


Abstract
Homozygous duplications contribute to genetic disease by altering gene dosage or disrupting gene regulation and can be more deleterious to organismal biology than heterozygous duplications. Intragenic exonic duplications can result in loss-of-function (LoF) or gain-of-function (GoF) alleles that when homozygosed, i.e. brought to homozygous state at a locus by identity by descent or state, could potentially result in autosomal recessive (AR) rare disease traits. However, the detection and functional interpretation of homozygous duplications from exome sequencing data remains a challenge. We developed a framework algorithm, HMZDupFinder, that is designed to detect exonic homozygous duplications from exome sequencing (ES) data. The HMZDupFinder algorithm can efficiently process large datasets and accurately identifies small intragenic duplications, including those associated with rare disease traits. HMZDupFinder called 965 homozygous duplications with three or less exons from 8,707 ES with a recall rate of 70.9% and a precision of 16.1%. We experimentally confirmed 8/10 rare homozygous duplications. Pathogenicity assessment of these copy number variant alleles allowed clinical genomics contextualization for three homozygous duplications alleles, including two affecting known OMIM disease genes EDAR (MIM# 224900), TNNT1(MIM# 605355), and one variant in a novel candidate disease gene: PAAF1.
Graphical Abstract
Graphical Abstract.
Introduction
Deletions and duplications of chromosomal segments (copy number variants, CNVs) are a major source of variability between personal genomes. Such CNVs can be a significant contributor to pathogenic alleles and rare variants that drive Mendelian disease traits and genomic disorders and can underlie both human gene and genome evolution (1). Intragenic or exonic duplications, particularly of small sizes (<10 kb), can result in null alleles, and their identification facilitates novel Mendelian gene discoveries for rare disease traits (2–4).
Currently available tools for the detection of CNVs (5–10) from exome sequencing (ES) data can mostly identify large CNVs encompassing three or more exons. Previous work focusing on homozygous and hemizygous CNV deletion alleles, i.e. deletions affecting both autosomes or the X chromosome in 46, XY males, enables further optimization of signal-to-noise ratios (11). This optimization captures the dynamic range of the copy number variation in a diploid genome and allows a significant reduction of the false positive exonic deletion calls by incorporating haplotype information. The haplotype information is captured by utilizing absence of heterozygosity (AOH) calculations as a surrogate measure for Runs of Homozygosity (ROH) and implementing joint calling from a large ES data set (11). The utility of ES for detecting homozygous duplication of intragenic CNV gain alleles remains unknown.
Herein, we extended the bioinformatic CNV calling algorithm HMZDelFinder and benchmarked it against experimental data to develop HMZDupFinder, which enables homozygous exon-level intragenic duplication calls. HMZDupFinder uses Pearson correlation coefficient to automatically group samples with the closest sequencing profile as the reference to increase the power of detecting true positive duplication calls with a reduced false discovery rate.
Materials and methods
Samples
Research samples were recruited as part of the Baylor Hopkins Center for Mendelian Genomics (BHCMG) and later the Baylor College of Medicine Genomics Research to Elucidate the Genetics of Rare disease (BCM-GREGoR) research center. Written informed consent was obtained under the Baylor Hopkins Center for Mendelian Genomics (BHCMG) protocol with approval by the institutional review board (IRB) at Baylor College of Medicine (BCM IRB H-29697). Blood derived genomic DNA samples were obtained through research and clinical collaborations and were subjected to research ES. Clinical ES data with same capture design were included for reanalysis as previously described (12).
Research exome sequencing harmonization and reprocessing
DNA capture and sequencing were performed as described at Baylor College of Medicine Human Genome Sequencing Center (HGSC) for the Mendelian Genomics initiative (13). Both HGSC core capture and VCRome capture designs were based on the reference genome GRCh37 (14). The FASTQ files were remapped to GRCh38; single nucleotide variants (SNVs) and small insertions and deletions variants (INDELs) were called with the xAtlas-0.2.1 pipeline (15,16).
Read count normalization
We calculated the raw read counts per individual Binary Alignment Map (BAM) file using the ‘Rsamtools::featureCounts’ function. The bed file of the region was based on the ‘liftover region’ of hg38 of the initial VCRome design as previously described (14). Afterward, we normalized these counts using the transcript per million (TPM) procedure, which allowed us to directly compare the relative read count between the proband and nearest references at the same locus.
Z-TPM, log2 ratio, and gene transcript visualization
For each sample, a bed file was generated which includes the genomic coordinates of probes, raw read count, TPM, Z-TPM, and log2 ratio of the nearest references. The TPM of exon was calculated with the below formula:
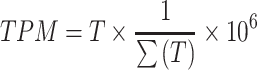 |
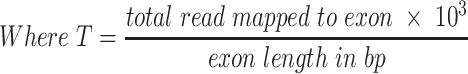 |
The Z-TPM was calculated as:
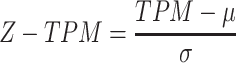 |
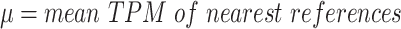 |
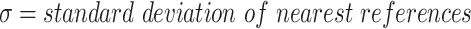 |
The log2 ratio was calculated as:
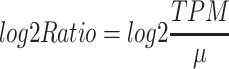 |
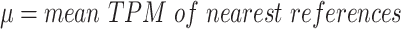 |
The log2 ratio was then segmented using ‘SLMSeg::HSLM’ model implemented in R (17) with the following parameters ‘omega = 0.7, FW = 0, eta = 1e-5, stepeta = 1,000’. For multi-exonic duplications, the mean Z-TPM was then calculated and used for downstream filtering.
The bed files were compressed and using tabix tool and indexed. Indexed files were then utilized as input for the Z-TPM and log2 ratio visualization. The probes were mapped to the Matched Annotation from NCBI and EMBL-EBI (MANE) transcript set (v1.0) to obtain consistent and well-represented transcript annotation.
Number of random targets required to select reference samples
Using closely correlated samples as the reference can minimize the experimental variance (i.e. noise) and aid in better estimation of a baseline read depth when analyzing a diploid genome. To reach optimal performance, the reference samples need to be run on a similar sequencing platform and with a similar capture design. A Pairwise Pearson similarity matrix calculated based on 1% (n = 2,000) of the total target using the ‘base::cor’ function from the R environment was used to select reference samples.
