulation (e.g., Brugada syndrome (Park et al. 2015)) make pigs an ideal model, pigs are prone to systolic HF after ATP-induced AF (Lugenbiel et al. 2015). Horses have been reported to exhibit spontaneous AF (Hesselkilde et al. 2019) and have human-like structural and electrical remodeling with AF-associated ectopy originating in the PVs.
Small animal models
Small animal models include mice, rats, guinea pigs, and rabbits. These models are ideal for hypothesis generation, but the requirement for significant physiologic perturbations to induce AF may limit their translatability (Fu et al. 2022). Nonetheless, small animal models are advantageous over large animal models in terms easy generation, lower cost, and possibility of genetic manipulation (Dobrev and Wehrens 2018). Guinea pigs and rabbits are advantageous over mouse models as the presence of a calcium-driven plateau phase during the action potential makes them suitable models for studying repolarization (Clauss et al. 2019). Moreover, the relatively larger atrial size makes experimental manipulation easier. Disadvantages of guinea pigs and rabbits include increased costs, more difficult genetic manipulation, and short AF duration on the order of seconds (Suzuki et al. 2014).
Mice are the most widely used animal model of AF. Most murine AF models require generation of an AF-predisposing substrate (e.g., TGF-β1 overexpression) (Verheule et al. 2004). Advantages of mouse models include wide accessibility, affordable genetic manipulation, genetic homology with humans, and human-like phenotypic effects of monogenic conditions, i.e., monogenic channelopathies. Disadvantages of mouse models include a high resting heart rate, different ion channel expression and APD compared to humans, limited EP instrumentation (e.g., catheters), and low intracardiac mapping resolution. Here, we discuss four murine models of spontaneous AF in detail, as they recapitulate the pathophysiology of human AF by developing a vulnerable substrate and progressing to spontaneously induced AF episodes.
Mouse models of spontaneous AF
Spontaneous AF, or AF not requiring an exogenous electrical stimulus, has been reported in several mouse models (Schuttler et al. 2020). Here, we describe all spontaneous AF mouse models to our knowledge and present in greater detail the four main spontaneous AF models that have been more extensively characterized—liver-kinase B1 (LKB1) knockout (KO), cyclic AMP-responsive element modulator (CREm)-transgenic (Tg) overexpression, angiotensin-converting enzyme (ACE)-Tg overexpression, and tumor necrosis factor alpha (TNF-α) Tg overexpression. In Table 1, we summarize the relevant pathophysiologic features present in each spontaneous AF model (i.e., fibrosis, changes in ERP, slowed conduction, DADs, EADs, and calcium mishandling).
Table 1.
Mechanisms contributing to AF development in mouse models of spontaneous AF
| Model | Altered substrate |
Ectopy |
References | ||||
|---|---|---|---|---|---|---|---|
| Atrial fibrosis/ dilatation | ERP changes | Slowed conduction | DADs | EADs | Calcium mishandling | ||
|
| |||||||
| Lkb1-KO | + + + | - | + | Not reported | Not reported | Not reported | (Ikeda et al. 2009; Ozcan et al. 2015; Hulsurkar et al. 2021) |
| CREm-Tg | + + | + | + | + | + | + + + | (Muller et al. 2005; Stumpel et al. 2018; Kirchhof et al. 2013; Schulte et al. 2016; Li et al. 2014) |
| Ace-Tg | + + | + | + | Not reported | Not reported | + | (Xiao et al. 2004; Demers et al. 2022) |
| Tnf-α-Tg | + + + | + | + | + | Not reported | + | (Saba et al. 2005) |
| Ankyrin B KO | - | + | + | + + | + + | + + | (Mohler et al. 2003; Cunha et al. 2011) |
| Junctin-Tg | + + | + | Not reported | Not reported | Not reported | + | (Hong et al. 2002) |
| Sarcolipin KO | + | + | Not reported | + | Not reported | + + | (Xie et al. 2012) |
| MURC-Tg | + + | + | + | Not reported | Not reported | Not reported | (Sah et al. 1999) (Ogata et al. 2008) |
| PI3K KO | + + | + | Not reported | Not reported | Not reported | Not reported | (Pretorius et al. 2009) |
| Rac1-Tg | + + + | Not reported | Not reported | Not reported | Not reported | Not reported | (Adam et al. 2007) |
| Nucleoporin KO | Not reported | + | Not reported | Not reported | Not reported | + | (Zhang et al. 2008) |
| miR-328-Tg | + | + + | Not reported | Not reported | Not reported | + + | (Lu et al. 2010; Liu et al. 2020) |
| miR-208a KO | + | Not reported | + | Not reported | Not reported | Not reported | (Callis et al. 2009) |
ACE angiotensin-converting enzyme, CREm cyclic AMP-responsive element modulator, DADs delayed afterdepolarizations, EADs early after-depolarizations, ERP effective refractory period, Lkb1 liver kinase B1, KO knockout, miR microRNA, MURC Muscle-restricted putative coiled-coil protein, PI3K Phosphoinositide 3-kinase, Tg transgenic, TNF-α tumor necrosis factor alpha
Lkb1-KO
LKB1 is a tumor suppressor, originally discovered as the causal loss-of-function mutation in Peutz-Jeghers syndrome (Hemminki et al. 1998), a premalignant syndrome characterized by gastrointestinal (GI) hamartomatous polyps and predisposition to solid organ cancers in the GI tract, breast, testis, and ovary (Giardiello et al. 1987). AMP-activated kinases (AMPKs) are important downstream mediators of LKB1 signaling (Shackelford and Shaw 2009). Loss of LKB1 in most tissues is characterized by hyperplasia while loss of LKB1 in the heart results in hypertrophy due to the non-dividing nature of cardiomyocytes (Ikeda et al. 2009). To this end, cardiomyocyte-specific Lkb1-KO (Lkb1-CKO) mice were generated by crossing the alpha-myosin heavy chain (αMHC)-Cre allele with the Lkb1-floxed allele (Ikeda et al. 2009). Lkb1-CKO mice developed bi-atrial enlargement with 100% AF incidence at five weeks of age. In Lkb1-CKO mice, collagen expression was increased three-fold by four weeks of age and histological evidence of atrial fibrosis was apparent at 12 weeks of age, followed by left ventricular (LV) hypertrophy. Ozcan et al. (2015) built upon the study by Ikeda et al. (2009) by performing a more detailed characterization of the arrhythmic phenotype observed in Lkb1-CKO mice. The prevalence of spontaneous AF increased with age, from 47% at four weeks-of-age to 74% at six weeks and 95% at 12 weeks (Fig. 2A). In Lkb1-CKO mice, arrhythmia progression began with paroxysmal AF without changes in baseline heart rate (Ozcan et al. 2015). Mice then became more bradycardic as they progressed into persistent AF. PR, QRS, and QT intervals were unchanged while Lkb1-CKO mice were in sinus rhythm, but PR and QRS intervals increased at the onset of paroxysmal AF. Mice in persistent AF also gained weight and showed concomitant signs of HF (Ozcan et al. 2015). While LV systolic function was preserved in mice with sinus rhythm and paroxysmal AF, LV ejection fraction (EF) decreased in mice with persistent AF. Despite these ventricular changes, however, an early morphologic hallmark of loss of cardiac Lkb1 was bi-atrial dilation, beginning prior to the onset of paroxysmal AF. Biatrial dilation was accompanied by fibrosis and infiltration of pro-inflammatory immune cells including macrophages, neutrophils, and lymphocytes. Strikingly, these morphologic changes were observed in Lkb1-CKO mice while they were still in sinus rhythm; fibrosis and immune cell infiltration increased as mice progressed into paroxysmal and persistent AF (Ozcan et al. 2015). Beginning prior to the onset of paroxysmal AF onset, Lkb1-CKO mice also demonstrated signs of decreased exercise capacity, which became progressively worse as mice progressed into paroxysmal and persistent AF. Survival in Lkb1 KO mice was 94% at six weeks, 82% at three months, and 65% at six months; common causes of death included HF and sudden cardiac death. In mice with AF, ventricular fibrillation (VF) was the most common cause of death (Ozcan et al. 2015). Interestingly, 18.8% of Lkb1 KO female mice died during pregnancy secondary to peripartum cardiomyopathy. Lkb1 KO mice also had decreased phosphorylated AMPK with concomitant evidence of atrial metabolic and oxidative stress. Indeed, a recent study demonstrated that administration of AMPK activators metformin and aspirin attenuated AF incidence by eightfold in Lkb1 KO mice with concomitant improvement in mitochondrial function, gap junction protein expression, and attenuated fibrosis (Ozcan et al. 2021). Altogether, Lkb1 KO mice have a characteristic human-like arrhythmia progression from sinus rhythm into paroxysmal and persistent AF with concomitant electrical and structural remodeling. Moreover, like in humans, premature atrial complexes (PACs), atrioventricular (AV) block, and atrial flutter predicted risk of progression into AF. Thus, loss of Lkb1 in the heart is a robust and translatable murine model of AF with mechanistic findings like human AF.
