

Abstract

Peptide-based covalent inhibitors targeted to nucleophilic protein residues have recently emerged as new modalities to target protein–protein interactions (PPIs) as they may provide some benefits over more classic competitive inhibitors. Covalent inhibitors are generally targeted to cysteine, the most intrinsically reactive amino acid residue, and to lysine, which is more abundant at the surface of proteins but much less frequently to histidine. Herein, we report the structure-guided design of targeted covalent inhibitors (TCIs) able to bind covalently and selectively to the bacterial sliding clamp (SC), by reacting with a well-conserved histidine residue located on the edge of the peptide-binding pocket. SC is an essential component of the bacterial DNA replication machinery, identified as a promising target for the development of new antibacterial compounds. Thermodynamic and kinetic analyses of ligands bearing different mild electrophilic warheads confirmed the higher efficiency of the chloroacetamide compared to Michael acceptors. Two high-resolution X-ray structures of covalent inhibitor–SC adducts were obtained, revealing the canonical orientation of the ligand and details of covalent bond formation with histidine. Proteomic studies were consistent with a selective SC engagement by the chloroacetamide-based TCI. Finally, the TCI of SC was substantially more active than the parent noncovalent inhibitor in an in vitro SC-dependent DNA synthesis assay, validating the potential of the approach to design covalent inhibitors of protein–protein interactions targeted to histidine.
Keywords: covalent inhibitors, histidine alkylation, protein−protein interactions, X-ray structures, DNA replication
Introduction
Over the past decade, the development of covalent drugs has gained momentum with the approvals of several targeted covalent inhibitors (TCIs) in oncology and as antivirals.1−5 Potential benefits of TCIs over noncovalent inhibitors are manifold.6−8 When an irreversible bond is formed, the inhibitor is no longer in competition with natural binding proteins. Additionally, covalent inhibition can potentially improve the inhibitory potency and/or selectivity, increase the duration of action, and reduce the emergence of resistance.9 Moreover, targeting extended protein–protein interaction (PPI) interfaces with competitive small-molecule inhibitors is not always achievable and peptide-based inhibitors with an irreversible mode of action can be considered as emerging modalities for such targets (recently reviewed).8,10 Most often, covalent inhibitors of PPIs are targeted to cysteine and less frequently to other amino acids (Ser, Lys), but there is only a handful of studies on the use of histidine as a nucleophile, and as highlighted in a recent account,11 its potential interest in drug discovery is largely overlooked. The fact that histidine is much less frequently targeted by covalent inhibitors in comparison to cysteine and even lysine12,13 may be attributed to its lower nucleophilicity,14 making it more challenging to target. Nevertheless, with lower pKa values in proteins compared to lysine,15 histidine generally remains unprotonated and thus intrinsically reactive at physiological pH, suggesting an interesting—yet largely unexplored—potential for the design of TCIs.11 The search of databases dedicated to high-resolution three-dimensional (3D) structures of biologically relevant covalent protein–ligand complexes16,17 revealed a paucity of structural data on covalent inhibitors targeting histidine, with crystal structures available being limited to non-reactive probes blocking enzymatic catalytic sites and to rare structures of covalent protein adducts with natural products (e.g., zampanolide, a microtubule-stabilizing agent in complex with αβ-tubulin, PDB ID 4I4T).18 This lack of structural studies strongly limits the knowledge of how to design covalent inhibitors of PPI targeted to noncatalytic histidine residues using drug-like mild electrophiles.
Over the last years, our groups have been exploring the potential of inhibiting the bacterial SC,19,20 an essential component of the bacterial replisome (a multiprotein machinery that ensures the replication of chromosomal DNA21,22) to discover antibacterial compounds against Gram-negative bacteria. SC is a homodimeric protein (Figure 1) serving as an anchoring platform for DNA polymerases and other proteins involved in DNA replication and repair.23−26 SC is engaged in numerous PPIs through a well-conserved binding site that can accommodate short linear peptide motifs. Efforts from several research groups including ours have led to the discovery of various SC ligands (small molecules,27−30 natural products,31 and optimized peptides such as 1(32−41)) with the ability to inhibit DNA replication. Importantly, the Escherichia coli (E. coli) SC (EcSC) contains a histidine residue lining the binding site, largely conserved among Gram-negative and Gram-positive pathogens,37,42 which we thought could be exploited for covalent inhibition. In this work, we anticipated that the lower reactivity of histidine could also be counterbalanced by the precise positioning of mild electrophilic warheads proximal to the imidazole group of histidine.