Selecting optimal number of reference samples
We empirically tested the difference between the read count distribution of the heterozygous triplication state (Copy Number = 4) and the control ‘baseline’ diploid state, i.e. gene copy number CN = 2, when including an increased number of samples as reference. In our analyses, we observed that as we increased the number of reference samples, the accuracy of Z-TPM cutoff separating CN = 2 and CN = 4 improved (Supplementary Figure S1A, Supplementary Table S1). However, between using 60–65 highly correlated reference samples, the improvement plateaued, indicating diminishing returns in accuracy with the addition of more reference samples. In parallel, when assessing the distribution in the BHCMG-GREGoR cohort, a reference count of 55 ensured that most samples retained enough references for accurate normalization (Supplementary Figure S1C). With a reference number of 55, Z-TPM cutoffs of 3.5, 4.0 and 4.5 were able to recall 88.52%, 78.09% and 61.39% of heterozygous triplication (Supplementary Figure S1B). Synthesizing these observations with metrics of sensitivity and overall performance, 55 emerged as the ideal reference sample count that best balanced accuracy with computational demands.
CNV call with XHMM
XHMM was performed on all ES data using default parameters (5). The same bed file of target design region was used to call CNV. The small duplication call from output file ‘DATA.xcnv’ were overlapped with AOH region. The duplication with at least 1bp overlapping were kept for further evaluation.
Precision and recall calculation
The detail of identifying potential homozygous duplication region is described in results section. We extracted the Z-TPM and log2 ratio values from the potential homozygous duplication region of both parents. The precision and recall were then calculated based on the following criteria:
The precision is calculated using the following formula:
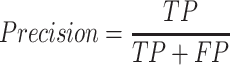 |
In this context, ‘TP’ denotes the number of homozygous duplication calls where both parents display a Z-TPM value exceeding 2.0 and a ratio ranging from 1.3 to 1.7 (log2 ratio between 0.38 and 0.77 potentially corresponding to heterozygous duplication). Conversely, ‘FP’ represents the instances of homozygous duplication calls where one or both parents do not meet the same Z-TPM and ratio criteria.
The recall is calculated using the formula:
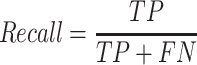 |
In this context, ‘TP’ refers to exons located in known triplicated regions that meet the expected Z-TPM and ratio criteria. ‘FN’, on the other hand, signifies the exons in the same regions that do not fulfill the expected Z-TPM or ratio requirements.
Segregation analysis of homozygous duplications with ddPCR
The genomic DNA of proband and parents were digested with RsaI or AluI (New England Biolab Inc.) restriction endonucleases before performing droplet-digital polymerase chain reaction (ddPCR) to break the potential linkage within tandem duplications. ddPCR was performed using the QX200™ AutoDG™ Droplet Digital™ PCR System from Bio-Rad, following the manufacturer's protocols. In brief, a 20 μl mixture was constructed for each PCR reaction, containing 10 μl of 2x Q200 ddPCR EvaGreen Supermix or 2x ddPCR supermix for probes (no dUTP), 0.25 μl of each primer (10 μM), 250 nM of probe (for ddPCR hydrolysis probe reactions), and 50 ng of restriction enzyme digested genomic DNA. The reaction mixture was subjected to automatic droplet generation, PCR reaction, and droplet reading. Cycling conditions for PCR were: 5 min at 95°C, 40 cycles of 30 s at 95°C/1 min at 61.2°C/1 min at 72°C, 5 min at 4°C, 5 min at 90°C, and finally infinite hold at 4°C. The ramp rate was set at 2°C/s for all steps. These data were analyzed using QuantaSoft™ Software from Bio-Rad, and concentrations of positive droplets (number of positive droplets per μl of reaction) were obtained for each PCR reaction. Primers to a control gene, RPPH1, RPP30 or TERT and affected genes were included (Supplementary Table S2).
Orthogonal validation
During the algorithm's development, two distinct sets of calls were chosen for wet bench validation based on different selection criteria. The first set, comprised of 25 calls, was selected solely based on the proband's Z-TPM values, which ranged from 3.54 to 7.83. While the AOH size (ranging from 0 to 21 Mb) was annotated, it wasn’t a decisive factor in the selection. The second set, consisting of 12 duplications, was chosen with consideration of both proband and parent Z-TPM values. The proband's Z-TPM varied between 3.69 and 6.59, while the parent's Z-TPM ranged from 1.79 to 4.88. Here again, the AOH size (0–28 Mb) was annotated but not treated as a strict selection criterion. From the first set, ddPCR relative positive droplet ratios in five cases (5/25) were in line with potential homozygous duplications. In the second set, aCGH log2 ratios for three cases (3/12) aligned with potential homozygous duplications. After refining the parameters (Z-TPM > 4.0, log2 ratio between 0.85–1.15, and AOH size exceeding 100 kb), 10 out of 37 regions were re-classified as potential homozygous duplications. Notably, all eight wet bench-validated homozygous duplications (8/10) were accurately reclassified using the adjusted parameters. Among these eight verified duplications, six (75%) represented CNV alleles that comprised three or fewer exons.
Absence of heterozygosity (AOH) genomic intervals
The AOH genomic intervals were identified based on the previously developed in-house tool BafCalculator (Eldomery et al. 2017) (https://github.com/BCM-Lupskilab/BafCalculator). Genomic segments with a mean signal > 0.47 and size > 100 kb were classified as AOH regions and were used to identify potential homozygous duplications (Supplementary Figure S2).
Custom-designed array comparative genomic hybridization (aCGH)
Validation and characterization of the genomic architecture of each predicted potential pathogenic CNV was experimentally investigated in probands by high-resolution array-based comparative genomic hybridization (aCGH). A custom 8 × 60K Agilent high-resolution oligonucleotide microarray (AMADID 086718) spanning 17.344 Mb targeting 59 genes mapping within predicted CNVs and their flanking regions with an average probe spacing of 245 bp was designed using (https://earray.chem.agilent.com/suredesign/). Microarray protocols, including DNA digestion, probe labeling, gender-matched hybridization, and post-washing, were performed as described previously with minor modifications (18). Agilent SureScan and Feature Extraction software were utilized to achieve the image-to-digital transition, with further data analysis and visualization on the Agilent Genomic Workbench. Genomic coordinates were described in reference to GRCh37/hg19 assembly.
Genomic feature analysis on Genome Aggregation Database (gnomAD) CNV alleles
We utilized the genomic coordinates and CNV calling method from gnomAD SVs v2.1, which is based on the GRCh37/hg19 reference (19). To convert these coordinates to the GRCh38 reference, we referred to the liftover data available at NCBI’s dbVar study nstd166. After merging the datasets, we filtered the gnomAD alleles based on several criteria: a ‘Remap Score’ of 1, ‘Variant Call type’ being either duplication or deletion, and having both read depth (RD) and split read (SR) evidence supporting the breakpoint. For further refinement, the breakpoints of the gnomAD intragenic alleles were aligned with the MANE v1.0 transcript structure to ascertain the related introns (Supplementary Table S3).To compute the density of repetitive elements, we sourced the genomic elements from the RepeatMasker file available on the UCSC genome browser, specifically for the GRCh38 reference build.