Fig. 2.
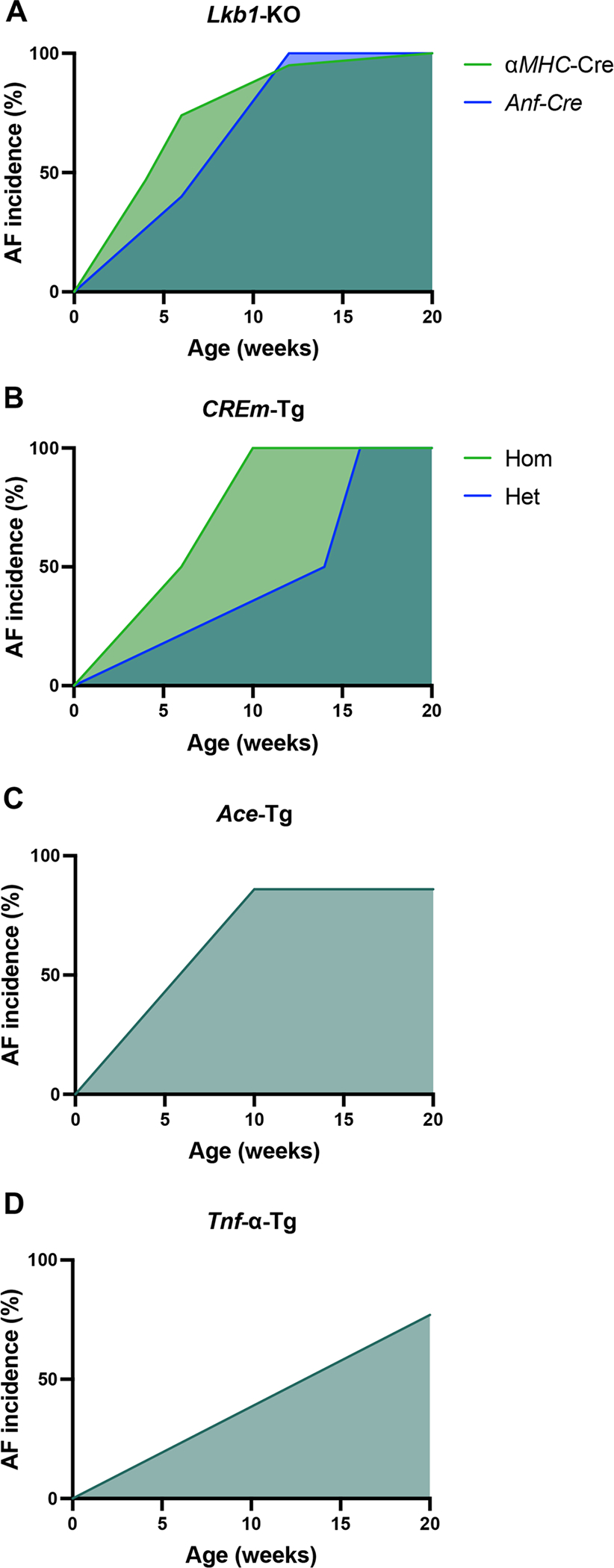
AF incidence by age in the Lkb1-CKO, CREm-Tg, Ace-Tg, and Tnf-α-Tg mouse models. Incidence of spontaneous AF is plotted against age for Lkb1-KO (Ozcan et al. 2015; Hulsurkar et al. 2021) (A), CREm-Tg (Muller et al. 2005; Stumpel et al. 2018) (B), ACE-Tg (Xiao et al. 2004) (C), and Tnf-α-Tg (Saba et al. 2005) (D). Age was plotted through 20 weeks for all models. Ace angiotensin-converting enzyme; Anf atrial natriuretic factor; CREm cyclic AMP-responsive element modulator; Hom homozygous; Het heterozygous; Lkb1 liver kinase B1; α-MHC myosin heavy chain; KO knockout; Tg transgenic; Tnf-α tumor necrosis factor alpha
Recently, Fu et al. (2022) studied the AF phenotype of the cardiomyocyte-specific Lkb1-CKO mouse in detail and reported that the electrocardiographic findings were representative of human AF (i.e., irregularly irregular R-R intervals without discernable P waves) (Fu et al. 2022). However, this study found that the electrocardiographic phenotype could be secondary to severe atrial cardiomyopathy, which prevented the conduction of sinoatrial node impulses through the severely remodeled atrial myocardium. They concluded that this pathophysiology differs from human AF, which is triggered by ectopic foci originating near the pulmonary veins. It is important to note, however, that the study by Fu et al. (2022) used a whole-heart cardiomyocyte-specific Lkb1-CKO allele (via crossing aMHC-Cre with Lkb1fl/fl mice). Thus, ventricular dysfunction, whether subclinical or overt, could have independently contributed to atrial myopathy due to volume and pressure overload.
One major limitation of the studies by Ozcan et al. (2015) and Fu et al. (2022) is that the Lkb1-CKO mice developed LV systolic dysfunction secondary to loss of Lkb1 in the ventricles, which might have promoted the development of AF in these mice. To address this, Hulsurkar et al. (2021) knocked out Lkb1 selectively in the atria (Lkb1-aKD) by injecting an adeno-associated virus (AAV) containing atrial natriuretic factor (ANF) promoter-driven Cre expression into 5-day-old Lkb1FL/FL mice (Hulsurkar et al. 2021). At six weeks, the AF incidence was 40% in Lkb1-aKD mice (vs 0% in controls, Fig. 2A). At 12 weeks, AF incidence was 100% in Lkb1-aKD mice (vs 0% in controls, Fig. 2A). AF duration and AF burden were also statistically significantly greater in Lkb1-aKD mice vs controls, with the magnitude of differences increasing with age. Importantly, 12-week-old Lkb1-aKD mice had normal LV EF, end-diastolic diameter, and end-systolic diameter. Moreover, this study showed that the Lkb1-aKD mice develop AF that progresses with time, as well as atrial fibrosis, atrial dilation, and reduced atrial function, which closely mimics the human pathophysiology. Altogether, atrial-specific Lkb1 KO mice are a robust model of spontaneous AF without a concomitant and potentially confounding ventricular phenotype.
Using the same Lkb1-aKD model, Moreira et al. (2020) studied the anti-fibrotic role of atrial paracrine calcitonin signaling in AF. Loss of atrial calcitonin signaling in Lkb1-aKD mice increased atrial fibrosis by ~ 2.5-fold compared to Lkb1-aKD mice with intact atrial calcitonin signaling, showing that the Lkb1-aKD mice could be used as a model to study the role of other genes in AF progression. Electrical remodeling also seems to play a role in spontaneous AF seen in mice with loss of Lkb1, as evidenced by the decreases in Nav1.5, Cacna1h, and Kcnq1 expression observed prior to atrial structural remodeling (Kim et al. 2015).
