

Abstract
Clinical and genetic features of five unrelated patients with de novo pathogenic variants in the synaptic vesicle associated membrane protein VAMP2 reveal common features of global developmental delay, autistic tendencies, behavioral disturbances, and a higher propensity to develop epilepsy. For one patient, a cognitively impaired adolescent with a de novo stop-gain VAMP2 mutation, we tested a potential treatment strategy, enhancing neurotransmission by prolonging action potentials with the aminopyridine family of potassium channel blockers, 4-aminopyridine and 3,4-diaminopyridine, in vitro and in vivo. Synaptic vesicle recycling and neurotransmission were assayed in neurons expressing three VAMP2 variants by live cell imaging and electrophysiology. In cellular models, two variants decrease both the rate of exocytosis and the number of vesicles released from the recycling pool, compared to wild type. Aminopyridine treatment increases the rate and extent of exocytosis and total synaptic charge transfer, and desynchronizes GABA release. The clinical response of the patient to two years of off-label aminopyridine treatment includes improved emotional and behavioral regulation by parental report, and objective improvement in standardized cognitive measures. Aminopyridine treatment may extend to patients with pathogenic variants in VAMP2 and other genes influencing presynaptic function or GABAergic tone, and tested in vitro prior to treatment.
Keywords: neurodevelopmental disorder, synaptic vesicle, synaptic transmission, VAMP2, aminopyridine
Graphical Abstract
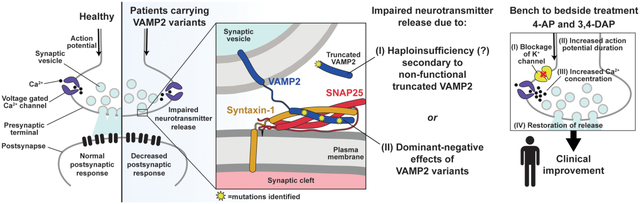
1. INTRODUCTION
Fast communication between neurons relies on precise and highly regulated fusion of synaptic vesicles (SVs) with the presynaptic plasma membrane, resulting in neurotransmitter exocytosis (Sudhof, 2013). Fusion of the two membranes is driven by the zippering of highly conserved hydrophobic side chain SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) motifs of the vesicular v-SNARE, vesicle associated membrane protein (VAMP2, MIM# 185881) with the plasma membrane target t-SNAREs, SNAP-25 and syntaxin-1, to form the SNARE complex (Jahn & Scheller, 2006). The SNARE complex interacts with accessory proteins, such as the synaptotagmin family of Ca2+ sensors. Synaptotagmins sense the local increase in Ca2+ concentration caused by opening of voltage-gated Ca2+ channels (VGCCs) upon action potential induced membrane depolarization. The resultant local increase in Ca2+ concentration is signaled to the SNARE complex mainly by synaptotagmin-1 for synchronous release and by synaptotagmin-7 for asynchronous release. Simultaneously, inward-rectifying K+ channels and the Na+/K+-ATPase repolarizes the membrane, halting VGCC activity, while active pumps clear local Ca2+ away, terminating neurotransmitter release. VAMP2 is also vital for the fast endocytosis of neuronal membrane to replenish the pool of SVs for the next round of exocytosis (Deak, Schoch, Liu, Sudhof, & Kavalali, 2004).
VAMP2 is the most abundant SV protein, with approximately 70 copies per vesicle, while only 2–3 are necessary to mediate fusion (Mohrmann & Sorensen, 2012; Takamori et al., 2006). Given the high conservation of the SNARE motif (Fasshauer, Eliason, Brunger, & Jahn, 1998), a mutation in this region would be expected to disrupt endocytosis or exocytosis of SVs, impairing neurotransmission and causing neurological disorders. Indeed, experimental SNARE mutations cause dominant negative disruption of fusion (Grote & Kelly, 1996; Koo et al., 2011), while VAMP2 heterozygosity loss of function in mice causes only a mild phenotype (Monteggia, Lin, Adachi, & Kavalali, 2018). Human VAMP2 SNARE mutations can impair fusion of reconstituted membranes. A clinical phenotype for VAMP2 mutations was recently described in five patients as hypotonia, intellectual disability, and autistic features (MIM# 618760, Salpietro et al., 2019). Here we report five unrelated patients, doubling the number identified with pathogenic de novo VAMP2 variants. Currently there is no effective treatment targeting the underlying impairment of neurotransmitter release. Patients likely make wild type VAMP2 from their functional allele, so we tested a strategy to overcome exocytosis defects by prolonging the action potential (AP) by delaying neuronal repolarization with the potassium channel inhibitors 4-aminopyridine (4-AP) and 3,4-diaminopyridine (DAP) to increase calcium entry and SV release probability. 4-AP and DAP have expanding roles in the treatment of multiple sclerosis, cerebellar ataxias, and Lambert-Eaton and congenital myasthenic syndromes (Claassen, Teufel, Kalla, Spiegel, & Strupp, 2013; Palace, Wiles, & Newsom-Davis, 1991; Strupp et al., 2017). Taken together, this suggests enhancement of SV release could improve cognitive function in patients with single allele VAMP2 pathogenic variants.
2. METHODS
2.1. Editorial policies and ethical considerations
Work with animals was conducted under the supervision of the Institutional Care and Use Committees of the University of California, San Francisco and Vanderbilt University Medical Center. Parents provided written consent prior to participation through a UCSF committee on human research approved protocol.
2.2. Clinical information
Patients with VAMP2 variants were assessed by chart review and caretaker phone interviews. Parents provided written consent prior to participation. Variants were assessed for clinical significance and pathogenicity by use of web sources including clinical data obtained from GeneDx, as well as predicted results from ClinVar, Poly-Phen2, and gnomAD. Thus, while variants of these patients were initially reported by GeneDx as “uncertain significance,” they were determined to be pathogenic based on clinical phenotype and predictive algorithms. Ancillary studies, including EEG, EMG, MRI, and neuropsychologic testing were reviewed by a physician to determine clinical relevance. After receiving consent from parents and the patient, Patient 1 was treated with low dose 4-AP that was gradually increased over the course of several months. Tolerability of 4-AP was assessed by parental report of worsening anxiety or insomnia. Effects of 4-AP were measured by qualitative assessments via subjective parental reports, and quantitatively via neuropsychological testing. Neuropsychological testing pre- and post-treatment were compared by converting scaled scores to z-scores.
2.3. Molecular biology and lentivirus preparation
VAMP2-mOrange2 fusions were constructed by fusing synthetic mOrange 2 (mOr2) (Shaner et al., 2008) to the C-terminus of human wild type (RefSeq NM_014232.3) or variant VAMP2 cDNAs with a linker (SGGSGGTG). Disease-associated point mutations Arg56Leu (R56L) or Gly73Trp (G73W) were generated in VAMP2 using Quikchange-XL2 Site Directed Mutagenesis (Agilent) using the following primer pairs: For Arg56Leu: 5’-GACAACTTCTGGTCCAGCTCCAGGACCTTGT-3’ and 5’-ACAAGGTCCTGGAGCTGGACCAGAAGTTGTC.