PacBio long read genome sequencing solves the breakpoint of insertional duplication
Whole genome sequencing was performed on genomic DNA extracted from four family members at HGSC, using the Pacific Biosciences platform. DNA samples were quantified with the DropSense96 system, and the DNA fragment sizes were accessed with Agilent Femto Pulse system. Although DNA quality was suboptimal for long-read sequencing, we proceeded with the construction of a sequencing library using 6.5 micrograms of DNA and the SMRTbell Express Template Preparation Kit 3.0. The sequencing was executed on the PacBio Sequel II system. Each individual genome was sequenced in a single SMRTcell due to the initial DNA quality concerns. This still resulted in varying coverage depths: 2.3× for the proband, 9.4× for the unaffected sibling, 21.5× for the mother, and 2.1× for the father. The data were processed with SMRTLink software version 12 and aligned using the pbmm2 aligner(version 1.10.0). Despite the suboptimal coverage, inspection of the low-coverage long-read data in the Integrative Genomics Viewer (IGV) facilitated the redesign of breakpoint sequencing primers, which confirmed the junctions.
Results
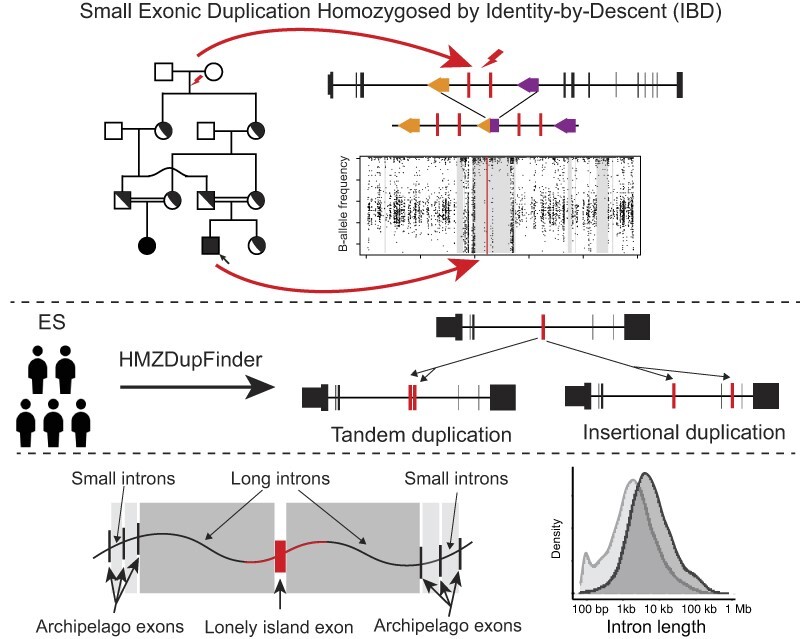
Overview of the homozygous duplication detection analysis pipeline
With a focus on detecting small exonic homozygous duplications of gene segments, we fine-tuned parameters to optimize computational calling of CNVs. The HMZDupFinder algorithm uses three steps to detect rare homozygous duplications (Figure 1): (i) selection of reference samples, (ii) normalization of TPM values and log2 ratio for each exon, and (iii) joint calling of potential duplications within AOH regions. In the first step, we calculate the pairwise Pearson correlation coefficient between all input samples based on the TPM value of 2000 targets that passed quality control (0.2 < GC content < 0.8, mappability > 0.8). The samples with the most correlated TPM profiles are selected as reference samples. The Z-score (Z-TPM) and log2 ratio of each target is calculated based on the distribution of the TPM values on the same targets from the selected references. Next, potential duplication targets are selected by setting a cutoff of Z-TPM value determined using a control set (Supplementary Figure S1). The computational output of this step contains the list of potential exonic CNVs (exCNV) for each sample. Next, exCNV calls in the given sample are intersected with the AOH region detected in the same sample. The calls that do not overlap with AOH regions are removed from further analysis. To compare the performance of HMZDupFinder and XHMM on small duplications, with less than three targets, we also overlapped small duplication calls, from XHMM with the AOH region in the same sample. This overlap was considered as the candidate region for homozygous duplication. The precision for both methods was evaluated as described in Methods.
Figure 1.

The computational framework for HMZDupFinder. (A–D) The HMZDupFinder workflow demonstrates the harmonization of exome capture design, nearest reference selection, Z-TPM normalization, and homozygous duplication enrichment process. (E) Estimated numbers of candidate CNVs after each ES data computational step. (F, G) An example of combined visualization of Z-TPM and log2 ratio validating a homozygous intragenic duplication. (F) The line plot displays Z-TPM values for exons that match the annotations from the MANE transcript of the investigated gene, which is shown at the bottom. A red, jagged horizontal line signifies the value from the subject, with grey lines representing the closest reference samples. The segmented log2 ratio of the TPM value in the subject is illustrated as a blue dashed line in panel (G).
AOH size of BHCMG-GREGoR cohort
We calculated the cumulative size of autosomal AOH regions for each sample using the defined parameters described in Methods. Overall, we identified 1,755,749 autosomal AOH blocks in 8,707 individuals (probands and parents) with a combined size of 1,046 Gb, resulting in an average AOH size of 120 Mb per individual. Based on the distribution of total autosomal size, we classified a subset of individuals (n = 1,479) as being from potential consanguineous families, as they displayed a high number (>150 Mb) of total AOH segments across the genome.
Homozygous duplication calls from the BHCMG-GREGoR trios
We evaluated the performance of HMZDupFinder on 1,208 BCM-GREGoR trios (Figure 2A). HMZDupFinder achieves an optimal precision and recall using a Z-TPM cutoff of 4.0 and log2 ratio between 0.85 and 1.15 as indicated by the highest F1-score (Supplementary Figure S3A, Supplementary Table S4) and AUC-PR (Supplementary Figure S3B). With these fine-tuned parameters, HMZDupFinder identified:
Figure 2.

Schematic graph illustrating parameter tuning of HMZDupFinder (A) and intragenic homozygous duplication call from BHCMG-GREGoR cohort (B).
93 homozygous duplications involving three or fewer exons. Of these, 15 duplications (a precision of 16.1% (15/93) displayed the expected heterozygous duplication Z-TPM (exceeding 2.0) and log2 ratio [ranging from log2(1.3) and to log2(1.7)] in both parental samples.
30 homozygous duplications spanning more than three exons, among these, 17 met the expected heterozygous duplication criteria in both parental samples, resulting in a precision of 56.7% (17/30).
In contrast, XHMM detected:
290 homozygous duplications involving three or fewer exons, but only six, met the expected heterozygous duplication Z-TPM and log2 ratio criteria in both parental samples, a precision of 2.1% (6/290).