Altogether, the loss of Lkb1 in the heart is a robust and well-characterized model of AF recapitulating many of the essential electrocardiographic and morphologic findings seen in human AF. Evidence points toward substrate remodeling (e.g., fibrosis, delayed/impaired intra-atrial conduction) as the major AF mechanism, with unclear evidence for a role for ectopy in this model.
CREm-Tg overexpression
cAMP-response element (CRE) and transcription factors of the CREB/CREm family function in the heart to maintain sarcoplasmic reticulum calcium ATPase 2a (SERCA2a) and beta-1 adrenergic receptors expression. While protective under physiologic conditions, CREm-mediated cAMP signaling becomes pathologic in diseases such as HF due to overstimulation and overexertion of the heart. In 2005, Muller et al. (2005) described hemizygous transgenic mice with cardiomyocyte-specific expression of a human cardiac CREm isoform (CREM-1b ΔC-X). CREm-overexpressing transgenic (CREm-Tg) mice displayed atrial enlargement at seven weeks and AF with rapid ventricular response at eight weeks-of-age. CREm-Tg mice also demonstrated increased maximum rates of LV contraction, 32% greater beta-1 adrenergic receptor density, and 127% higher ventricular SERCA2a expression. Expression of phospholamban and RyR2 binding partners calsequestrin and junctin was unchanged. Interestingly, phospholamban phosphorylation was decreased at Ser16 and Thr17, targets of beta-1 adrenergic signaling and CaMKII, respectively. This finding was hypothesized to be a compensatory mechanism to attenuate the CREm-mediated pathologic increases in LV function.
In 2018, Stumpel et al. (2018) built upon the study by Muller et al. (2005) by designing homozygous transgenic CREm-Tg mice (Stumpel et al. 2018). This was accomplished by mating pairs of hemizygous transgenic mice and identifying homozygous transgenic mice via Southern blotting and real-time polymerase chain reaction. Transgene DNA content was roughly two-fold higher in homozygous compared to heterozygous CREm-transgenic mice. AF incidence was 50% by 6 weeks-of-age and 100% by 10-weeks-old (Fig. 2B). At 11 weeks-of-age, treatment with the Vaughn-Williams class III anti-arrhythmic esmolol reduced overall HR and increased HR variability, reflective of the response seen in humans. However, treatment of 6–7-week-old mice with class Ic anti-arrhythmic, flecainide failed to convert the mice back into sinus rhythm, indicative of potential RyR2 and/or sodium channel dysregulation (Kryshtal et al. 2021; Yang et al. 2016). Morphologically, homozygous mice had mild ventricular hypertrophy while hemizygous mice exhibited no hypertrophy. However, no differences in ventricular collagen content were reported in either CREm-overexpressing strain compared to WT mice. While the pathophysiologic mechanism of AF in CREm-transgenic mice is not fully understood, one hypothesis is that atrial dilatation occurs secondary to systolic LV failure as LV hypertrophy was seen by 14 weeks-of-age (Stumpel et al. 2018). However, this cannot fully explain the atrial dilatation as atrial dilatation occurred prior to ventricular dysfunction, and end-diastolic aortic and LV pressures were not increased (Muller et al. 2005).
Thus, ectopy in the form of EADs and calcium mishandling likely plays a role in CREm-mediated spontaneous AF. Indeed, Kirchhof et al. (2013) demonstrated in the atria of > 15-week-old CREm-Tg heterozygous mice that interatrial conduction block with concomitant SR calcium leak is associated with decreased SR calcium load despite normal SERCA2a, phospholamban, and RyR2 protein levels. Similar findings have been demonstrated in ventricular cardiomyocytes by Schulte et al. (2016), i.e., accelerated decay of the calcium transient, mediated by increased NCX and SERCA2a transport. Increased NCX transport was associated with prolongation of the action potential and EADs. Complementing these findings, Li et al. (2014) demonstrated that enhanced atrial SR calcium leak in CREm-Tg mice was partially mediated by hyperphosphorylation of RyR2 at Serine (Ser)2814, mediated by calcium/calmodulin-dependent protein kinase II (CaMKII) and an important factor in AF initiation (Chelu et al. 2009). Importantly, Li et al. (2014) demonstrated that enhanced RyR2-mediated SR calcium leak not only provided an AF trigger but also resulted in calcium-dependent fibrotic and hypertrophic remodeling mediated via the calcineurin-nuclear factor of activated T cells (NFAT)-regulator of calcineurin (RCAN) pathway. Crossing CREm-Tg mice with mice carrying a mutated RyR2 phosphorylation site (Ser2814Ala) resulted in abrogation of fibrosis, indicating a fundamental role of RyR2 phosphorylation in calcium-dependent remodeling. Indeed, NFAT activity is increased in the atria of patients with chronic AF, and NFAT inhibition abrogates atrial ectopy and spontaneous AF in CREm-Tg mice, likely mediated by attenuated atrial remodeling and normalized calcium handling (Ni et al. 2021).
In addition to ectopy and calcium-dependent remodeling, inflammation has been shown to play a causal role in spontaneous CREm-mediated AF. At 7 months (~ 30 weeks) of age, double-mutant (DM) CREm-Tg/NLRP3−/− mice exhibited no spontaneous AF compared to 67% spontaneous AF incidence in CREm-Tg single mutant mice. (Yao et al. 2018) LA dilatation was concomitantly reduced in DM mice, indicating that the pro-arrhythmogenic effects of NLRP3 are, at least in part, mediated by atrial remodeling. Altogether, calcium mishandling seems central to the pathophysiology of the CREm-Tg model of spontaneous AF as dysregulated calcium provides the ectopic triggers and pathologic atrial remodeling required for AF triggering and propagation. In addition, enhanced NLRP3 inflammasome activation appears to play a role in AF development, while the role of fibrosis is less certain in this model.
Ace-Tg overexpression
The renin–angiotensin–aldosterone system (RAAS) is central to the pathophysiology of many cardiovascular diseases such as HF and hypertension. Increased RAAS activation is the major pathophysiologic mechanism driving systolic HF due to angiotensin II-mediated vasoconstriction and aldosterone-mediated salt and water retention. Most drugs that portend a mortality benefit in systolic HF antagonize RAAS (Stolfo and Savarese 2019). Increased RAAS is also linked with human AF, and treatment of systolic HF with RAAS blockers decreases risk of new-onset AF by 21–50% (Nair et al. 2014). To further explore the role of RAAS in AF, Xiao et al. (2004) created homozygous transgenic ACE overexpression under the αMHC promoter (ACE-Tg). Consistent with unchanged renal and vascular ACE expression, ACE-Tg mice had an ~ 8.5% reduction in BP compared to WT controls (111.3 mmHg control vs 101.8 mmHg ACE-Tg). Mild atrial enlargement was observed starting at two weeks-of-age, progressing to severe atrial enlargement by three weeks-of-age. These findings occurred concomitantly with increased risk of sudden death despite a normal electrolyte profile. Ventricular size and function were also normal, suggesting that ventricular dysfunction was not the major cause of AF. Ambulatory EKGs were used to investigate the increased incidence of sudden death and revealed low QRS voltages, prolonged PR interval, and an 86% incidence (6/7) of spontaneous AF among ACE-Tg mice (Fig. 2C). By 10–16 weeks old, atrial weight was three-fold greater in ACE-Tg mice compared to controls; however, only focal atrial fibrosis was observed with no ventricular fibrosis. Altogether, the study by Xiao et al. (Xiao et al. 2004) demonstrated that spontaneous AF in mice with cardiac-specific ACE overexpression likely results, in part, from atrial myopathy with slowed conduction.