For Gly73Trp: 5’-GGAGGCCCATGCCTGGAGGGCATC-3’ and 5’-GATGGCCTCCAGGVATGGGCCTCC-3’. To mimic the truncated human VAMP2 Arg56* found in patients, the Arg56X (R56X) VAMP2-mOr2 construct was made by fusing the VAMP2 coding sequence for the first 55 amino acids to the N-terminus of mOr2 with the same linker as above, since introduction of a stop codon into the full length VAMP2 cDNA would not allow expression of the downstream mOr2. All PCR generated VAMP2 sequences were verified by sequencing, then subcloned into the wild type pCAGGS vector by EcoRI and Xho I using standard molecular biology methods. Synaptophysin-pHluorin (syp-pH) was made by inserting pHluorin flanked by a PCR-generated 5’ linker (SGGTGGSGGTGGSGGTGSTSGGSGGTGG) and 3’ linker (SGGTGGSGGTGGSGGTGGSGGTGGSGGTGGSG) into an engineered Age1 site between amino acids 181T and 182G in the second luminal loop of rat synaptophysin (gift of R. Edwards, UCSF), generated by PCR mediated mutagenesis, confirmed by sequencing, and subcloned into a pCAGGS vector.
To overexpress WT as well as variant VAMP2 in primary culture for electrophysiological experiments, lentiviral constructs carrying the corresponding cDNA sequences were subcloned into a pFUGW lentiviral vector using standard molecular biology techniques and verified by sequencing. Lentiviral particles were produced by transfecting HEK293T cells with the corresponding pFUGW transfer vector and 3 packaging plasmids (pVSVg, pMdLg/pPRE and pRSV-Rev) using FuGENE 6 transfection reagent (Promega). 24 hours after transfection, the HEK293T culture media was replaced by neuronal growth media. Lentiviral particles were released into the media over 48 hours and harvested by low-speed centrifugation on the day of infection.
2.4. Primary hippocampal culture, transfection and lentiviral infection
For live cell imaging experiments, hippocampi from embryonic day 19–20 rats of either sex were dissociated as previously described (Li, Santos, Park, Dobry, & Voglmaier, 2017). Neurons were co-transfected with 0.8 ug syp-pH and 0.2 ug VAMP2-mOr2 in pCAGGS, using the Basic Neuron SCN Nucleofector kit according to manufacturer’s directions (Lonza). DNA amounts were optimized to produce relatively equal, moderate expression and punctate localization consistent with synaptic delivery (data not shown). All co-transfected VAMP2-mOr2 proteins co-localize with syp-pH in synaptic boutons (data not shown). Cells were maintained in Neurobasal media supplemented with 1% heat inactivated fetal bovine serum, B27 (Gibco), 2 mM GlutaMax, 15 mM NaCl, and 10 μg/ml MycoZap antibiotic (Lonza) and imaged at 14–19 days in vitro (DIV). 5-fluoro-2’-deoxyuridine (10 μM final concentration) was added at DIV3–5 as a mitotic inhibitor to control glial growth. This work with animals was conducted under the supervision of the Institutional Care and Use Committee of the University of California, San Francisco.
For electrophysiology experiments, dissociated hippocampal cultures were prepared using postnatal 2–4 days old Sprague Dawley rats of either sex as previously described (Kavalali, Klingauf, & Tsien, 1999). On DIV4, neurons were infected by corresponding lentiviral particles per well in 24 well plates. Electrophysiology experiments were performed after DIV14, when synapses reach maturity and overexpression of the target protein had plateaued. This work with animals was conducted under the supervision of the Vanderbilt University Medical Center Institutional Care and Use Committee.
2.5. Live cell imaging and data analysis
Assessment of SV recycling by live cell imaging was performed essentially as described previously (Li et al., 2017). Coverslips with transfected hippocampal neurons were mounted in a rapid switching, laminar-flow perfusion and stimulation chamber (Warner) on an inverted epifluorescence microscope (Nikon) and imaged at room temperature using a 63X oil objective (NA=1.4). Cells were imaged in modified Tyrode’s solution pH 7.4 (in mM: 119 NaCl, 10 HEPES-NaOH, 30 glucose, 2.5 KCl, 2 CaCl2, 2 MgCl2) containing 10 μM each of the glutamate receptor inhibitors CNQX and CPP. Electrical stimulation to elicit action potentials (APs) was applied using an A310 Accupulser (WPI) at 5–100 Hz with 1 ms bipolar current pulses through platinum-iridium electrodes, to yield fields of 5–10 V/cm across the chamber. Cells were illuminated using a Xenon lamp (Sutter Instruments) with either a 470/40-nm excitation and a 525/50-nm emission filter for GFP, or a 545/25 nm excitation and 605/70 nm emission filter for mOr2 (Chroma). Images were acquired on a QuantEM CCD camera (Photometrics), exposing each fluorophore for 300 ms for images collected every 3 s. MetaMorph software was used to control data collection and to perform offline analysis (Molecular Devices). The total pool size was determined using Tyrode’s solution with 50 mM NH4Cl (NaCl reduced by 50 mM). To measure exocytosis alone, cultures were incubated in modified Tyrode’s medium containing 0.5–1 uM bafilomycin A for 30 s before imaging in the same medium. Neurons were incubated with 500 uM 3,4-diaminopyridine (DAP, Sigma) in media for 30 m before imaging in Tyrode’s solution containing 200 uM DAP.
MetaMorph software was used to quantify the average fluorescence of regions of interest (ROI) at synaptic sites at manually selected 4 × 4 pixel boxes placed over the center of boutons. The average fluorescence of three 4 × 4 pixel ROIs without cellular elements was subtracted as background. Baseline values from the first 5 frames (before stimulation) were averaged as initial fluorescence F0, and the dynamics of fluorescence intensity expressed as fractional change (F) over initial fluorescence. For normalized measurements, the average pHluorin fluorescence over individual boutons was normalized to the total fluorescence visualized by application of modified Tyrode’s solution containing 50 mM NH4Cl to alkalinize all synaptic compartments. Fluorescence measurements from 22 to 145 boutons per coverslip were averaged and the means from 7 to 12 coverslips from at least two independent cultures were averaged. The fraction of transporter that undergoes exocytosis (recycling pool, RP) was measured as the fraction of the total pool that undergoes exocytosis in response to 10 Hz 90 s stimulation (Foss, Li, Santos, Edwards, & Voglmaier, 2013). The rate of exocytosis [(F/F0) / s] was estimated from a linear fit to the increase in pHluorin fluorescence during the initial 15 s of stimulation in the presence of bafilomycin.
2.6. Electrophysiology and data analysis
Whole-cell patch clamp recordings were performed on pyramidal cells using a CV203BU headstage, Axopatch 200B amplifier, Digidata 1320 digitizer and Clampex 8.0 software (Molecular Devices). Recordings were filtered at 1 kHz and sampled at 100 s. For external bath solution, a modified Tyrode’s solution containing the following was used: (in mM): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES at pH 7.4. To isolate mEPSCs, 1 μm TTX 50 μm PTX (picrotoxin), and 50 μm D-AP5 were added. To isolate evoked IPSCs, 50 μm D-AP5, and 10 μm CNQX were added. For evoked IPSC recordings, field stimulation was provided using a parallel bipolar electrode (FHC) immersed in the external bath solution, delivering 35 mA pulses. The 3–5 Mohm borosilicate glass patch pipettes were filled with the internal solution contained the following (in mM): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na3GTP, and 10 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide] at pH 7.35 and 300 mOsm. For all recordings included for the analysis, membrane resistance was greater than 100 M, access resistance was less than 20 Mohm and time constant (τ) was less than 3 ms. mEPSC frequencies and amplitudes were analyzed using Mini Analysis software (Synaptosoft). Evoked IPSC peak amplitudes and cumulative charge transfer were analyzed by using Clampfit (Molecular Devices).