32 homozygous duplications spanning more than three exons, among these, 11 met the expected heterozygous duplication criteria in both parental samples, resulting in a precision of 34.4% (11/32).
When comparing the results of both tools (XHMM and HMZDupFinder), they collectively detected 25 homozygous duplications. Of these, 14 (three homozygous duplication spanning three or less exons) met the expected heterozygous duplication Z-TPM and log2 ratio criteria in both parental samples, a precision of 56.0% (14/25). These findings suggest that HMZDupFinder precision outperforms XHMM in detecting homozygous duplication alleles.
Homozygous duplication calls from all BHCMG-GREGoR samples
Utilizing optimized parameters (Z-TPM cutoff of 4.0 and log2 ratio between 0.85 and 1.15) which was fine-tuned from BHCMG-GREGoR trios, we applied HMZDupFinder to all BHCMG-GREGoR samples which have sufficient reference samples (n = 7,808) (Figure 2B). In total, we identified 1,230 homozygous duplications across 764 subjects. The median homozygous duplication call per subject was one. Based on the distribution, we observed nine samples with extremely high number of homozygous calls which may result from sequencing of potential degraded DNA. We excluded calls from these nine samples from further analysis. Ultimately, 965 homozygous duplications were computationally filtered in 755 subjects. Of the 965 high quality homozygous duplication calls, we identified 842 unique alleles, including 723 singletons that were only identified in one individual. Of these unique alleles, 83 of them have at least 50% reciprocal overlapping with the gnomAD SVs v2.1 duplication alleles. Of all predicted homozygous duplication alleles, 85.0% (716/842) of these homozygous duplication alleles affected three or fewer exons; 44.4% of these (374/842) were predicted to be intragenic. Wet bench confirmation was performed on ten cases and eight of them confirmed true homozygous duplications (Method section), with a benchmark precision of 80%. Six of them were CNV alleles encompassing three or less exons.
Contextualizing potential pathogenicity of homozygous duplication variant alleles
After considering the clinical phenotype of the cases, three out of eight confirmed homozygous duplications were considered to potentially contribute to the patient phenotype (Table 1). The remaining duplication alleles were not interpreted as potentially pathogenic since the duplication involves either 5′ or 3′ exons, which under a model of tandem duplication retain two normal copies of the gene, and lack of literature support of phenotype and genotype association. Two of pathogenic duplication alleles involved known disease genes (EDAR [MIM# 224900] and TNNT1 [MIM# 605355]), and the third represented a potential novel ‘disease gene’ variant allele of PAAF1. Of note, the three genes had a genomic instability susceptibility relative risk score for AAMR (Alu-Alu mediated rearrangement) of 0.465 (EDAR, rank 47% of the RefSeq genes), 0.587 (TNNT1, rank 22% of the RefSeq genes), and 0.781 (PAAF1, rank 6% of the RefSeq genes) based on AluAluCNVpredictor (20).
Table 1.
BHCMG-GREGoR cases solved/explained by pathogenic homozygous duplications
| Family | Proband | Orthogonal methods for CNV assessment | Variant allele |
|---|---|---|---|
| HOU2049 | BAB5192 | ddPCR segregation; breakpoint PCR | TNNT1, NM_003283.6; hmz dup exon 10–11 |
| HOU2022 | BAB5092, BAB5094 | ddPCR segregation; breakpoint PCR | EDAR, NM_022336.4; hmz dup exon 6–8 |
| HOU3122 | BAB8590 | ddPCR segregation; aCGH | PAAF1, NM_025155.3; hmz dup exon 5–6 |
hmz, homozygous; ddPCR, digital droplet polymerase chain reaction; dup, duplication.
In family 1, a homozygous duplication was called in BAB5092 and affected cousin BAB5094, each of whom had a clinical diagnosis of ectodermal dysplasia. The duplication segregates within the consanguineous family and is surrounded by an AOH block of different size in the affected siblings, consistent with identity-by-descent (IBD) from a common ancestor (Figure 3). The carrier status of the duplication allele was confirmed in the parental and unaffected sibling samples using ddPCR (Figure 3D). The observed ratio in these samples aligns with the expected ratio for heterozygous duplication. Sanger dideoxy sequencing of the breakpoint junction sequence indicate a 9.2 kb tandem duplication predicted to disrupt the reading frame of the transcript (EDAR), resulting in a loss-of-function (LoF) allele. Sequence microhomology was identified at breakpoint junctions (Figure 3), suggesting replicative mechanisms, such as fork stalling and template switching/microhomology-mediated break-induced replication (MMBIR), as a mechanism for the formation of duplication (21).
Figure 3.

De novo ancestral pathogenic duplication CNV in clan and identity-by-descent (IBD). (A) Pedigree of the family HOU2022 with standard pedigree symbols used; historical evidence of consanguinity depicted by horizontal double black lines. (B) Homozygous duplication observed at EDAR locus with an apparent read count change demarcated by normalized read depth plot (horizontal jagged red line) in the proband BAB5092 and affected brother BAB5094 and comparison controls (black horizontal lines) of nearest reference samples. The red rectangle shows the predicted homozygous duplicated region. (C) Scatter plot displays the predicted AOH region of proband BAB5092 and affected brother BAB5094; the vertical line in red indicates the center point of homozygous duplication allele and grey rectangle denotes the AOH region. (D) Segregation of homozygous duplication confirmed by ddPCR; red vertical bar showing homozygous exonic duplication CNV whilst heterozygous father (black), heterozygous mother (grey) and control normal diploid copy number (blue) are shown. The relative droplet ratio of two is consistent with homozygous duplication. (E) Breakpoint junction sequence aligned with the distal (red) and proximal reference sequence (blue), revealing the 3 bp microhomology (teal) at the breakpoint junction.
In family 2, a homozygous duplication affecting exons 10–11 of TNNT1 was called in the proband only by HMZDupFinder. The subject had a clinical diagnosis of neuromuscular disease. The duplication allele segregated with the proband's phenotype in accordance with Mendelian expectations (Figure 4D). Sanger dideoxy sequencing revealed breakpoint junction sequence located within directly oriented Alu elements, suggesting AAMR (20) as a potential mechanism fomenting the genomic duplication allele (Figure 4).
Figure 4.

Pathogenic intragenic homozygous duplication of TNNT1 caused by Alu/Alu-mediated rearrangement (AAMR). (A) Pedigree of family HOU2049. (B, C) Read depth and AOH plot visualization indicate homozygous intragenic duplication in proband BAB5192. (D) Segregation of homozygous variant supported by ddPCR relative positive droplet ratio in the proband (red vertical bar) and heterozygous father (black) versus control (blue). (E) AAMR risk score of TNNT1 predicted with AluAluCNVpredictor (20). (F) Patient photograph and chest x-ray shows pectus carinatum, joint contracture and scoliosis consistent with Amish type nemaline myopathy. (G) Breakpoint junction sequence aligned with the distal (red) and proximal reference sequence (blue), revealing the novel chimeric Alu sequence.