Other studies have explored AF mouse models of RAAS overactivity. While these studies have looked at inducible AF, the histologic, electrophysiologic, and molecular findings provide insight into the underlying pathophysiological role of RAAS dysregulation in the ACE-Tg mouse model of spontaneous AF. Han et al. (Han et al. 2020) demonstrated that subcutaneous angiotensin II infusion at 2,000 ng/kg/min for three weeks results in LA dilatation, fibrosis, and inflammation in 8-to-10-week-old mice. Evidence for ectopy in the pathophysiology of RAAS-mediated AF in mice comes from a study of cardiac-specific angiotensin II receptor type 1 (AT1R) overexpression (Demers et al. 2022). Six-month-old mice had a 60% reduction in inward sodium current and 50% reduction in LTCC current compared to controls. Similar to prior studies, AT1R overexpressing mice had delayed atrial conduction time with prolonged PR interval despite normal APD. Altogether, pathologic atrial structural remodeling seems to be central to the pathophysiology of the ACE-Tg mouse model of spontaneous AF, with evidence that ion channel dysregulation may provide additional ectopic electrical triggers.
Cardiac-specific Tnf-α-Tg overexpression
Saba et al. (Saba et al. 2005) created heterozygous cardiomyocyte-specific transgenic mice overexpressing Tnf-α under the αMHC promoter (Tnf-α-Tg). By 10 week-of-age, Tnf-α-Tg mice had a 20-fold greater atrial collagen deposition by histology, concomitant with bi-atrial dilation. These changes occurred prior to ventricular enlargement, which occurred at 6–9 months-of-age. Spontaneous AF by telemetry was observed starting at 5 months-of-age, with 77% incidence in Tnf-α-Tg mice (mean age 7 ± 4 months old) compared to 10% in the control mice (Fig. 2D). Interestingly, mean heart rate (HR) was lower in Tg mice over 3 months old. These results were driven by Tg mice with spontaneous AF, indicating likely slowed atrial conduction perhaps mediated via decreased connexin 40 expression (Sawaya et al. 2007). Indeed, Tnf-α-Tg mice had prolonged PR, QT, and QRS intervals, and optical mapping demonstrated reentrant atrial tachycardia with variable AV conduction in response to intra-atrial pacing (Saba et al. 2005). APD was similar between Tnf-α-Tg mice and controls. In addition to atrial structural remodeling, slowed conduction, and reentrant-prone substrate, Saba et al. (Saba et al. 2005) demonstrated evidence for calcium mishandling in the pathogenesis of spontaneous AF in the Tnf-α-Tg mice. While diastolic intracellular calcium did not differ between Tg and control mice, atrial cardiomyocytes from six-month-old Tnf-α-Tg mice had reduced calcium transient amplitude and prolonged calcium transient decay, indicative of impaired SR calcium release and reuptake, respectively. Moreover, spontaneous calcium transients were three times more likely Tnf-α-Tg atrial cardiomyocytes, indicating that DADs likely provide a trigger for the spontaneous AF in Tnf-α-Tg mice. A role of systemic Tnf-α in AF is evidenced by the attenuation in AF after inhibition of soluble Tnf-α in a mouse model of intense exercise (Lakin et al. 2019). Altogether, the Tnf-α-Tg mouse develops spontaneous AF due to ectopy in the form of DADs, which propagate to form a reentrant circuit in the setting of TNF-α-mediated abnormal atrial structure, conduction, and fibrotic remodeling.
Other AF mouse models with reported spontaneous AF
Ankyrin B KO: Mohler et al. (Mohler et al. 2003) reported a loss-of-function mutation in the adapter protein, ankyrin B, on chromosome 4q25–27 (E1425G) found in patients presenting with long QT syndrome, sinus bradycardia, and early-onset AF. A nkyrin± mice were generated and exhibited bradycardia with high heart-rate variability and intermittent AV dissociation on ECG (Mohler et al. 2003). No structural heart defects were seen in a nkyrin± mice. Cardiomyocytes isolated from a nkyrin± mice had delayed calcium transient decay and increased calcium transient amplitude—likely mediated by loss of the Na+/K+-ATPase in a manner similar to the mechanism of digoxin. In response to 1 μM isoproterenol, ankyrin± cardiomyocytes exhibited DADs and EADs, likely mediated due to RyR2 hyperactivity. Altogether this study Mohler et al. (Mohler et al. 2003) demonstrates a role of ectopy in AF in ankyrin± mice.
Cunha et al. (Cunha et al. 2011) further explored atrial function in ankyrin± mice. Telemetry revealed spontaneous AF with bradycardia in a nkyrin± mice. Ankyrin± atrial cardiomyocytes had reduced APD likely mediated by reduced LTCC and NCX currents. The ventricular cardiomyocyte APD was unchanged. There were no changes in inward sodium current or outward potassium current in ankyrin± cardiomyocytes. Studies have also implicated CaMKII-mediated RyR2 phosphorylation at Ser2814 in AF development in ankyrin± mice as inhibition of CaMKII attenuates the abnormal EP phenotype in cardiomyocytes from ankyrin± mice (DeGrande et al. 2012).
Altogether, loss-of-ankyrin increases AF susceptibility due to ectopy and a reentrant-prone substrate characterized by abbreviated APD likely predisposing to DADs.
Junctin overexpression
Junctin is a transmembrane protein located on the SR that modulates RyR2 activity and regulates SR calcium release (Hong et al. 2002). To study the role of junctin in excitation–contraction coupling, Hong et al. (Hong et al. 2002) transgenically overexpressed canine cardiac junctin in mice (junctin-Tg). Six-to-eight-week-old junctin-Tg mice had biventricular and bi-atrial dilation with fibrosis and thinning of the atrial walls. Thrombi were found in both atria, indicative of stasis likely secondary to AF. Indeed, six-to-eight-week-old junctin-Tg mice had lower HR compared with WT controls, and their ECGs showed spontaneous AF. Mice died from HF between nine and 11 weeks-of-age. These findings, together with prolonged APD and increased LTCC current in junctin-Tg atrial cardiomyocytes, suggest that junctin overexpression causes AF due to atrial structural and electrical remodeling, creating a reentrant-prone substrate. The role of ectopy (i.e., DADs and EADs) is less clear in this model. While the AF incidence was not reported in the study, the relative early-onset and severity of the electrophysiological findings indicate that this model induces a severe AF phenotype.
Sarcolipin (SLN) KO
SLN, like phospholamban, is a negative regulator of SERCA2a activity expressed in cardiac and skeletal muscle (Bhupathy et al. 2007), present more in the atria than in ventricular myocardium. SLN is phosphorylated via CaMKII and inhibits SERCA2a-mediated SR calcium uptake. Right atrial SLN levels have been shown to be decreased in chronic AF, with the degree of decrease proportional to right atrial pressure (Uemura et al. 2004). To this end, Xie et al. (Xie et al. 2012) created homozygous cardiac Sln−/− mice. Atrial cardiomyocytes from Sln−/− mice had increased SR calcium content and calcium transients. APD was prolonged, mediated by a greater forward mode NCX current. DADs were observed along with increasing LTCC current. Phosphorylation of RyR2 at Ser2808 and Ser2814 were both increased in the atria of Sln−/− mice. No atrial hypertrophic effect was observed, but trichrome staining revealed atrial fibrosis. ECGs were normal at five months-of-age, and spontaneous AF was seen starting at 12 months-of-age with 66.7% incidence (Xie et al. 2012). Altogether, calcium mishandling plays a key role in AF in Sln−/− mice, with fibrosis and prolonged APD creating a reentry-prone substrate.