Figure 1.
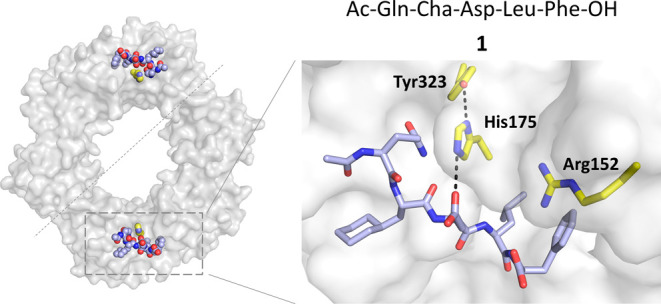
X-ray cocrystal structure of EcSC-1 complex (PDB ID:6FVL).39
Results
Structure-Guided Design and Synthesis of Covalent Inhibitors
Examination of the X-ray crystal structures of EcSC in complex with optimized peptide ligands such as pentapeptide 1(34) (PDB ID: 6FVL(39)), identified His175 (Figure 1) in close vicinity of the ligand binding site. The imidazole group of His175 is adjacent and sometimes H-bonded to the side chain of the central residue (e.g., Asp3 in 1) in peptide ligands (distance between Nε(His175) and O=C(Asp3 in 1) ∼2.9–3.8 Å) suggesting that it could be engaged by peptide inhibitors incorporating an electrophile on the appropriate side chain. We thus designed and synthesized a series of SC TCIs based on the X-ray cocrystal structure of peptide ligand 1 bound to EcSC.39 To promote the formation of a covalent bond with the imidazole and ensure minimal geometric distortion upon reaction, it is essential for the electrophilic atom to be optimally positioned adjacent to the imidazole group. We thought to substitute the −CH2–C(=O)–O– chain of the Asp residue with a slightly longer chain, placing the electrophilic atom e at either the fourth or fifth position. To allow the attachment of electrophilic groups on the peptide, we replaced Asp3 in 1 by a 2,3-diamino-propionic acid (Dap) residue temporally protected by an allyloxycarbonyl (alloc) group (Figure 2). Different warheads with a gradual variation of reactivity43 and with different geometries and rigidities were selected and grafted on the Dap side chain by acylation following removal of the alloc group (Figure 2, compounds 2–5). These weak electrophiles (i.e., haloacetamides and Michael acceptors including acrylamides) are versatile warheads frequently found in covalent drugs and drug candidates targeting cysteine.44−46 All His175-targeting peptides 2–5 were synthesized by solid-phase peptide synthesis in good overall yield (16–51%) following reversed-phase high-performance liquid chromatography (HPLC) purification (see SI.1 and SI.2).
Figure 2.

Chemical structures of noncovalent inhibitor 1 and covalent inhibitors 2–5.
Determination of Dissociation and Kinetic Constants
Next, we used complementary biophysical methods, namely, isothermal titration calorimetry (ITC; SI.3) and ESI+ mass spectrometry (SI.4 and SI.5) to evaluate the ability of these compounds to bind and react with recombinant EcSC. ITC experiments confirmed that the substitution of Asp3 with acetylated Dap did not affect the binding affinity, nor did the incorporation of the electrophilic warheads in the whole inhibitor series (see KD values in Table 1 and thermodynamic parameters in Tables S1–S5 and Figures S1–S4).
Table 1. Dissociation Constants, Ratio of Covalent Bond Formation, and Second-Order Rate Constants of Compounds 1–5a.
|
KD (nM) |
% covalent adduct | kapparent (M–1 s–1) | |||
|---|---|---|---|---|---|
| compd | 25 °C Hepes | 37 °C Hepes | 37 °C NH4OAc | 1 h, 37 °C NH4OAc | 37 °C NH4OAc |
| 1 | 173 ± 67 | 546 ± 12 | 486 ± 22 | 0 | |
| 2 | 221 ± 80 | 452 ± 183 | 580 ± 315 | 39.9 | 1.04 ± 0.01 |
| 3 | 191 ± 22 | 446 ± 163 | 482 ± 89 | 16 | 0.236 ± 0.006 |
| 4 | 166 ± 10 | 402 ± 113 | 475 ± 46 | 11 | n.d. |
| 5 | 66.4 ± 9.8 | 74 ± 3 | n.d. | 0 | n.d. |
See SI.3, SI.4, and SI.5 for experimental details. Buffers were adjusted to a pH of 7.4.