In family 3, HMZDupFinder called the homozygous duplication affecting exons 5–6 of PAAF1 within an AOH block (Figure 5). The duplication allele was confirmed segregating within the family using both aCGH and ddPCR (Figure 5). PacBio long read sequencing revealed the nature of this duplication as an insertional duplication (Supplementary Figure S4). Upon mapping of the inserted duplication and the breakpoint junction sequences, we found that the duplication spans exons 5–6 that were inserted into the intron 10–11 (Supplementary Figure S4A), resulting in a predicted out-of-frame transcript (Supplementary Figure S4B), hinting at a likely loss of function (LoF) allele of PAAF1. Of note, we identified two breakpoint junctions flanking the inserted duplication and both junctions were mapped to an Alu element suggesting Alu-Alu mediated rearrangement (AAMR) (Supplementary Figure S4C). The phenotype of proband BAB8590 includes albinism, uterine hypoplasia with hypogonadism and primary amenorrhea, intellectual disability, kyphosis, crowded teeth, hypothyroidism, congenital hearing loss, and nystagmus. PAAF1 encodes proteasomal ATPase-associated factor 1 which functions as a negative regulator of proteasome assembly (22). Loss of function of PAAF1 could potentially enhance proteasome assembly, and thereby disrupt protein homeostasis (23). Dysfunctions in the proteasome have been linked to various rare disease traits including conditions involving the hair, skin, and ocular pigmentation (24), congenital hearing loss (25), and female reproductive tract development (26). The finding of a homozygous intragenic duplication of PAAF1 in the proband with albinism and developmental abnormalities of the female reproductive tract warrants further investigation. In addition to PAAF1, BAB8590 was found to have homozygous variants affecting TYR (MIM:606933, NM_000372:exon1:c.715C > T:p.R239W, gnomAD MAF = 2.85 × 10−5, CADD = 24.1) and MYO15A (MIM:602666, NM_016239:exon42:c.8067G > A:p.W2689*, gnomAD MAF = 0, CADD = 44), which potentially contribute to albinism and congenital hearing loss phenotypes, respectively.
Figure 5.

Homozygous exonic duplication of PAAF1. (A–C) AOH plot and Z-TPM and log2 ratio visualization indicate homozygous intragenic duplication in proband BAB8590. (D) aCGH confirms the segregation of the duplication in unaffected family members. (E) Segregation of HMZ duplication supported by probe-based ddPCR.
Genomic features of exonic CNVs
Repetitive elements are known to cause genomic instability. To investigate the potential contribution of repetitive elements, such as Alu elements, to small exonic CNV allele formation, we collected all intragenic homozygous alleles identified in BHCMG-GREGoR cohort so far, this includes the four intragenic duplication alleles (one from previous report (2), and three new alleles reported in this report) that have Sanger validated breakpoints and 30 intragenic deletion alleles from our previous report (11) (Supplementary Table S5). We used the Integrative Genomics Viewer (IGV) to manually inspect the exons neighboring the CNV alleles, ensuring that the breakpoint is situated within the respective intron. The median length of introns where breakpoints were mapped is 7.5 kb (n = 30), which is significantly greater (Wilcoxon tests p= 0.0015) than the median length of introns within the same genes (2.6 kb; n = 516) or the median length of the introns genome-wide (1.6 kb, Wilcoxon tests p= 8.1 × 10^-08) (Figure 6A). A similar trend was also observed when mapping 2,833 intragenic deletion and duplication CNV alleles from the gnomAD database (regardless of genotype): the intron lengths associated with a breakpoint were significantly longer (Wilcoxon test, p= 2.2 × 10^-22) when compared to other introns. This observation (Figure 6C) implies that exons flanked by longer introns might be more prone to structural variant mutagenesis. The density distribution of repetitive element families mapping to introns slightly differ between introns containing breakpoint junctions compared to introns genome-wide for BHMG-GREGoR alleles (Figure 6B). This difference is not reproduced on analysis of gnomAD intragenic alleles (Figure 6D). The average count of Alu elements per kilobase in introns encompassing breakpoints was 0.548 (95% CI [0.360, 0.950]) for BHCMG-GREGoR alleles, which is marginally higher than the density of Alu elements (0.323, 95% CI [0.383, 0.371]) in the rest of introns. Other repetitive element families (e.g. L1, L2, MIR) show no evidence for enrichment in both BHCMG-GREGoR alleles and gnomAD alleles (Figures 6B and D).
Figure 6.

The genomic features of exonic CNVs. The dark grey color denotes the intron associated with an exonic CNVs breakpoint, while the light grey signifies the introns that do not associate with a breakpoint. (A) The curves display the distribution of intron lengths for BHCMG-GREGoR CNV alleles. Breakpoint locations for exonic CNV alleles (n = 15) were extracted from both this report and previous publications (2,11). (B) The bar graph shows the density of repetitive element count per kilobase within an intron (n = 516) involved in BHCMG-GREGoR CNV alleles. The color denotes the group of introns: dark grey represents the introns that have a breakpoint mapped, and light grey represents the rest of the introns. (C) The curves display the distribution of intron lengths for gnomAD SVs v2.1 CNV alleles. (D) The bar graph shows the density of repetitive element count per kilobase within an intron (n = 22,888) involved in gnomAD SVs v2.1 CNV alleles.
Discussion
Exonic CNVs have been increasingly associated with rare disease traits and genomic disorders. Detecting such CNVs as structural variant alleles is relevant to the functional annotation of genes and can be important for clinical genomics contextualization and molecular diagnosis (27–30). Adapting exon-targeted design in chromosome microarray analysis (CMA) enables the detection of small exonic CNVs involving as little as a single exon (31). Incorporating the detection of ROH and CNV analysis using cSNP array further improved molecular diagnosis with clinical exome cases (32).