RhoGTPase inhibition and Muscle-restricted putative coiled-coil protein (MURC) overexpression
The Rho GTPase family, which includes RhoA, Rac1, and Cdc42, mediates pressure and volume overload cardiac hypertrophy (Clerk and Sugden 2000). Overexpression of RhoA in the heart results in HF with concomitant SA and AV nodal dysfunction (Sah et al. 1999). On the other hand, inhibition of cardiac-specific RhoA signaling is also pathologic, resulting in atrial and ventricular enlargement by 4 months-of-age in mice. Surface ECGs showed 100% incidence of first-degree AV block by 4 weeks old with normal QRS and QTc intervals. At 7 months old, atrial arrhythmias were seen in mice with deficient cardiac RhoA signaling—transient sinus arrest (60%), sinus bradycardia (60%), atrial tachycardia (60%), and AF (20%). RhoA-deficient mice also had prolonged atrial ERP, indicative of impaired atrial recovery from electrical triggers. While the association between deficient cardiac RhoA signaling and AF is rather weak (20% incidence), atrial arrhythmias like resulted from an AF-prone substrate characterized by hypertrophy and slowed conduction.
Building upon these findings, Ogata et al. (Ogata et al. 2008) studied MURC, a molecule localized to the Z-line of sarcomeres that functions as a positive modulator of Rho GTPase signaling in the heart via augmentation of atrial natriuretic peptide (ANP) expression (Ogata et al. 2008). MURC was overexpressed in the murine heart by cloning mouse MURC into the 5’ untranslated region of the αMHC promoter (Ogata et al. 2008). At 13 weeks-of-age, MURC-overexpressing mice had atrial enlargement with intra-atrial thrombi and ventricular wall thinning. Fibrosis was seen histologically in the atria and ventricles bilaterally. Echocardiography revealed LV systolic dysfunction, likely mediated by ventricular fibrosis. At nine weeks-of-age, surface ECGs revealed that 16.7% of the MURC-overexpressing mice had AF, 8.3% had 3rd degree AV block, and 75% had prolonged PR intervals. At 20 weeks-of-age, 42% of MURC-overexpressing mice had either AF or AV block. Altogether, the RhoA GTPase pathway predisposes to primarily via structural remodeling, as evidenced by fibrosis, atrial dilatation, and concomitant LV systolic dysfunction. A role for calcium mishandling and other ectopic triggers remains elusive in this model.
Phosphoinositide 3-kinase (PI3K) KO
PI3K is a key regulator of physiologic cardiac hypertrophic growth (McMullen et al. 2003) that has been shown to inhibit pathologic G-protein coupled receptor signaling cascades (McMullen et al. 2007) and exhibits crosstalk with the AMPK metabolic stress pathway (Walkowski et al. 2022; Harada et al. 2012). Inhibition of PI3K exacerbates systolic dysfunction in a mouse model of dilated cardiomyopathy (DCM) (McMullen et al. 2007). Based on several studies demonstrating clinical and biochemical evidence of a link between PI3K and AF (van der Hooft et al. 2004; Kampinga et al. 2007), Pretorius et al. (Pretorius et al. 2009) crossed a transgenic mouse model of DCM (Yamamoto et al. 2003) with cardiac-specific transgenic mice expressing a dominant negative mutant of PI3K (Pretorius et al. 2009). Expression of the mutant PI3K transgene in the DCM Tg mice increased the PR interval and resulted in spontaneous AF occurred in 40% of 4.5-month-old mice. Atrial fibrosis was 5.5-fold higher by histology in the double transgenic mice compared to WT controls. Atrial expression of potassium channels Kcnv2, Kcnd3, and Kcnj2 were also decreased in the DCM/PI3K-Tg mice compared to DCM-Tg and WT control mice. Human atrial samples showed lower PI3K in patients who developed paroxysmal AF following cardiothoracic surgery—indicating that lower PI3K levels, whether due to aging or other pathology, may predispose patients to post-operative AF. It is important to note with this model of spontaneous AF, however, that HF exacerbation could have caused and/or contributed to spontaneous AF. Nonetheless, aged (15-month-old) DCM single Tg mice did not exhibit AF nor the decrease in Kcnj2 and Kcnv2 seen in the DCM/PI3K-Tg mice. Thus, PI3K seems to play an independent role in AF generation apart from the HF also seen with loss of PI3K. Altogether, Pretorius et al. (Pretorius et al. 2009) provides evidence that the increase in AF susceptibility may be mediated through changes in substrate characterized by structural, electrical, and fibrotic remodeling.
Rac1 overexpression
AF is associated with oxidative stress, in which nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in the left atrium plays an important role (Dudley et al. 2005). Rac1 is a Rho GTPase regulator of NADPH oxidase activity that has been shown to mediate oxidative stress and hypertrophic responses in the heart (Custodis et al. 2006). To this end, Adam et al. (Adam et al. 2007) created homozygous mice with overexpression of active Rac1 under control of the alpha-MHC promoter (Adam et al. 2007). Because concentric ventricular hypertrophy was seen as early as three weeks old, transverse aortic constriction (TAC) was performed to differentiate the atrial effects of Rac1 activation secondary to ventricular hypertrophy from the atrial effects of Rac1 overexpression itself. Homozygous mice overexpressing Rac1 developed spontaneous AF, with incidences of 44% at 10 months and 75% at 16 months. Atrial fibrosis was increased in Rac1-Tg mice compared to WT mice who underwent TAC; atrial fibrosis was more pronounced than ventricular fibrosis. AF patient samples similarly showed greater Rac1 protein expression. Altogether, spontaneous AF in Rac1 overexpressing mice was mediated by substrate remodeling, as evidenced by atrial fibrosis, with potential contribution from concomitant ventricular dysfunction.
Nucleoporin (NUP) deletion
Mutations in chromosome 5p13, which encodes the NUP155 gene, have been linked to autosomal-recessive AF (Oberti et al. 2004). NUP155 encodes for nucleoporin, a component of the nuclear pore complex. One mutation in the 5p13 region, R391H, co-segregates with AF, and has been shown to decrease nuclear envelope permeabilization (Zhang et al. 2008). To further explore this genetic evidence, Zhang et al. (Zhang et al. 2008) made heterozygous NUP155 deletions in mice. Loss of NUP155 resulted in spontaneous AF with shortened APD. AF pathophysiology in this model was hypothesized to be due to attenuation of mRNA nuclear export of key atrial genes such as calcium handling proteins and ion channels, as evidenced by the shortened APD seen in NUP155± atrial cardiomyocytes.
Micro-RNAs (miR)
The role of micro-RNAs as therapeutic and diagnostic targets is an emerging field of research in cardiovascular disease (Romaine et al. 2015). miR-382, which targets the alpha 1c and beta-1 subunits of the cardiomyocyte LTCC, is upregulated in AF patients with rheumatic heart disease (Lu et al. 2010). To explore the implications of this finding, Lu et al. (Lu et al. 2010) transgenically overexpressed miR-328 in the hearts of mice under the αMHC promoter. Spontaneous AF was observed in 100% of Tg mice starting at 28 days-of-age. Consistent with Cacna1c and Cacnb1 being targets of miR-328, the LTCC current was decreased in miR-328 Tg mice, resulting in shortened atrial APD. On the other hand, potassium currents were unaltered. Altogether, miR-328 induces spontaneous AF due to atrial electrical remodeling, characterized by shortened APD secondary to decreased LTCC current. Evidence for the role of fibrotic remodeling comes from a study of miRs collected from exosomes of pericardial fluid that linked miR-328 with cardiac fibrosis (Liu et al. 2020).
miR-208a is encoded by an intron of the alpha-myosin heavy chain (Myh6) and has been implicated in stress-related cardiac growth and remodeling (van Rooij et al. 2007). Callis et al. (Callis et al. 2009) demonstrated that miR-208a overexpression causes ventricular hypertrophy and dilatation with decreased EF by three months of age. miR-208a overexpression also resulted in AV block in four-month-old mice. In contrast to overexpression, loss of miR-208a, which also prolonged the PR interval, resulted in spontaneous AF in 80% of mice with concomitant decreased connexin 40 expression. These seemingly contradictory results of miR-208a overexpression and knockout could perhaps be explained by the fact that miR-208a directly targets key cardiac transcription factor GATA4, and therefore both overexpression and loss-of-function results in pathologic GATA4 dysregulation. Altogether, the spontaneous AF seen in miR-208a−/− mice results from structural remodeling and aberrant conduction.