Derivatives 2–4 were found to bind SC with an affinity in the same range as that measured for the parent noncovalent inhibitor 1 (KD = 173 nM at 25 °C in Hepes buffer), whereas compound 5 with a 2-butynamide warhead shows a substantial increase in affinity (KD = 66 nM at 25 °C). A similar trend was observed with a SC mutated at the His175 position (EcH175GSC), but with a 3.5- to 14-fold reduction of the affinities (except for 5), reflecting the important contribution of His175 to the binding (Table S6 and Figure S5). The relative ability of compounds 2–5 to form covalent adducts with the protein was evaluated by mass spectrometry following incubation with recombinant EcSC (Figure 3 and SI.4 and Figures S6–S10). Notably, compounds 2, 3, and 4 were all found to form a covalent bond with EcSC under these conditions but in different proportions, the highest ratio of covalent adduct being obtained with 2 and 3 at 37 °C (Table 1 and Figure S10). However, no covalent adduct was observed with 5 under these conditions. Then, we selected compounds 2 and 3 and, using MS analysis for TCI-SC complex detection, we conducted kinetic studies to determine the corresponding rate constants at 22 and 37 °C (SI.5). Experimental data were fitted to a one-step model to yield second-order rate constants (kapparent) of 1.04 and 0.236 M–1·s–1 for 2 and 3, respectively, at 37 °C. Values were 5- and 4-fold lower at 22 °C (Figure S11). In all cases, the rate constants for compound 3 were found to be about 4-fold lower than for 2 (SI.5 and Figure S11). The global activation energies (Eatot) of the peptide-SC interaction were calculated to be 20.3 ± 0.3 and 16.9 ± 0.5 kcal·mol–1 for 2 and 3, respectively (SI.5 and Table S7). We calculated that Eacov, the activation energy for the formation of the covalent bond, is similar to Eatot. This rather low energy for the formation of a C–N bond probably results from the prepositioning of the warheads close to the His target, due to the peptide interaction within the SC pocket.
Figure 3.
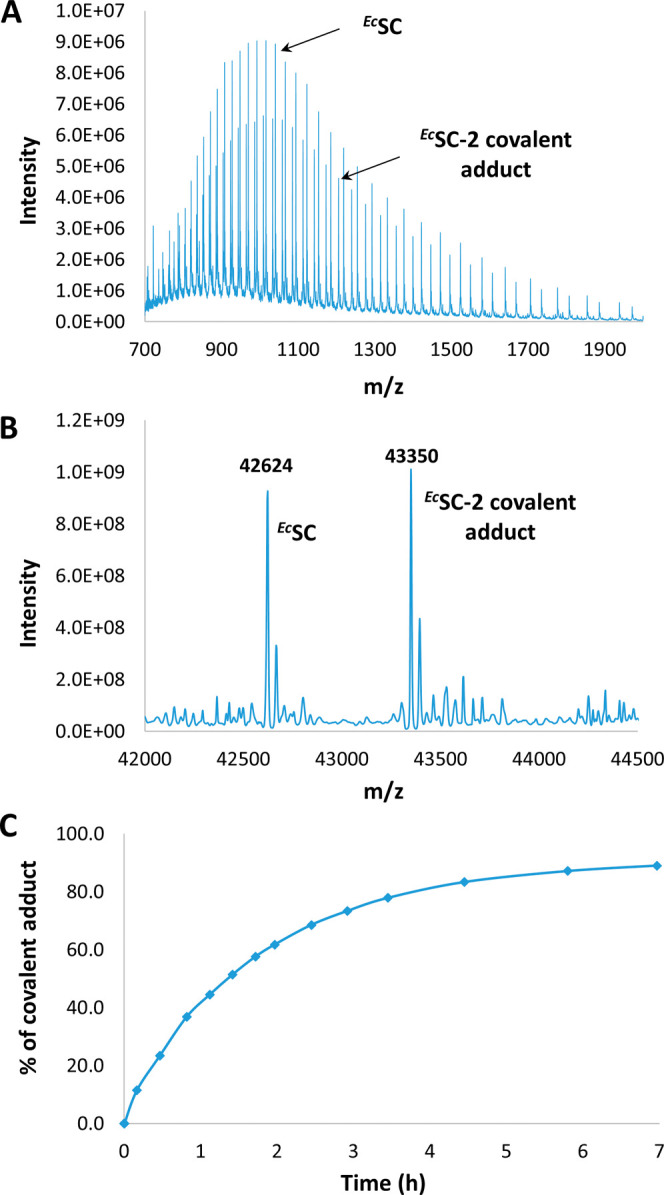
(A) Representative example of a mass spectrum obtained after incubating ECSC with a covalent inhibitor (here, compound 2 was incubated at 37 °C for 49 min). (B) Corresponding deconvoluted mass spectrum. (C) Percentage of covalent adduct formed over time determined from deconvoluted spectra (see SI.5).