2.7. Statistical analysis
Data is presented as mean +/− standard error of the mean. Graphpad Prism 8 was used for statistical analysis. The effects of variants were compared against WT VAMP2 by t-test. The mean difference was accepted as significant at P <0.05.
3. RESULTS
In this study, we describe five previously unreported, unrelated patients with novel de novo heterozygous VAMP2 pathogenic variants and their clinical characteristics (Table 1). Patients with these VAMP2 pathogenic variants were assessed by chart review and caretaker phone interviews. Parents provided written consent prior to participation. Common features include global developmental delay, autistic tendencies, and behavioral disturbances. The SNARE motif VAMP2 variants of Patients 1–3 were studied in vitro. Patients 4 and 5 were subsequently referred to our study.
TABLE 1.
Clinical features of patients with de novo VAMP2 mutations
| Patient number | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Variant | Heterozygous de novo C.166C>T, p.Arg56X | Heterozygous de novo c.217G>T, p.Gly73Trp | Heterozygous de novo c.167G>T, p.Arg56Leu | Heterozygous de novo c.337_341 deletion TACTT p.Tyr113Gln frameshift, creating stop codon 12 | Heterozygous de novo c.1A>G, p. Met1? |
| Predicted change | Premature truncation, causing haploinsufficiency | Missense mutation, causing dominant-negative effect | Missense mutation, causing dominant-negative effect | Frameshift deletion creating a premature stop codon, causing haploinsufficiency | Elimination of initiator methionine could result in no protein, truncated protein, most likely haploinsufficiency |
| Age | 20 | 39 | 5 | 6 | 5 |
| Gender | F | M | M | M | M |
| Age of presentation | Infancy | 3 Years | Unknown | 18 Months | 1 Year |
| Clinical seizures | No | Generalized convulsions | Infantile spasms, focal, and tonic seizures | No | No |
| EEG | Reduced organization or normal | Excessive generalized slowing | Burst suppression, hypsarrhythmia | Not done | Not done |
| Visual deficits | Visual acuity deficits | Retinitis pigmentosa | CVI | None | Hypermetropia and astigmatism |
| Speech impairment | No | Echolalia | Nonverbal | Speech delay | Speech delay |
| Movement disorder | Catatonia | Nystagmus, progressive ataxia, tremor | No | No | No |
| Dysautonomia | Autonomic and small fiber neuropathy | Not tested | Frequent unexplained fevers, no formal testing | Not tested | Not tested |
| Psychiatric features | Hallucinations, delusions, anxiety, depression | Obsessive-compulsive tendencies | Unable to assess | Hyperactivity, impulsivity | Hyperactivity, impulsivity |
| Behavioral disturbances | Aggressive outbursts, self-injurious behavior | No | Unable to assess due to severe ID | Aggressive outbursts, self-injurious behavior | Aggressive outbursts |
| Autistic features | Yes | Yes | Unable to assess due to severe ID | Yes | No |
| Brain imaging | Normal | Periventricular FLAIR hyperintensities | Hypoplastic corpus callosum | Not done | Normal |
| Ancillary studies | EMG with reduced sympathetic skin responses | None | None | None | None |
| Global DD | Yes | Yes | Yes | Yes | Yes |
| Dysmorphisms | Mild hypertelorism | None | Growth restricted | Short stature | None |
| Level of independence | Performs all ADLs | Independent in most ADLs | Completely dependent | Age appropriate | Age appropriate |
Note: Variants were reported through whole exome testing by GeneDx, where c. designates a location in the cDNA and p. designates a location in the predicted protein.
Abbreviations: ADL, activities of daily living; DD, developmental delay; EEG, electroencephalography; EMG, electromyography; ID, intellectual disability.
3.1. Case histories
Patient (Pt.) 1 (BRDP ID 2355–0) exhibited developmental delay and social impairments beginning in infancy. In adolescence she developed worsening behavioral problems, aggression, emotional lability, anxiety, hallucinations and delusions. She was presumptively diagnosed with Hashimoto’s encephalopathy and received empiric treatment with plasmapheresis, steroids and rituximab, with some degree of improvement in her mood and function with improved thyroid antibody titers, however she still exhibited slowed psychomotor responsiveness and cognitive processing. Comprehensive work-up included CSF autoimmune encephalopathy panel (negative) and neurotransmitter metabolites (normal). MRI showed normal brain structure. Multiple extended EEGs had either moderate background slowing or were normal.
She came to UCSF as an adolescent after whole exome sequencing revealed a heterozygous de novo VAMP2 mutation (c.166 C>T, p.Arg56X, where c. designates a location in the cDNA and p. designates a location in the predicted protein) predicting a premature truncation and haploinsufficiency from nonsense mediated decay. Although not directly tested, this variant could also result in a dominant negative effect. On initial evaluation, she had catatonia and was largely non-verbal with minimal motor activity. A nerve conduction study was undertaken to evaluate autonomic nerve function, where VAMP2 protein is expressed and hence activity is assayable. Sympathetic skin responses were very low amplitude, with relative preservation of response latencies, providing confirmation that VAMP2 haploinsufficiency caused symptoms.
Patient 2 (BDRP 2362–0) is a 39-year-old male with cognitive impairment, autism spectrum disorder (ASD), epilepsy, and retinitis pigmentosa. Parents noticed nystagmus during infancy, and he was evaluated at age 3 years for developmental delay. Whole exome testing revealed a VAMP2 variant (heterozygous de novo c.217G>T, p.Gly73Trp). This is predicted to be a missense mutation causing a dominant negative effect. He independently performs some activities of daily living, such as showering, toileting, and dressing, and participates in a sheltered workshop program. He uses basic appliances and repairs simple things. He speaks in complete sentences and converses, but he displays obsessive compulsive tendencies, restricted interests, and echolalia. Progressively worsening ataxia and tremor impair his ability to walk and write. He developed epilepsy at 3 years of age, described as full body convulsions, which has been relatively well controlled on oxcarbazepine and more recently lamotrigine. Parents describe him as mellow, helpful and cooperative; he does not have behavioral aggression or hallucinations.
Patient 3 was identified by a clinical laboratory by whole exome testing. He carries a de novo VAMP2 variant (heterozygous de novo c.167 G>T, p.Arg56Leu) predicted to cause an amino acid missense mutation predicted to result in a dominant negative effect. He is a 6-year-old child with refractory infantile spasms and global developmental delay. EEG showed burst-suppression and hypsarrhythmia. MRI of the brain demonstrated a mildly hypoplastic corpus callosum. Severe intellectual disability confounds any behavioral or autistic features that might be present.