Nav1.5 overexpression
Based on the observation that gain-of-function variants in SCNA5 are associated with increased AF incidence, a prior study generated Tg mice overexpressing human Nav1.5-F1759A, which causes a persistent sodium current (Wan et al. 2016). Nav1.5-F1759A Tg mice exhibited biatrial enlargement by 1 month old, with histologic evidence of fibrosis as well as ventricular dilation and systolic HF by 3–4 months of age (Wan et al. 2016). Nav1.5-F1759A mice had prolonged QT intervals, indicative of persistent inward sodium current causing APD prolongation. Spontaneous AF was observed in 25% of mice by 5–6 weeks of age. By 10 weeks of age, spontaneous AF incidence was reported in 80% of mice (Wan et al. 2016). A more recent study of the same model reported that oxidative stress due to mitochondrial reactive oxygen species could a mechanistic driver of AF in the Nav1.5-F1759A mouse model (Avula et al. 2021). Phosphorylation of RyR2 by protein kinase A and CaMKII at Ser2808 and Ser2814, respectively, as well as calcium spark frequency were greater in Nav1.5-F1759A mice compared to controls (Avula et al. 2021). These changes were abrogated after catalase overexpression, providing evidence of causality for mitochondrial reactive oxygen species in the Nav1.5-F1759A mouse model (Avula et al. 2021).
Conclusion
While there are many mouse models of AF, only a handful of these models exhibit spontaneous AF as the majority of mouse models require supraphysiological stimuli to uncover (inducible) AF. Among the spontaneous mouse models of AF, atrial dilatation and fibrosis seem to be common to most. Indeed, atrial dilatation and fibrosis are two of the most well-known risk factors for AF in humans and mice (Li et al. 2021; Schotten et al. 2003). In line with known AF pathophysiology (Heijman et al. 2014), calcium mishandling is also prevalent among spontaneous AF mouse models, particularly the CREm-Tg model. The most robust and well-studied models of spontaneous AF, Lkb1-CKO and CREm-Tg, exhibit characteristics of fibrosis and calcium dysregulation, with the Lkb1-KO model appearing to be driven more by fibrosis and CREm-Tg driven more by calcium mishandling. Taken together, spontaneous mouse models of AF are useful to study the effects of molecular, biochemical, and genetic manipulation on electrophysiologic phenotype and for gene therapy in targeting specific arrhythmia-promoting pathways (Dobrev and Wehrens 2018). Results must be interpreted in context, however, as human AF differs from mouse AF. For instance, human AF arises from ectopic foci near the pulmonary veins (Khan 2004), but this finding is not typically seen in mouse AF, which could be due to phenotyping limitations (Schuttler et al. 2020). Thus, understanding the underlying pathophysiology of spontaneous mouse models of AF is key to enable clinical translation.
Funding
This work was supported by National Institutes of Health grants R01-HL147108, R01-HL153350, and R01-HL089598 (to X.H.T.W.), the Robert and Janice McNair Foundation McNair MD/PhD Scholars Program (J.A.K.), the Baylor College of Medicine Medical Scientist Training Program (J.A.K.), and the American Heart Association Career Development Award (M.M.H.).
Footnotes
Conflict of interest On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Adam O, Frost G, Custodis F, Sussman MA, Schafers HJ, Bohm M, Laufs U (2007) Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol 50(4):359–367 [DOI] [PubMed] [Google Scholar]
- Anne W, Willems R, Holemans P, Beckers F, Roskams T, Lenaerts I, Ector H, Heidbuchel H (2007) Self-terminating AF depends on electrical remodeling while persistent AF depends on additional structural changes in a rapid atrially paced sheep model. J Mol Cell Cardiol 43(2):148–158 [DOI] [PubMed] [Google Scholar]
- Avula UMR, Hernandez JJ, Yamazaki M, Valdivia CR, Chu A, Rojas-Pena A, Kaur K, Ramos-Mondragon R, Anumonwo JM, Nattel S, Valdivia HH, Kalifa J (2018) Atrial infarction-induced spontaneous focal discharges and atrial fibrillation in sheep: role of dantrolene-sensitive aberrant ryanodine receptor calcium release. Circ Arrhythm Electrophysiol 11(3):e005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avula UMR, Dridi H, Chen BX, Yuan Q, Katchman AN, Reiken SR, Desai AD, Parsons S, Baksh H, Ma E, Dasrat P, Ji R, Lin Y, Sison C, Lederer WJ, Joca HC, Ward CW, Greiser M, Marks AR, Marx SO, Wan EY (2021) Attenuating persistent sodium current-induced atrial myopathy and fibrillation by preventing mitochondrial oxidative stress. JCI Insight 6(23):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson MV, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita S, Moran AE, Mussolino ME, O'Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, E. American Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics (2019) Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139(10):e56–e528. [DOI] [PubMed] [Google Scholar]
- Bhupathy P, Babu GJ, Periasamy M (2007) Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol 42(5):903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan JW (1965) Spontaneous arrhythmias and conduction disturbances in domestic animals. Ann N Y Acad Sci 127(1):224–238 [DOI] [PubMed] [Google Scholar]
- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ (2009) MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest 119(9):2772–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin S, Libby E, Pelletier P, Le Bouter S, Shiroshita-Takeshita A, Le Meur N, Leger J, Demolombe S, Ponton A, Glass L, Nattel S (2007) Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res 100(3):425–433 [DOI] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH (2009) Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 119(7):1940–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Q, Dong R, Gao H, Peng H, Wu Y (2014) The effect of the Ras homolog gene family (Rho), member A/Rho associated coiled-coil forming protein kinase pathway in atrial fibrosis of type 2 diabetes in rats. Exp Ther Med 8(3):836–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss S, Bleyer C, Schuttler D, Tomsits P, Renner S, Klymiuk N, Wakili R, Massberg S, Wolf E, Kaab S (2019) Animal models of arrhythmia: classic electrophysiology to genetically modified large animals. Nat Rev Cardiol 16(8):457–475 [DOI] [PubMed] [Google Scholar]
- Clerk A, Sugden PH (2000) Small guanine nucleotide-binding proteins and myocardial hypertrophy. Circ Res 86(10):1019–1023 [DOI] [PubMed] [Google Scholar]
- Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, Koval O, Li J, Gudmundsson H, Gumina RJ, Karck M, Schott JJ, Probst V, Le Marec H, Anderson ME, Dobrev D, Wehrens XH, Mohler PJ (2011) Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124(11):1212–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodis F, Eberl M, Kilter H, Bohm M, Laufs U (2006) Association of RhoGDIalpha with Rac1 GTPase mediates free radical production during myocardial hypertrophy. Cardiovasc Res 71(2):342–351 [DOI] [PubMed] [Google Scholar]
- DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XH, Anderson ME, Hund TJ, Mohler PJ (2012) CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm 9(12):2034–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers J, Ton AT, Huynh F, Thibault S, Ducharme A, Paradis P, Nemer M, Fiset C (2022) Atrial electrical remodeling in mice with cardiac-specific overexpression of Angiotensin II Type 1 Receptor. J Am Heart Assoc 11(8):e023974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D, Wehrens XHT (2018) Mouse models of cardiac arrhythmias. Circ Res 123(3):332–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosdall DJ, Ranjan R, Higuchi K, Kholmovski E, Angel N, Li L, Macleod R, Norlund L, Olsen A, Davies CJ, Marrouche NF (2013) Chronic atrial fibrillation causes left ventricular dysfunction in dogs but not goats: experience with dogs, goats, and pigs. Am J Physiol Heart Circ Physiol 305(5):H725–H731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley SC Jr, Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, Harrison DG, Dikalov SI, Langberg J (2005) Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112(9):1266–1273 [DOI] [PubMed] [Google Scholar]