X-ray Crystallographic Studies of EcSC Covalent Adducts
To confirm covalent bond formation with the imidazole side chain of His175 and gain additional structural insight, we co-crystallized EcSC with compounds 2 and 3 and solved the structures at 1.22 and 1.37 Å, respectively (SI.6 and Tables S8 and S9). In the EcSC-2 structure (PDB ID: 8PAY, Figure 4A), the electron density of the (2Fo-Fc)-type map between the Nε of His175 and the acetamide moiety on the Dap side chain is well defined, thus unambiguously supporting the covalent bond between 2 and His175. Similarly, the electron density in the EcSC-3 structure (PDB entry 8PAT, Figure 4B) clearly shows that His175 is within a bond length of the electrophilic carbon. Interestingly, in the two structures, the carbonyl group of the acylated Dap is involved in a bidentate hydrogen bond (HB) with Arg152. This additional HB could compensate for the loss of HB between the Asp residue in 1 and His175, potentially explaining that the affinity for EcSC was retained when going from 1 to 2 and 3. Moreover, this new interaction might also contribute positively to the kinetics of covalent bond formation by controlling the conformation and by activating the electrophilic carbon. Peptides 2 and 3 in these structures superimpose very well with the structure of 1 bound to EcSC, indicating that the structure of the complex is not affected by the formation of the covalent bond (Figure 4C), in good agreement with the dissociation constants (for the noncovalent prefixation) of peptides 1–3 being in the same range of magnitude as discussed above (Table 1). This overlay also reveals that the imidazole ring of His175 in the EcSC-2 but not in the EcSC-3 structure is slightly rotated compared to EcSC-1 (variation of χ2 from −26.3 to −54.4°), presumably to better accommodate the geometrical constraints imposed by the shorter chloroacetamide electrophile (Figure 4D).
Figure 4.

Crystal structures of EcSC-2 adduct (A) solved at 1.22 Å resolution (PDB ID: 8PAY) and EcSC-3 adduct (B) solved at 1.37 Å resolution (PDB ID: 8PAT); the electron density is shown in gray around the covalent bond formed with His175; the carbon atoms from the warheads are highlighted in magenta and orange, respectively. (C) Superimposition of the crystal structures of EcSC-1 complex (light blue), EcSC-2 (pink, RMSD of atomic positions with EcSC-1 complex = 0.498 Å (316 to 316 atoms)), and EcSC-3 (beige, RMSD of atomic positions with EcSC-1 = 0.461 Å (334 to 334 atoms)) adducts. (D) Zoom on the covalent bonds showing the rotation of His175 on EcSC-2 compared to EcSC-1 and EcSC-3 crystal structures.