Patient 4 was born at term without complications after an uneventful pregnancy. At 18 months of age, parents noticed issues with speech and gross motor skills. He was evaluated through their regional center and began receiving services (speech, occupational and physical therapies). Whole exome sequencing at 4 years of age revealed a variation in VAMP2 (heterozygous de novo c.337_341 deletion TACTT p.Tyr113Gln frameshift, predicted to create a stop codon 12 (p.Tyr113GlnfsX12)), and resulting haploinsufficiency. Later he was diagnosed with ASD and attention deficit hyperactivity disorder (ADHD). He also carries a maternally inherited deletion of unclear significance (15q13.3 between D-CHRNA7 to BP5 arr[GRCh37] 15q13.3 (32024132_32509926; size 486 Kb)). The patient’s mother is cognitively normal. Reports show incomplete penetrance of this deletion, ranging from mildly affected to normal individuals (Shinawi et al., 2009). While an additive contribution of this deletion to the VAMP2 phenotype in this patient cannot be excluded, it remains uncertain given that the patient’s mother is phenotypically normal. Currently 6 years old, he is making developmental progress in pre-kindergarten with services. He can speak in short sentences and can repeat phrases however does not know primary colors or ABCs. For fine motor skills, he can color. He walks independently but is clumsy and falls frequently. He exhibits aggressive outbursts and self-injurious behavior. He is hyperactive and impulsive, for which he takes stimulant medications. He does not have a history of seizures or movement disorders.
Patient 5 was born at term, and the pregnancy was complicated by polyhydramnios and decreased fetal movements. Parents first noticed developmental differences at 1 year of age, mainly involving speech but eventually including low muscle tone and frequent falls. At 1 year of age he spoke no words. Audiometry showed mild hearing loss, likely due to frequent ear infections, which resolved with tympanostomy tubes. His family noticed frequent falls (upwards of 30 falls per day), trouble climbing stairs, and difficulty transitioning from different positions. With therapies (speech, occupational and physical), he saw slow improvement in these domains, however his mother estimates he continues functioning several years behind expected milestones. Currently 5 years old, he speaks in 5-word sentences, but has trouble understanding multistep commands and is slow to process verbal information. He can run, walk slowly upstairs, and scribble with a coarse grasp. He continues to fall several times per day and has trouble with fine coordination, most noticeable when attempting to feed himself with utensils. Parents say he is very cautions and has trouble with depth perception. His mother describes him as sweet and soft-spoken but does have aggressive outbursts when overwhelmed or over stimulated. Whole exome sequencing was performed and revealed a heterozygous de novo c.1 A>G, p. Met1? VAMP2 mutation. This change is thought to cause elimination of initiator methionine, which could result in no protein formation, or a truncated protein with a different initiator amino acid. For additional workup, brain MRI was obtained and normal. He received an autism evaluation at age 3, and did not meet criteria for autism, but displayed ADHD tendencies. There have been no concerns for seizures. The patient has an older brother with ADHD and dyslexia, and a healthy younger sister. There is a first cousin with autism.
3.2. VAMP2 variants decrease the rate and extent of SV exocytosis
To investigate potential dominant negative effects of disease-associated VAMP2 SNARE motif variants on SV protein trafficking in cellular models, we used live cell imaging of an optical reporter of SV recycling, synaptophysin-pHluorin (syp-pH). Syp-pH is a fusion of the pH-sensitive green fluorescent protein pHluorin to a luminal domain of synaptophysin, an integral membrane protein that associates with VAMP2 on SVs (Figure 1b) (Calakos & Scheller, 1994; Li et al., 2017). Transfected into hippocampal neurons in culture, syp-pH fluorescence is quenched at the acidic pH of SVs. Exocytosis induced by electrical stimulation increases syp-pH fluorescence as quenching is relieved upon exposure of the SV lumen to the higher external pH during fusion. After stimulation, fluorescence decreases due to internalization of the reporter and reacidification of SVs upon endocytosis (Figure 1a,b). Since VAMP2 mediates vesicle fusion, we first measured syp-pH exocytosis in neurons co-transfected with WT or variant VAMP2-mOr2 in the presence of the vacuolar H+-ATPase inhibitor bafilomycin. Bafilomycin blocks reacidification of SVs that have taken up the drug after exocytosis, eliminating fluorescence changes due to endocytosis. Compared to WT, co-transfection of Gly73Trp (G73W, Pt. 2) or Arg56Leu (R56L, Pt. 3) VAMP2 variants results in a slower rate of fluorescence increase, corresponding to slower exocytosis, in response to 900 APs, which releases the recycling pool (RP) of SVs (Figure 1c,d). The extent of exocytosis, reflecting the number of SVs released from the RP, is also decreased for Gly73Trp (G73W, Pt. 2) and Arg56Leu (R56L, Pt. 3) compared to WT. An effect of the Arg56X mutation (R56X, Pt. 1) was not detected, perhaps because the truncation lacks the transmembrane domain responsible for synaptophysin interaction and membrane localization (Edelmann, Hanson, Chapman, & Jahn, 1995).