A comparison between ES, CMA and cSNP array in a recent study suggests that ES is more sensitive in detecting homozygous CNV deletions with an optimized algorithm than CMA (11,30). Implementing CNV detection in ES or combining ES with exon-target CMA can provide an additional 2–10% increase in the molecular diagnostic rate (29,32,33). Multiple computational algorithms have been developed to call CNVs directly from ES data (5,6,9); running these algorithms at a suboptimal setting can compromise performance, resulting in reduced sensitivity and specificity. The ‘best reference’ method employed herein uses read count data derived from mapped BAM files to automatically group samples without prior knowledge, e.g. capture design and sequence platform, therefore optimizing the performance of the algorithm. Such reference sample set selection has shown improved sensitivity and precision for most current available CNV callers (34). We fine-tuned the number of reference samples used for normalization, Z-TPM and log2 ratio cutoff for copy number N = 4, i.e. homozygous duplication or heterozygous triplication, based on orthogonal experimental wet bench validation. The optimal parameters were based on a subset of the BHCMG-GREGoR samples. The same parameters were applied to the rest of BHCMG-GREGoR individuals, and we were able to achieve a similar median number of homozygous duplication calls, suggesting robustness of the procedure. While we have not applied HMZDupFinder to other cohorts, we do anticipate the population structure, (e.g. rate of consanguinity), and data quality among other cohorts may require different parameter settings for optimal performance. With that in mind, HMZDupFinder was designed with adaptability, allowing researchers to easily customize parameters e.g. the Z-TPM, log2 Ratio, and number of references used to normalize. For those wishing to apply the tool to new cohorts, we recommend a preliminary analysis (Method section) on orthogonally validated true positive set to optimize and adjust the parameters specific to their dataset.
One of the major aims of HMZDupFinder is to detect homozygous duplications, particularly those spanning three or less exons, in familial genomic studies, where sensitivity takes precedence over high precision. With the optimized parameters, HMZDupFinder achieves an estimated 70.9% recall and 16.1% precision. Notably, the latest benchmarking revealed that the sole method achieving a recall rate exceeding 60% for all CNV categories reported a precision rate of merely 5% (35). Our approach also included a rigorous validation process, such as phenotype-genotype assessments, ddPCR confirmation, and breakpoint sequencing, which minimizes the risk of false positives carrying through to the results and interpretation of homozygous duplication.
Secondary methods such as aCGH and multiplex ligation-dependent probe amplification (MLPA) are used in clinical laboratories for exonic CNV validation (36). ddPCR is a robust experimental method to measure copy number states and confirm the presence of an exonic duplication (37). Read-depth visualizations with a clear separation of the subject from the best reference samples could further rule out some of the false positive calls and help reduce the number of candidates for secondary confirmation. Mapping parents' depth information of trio ES samples added additional evidence for potential homozygous duplication call.
There remain many limitations in detecting CNVs from ES data. Read depth is significantly distorted at genomic regions with repetitive sequencing characteristics, e.g. pseudogenes and segmental duplications, which make such regions ‘invisible’ to a read-depth based CNV detection method. While the terminal duplication might mildly contribute to clinical phenotype by disrupting the gene expression (38), interpreting the impact of duplication proves more challenging than interpreting deletions. Through parameter tuning, our method can call single exon heterozygous duplication and deletion. However, additional information e.g. phenotype or potentially high-impact variant on the allele, will be needed to prioritize computational calls for further validation. For scenarios where a loss of function (LoF) SNV/INDEL allele is identified for an autosomal recessive (AR) rare disease trait, single exon duplication/deletion predicted by normalized Z-TPM may trigger an alert for a compound heterozygous call.
Potential sources of error, such as the false positive call of AOH regions or the established Z-TPM and log2 ratio cutoffs, can cause misclassification of heterozygous duplication or triplication. To elucidate the contribution of each factor, we systematically examined the HMZDupFinder calls from the trio analysis: Model A (Heterozygous triplications inherited from a single parent): Of the homozygous duplication calls, four (4/93) instances were identified where one parent showed a Z-TPM surpassing 4.0 and a log2 ratio between 0.85 and 1.15, indicative of a potential heterozygous triplication; Model B (Heterozygous proband duplications mistakenly called as homozygous within true AOH blocks): No instances fitting this model were found in our dataset. This outcome results from our filter application that uses a log2 ratio between 0.85 and 1.15 and Z-TPM > 4.0, thus excluding potential heterozygous duplications; Model C (false positive AOH blocks): Out of the 15 homozygous duplication calls supported by both parents' coverage, five of the 30 alleles were detected within an AOH block over 100 kb, suggesting a false positive rate of 16.7% for AOH blocks; Models D and E (false negative heterozygous duplications in parents or false positive duplication calls in probands): 12 out of 93 calls showed only one parent with the expected heterozygous duplication based on Z-TPM and log2 ratio, aligning with either of these models.
Identifying genomic regions and specific genes that are susceptible to structural variant mutagenesis and result in exonic CNVs can assist gene variant-rare disease trait association research and candidate disease gene discovery. The reason for genomic architecture predisposing to CNV is also recognized at the gene level to have an evolutionary role, that is the duplication of genes to contribute to genomic plasticity and the creation of paralogs with unique functions (39,40). Understanding genomic instability and loss of genome integrity can further provide an adjuvant analytical tool for clinical genomic laboratories working to identify potential disease-contributing variations and molecular diagnoses.
In previous research conducted by Song et al., a ranked list of genes that are susceptible to Alu-Alu mediated rearrangement (AAMR) (20) was predicted with a machine learning model trained on wet-bench, experimentally studied AAMR events. Our analysis of intragenic CNVs in the current study reinforces the hypothesis that ‘lonely island exons,’ or exons surrounded by extended intronic sequences, are more vulnerable to intragenic CNVs compared to ‘archipelago exons,’ which are flanked by shorter intronic sequences. Enrichment of Alu elements in introns flanking lonely island exons points to one possible structural variant mutagenesis mechanism for the formation of these intragenic CNVs. Recent research in cancer genomics has indicated a higher prevalence of Alu elements in tandem duplications (41). Our assessment of BHCMG-GREGoR intragenic CNV alleles did reveal a modest increase in Alu count in intron-associated breakpoints when compared with other introns. However, the same observation was not made for intragenic alleles from gnomAD database. Further comprehensive analysis of genome-wide germline exonic CNV events is essential to better comprehend the ‘susceptibility to genomic instability score’ pertaining to SV mutagenesis of the human genome.
Conclusions
Herein, we extended the bioinformatic CNV calling algorithm HMZDelFinder and benchmarked it against experimental data from orthogonal genome technologies to develop HMZDupFinder, which provides a framework for discovering intragenic pathogenic homozygous exonic duplication CNV alleles that are likely to be missed by standard analytical workflows and can potentially improve the molecular diagnostic yield of research and clinical ES studies.
Supplementary Material
Acknowledgements
We thank the families and collaborators for their participation in this study.