Selective Engagement of EcSC His175 by Covalent Inhibitors
To further investigate the ability of TCIs to covalently modify EcSC in various competitive environments including cell lysates, we prepared peptide 6 (Figure 5A), a biotinylated chloroacetamide-bearing TCI probe for pull-down experiments. Its sequence derives from a high-affinity noncovalent EcSC binder (P14,34,41KD = 64 nM). For comparison, we also used noncovalent inhibitors 7, a strong SC binder with KD = 39 nM,41 and 8, the cognate biotin-labeled noncovalent version of 6. Compound 6 was first tested with purified recombinant wild-type His-tagged EcSC and the His175 mutant protein (H175G). After incubation for 3 h at a concentration of 10 μM at 37 °C (see SI. 7), 6 was found to bind covalently to wild-type EcSC but not to H175G SC (Figures 5B and S12). The noncovalent peptide ligand 8 was not effective in this assay. Co-incubation with increasing concentrations of a noncovalent 7 from 10 to 200 μM (see SI. 8) resulted in a concentration-dependent loss (up to ∼70%) of SC pull-down mediated by 6 (Figures 5C and S13). This result is consistent with both inhibitors competing for the same binding site on SC. In a second set of experiments, we evaluated target engagement in the E. coli whole cell lysate (SI. 9). Cell lysates were treated for 1 h with increasing concentrations of 6. SDS-PAGE electrophoresis after pull-down and denaturation revealed a band at ca. 40–45 kDa compatible with the molecular weight of the EcSC-6 adduct, whose intensity increases with the concentration of 6 (Figure 5D). The absence of any other clearly visible band at low concentrations of 6 (i.e., 0.17 and 0.83 μM) supports a selective engagement of the target in a physiological environment. The selectivity of 6 was further assessed by proteomic analysis (SI. 10 and Figure 5E) using the noncovalent peptide 8 and the scrambled sequence 9 (Figure 5A) for direct comparison. Following incubation of the peptides with the whole cell lysate for 1 h at 37 °C, enrichment with streptavidin-coated beads, mild washing, and classical preinjection processing including trypsin digestion (see SI. 10), the proteins associated with the beads (bound to the peptide inhibitors, covalently or not) were analyzed by LC-MS/MS. Figure 5E displays a Volcano plot representation showing the distribution of the proteins specifically enriched by either peptide 6 or 8 compared to the experiment with peptide 9. The EcSC protein (the dnaN gene product) was highly enriched with both peptides 6 and 8 (log2 FC > 9), with very significant adjusted p-values (adj p < 5 × 10–17) (see Table S10). In the pull-down mediated by 6, as compared to 9, three additional proteins (i.e., NagC, TrxA, and MoaB) were detected but with a much lower significance (4 × 10–3 < adjp < 1 × 10–2) compared to EcSC (Table S10). Direct comparison of the proteins specifically enriched with 6 compared to 8 (Table S11) revealed two additional potential off-targets of 6 (LuxS; adjp = 2.48 × 10–2 and YqjG, adjp = 3.16 × 10–2). At best, these proteins are 15 times less enriched than EcSC denoting excellent selectivity of the covalent binder 6. The AlphaFold structures of NagC (N-acetylglucosamine repressor) and LuxS (S-ribosylhomocysteine lyase) and the X-ray structures of TrxA (Thioredoxin 1, PDB ID: 1KEB), MoaB (Molybdenum cofactor biosynthesis protein B, PDB ID: 1MKZ), and YqjG (Glutathionyl-hydroquinone reductase, PDB ID: 3R3E) reveal potentially accessible cysteine residues that could react with the chloroacetamide of 6. This could account for the little enrichment of these proteins.
Figure 5.

(A) Structure of compounds 6–9. The spacer Gly-Pro-Arg was used for biotinylated peptides 6, 8, and 9. (B) Histidine 175 is essential for the formation of a covalent adduct with 6. His-tag purified wild-type or H175G EcSC were incubated with or without compound 6 or the cognate biotin-labeled noncovalent peptide ligand 8. SDS-PAGE electrophoresis of the incubated SC and Coomassie blue staining (top) reveals a mobility shift only when 6 is incubated with the wild-type SC, but not with the H175G mutant. Staining of biotinylated products (bottom) identifies the shifted band as the covalent 6-EcwtSC complex (SI. 7 and Figure S12). (C) SC-binding competition assay. Purified His-Tag-SC was incubated with a fixed amount of 6 (10 μM) and increasing concentrations (0–200 μM) of the noncovalent high-affinity SC binder 7 as indicated. Covalently bound biotinylated peptides were revealed after SDS-PAGE separation and infrared dye coupled streptavidin labeling (see SI. 8). The relative fluorescence detected in each lane was plotted against the noncovalent competitor concentration. (D) Pull-down experiments following co-incubation of E. coli whole cell lysate with compound 6. Covalent reaction products were captured onto streptavidin-coated magnetic beads and analyzed as described above. The Mw of the most intense band detected (around 40 kDa) indicates that the major reaction product formed is likely the covalent 6-SC adduct. (E) Spectral-count-based proteomic analysis of pull-down experiments in whole cell lysate. The volcano plot displays the log2 fold change (x axis) against the–log10 adjusted p-value (y axis) for all proteins enriched when using peptide 9 (control, left part of the graph) compared with SC-binding peptides 6 (green circles) or 8 (red circles). Cut-offs (dashed lines) of log2 FC > 1 (ratio >2) and −log10 adj p value >1.3 (adj p value <0.05) were applied to highlight significantly enriched proteins (see SI.9 and Tables S10).