Figure 1. K+ channel blocker rescue of VAMP2 mutation-induced exocytosis defects in vitro.
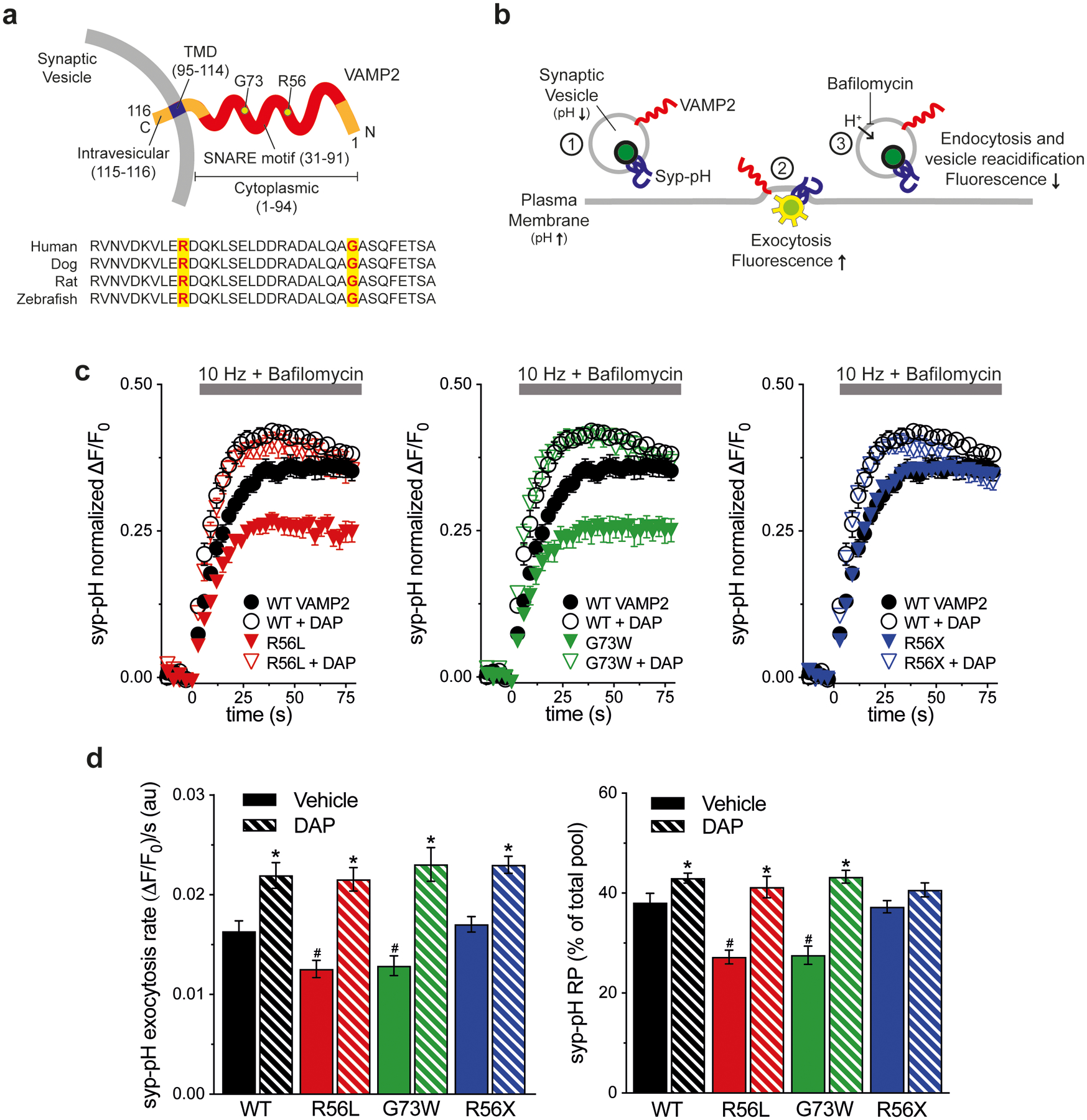
(a) Schematic of human VAMP2 depicts the C-terminal SV transmembrane domain (TMD), cytoplasmic SNARE motif, and variant amino acids. Alignment shows sequence conservation of SNARE across species. (b) Schematic of live cell imaging of SV recycling. Syp-pH fluorescence is quenched at low SV pH (1). Electrical stimulation to elicit exocytosis relieves fluorescence quenching upon exposure of the luminal syp-pH to higher external pH (2). After stimulation, syp-pH fluorescence decreases upon endocytosis and reacidification of SVs by the vacuolar H+-ATPase (3). The vacuolar H+-ATPase inhibitor bafilomycin in the external media blocks reacidification of SVs that have taken up the drug, eliminating fluorescence changes due to endocytosis (3). (c) Time course of exocytosis in response to 10 Hz stimulation in bafilomycin, in neurons co-transfected with the indicated VAMP2-mOr2 constructs, with vehicle or DAP. (d) Quantification of the rate and extent of exocytosis from the recycling SV pool (RP), as a percent of the total pool. Arg56Leu (p.R56L) and Gly73Trp (p.G73W) variants decrease exocytosis rate and extent compared to WT (#P<0.05 for each). Arg56X (p.R56X) truncation is similar to WT. DAP increases the extent of exocytosis with WT, p.R56L, and p.G73 variants, compared to control (*P<0.05). Exocytosis rate is increased with all VAMP2 constructs (*P<0.01 each). Data are means ± SEM of the change in fluorescence (ΔF) normalized to initial fluorescence (F0) over at least 23 boutons per coverslip from 9–12 coverslips from at least three independent cultures. Significance determined by t-tests.
3.3. K+ efflux blocker DAP increases SV exocytosis
To test whether prolonging the action potential by blocking K+ outflow affects exocytosis, we incubated neurons with the K+ channel inhibitor DAP. Compared to control, DAP treatment of neurons co-transfected with WT VAMP2 significantly increases the rate and extent of syp-pH exocytosis (Figure 1c,d). DAP also significantly increases the rate and extent of syp-pH exocytosis in neurons co-transfected with Arg56X (R56X, Pt. 1), Gly73Trp (G73W, Pt. 2) or Arg56Leu (R56L, Pt. 3) to a level similar to WT + DAP.
Changes in exocytosis could also result from differences in endocytosis or release site clearance. Since several residues in the SNARE motif of VAMP2 have been implicated in SV endocytosis (Grote & Kelly, 1996), we also tested the effects of the VAMP2 variants on endocytosis of syp-pH in the absence of bafilomycin. The post-stimulus fluorescence decay of syp-pH, representing endocytosis, is not significantly altered by any of the VAMP2 variants compared to WT (Figure 2). Addition of DAP does not significantly affect the endocytosis rate, but does increase peak fluorescence levels, consistent with the demonstrated effects of DAP on exocytosis. Data quantifications are reported in Supp. Table S1.
Figure 2. No effect of DAP on endocytosis.
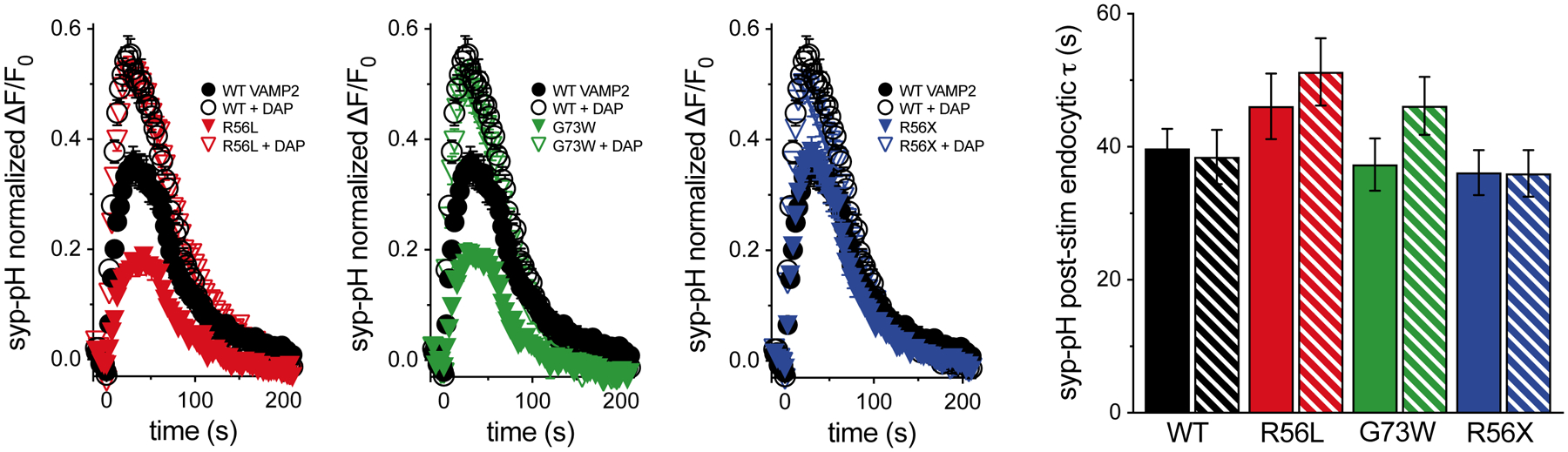
In the absence of bafilomycin, there are no significant differences in post-stimulus endocytosis rates between WT VAMP2 and any of the variants, or with DAP treatment. Data are means ± SEM of the change in fluorescence (ΔF) normalized to initial fluorescence (F0) over at least 22 boutons per coverslip from 7 to 9 coverslips and at least two independent cultures. Significance determined by t-tests.
3.4. Effects of VAMP2 variants and DAP on action potential evoked release
To investigate effects of VAMP2 variants on action potential triggered evoked neurotransmitter release, whole cell patch clamp electrophysiology was used to record inhibitory currents from primary hippocampal neurons expressing WT VAMP2 as well as VAMP2 variants. We recorded inhibitory postsynaptic currents to avoid the high frequency spontaneous AP firing and recurrent excitation that contaminates excitatory recordings (Maximov, Shin, Liu, & Sudhof, 2007). In response to a single AP, Gly73Trp (G73W, Pt. 2) and Arg56Leu (R56L, Pt. 3) VAMP2 variants respond less than WT, measured by synaptic charge transfer (Figure 3a). In this assay, Arg56X (R56X, Pt. 1) responds similarly to WT. No variants changed miniature current amplitudes, suggesting an effect on the presynaptic release machinery without changing the number of postsynaptic receptors (data not shown).