Authors contributions: Conceptualization: H.D., Z.D, A.J., J.R.L.; Data curation: S.N.J., Z.D, J.M.F., C.M.G., H.L., D.M., G.Y., N.H.E., A.G., D.P, D.C; Formal analysis: H.D., Z.D., and A.J.; Funding acquisition: J.R.L. and J.E.P; Visualization: H.D., Z.D. A.J; Methodology: H.D., T.G., and Z.C.A.; Resources: J.R.L. and J.E.P.; Supervision: J.R.L.; Writing original draft: H.D. and JRL; Writing review and editing: H.D., Z.D., A.J., D.P., C.C, T.G., Z.C.A., J.E.P., D.M., J.R.L. All author(s) read and approved the final manuscript.
Contributor Information
Haowei Du, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Zain Dardas, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Angad Jolly, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Christopher M Grochowski, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Shalini N Jhangiani, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
He Li, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Donna Muzny, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Jawid M Fatih, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Gozde Yesil, Department of Medical Genetics, Istanbul Medical Faculty, Istanbul 34093, Turkey.
Nursel H Elçioglu, Department of Pediatric Genetics, Marmara University Medical Faculty, Istanbul and Eastern Mediterranean University Faculty of Medicine, Mersin 10, Turkey.
Alper Gezdirici, Department of Medical Genetics, University of Health Sciences, Basaksehir Cam and Sakura City Hospital, 34480 Istanbul, Turkey.
Dana Marafi, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Department of Pediatrics, Faculty of Medicine, Kuwait University, Kuwait.
Davut Pehlivan, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Section of Pediatric Neurology and Developmental Neuroscience, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; Texas Children's Hospital, Houston, TX 77030, USA.
Daniel G Calame, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Section of Pediatric Neurology and Developmental Neuroscience, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; Texas Children's Hospital, Houston, TX 77030, USA.
Claudia M B Carvalho, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Pacific Northwest Research Institute, Seattle, WA 98122, USA.
Jennifer E Posey, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Tomasz Gambin, Institute of Computer Science, Warsaw University of Technology, Warsaw, Poland; Department of Medical Genetics, Institute of Mother and Child, Warsaw, Poland.
Zeynep Coban-Akdemir, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Human Genetics Center, Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
James R Lupski, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA; Texas Children's Hospital, Houston, TX 77030, USA; Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030, USA.
Data availability
The code used for HMZDupFinder is available on GitHub along with a description of the necessary steps accompanying this manuscript https://github.com/BCM-Lupskilab/HMZDupFinder. Online resource: AluAluCNVpredictor: http://alualucnvpredictor.research.bcm.edu:3838/, BafCalculator: https://github.com/BCM-Lupskilab/BafCalculator.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
US National Institutes of Health, National Human Genome Research Institute (NHGRI)/National Heart, Lung and Blood Institute (NHLBI) [UM1 HG006542 to the Baylor Hopkins Center for Mendelian Genomics] (in part); NHGRI [U54 HG003273]; NHGRI [UM1 HG008898]; NHGRI Genomic Research Elucidates Genetics of Rare disease (GREGoR) consortium [U01 HG011758, NHGRI K08 HG008986 to J.E.P.]; National Institute of General Medical Sciences (NIGMS) [R01 GM132589 to C.M.B.C. and R01 GM106373]; National Institute for Neurological Disorders and Stroke (NINDS) [R35 NS105078]; D.P. is supported by Rett Syndrome Research Trust (RSRT); International Rett Syndrome Foundation (IRSF) [3701-1]; Doris Duke Charitable Foundation [2023-0235]; NINDS [K23 NS125126-01A1]; D.M. was supported by a Medical Genetics Research Fellowship Program through the United States National Institute of Health [T32 GM007526-42]; D.G.C. was supported by the Child Neurologist Career Development Program K12 and MDA Development Grant [873841]. Funding for open access charge: NHGRI Genomic Research Elucidates Genetics of Rare disease (GREGoR) consortium [U01 HG011758, NHGRI K08 HG008986 to J.E.P.].
Conflict of interest statement. Baylor College of Medicine (BCM) and Miraca Holdings have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs clinical chromosome microarray analysis (CMA) and other genomic studies (ES, genome sequencing) for patient/family care. J.R.L. serves on the Scientific Advisory Board of BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for Genomics International, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The remaining authors declare that they have no competing interests.
References
- 1. Lupski J.R. Structural variation mutagenesis of the human genome: impact on disease and evolution. Environ. Mol. Mutagen. 2015; 56:419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Okamoto Y., Goksungur M.T., Pehlivan D., Beck C.R., Gonzaga-Jauregui C., Muzny D.M., Atik M.M., Carvalho C.M.B., Matur Z., Bayraktar S. et al. Exonic duplication CNV of NDRG1 associated with autosomal-recessive HMSN-Lom/CMT4D. Genet. Med. 2014; 16:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Merico D., Pasternak Y., Zarrei M., Higginbotham E.J., Thiruvahindrapuram B., Scott O., Willett-Pachul J., Grunebaum E., Upton J., Atkinson A. et al. Homozygous duplication identified by whole genome sequencing causes LRBA deficiency. Npj Genomic Med. 2021; 6:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duan R., Hijazi H., Gulec E.Y., Eker H.K., Costa S.R., Sahin Y., Ocak Z., Isikay S., Ozalp O., Bozdogan S. et al. Developmental genomics of limb malformations: allelic series in association with gene dosage effects contribute to the clinical variability. HGG Adv. 2022; 3:100132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fromer M., Moran J.L., Chambert K., Banks E., Bergen S.E., Ruderfer D.M., Handsaker R.E., McCarroll S.A., O’Donovan M.C., Owen M.J. et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012; 91:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krumm N., Sudmant P.H., Ko A., O’Roak B.J., Malig M., Coe B.P., Exome Sequencing Project N.H.L.B.I., Quinlan A.R., Nickerson D.A., Eichler E.E Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012; 22:1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plagnol V., Curtis J., Epstein M., Mok K.Y., Stebbings E., Grigoriadou S., Wood N.W., Hambleton S., Burns S.O., Thrasher A.J. et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012; 28:2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magi A., Tattini L., Cifola I., D’Aurizio R., Benelli M., Mangano E., Battaglia C., Bonora E., Kurg A., Seri M. et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013; 14:R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang Y., Oldridge D.A., Diskin S.J., Zhang N.R. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015; 43:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Packer J.S., Maxwell E.K., O’Dushlaine C., Lopez A.E., Dewey F.E., Chernomorsky R., Baras A., Overton J.D., Habegger L., Reid J.G. CLAMMS: a scalable algorithm for calling common and rare copy number variants from exome sequencing data. Bioinformatics. 