- Ehrlich JR, Cha TJ, Zhang L, Chartier D, Melnyk P, Hohnloser SH, Nattel S (2003) Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol 551(Pt 3):801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG (1995) Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med 155(5):469–473 [PubMed] [Google Scholar]
- Frydrychowski P, Michalek M, Slawuta A, Noszczyk-Nowak A (2020) Large animals as models of atrial fibrillation. Adv Clin Exp Med 29(6):757–767 [DOI] [PubMed] [Google Scholar]
- Fu F, Pietropaolo M, Cui L, Pandit S, Li W, Tarnavski O, Shetty SS, Liu J, Lussier JM, Murakami Y, Grewal PK, Deyneko G, Turner GM, Taggart AKP, Waters MG, Coughlin S, Adachi Y (2022) Lack of authentic atrial fibrillation in commonly used murine atrial fibrillation models. PLoS ONE 17(1):e0256512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemel J, Su Z, Gileles-Hillel A, Khalyfa A, Gozal D, Beyer EC (2017) Intermittent hypoxia causes NOX2-dependent remodeling of atrial connexins. BMC Cell Biol 18(Suppl 1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, Krush AJ, Yardley JH, Luk GD (1987) Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 316(24):1511–1514 [DOI] [PubMed] [Google Scholar]
- Han D, Zhang QY, Zhang YL, Han X, Guo SB, Teng F, Yan X, Li HH (2020) Gallic acid ameliorates Angiotensin II-induced atrial fibrillation by inhibiting immunoproteasome- mediated PTEN degradation in mice. Front Cell Dev Biol 8:594683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M, Nattel SN (2012) Nattel S (2012) AMP-activated protein kinase. Circulation 5(4):860–867 [DOI] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Wehrens XH, Dobrev D (2014) Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front Pharmacol 5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S (2016) The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res 109(4):467–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391(6663):184–187 [DOI] [PubMed] [Google Scholar]
- Hesselkilde EZ, Carstensen H, Flethoj M, Fenner M, Kruse DD, Sattler SM, Tfelt-Hansen J, Pehrson S, Braunstein TH, Carlson J, Platonov PG, Jespersen T, Buhl R (2019) Longitudinal study of electrical, functional and structural remodelling in an equine model of atrial fibrillation. BMC Cardiovasc Disord 19(1):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Cho MC, Kwak YG, Song CH, Lee YH, Lim JS, Kwon YK, Chae SW, Kim DH (2002) Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J 16(10):1310–1312 [DOI] [PubMed] [Google Scholar]
- Hulsurkar MM, Lahiri SK, Moore O, Moreira LM, Abu-Taha I, Kamler M, Dobrev D, Nattel S, Reilly S, Wehrens XHT (2021) Atrial-specific lkb1 knockdown represents a novel mouse model of atrial cardiomyopathy with spontaneous atrial fibrillation. Circulation 144(11):909–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Sato K, Pimentel DR, Sam F, Shaw RJ, Dyck JR, Walsh K (2009) Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem 284(51):35839–35849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, Hirano M, Morrow JP (2016) Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE 11(1):e0145750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SW, Sun W, Griffiths LG, Kittleson MD (2016) Atrial fibrillation as a prognostic indicator in medium to large-sized dogs with myxomatous mitral valvular degeneration and congestive heart failure. J Vet Intern Med 30(1):51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Henning RH, van Gelder IC, Brundel BJ (2007) Beat shock proteins and atrial fibrillation. Cell Stress Chaperones 12(2):97–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Yamashita T, Sekiguchi A, Tsuneda T, Sagara K, Takamura M, Kaneko S, Aizawa T, Fu LT (2008) AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J Cardiovasc Electrophysiol 19(4):415–420 [DOI] [PubMed] [Google Scholar]
- Khan R (2004) Identifying and understanding the role of pulmonary vein activity in atrial fibrillation. Cardiovasc Res 64(3):387–394 [DOI] [PubMed] [Google Scholar]
- Kim GE, Ross JL, Xie C, Su KN, Zaha VG, Wu X, Palmeri M, Ashraf M, Akar JG, Russell KS, Akar FG, Young LH (2015) LKB1 deletion causes early changes in atrial channel expression and electrophysiology prior to atrial fibrillation. Cardiovasc Res 108(1):197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhof P, Marijon E, Fabritz L, Li N, Wang W, Wang T, Schulte K, Hanstein J, Schulte JS, Vogel M, Mougenot N, Laakmann S, Fortmueller L, Eckstein J, Verheule S, Kaese S, Staab A, Grote-Wessels S, Schotten U, Moubarak G, Wehrens XH, Schmitz W, Hatem S, Muller FU (2013) Overexpression of cAMP-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol 166(2):366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryshtal DO, Blackwell DJ, Egly CL, Smith AN, Batiste SM, Johnston JN, Laver DR, Knollmann BC (2021) RYR2 channel inhibition is the principal mechanism of flecainide action in CPVT. Circ Res 128(3):321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakin R, Polidovitch N, Yang S, Guzman C, Gao X, Wauchop M, Burns J, Izaddoustdar F, Backx PH (2019) Inhibition of soluble TNFalpha prevents adverse atrial remodeling and atrial arrhythmia susceptibility induced in mice by endurance exercise. J Mol Cell Cardiol 129:165–173 [DOI] [PubMed] [Google Scholar]
- Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Muller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XHT (2014) Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129(12):1276–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Zhang JR, Hu WN, Li SN (2021) Atrial fibrosis underlying atrial fibrillation (Review). Int J Mol Med 47(3):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Fu H, Li J, Yang W, Cheng L, Liu T, Li G (2012) Hyperglycemia aggravates atrial interstitial fibrosis, ionic remodeling and vulnerability to atrial fibrillation in diabetic rabbits. Anadolu Kardiyol Derg 12(7):543–550 [DOI] [PubMed] [Google Scholar]
- Liu L, Chen Y, Shu J, Tang CE, Jiang Y, Luo F (2020) Identification of microRNAs enriched in exosomes in human pericardial fluid of patients with atrial fibrillation based on bioinformatic analysis. J Thorac Dis 12(10):5617–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Zhang Y, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B (2010) MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 122(23):2378–2387 [DOI] [PubMed] [Google Scholar]
- Lugenbiel P, Wenz F, Govorov K, Schweizer PA, Katus HA, Thomas D (2015) Atrial fibrillation complicated by heart failure induces distinct remodeling of calcium cycling proteins. PLoS ONE 10(3):e0116395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S (2003) Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A 100(21):12355–12360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL (2007) Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 104(2):612–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira YE, Muhuyati, Lu WH, He PY, Liu ZQ, Yang YC (2013) TGF-beta1 signal pathway in the regulation of inflammation in patients with atrial fibrillation. Asian Pac J Trop Med 6(12):999–1003 [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421(6923):634–639 [DOI] [PubMed] [Google Scholar]
- Moreira LM, Takawale A, Hulsurkar M, Menassa DA, Antanaviciute A, Lahiri SK, Mehta N, Evans N, Psarros C, Robinson P, Sparrow AJ, Gillis MA, Ashley N, Naud P, Barallobre-Barreiro J, Theofilatos K, Lee A, Norris M, Clarke MV, Russell PK, Casadei B, Bhattacharya S, Zajac JD, Davey RA, Sirois M, Mead A, Simmons A, Mayr M, Sayeed R, Krasopoulos G, Redwood C, Channon KM, Tardif JC, Wehrens XHT, Nattel S, Reilly S (2020) Paracrine signalling by cardiac calcitonin controls atrial fibrogenesis and arrhythmia. Nature 587(7834):460–465 [DOI] [PubMed] [Google Scholar]
- Muller FU, Lewin G, Baba HA, Boknik P, Fabritz L, Kirchhefer U, Kirchhof P, Loser K, Matus M, Neumann J, Riemann B, Schmitz W (2005) Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem 280(8):6906–6914 [DOI] [PubMed] [Google Scholar]
- Nair GM, Nery PB, Redpath CJ, Birnie DH (2014) The role of renin angiotensin system in atrial fibrillation. J Atr Fibrillation 6(6):972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel S, Dobrev D (2016) Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol 13(10):575–590 [DOI] [PubMed] [Google Scholar]