In the same experiment, three database search algorithms (Mascot, Sequest, and MS-Amanda) successfully identified the covalently His175-modified EcSC peptide TVATDGHR, through y fragment ions and b fragment ions (Figure S14) and confirmed covalent bond formation at His175. In addition, the presence of chlorine atoms, from 6 (3,4-di-Cl-Phe), in the modified peptide was confirmed by the distortion of its isotopic pattern (Figure S14 and Table S12). The proportion of the covalently modified TVATDGHR peptide compared to nonmodified sequence was estimated by an accurate MS1 label-free quantification to be ≈86% (see SI.10.2, Table S13 and Figure S14), indicating that the large majority of the captured SCs were covalently modified by the TCI.
Inhibition of SC-Dependent DNA Synthesis upon Covalent Bond Formation
Finally, we evaluated the activity of TCI 6 relative to that of the cognate noncovalent ligand 8 by measuring the ability of the two compounds to inhibit the SC-dependent DNA synthesis activity of the replicative E. coli DNA polymerase III (PolIII) in vitro (SI. 11 and Figures 6 and S15–S17).34 We first determined IC50 values as a function of preincubation time of EcSC with 6 or 8 prior to the addition of the DNA substrate and PolIII (Figure S15). Although little variations of IC50 values were observed with compound 8 at the different incubation times tested, IC50 values measured with TCI 6 decrease substantially (up to 3-fold) with increasing incubation time. The time-dependent inhibitory effect is consistent with a mechanism of action involving the formation of a covalent adduct. Furthermore, we show that the enhanced ability of TCI 6 to inhibit SC-dependent DNA synthesis in comparison to 8 relies on the presence of the H175 residue in the SC (Figure 6). Collectively, these results establish that the increased potency of TCI 6 over the noncovalent inhibitor 8 stems from the covalent interaction with H175 residue of the SC. Importantly, this increased potency persists even in the presence of a high concentration of reduced glutathione (a natural strong nucleophile), reflecting a more physiological environment (Figure S17). The next step will be to investigate whether SC TCIs would manifest enhanced antimicrobial activity compared to the noncovalent version using, e.g., conjugation to bacteria-penetrating molecules such as proline-rich antimicrobial peptides (PrAMPs).40
Figure 6.
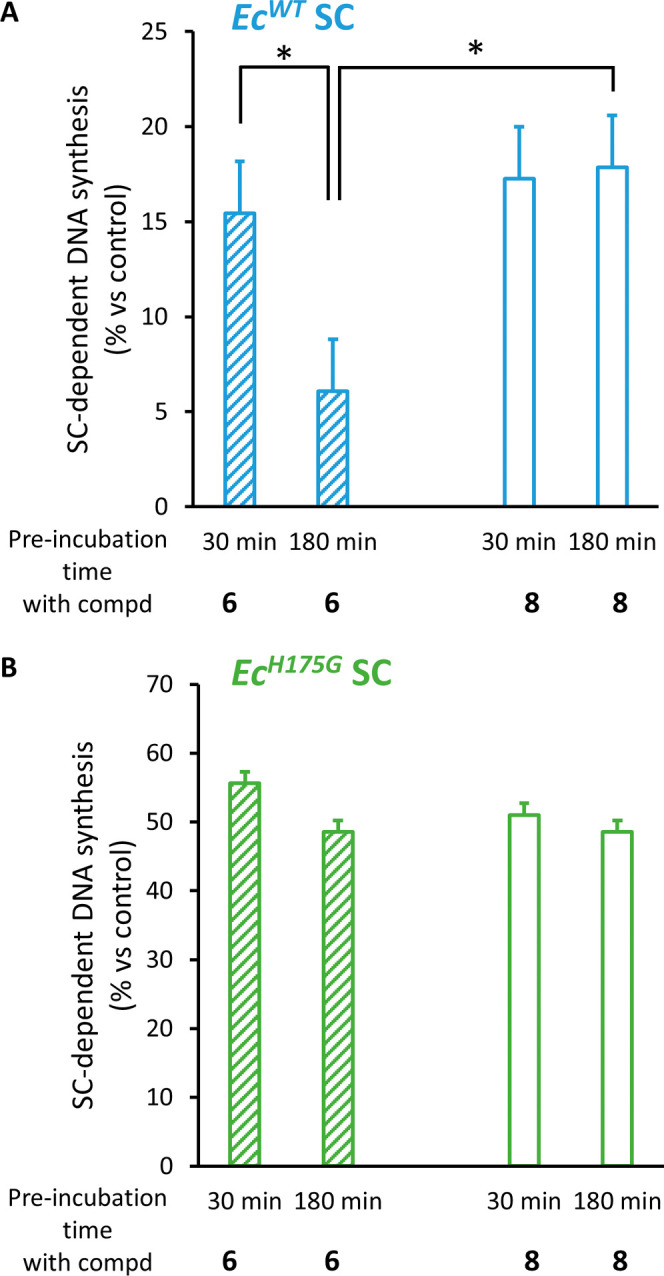
Time-dependent increased inhibitory effect on SC-dependent DNA synthesis of compound 6 as compared to 8 necessitates the presence of H175 on the SC. SC-dependent DNA synthesis was measured after preincubation of the indicated E. coli SC variants (either wild-type or H175G mutant) with or without compound 8 or 6 for the indicated period. Results presented are the mean (±sd) of 3 independent assays. Statistically different results are indicated (*: p value <0.05). See SI. 11 and Figure S16 for experimental details and gel images.
Conclusions