2016; 32:133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gambin T., Akdemir Z.C., Yuan B., Gu S., Chiang T., Carvalho C.M.B., Shaw C., Jhangiani S., Boone P.M., Eldomery M.K. et al. Homozygous and hemizygous CNV detection from exome sequencing data in a mendelian disease cohort. Nucleic Acids Res. 2017; 45:1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eldomery M.K., Coban-Akdemir Z., Harel T., Rosenfeld J.A., Gambin T., Stray-Pedersen A., Küry S., Mercier S., Lessel D., Denecke J. et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017; 9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitani T., Isikay S., Gezdirici A., Gulec E.Y., Punetha J., Fatih J.M., Herman I., Akay G., Du H., Calame D.G. et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 2021; 108:1981–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bainbridge M.N., Wang M., Wu Y., Newsham I., Muzny D.M., Jefferies J.L., Albert T.J., Burgess D.L., Gibbs R.A. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011; 12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H., Dawood M., Khayat M.M., Farek J.R., Jhangiani S.N., Khan Z.M., Mitani T., Coban-Akdemir Z., Lupski J.R., Venner E. et al. Exome variant discrepancies due to reference-genome differences. Am. J. Hum. Genet. 2021; 108:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farek J., Hughes D., Salerno W., Zhu Y., Pisupati A., Mansfield A., Krasheninina O., English A.C., Metcalf G., Boerwinkle E. et al. xAtlas: scalable small variant calling across heterogeneous next-generation sequencing experiments. Gigascience. 2022; 12:giac125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orlandini V., Provenzano A., Giglio S., Magi A. SLMSuite: a suite of algorithms for segmenting genomic profiles. BMC Bioinf. 2017; 18:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carvalho C.M.B., Zhang F., Liu P., Patel A., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J. et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009; 18:2188–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collins R.L., Brand H., Karczewski K.J., Zhao X., Alföldi J., Francioli L.C., Khera A.V., Lowther C., Gauthier L.D., Wang H. et al. A structural variation reference for medical and population genetics. Nature. 2020; 581:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song X., Beck C.R., Du R., Campbell I.M., Coban-Akdemir Z., Gu S., Breman A.M., Stankiewicz P., Ira G., Shaw C.A. et al. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018; 28:1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009; 5:e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park Y., Hwang Y.-P., Lee J.-S., Seo S.-H., Yoon S.K., Yoon J.-B. Proteasomal ATPase-associated factor 1 negatively regulates proteasome activity by interacting with proteasomal ATPases. Mol. Cell. Biol. 2005; 25:3842–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zavodszky E., Peak-Chew S.-Y., Juszkiewicz S., Narvaez A.J., Hegde R.S. Identification of a quality-control factor that monitors failures during proteasome assembly. Science. 2021; 373:998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ando H., Ichihashi M., Hearing V.J. Role of the ubiquitin proteasome system in regulating skin pigmentation. Int. J. Mol. Sci. 2009; 10:4428–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kröll-Hermi A., Ebstein F., Stoetzel C., Geoffroy V., Schaefer E., Scheidecker S., Bär S., Takamiya M., Kawakami K., Zieba B.A. et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 2020; 12:e11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zangen D., Kaufman Y., Zeligson S., Perlberg S., Fridman H., Kanaan M., Abdulhadi-Atwan M., Abu Libdeh A., Gussow A., Kisslov I. et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am. J. Hum. Genet. 2011; 89:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lupski J.R. Genomic disorders ten years on. Genome Med. 2009; 1:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boone P.M., Bacino C.A., Shaw C.A., Eng P.A., Hixson P.M., Pursley A.N., Kang S.-H.L., Yang Y., Wiszniewska J., Nowakowska B.A. et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010; 31:1326–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Retterer K., Scuffins J., Schmidt D., Lewis R., Pineda-Alvarez D., Stafford A., Schmidt L., Warren S., Gibellini F., Kondakova A. et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet. Med. 2015; 17:623–629. [DOI] [PubMed] [Google Scholar]
- 30. Yuan B., Wang L., Liu P., Shaw C., Dai H., Cooper L., Zhu W., Anderson S.A., Meng L., Wang X. et al. CNVs cause autosomal recessive genetic diseases with or without involvement of SNV/indels. Genet. Med. 2020; 22:1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gambin T., Yuan B., Bi W., Liu P., Rosenfeld J.A., Coban-Akdemir Z., Pursley A.N., Nagamani S.C.S., Marom R., Golla S. et al. Identification of novel candidate disease genes from de novo exonic copy number variants. Genome Med. 2017; 9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dharmadhikari A.V., Ghosh R., Yuan B., Liu P., Dai H., Al Masri S., Scull J., Posey J.E., Jiang A.H., He W. et al. Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Med. 2019; 11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bergant G., Maver A., Lovrecic L., Čuturilo G., Hodzic A., Peterlin B. Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: a retrospective survey in 1,059 cases. Genet. Med. 2018; 20:303–312. [DOI] [PubMed] [Google Scholar]
- 34. Kuśmirek W., Szmurło A., Wiewiórka M., Nowak R., Gambin T. Comparison of kNN and k-means optimization methods of reference set selection for improved CNV callers performance. BMC Bioinf. 2019; 20:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordeeva V., Sharova E., Babalyan K., Sultanov R., Govorun V.M., Arapidi G. Benchmarking germline CNV calling tools from exome sequencing data. Sci. Rep. 2021; 11:14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yao R., Zhang C., Yu T., Li N., Hu X., Wang X., Wang J., Shen Y. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol. Cytogenet. 2017; 10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Q., Karolak J.A., Grochowski C.M., Wilson T.A., Rosenfeld J.A., Bacino C.A., Lalani S.R., Patel A., Breman A., Smith J.L. et al. Parental somatic mosaicism for CNV deletions – A need for more sensitive and precise detection methods in clinical diagnostics settings. Genomics. 2020; 112:2937–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stankiewicz P., Pursley A.N., Cheung S.W. Challenges in clinical interpretation of microduplications detected by array CGH analysis. Am. J. Med. Genet. A. 2010; 152A:1089–1100. [DOI] [PubMed] [Google Scholar]
- 39. Kondrashov F.A., Koonin E.V. Origin of alternative splicing by tandem exon duplication. Hum. Mol. Genet. 2001; 10:2661–2669. [DOI] [PubMed] [Google Scholar]
- 40. Martinez-Gomez L., Cerdán-Vélez D., Abascal F., Tress M.L. Origins and evolution of Human tandem duplicated exon substitution events. Genome Biol. Evol. 2022; 14:evac162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Roberts N.D., Wala J.A., Shapira O., Schumacher S.E., Kumar K., Khurana E., Waszak S., Korbel J.O., Haber J.E. et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020; 578:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used for HMZDupFinder is available on GitHub along with a description of the necessary steps accompanying this manuscript https://github.com/BCM-Lupskilab/HMZDupFinder. Online resource: AluAluCNVpredictor: http://alualucnvpredictor.research.bcm.edu:3838/, BafCalculator: https://github.com/BCM-Lupskilab/BafCalculator.