Figure 3. Effects of VAMP2 variants and DAP on evoked release.
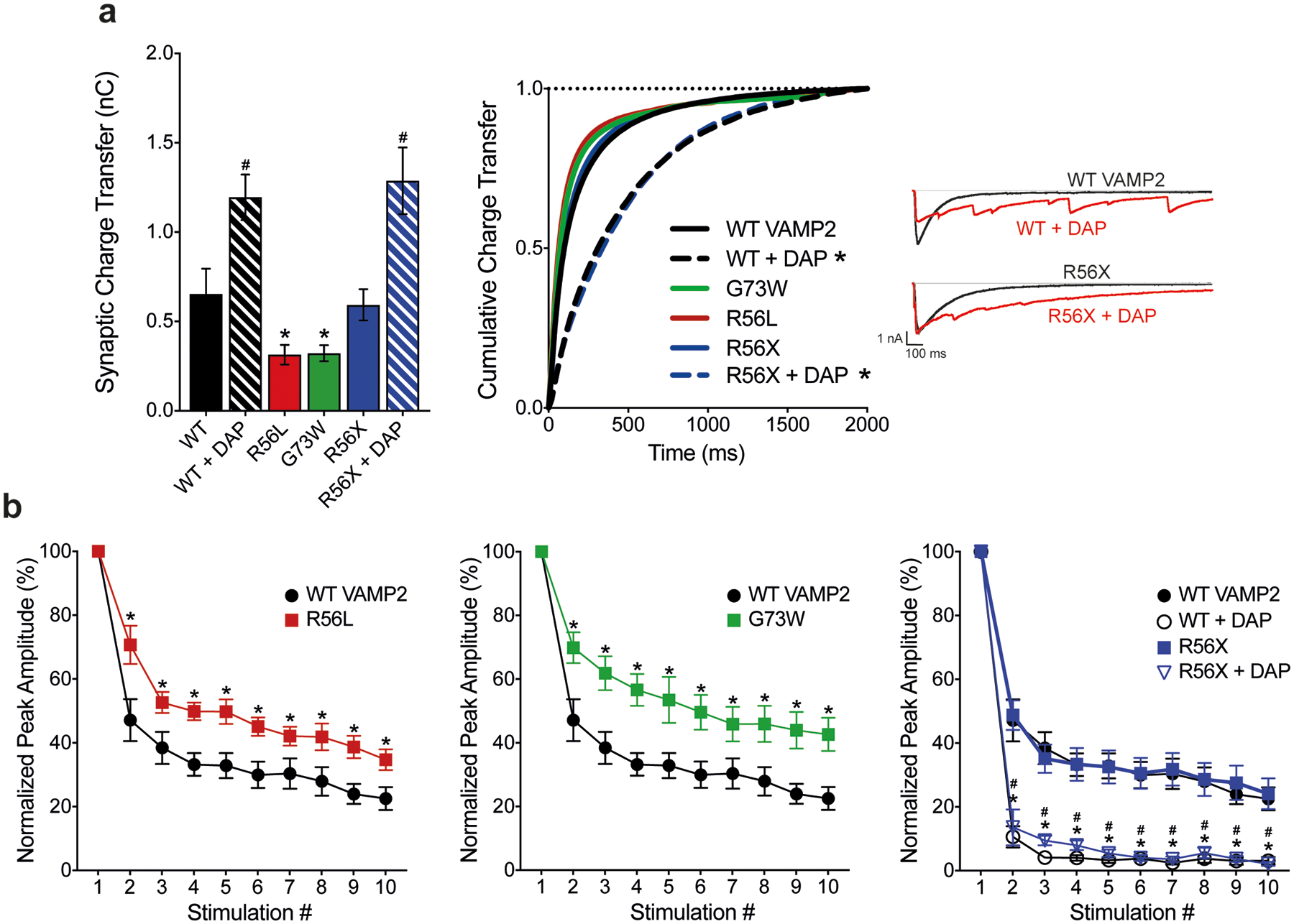
(a) p.R56L and p.G73W variants decrease the overall synaptic charge transfer, measured as the area under the curve of an evoked inhibitory postsynaptic current (eIPSC). DAP treatments increase synaptic charge transfer (WT 0.655 ± 0.141 nC, WT+DAP 1.195 ± 0.128 nC, p.R56L 0.315 ± 0.055 nC, p.G73W 0.323 ± 0.045 nC, p.R56X 0.593 ± 0.088 nC, p.R56X+DAP 1.287 ± 0.187 nC, *P<0.05 for WT vs p.R56L and WT vs p.G73W, #P<0.05 for WT vs WT+DAP and p.R56X vs p.R56X+DAP, two-tailed t-test). Variants do not change the synchronicity of the release (*P>0.05 for WT vs p.R56L, p.G73W and p.R56X, Kolmogorov-Smirnov test), but corresponding 3,4-DAP treatments result in more asynchronous release (*P<0.001 for both WT vs WT+DAP and p.R56X vs p.R56X+DAP, Kolmogorov-Smirnov test). (b) Normalized peak amplitudes of eIPSCs in response to 10 consecutive stimulations at 10 Hz. p.G73W and p.R56L variants cause less depression after initial stimulation compared to WT VAMP2 (*P<0.05 for WT VAMP2 vs p.G73W and WT VAMP2 vs p.R56L, multiple t-tests for each stimulation). Although p.R56X responds similarly as WT, corresponding DAP treatments cause more robust depression in response to stimulation (P<0.05 for *WT vs WT+DAP and #p.R56X vs p.R56X+DAP, multiple t-tests for each stimulation).
When multiple APs are applied at short time intervals causing SV depletion, successive postsynaptic responses decrease in amplitude. In addition, higher release probability is associated with more robust synaptic depression, since the initial stimulation causes release of more SVs, leaving only a few to be released in response to the following stimulations. As expected, WT VAMP2 depressed to 22 ± 3.57% of the first response in response to the 10th AP at 10 Hz (Figure 3b). Consistent with live imaging results, Gly73Trp (G73W, Pt. 2) and Arg56Leu (R56L, Pt. 3) depress less than WT, suggesting that the release probability in the presence of these variants is lower compared to WT. Arg56X (R56X, Pt. 1) does not affect synaptic depression. DAP causes release to become more asynchronous, thus increasing overall synaptic charge transfer (Figure 3a). This increase in charge transfer is also associated with higher release probability, as suggested by more robust synaptic depression (Figure 3b).
3.5. Clinical course
Extrapolating from in vitro studies, we hypothesized 4-AP could improve clinical outcomes in patients harboring VAMP2 pathogenic variants. After minimal efficacy with prior treatments using plasmapheresis, steroids and rituximab, Patient 1 was begun on an off-label treatment with 4-AP (Ampyra, Acorda Therapeutics, Ireland) beginning March 2018 beginning when she was 18 years of age, starting at low-dose (2.5 mg TID) and increasing over several months to maximal clinical efficacy and tolerability. Benefits were measured by qualitative assessments (subjective parental reports) and quantitatively (neuropsychological testing). Tolerability was assessed by parental report of worsening anxiety or insomnia. Within two months, she experienced dramatic improvement, most notably in social interaction. She seemed to “awaken”. She was calmer, attentive, socially engaging, with increased verbal and motor output. During a period of accidental half dosing, she returned to the pretreatment state; symptoms improved with correct dosing. In April 2019 she developed tolerance to 4-AP, so it was increased to 7.5 mg AM and 5 mg PM extended release (XR). She responded with additional improvements in sleep and mood.