In summary, we report the structure-guided design of effective and selective covalent peptide inhibitors of the bacterial SC by targeting a well-conserved histidine residue adjacent to the DNA polymerase-binding region. Remarkably, His175, despite its lower intrinsic reactivity compared to more reactive protein nucleophiles such as cysteine, is reactive enough to engage mild electrophiles, i.e., chloroacetamide and acrylamide Michael acceptors, attached on a central residue of the peptide ligand precisely positioned in close proximity to the imidazole side chain. The two high-resolution X-ray crystal structures of the covalent protein–ligand adducts we obtained are particularly revealing in showing that the canonical binding mode of the peptide ligand is fully retained upon covalent bond formation. Knowing that crystallographic structures of covalent adducts with histidine represent only a very small fraction (<3%) of all structures of covalent complexes in the pdb,14 the structural information collected here is even more notable. Taken together, these and other recent studies47 illustrate the potential of peptide-based covalent inhibitors of PPIs to block bacterial machinery. More generally, our results along with the higher prevalence of His residues at ligand binding sites compared to Lys and Cys residues48 underscore the “ligandability” of histidine as a nucleophile for the design of covalent inhibitors of PPIs.11
Acknowledgments
The authors thank the ANR for financial support (Projects FIIBRE ANR-22-CE18-0037 and MADNART ANR-21-ASTR-0002). A doctoral fellowship (to C.M.) from DGA (no. 2018600030) and Conseil Régional de Nouvelle-Aquitaine (no. 2018-1R30229) is gratefully acknowledged. X-ray data were collected at the Paul Scherrer Institut, Villigen-PSI, Switzerland, through several SLS proposals for the provision of synchrotron radiation beamtime, at beamline X06DA PXIII. The authors thank Johana Chicher for her help in the MS analyses and the IdEx 2015 program Equipements mi-lourds and the mass spectrometry platform from the Université de Strasbourg.
Data Availability Statement
Mass spectrometry proteomics raw data have been deposited to the ProteomeXchange Consortium via the PRIDE49 partner repository with the following project accession number PXD042384 and 10.6019/PXD042384.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00572.
Experimental procedures for the synthesis of peptides and characterization data, including HPLC chromatograms and LCMS; ITC titrations curves; TCI-EcSC adducts MS spectra used to determine kapparent; crystal data; SDS-PAGE gels; procedure for pull-down experiments; proteomic analyses; and DNA synthesis inhibition assays (PDF)
Proteomic raw data (XLS)
Author Contributions
G.C. and C.M. synthesized the compounds. K.B., P.W., C.M., and D.B. performed ITC and mass spectrometry experiments. P.D. performed mathematical analysis of the kinetics of covalent bond formation. J.W. and C.M. performed the competitive experiments with recombinant EcSC and in cell lysate. J.W. and C.M. performed SC-dependent DNA synthesis assays. J.B., P.H., and L.K. performed the proteomic analyses. D.B., I.M., S.E., and V.O. performed the X-ray diffraction analyses. G.C., J.W., D.B., and G.G. conceived the project and the experimental plan and wrote the manuscript. All of the authors commented on the draft of the manuscript and contributed to the analysis and interpretation of the data.