- Ni L, Lahiri SK, Nie J, Pan X, Abu-Taha I, Reynolds JO, Campbell HM, Wang H, Kamler M, Schmitz W, Muller FU, Li N, Wei X, Wang DW, Dobrev D, Wehrens XHT (2021) Genetic inhibition of nuclear factor of activated T-cell c2 (NFATc2) prevents atrial fibrillation in CREM transgenic mice, Cardiovasc Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Michael G, Dobrev D, Nattel S (2010) Animal models for atrial fibrillation: clinical insights and scientific opportunities. Europace 12(2):160–172 [DOI] [PubMed] [Google Scholar]
- Oberti C, Wang L, Li L, Dong J, Rao S, Du W, Wang Q (2004) Genome-wide linkage scan identifies a novel genetic locus on chromosome 5p13 for neonatal atrial fibrillation associated with sudden death and variable cardiomyopathy. Circulation 110(25):3753–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odening KE, Gomez AM, Dobrev D, Fabritz L, Heinzel FR, Mangoni ME, Molina CE, Sacconi L, Smith G, Stengl M, Thomas D, Zaza A, Remme CA, Heijman J (2021) ESC working group on cardiac cellular electrophysiology position paper: relevance, opportunities, and limitations of experimental models for cardiac electrophysiology research. Europace 23(11):1795–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Ueyama T, Isodono K, Tagawa M, Takehara N, Kawashima T, Harada K, Takahashi T, Shioi T, Matsubara H, Oh H (2008) MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol Cell Biol 28(10):3424–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan C, Battaglia E, Young R, Suzuki G (2015) LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J Am Heart Assoc 4(3):e001733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan C, Dixit G, Li Z (2021) Activation of AMP-activated protein kinases prevents atrial fibrillation. J Cardiovasc Transl Res 14(3):492–502 [DOI] [PubMed] [Google Scholar]
- Park DS, Cerrone M, Morley G, Vasquez C, Fowler S, Liu N, Bernstein SA, Liu FY, Zhang J, Rogers CS, Priori SG, Chinitz LA, Fishman GI (2015) Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest 125(1):403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccini JP Sr, Fonarow GC (2016) Preventing stroke in patients with atrial fibrillation-a steep climb away from achieving peak performance. JAMA Cardiol 1(1):63–64 [DOI] [PubMed] [Google Scholar]
- Pretorius L, Du XJ, Woodcock EA, Kiriazis H, Lin RC, Marasco S, Medcalf RL, Ming Z, Head GA, Tan JW, Cemerlang N, Sadoshima J, Shioi T, Izumo S, Lukoshkova EV, Dart AM, Jennings GL, McMullen JR (2009) Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am J Pathol 175(3):998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaine SP, Tomaszewski M, Condorelli G, Samani NJ (2015) MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart 101(12):921–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF, London B (2005) Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-{alpha}. Am J Physiol Heart Circ Physiol 289(4):H1456–H1467 [DOI] [PubMed] [Google Scholar]
- Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW 2nd, Ross J Jr, Chien KR, Brown JH (1999) Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest 103(12):1627–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Clancy CE, Bers DM (2017) Dynamics of sodium current mediated early afterdepolarizations. Heliyon 3(9):e00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS (2007) Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292(3):H1561–H1567 [DOI] [PubMed] [Google Scholar]
- Schnabel RB, Yin X, Gona P, Larson MG, Beiser AS, McManus DD, Newton-Cheh C, Lubitz SA, Magnani JW, Ellinor PT, Seshadri S, Wolf PA, Vasan RS, Benjamin EJ, Levy D (2015) 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet 386(9989):154–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotten U, Neuberger HR, Allessie MA (2003) The role of atrial dilatation in the domestication of atrial fibrillation. Prog Biophys Mol Biol 82(1–3):151–162 [DOI] [PubMed] [Google Scholar]
- Schulte JS, Fehrmann E, Tekook MA, Kranick D, Fels B, Li N, Wehrens XH, Heinick A, Seidl MD, Schmitz W, Muller FU (2016) Cardiac expression of the CREM repressor isoform CREM-IbDeltaC-X in mice leads to arrhythmogenic alterations in ventricular cardiomyocytes. Basic Res Cardiol 111(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttler D, Bapat A, Kaab S, Lee K, Tomsits P, Clauss S, Hucker WJ (2020) Animal models of atrial fibrillation. Circ Res 127(1):91–110 [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9(8):563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, Shaw RM (2010) Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest 120(1):266–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfo D, Savarese G (2019) Use of renin-angiotensin-aldosterone system inhibitors in older patients with heart failure and reduced ejection fraction. Card Fail Rev 5(2):70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpel FT, Stein J, Himmler K, Scholz B, Seidl MD, Skryabin BV, Muller FU (2018) Homozygous CREM-IbDeltaC-X overexpressing mice are a reliable and effective disease model for atrial fibrillation. Front Pharmacol 9:706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Matsumoto A, Nishida H, Reien Y, Maruyama H, Nakaya H (2014) Termination of aconitine-induced atrial fibrillation by the KACh-channel blocker tertiapin: underlying electrophysiological mechanism. J Pharmacol Sci 125(4):406–414 [DOI] [PubMed] [Google Scholar]
- Tse G (2016) Mechanisms of cardiac arrhythmias. J Arrhythm 32(2):75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura N, Ohkusa T, Hamano K, Nakagome M, Hori H, Shimizu M, Matsuzaki M, Mochizuki S, Minamisawa S, Ishikawa Y (2004) Down-regulation of sarcolipin mRNA expression in chronic atrial fibrillation. Eur J Clin Invest 34(11):723–730 [DOI] [PubMed] [Google Scholar]
- van der Hooft CS, Heeringa J, van Herpen G, Kors JA, Kingma JH, Stricker BH (2004) Drug-induced atrial fibrillation. J Am Coll Cardiol 44(11):2117–2124 [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316(5824):575–579 [DOI] [PubMed] [Google Scholar]
- Verheule S, Sato T, Everett TT, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE (2004) Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res 94(11):1458–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D (2012) Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125(17):2059–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkowski B, Kleibert M, Majka M, Wojciechowska M (2022) Insight into the role of the PI3K/Akt pathway in ischemic injury and post-infarct left ventricular remodeling in normal and diabetic heart. Cells 11(9) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO (2016) Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest 126(1):112–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA (1995) Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92(7):1954–1968 [DOI] [PubMed] [Google Scholar]
- Wit AL, Boyden PA (2007) Triggered activity and atrial fibrillation. Heart Rhythm 4(3 Suppl):S17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC Jr, Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR, Bernstein KE (2004) Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol 165(3):1019–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie LH, Shanmugam M, Park JY, Zhao Z, Wen H, Tian B, Periasamy M, Babu GJ (2012) Ablation of sarcolipin results in atrial remodeling. Am J Physiol Cell Physiol 302(12):C1762–C1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF, Sadoshima J (2003) Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest 111(10):1463–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PC, Moreno JD, Miyake CY, Vaughn-Behrens SB, Jeng MT, Grandi E, Wehrens XH, Noskov SY, Clancy CE (2016) In silico prediction of drug therapy in catecholaminergic polymorphic ventricular tachycardia. J Physiol 594(3):567–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha ID, Ghezelbash S, Reynolds CL, Shen YH, LeMaire SA, Schmitz W, Muller FU, El-Armouche A, Eissa NT, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N (2018) Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138(20):2227–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R, Wang L, Chen Q, Wang QK (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135(6):1017–1027 [DOI] [PubMed] [Google Scholar]