After optimal dosing of 5 mg TID XR, she completed a neuropsychological evaluation in May 2019. Compared to prior evaluations in 2017, speed of information processing and verbal memory recall improved by 133%. Neuropsychological testing pre- and post-treatment were compared by converting scaled scores to z-scores. Other cognitive functions appeared stable (Table 2). Parents reported improvement in emotional and behavioral regulation; she was less labile and more interactive. A worsening of pre-existing anxiety accompanied cognitive improvements, perhaps from increased insight into her limitations. This anxiety was responsive to subsequent introduction of low dose (10 mg TID) propranolol.
TABLE 2.
Comparison of neuropsychologic testing scores for patient 1
| 2017 | 2019 | 2017 | 2019 | Change in Z-score | |
|---|---|---|---|---|---|
| WRAML-2 | No. of correct/recalled | Scaled score | |||
| Sentence memory | 14 | 16 | 1 | 3 | 0.67 |
| Story memory (Immediate) | 5 | 12 | 1 | 4 | 1.00 |
| Story memory delay recall | 0 | 11 | 1 | 4 | 1.00 |
| Verbal learning (immediate) | 15 | 20 | 2 | 3 | 0.33 |
| Verbal learning delay recall | 7 | 10 | 7 | 9 | 0.67 |
| Design memory (immediate) | 13 | 14 | 1 | 2 | 0.33 |
| Design memory delayed recognition | 25 | 21 | 5 | 1 | −1.33 |
| Picture memory (immediate) | 18 | 23 | 4 | 6 | 0.67 |
| Picture memory delayed recognition | 30 | 29 | 6 | 5 | −0.33 |
| D-KEFS trail making test | Time to completion | Scaled score | |||
| Visual scanning | 31 | 34 | 6 | 4 | −0.67 |
| Number sequencing | 106 | 53 | 1 | 4 | 1.00 |
| Letter sequencing | 53 | 40 | 3 | 7 | 1.33 |
| Number-letter switching | 203 | 136 | 1 | 2 | 0.33 |
| Motor speed | 61 | 43 | 3 | 7 | 1.33 |
| D-KEFS verbal fluency | No. of correct within time limit | Scaled score | |||
| Letter fluency | 6 | 7 | 1 | 1 | 0.00 |
| Category fluency | 8 | 12 | 1 | 1 | 0.00 |
| Category switching total | 7 | 6 | 3 | 2 | −0.33 |
| Category Switching Accuracy | 5 | 4 | 4 | 3 | −0.33 |
| Golden stroop | No. of correct within time limit | T score | |||
| Word score | 52 | 60 | 18 | 25 | 0.70 |
| Color score | 35 | 41 | 16 | 22 | 0.60 |
| Color-word score | 23 | 20 | 33 | 31 | −0.20 |
| Interference score | 3 | −4 | 53 | 46 | −0.70 |
Note: Comparison between neuropsychological evaluations from May 2019 (UCSF) while on optimal dosing of 4‐AP to prior evaluations in 2017 (Sutter) before starting treatment. Results show speed of information processing and verbal memory recall improved by 133%. Other cognitive functions appeared stable. UCSF testers were blinded to prior results from 2017 but not to treatment with 4‐AP.
Abbreviations: 4‐AP, 4‐aminopyridine; UCSF, University of California, San Francisco.
4. DISCUSSION
In this study, we identified five individuals with novel de novo heterozygous pathogenic variants in VAMP2, a key synaptic vesicle protein. They presented with global developmental delay, autistic features, behavioral disturbances, and a higher propensity to develop epilepsy. Furthermore, we showed that the missense variants of VAMP2, Gly73Trp (Pt. 2) and Arg56Leu (Pt. 3), associated with a more severe phenotype with epilepsy in vivo, exert a dominant-negative effect on action potential-triggered SV fusion and neurotransmitter release in vitro. Consistent with these results, the recent report by Salpietro, et. al. suggested that an independently identified disease-associated VAMP2 variant in close proximity to the Gly73Trp (Pt. 2) variant described here, Ser75Pro, impairs vesicle fusion of reconstituted membranes in vitro in a dominant negative manner (Salpietro et al., 2019). In contrast, the nonsense variant (Arg56X, Pt. 1) does not cause dominant-negative effects, suggesting that haploinsufficiency underlies the disease mechanism for the R56X variant patient, who presented with a milder phenotype without epilepsy. Since both missense and nonsense variants are associated with impaired action potential-triggered neurotransmitter release, we hypothesized that prolonging local Ca2+ availability by inhibiting K+ channels with 4-AP could increase the probability of release, thus restoring synaptic transmission (Storm, 1987; Wheeler, Randall, & Tsien, 1996). As expected, we showed that treatment with the 4-AP analog DAP corrects the in vitro deficits associated with the VAMP2 variants. In particular, in prolonging the period in which local Ca2+ concentration is high enough to trigger neurotransmitter release, DAP switched the mode of release from fast-synchronous to asynchronous. This, in turn, increases overall synaptic charge transfer, thus rescuing the deficits in neurotransmitter release. Given that DAP functions through prolonging action potential duration and increasing release probability, the effect of DAP may vary in presynaptic terminals releasing different neurotransmitters. For example, GABAergic terminals, which comprise the main inhibitory system of CNS, typically have a higher probability of release than glutamatergic terminals, the main excitatory system. Disturbances of excitatory-inhibitory balance in neuronal circuits may underlie certain neuropsychiatric disorders and enhancement of inhibitory neurotransmission has been shown to ameliorate some behavioral deficits in mouse models of autism (Sohal & Rubenstein, 2019).
Extrapolating from in vitro studies, we started 4-AP (Ampyra) treatment for Patient 1, the Arg56X affected individual, in 2018. She responded dramatically, confirming that in vitro studies successfully predicted clinical response. With treatment for the last two years, Patient 1 shows remarkable improvement in cognitive processing speed and verbal memory recall. Treatment drastically improved the quality of life for our patient and her family given her enhanced ability to interact and decreased emotional lability. In this study, we demonstrate that augmentation of neurotransmitter release by aminopyridines can be a viable treatment option for VAMP2 associated disorders with impaired neurotransmitter release. Most importantly, we showed the first evidence of clinical improvement upon 4-AP treatment in a patient harboring a nonsense variant of VAMP2. 4-AP could treat other patients with VAMP2 or other SNARE protein mutations. Our results are in agreement with recent in vitro studies showing that DAP also can overcome release deficits associated with disease causing synaptotagmin-1 variants (Bradberry et al., 2020). Taken together, these observations confirm and expand our hypothesis that augmentation of release by DAP or 4-AP would be a viable treatment option for other SNAREopathies as well (Baker et al., 2018; Harper, Mancini, van Slegtenhorst, & Cousin, 2017; Salpietro et al., 2017; Verhage & Sorensen, 2020). This approach could be tested in vitro prior to clinical implementation. 4-AP should be used cautiously in patients with epilepsy given risk of lowering seizure threshold, which can limit its broad-based utility. Two of the patients with dominant, missense SNARE motif mutations had epilepsy (Pt. 2, Gly73Trp and Pt. 3, Arg56Leu) whereas two patients with heterozygous loss of function mutations (Pt. 1, Arg56X and Pt. 4, Tyr113GlnfsX12) did not have seizures, but did share the same range of behavioral deficits. Consistent with a prior study of five patients by Salpietro et al., the patients in our cohort also display global developmental delay, autistic features and behavioral disturbances. Additional patients will better and more fully characterize the VAMP2 phenotype.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (MH083691 to SMV; MH066198 to ETK; NS120667 to EHS) and the University of California, San Francisco.
Footnotes
CONFLICTS OF INTERESTS
The authors declare no competing interests.
WEB RESOURCES
GeneDx
ClinVar
http://www.ncbi.nlm.nih.gov/clinvar/
Poly-Phen2
http://genetics.bwh.harvard.edu/pph2/
GnomAD
Accession numbers from ClinVar for VAMP2 variants:
Pt. 1: SUB7621453; c.166C>T, p.Arg56X
http://www.ncbi.nlm.nih.gov/clinvar/variation/929460/
Pt. 2: SUB7630353; c.217G>T, p.Gly73Trp
http://www.ncbi.nlm.nih.gov/clinvar/variation/929461/
Pt. 3: SUB7630360; c.167G>T; p.Arg56Leu
http://www.ncbi.nlm.nih.gov/clinvar/variation/929463/
Pt. 4: SUB7630356; c.337_341delTACTT, p.Tyr113GlnfsX12
http://www.ncbi.nlm.nih.gov/clinvar/variation/929464/
Pt. 5: SUB7630357; c.1A>G, p. Met1?
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES:
- Baker K, Gordon SL, Melland H, Bumbak F, Scott DJ, Jiang TJ, … Raymond FL (2018). SYT1-associated neurodevelopmental disorder: a case series. Brain, 141(9), 2576–2591. doi: 10.1093/brain/awy209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradberry MM, Courtney NA, Dominguez MJ, Lofquist SM, Knox AT, Sutton RB, & Chapman ER (2020). Molecular Basis for Synaptotagmin-1-Associated Neurodevelopmental Disorder. Neuron, 107(1), 52–64 e57. doi: 10.1016/j.neuron.2020.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calakos N, & Scheller RH (1994). Vesicle-associated membrane protein and synaptophysin are associated on the synaptic vesicle. Journal of Biological Chemistry, 269(40), 24534–24537. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7929121 [PubMed] [Google Scholar]
- Claassen J, Teufel J, Kalla R, Spiegel R, & Strupp M (2013). Effects of dalfampridine on attacks in patients with episodic ataxia type 2: an observational study. J Neurol, 260(2), 668–669. doi: 10.1007/s00415-012-6764-3 [DOI] [PubMed] [Google Scholar]
- Deak F, Schoch S, Liu X, Sudhof TC, & Kavalali ET (2004). Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol, 6(11), 1102–1108. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15475946 [DOI] [PubMed] [Google Scholar]
- Edelmann L, Hanson PI, Chapman ER, & Jahn R (1995). Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. Embo Journal, 14(2), 224–231. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7835333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Eliason WK, Brunger AT, & Jahn R (1998). Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry, 37(29), 10354–10362. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9671503 [DOI] [PubMed] [Google Scholar]
- Foss SM, Li H, Santos MS, Edwards RH, & Voglmaier SM (2013). Multiple Dileucine-like Motifs Direct VGLUT1 Trafficking. The Journal of neuroscience : the official journal of the Society for Neuroscience, 33(26), 10647–10660. doi: 10.1523/JNEUROSCI.5662-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, & Kelly RB (1996). Endocytosis of VAMP is facilitated by a synaptic vesicle targeting signal. J. Cell Biol, 132, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CB, Mancini GMS, van Slegtenhorst M, & Cousin MA (2017). Altered synaptobrevin-II trafficking in neurons expressing a synaptophysin mutation associated with a severe neurodevelopmental disorder. Neurobiology of disease, 108, 298–306. doi: 10.1016/j.nbd.2017.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, & Scheller RH (2006). SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol, 7(9), 631–643. doi: 10.1038/nrm2002 [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, & Tsien RW (1999). Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proceedings of the National Academy of Sciences of the United States of America, 96(22), 12893–12900. doi: 10.1073/pnas.96.22.12893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SJ, Markovic S, Puchkov D, Mahrenholz CC, Beceren-Braun F, Maritzen T, … Haucke V (2011). SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proceedings of the National Academy of Sciences of the United States of America, 108(33), 13540–13545. doi: 10.1073/pnas.1107067108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Santos MS, Park CK, Dobry Y, & Voglmaier SM (2017). VGLUT2 Trafficking Is Differentially Regulated by Adaptor Proteins AP-1 and AP-3. Frontiers in cellular neuroscience, 11, 324. doi: 10.3389/fncel.2017.00324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, & Sudhof TC (2007). Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. Journal of Cell Biology, 176(1), 113–124. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17190793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, & Sorensen JB (2012). SNARE requirements en route to exocytosis: from many to few. J Mol Neurosci, 48(2), 387–394. doi: 10.1007/s12031-012-9744-2 [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Lin PY, Adachi M, & Kavalali ET (2018). Behavioral Analysis of SNAP-25 and Synaptobrevin-2 Haploinsufficiency in Mice. Neuroscience. doi: 10.1016/j.neuroscience.2018.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palace J, Wiles CM, & Newsom-Davis J (1991). 3,4-Diaminopyridine in the treatment of congenital (hereditary) myasthenia. J Neurol Neurosurg Psychiatry, 54(12), 1069–1072. doi: 10.1136/jnnp.54.12.1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpietro V, Lin W, Delle Vedove A, Storbeck M, Liu Y, Efthymiou S, … Houlden H (2017). Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann Neurol, 81(4), 597–603. doi: 10.1002/ana.24905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpietro V, Malintan NT, Llano-Rivas I, Spaeth CG, Efthymiou S, Striano P, … Houlden H (2019). Mutations in the Neuronal Vesicular SNARE VAMP2 Affect Synaptic Membrane Fusion and Impair Human Neurodevelopment. Am J Hum Genet, 104(4), 721–730. doi: 10.1016/j.ajhg.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, & Tsien RY (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods, 5(6), 545–551. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18454154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, … Stankiewicz P (2009). A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet, 41(12), 1269–1271. doi: 10.1038/ng.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, & Rubenstein JLR (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry, 24(9), 1248–1257. doi: 10.1038/s41380-019-0426-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF (1987). Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol, 385, 733–759. doi: 10.1113/jphysiol.1987.sp016517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strupp M, Teufel J, Zwergal A, Schniepp R, Khodakhah K, & Feil K (2017). Aminopyridines for the treatment of neurologic disorders. Neurol Clin Pract, 7(1), 65–76. doi: 10.1212/CPJ.0000000000000321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron, 80(3), 675–690. doi: 10.1016/j.neuron.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, … Jahn R (2006). Molecular anatomy of a trafficking organelle. Cell, 127(4), 831–846. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17110340 [DOI] [PubMed] [Google Scholar]
- Verhage M, & Sorensen JB (2020). SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron, 107(1), 22–37. doi: 10.1016/j.neuron.2020.05.036 [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, & Tsien RW (1996). Changes in action potential duration alter reliance of excitatory synaptic transmission on multiple types of Ca2+ channels in rat hippocampus. Journal of Neuroscience, 16(7), 2226–2237. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/8601803 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.