The authors declare no competing financial interest.
Supplementary Material
References
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Bauer R. A. Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies. Drug Discovery Today 2015, 20 (9), 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Ward R. A. Structure-Based Design of Targeted Covalent Inhibitors. Chem. Soc. Rev. 2018, 47 (11), 3816–3830. 10.1039/C7CS00220C. [DOI] [PubMed] [Google Scholar]
- Gehringer M.; Laufer S. A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62 (12), 5673–5724. 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Boike L.; Henning N. J.; Nomura D. K. Advances in Covalent Drug Discovery. Nat. Rev. Drug Discovery 2022, 21, 881–898. 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way J. Covalent Modification as a Strategy to Block Protein–Protein Interactions with Small-Molecule Drugs. Curr. Opin. Chem. Biol. 2000, 4 (1), 40–46. 10.1016/S1367-5931(99)00049-6. [DOI] [PubMed] [Google Scholar]
- Cheng S.-S.; Yang G.-J.; Wang W.; Leung C.-H.; Ma D.-L. The Design and Development of Covalent Protein-Protein Interaction Inhibitors for Cancer Treatment. J. Hematol. Oncol. 2020, 13 (1), 26 10.1186/s13045-020-00850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen F. M.; Grossmann T. N. Peptide-based Covalent Inhibitors of Protein–Protein Interactions. J. Pept. Sci. 2023, 29, e3457 10.1002/psc.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Samanta I.; Mondal A.; Liu W. R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14 (9), 889–906. 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob N. M.; Remarcik C.; Rössler S. L.; Wong J. Y. K.; Wang J. C. K.; Tao J.; Smith C. L.; Loas A.; Buchwald S. L.; Eaton D. L.; López M. P.; Pentelute B. L. Electrophile Scanning Reveals Reactivity Hotspots for the Design of Covalent Peptide Binders. ACS Chem. Biol. 2024, 19, 101. 10.1021/acschembio.3c00538. [DOI] [PubMed] [Google Scholar]
- Che J.; Jones L. H. Covalent Drugs Targeting Histidine – an Unexploited Opportunity?. RSC Med. Chem. 2022, 13 (10), 1121–1126. 10.1039/D2MD00258B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://cadd.zju.edu.cn/cidb/.
- Mukherjee H.; Grimster N. P. Beyond Cysteine: Recent Developments in the Area of Targeted Covalent Inhibition. Curr. Opin. Chem. Biol. 2018, 44, 30–38. 10.1016/j.cbpa.2018.05.011. [DOI] [PubMed] [Google Scholar]
- Zanon P. R. A.; Yu F.; Musacchio P.; Lewald L.; Zollo M.; Krauskopf K.; Mrdović D.; Raunft P.; Maher T. E.; Cigler M.; Chang C.; Lang K.; Toste F. D.; Nesvizhskii A. I.; Hacker S. M.. Profiling the Proteome-Wide Selectivity of Diverse Electrophiles. ChemRxiv 2021. 10.26434/chemrxiv.14186561.v1 [DOI] [Google Scholar]
- Pahari S.; Sun L.; Alexov E. PKAD: A Database of Experimentally Measured pKa Values of Ionizable Groups in Proteins. Database 2019, 2019, baz024 10.1093/database/baz024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Moumbock A. F. A.; Qaseem A.; Xu Q.; Günther S. CovPDB: A High-Resolution Coverage of the Covalent Protein–Ligand Interactome. Nucleic Acids Res. 2022, 50 (D1), D445–D450. 10.1093/nar/gkab868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H.; Gao J.; Weng G.; Ding J.; Chai X.; Pang J.; Kang Y.; Li D.; Cao D.; Hou T. CovalentInDB: A Comprehensive Database Facilitating the Discovery of Covalent Inhibitors. Nucleic Acids Res. 2021, 49 (D1), D1122–D1129. 10.1093/nar/gkaa876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota A. E.; Bargsten K.; Zurwerra D.; Field J. J.; Díaz J. F.; Altmann K.-H.; Steinmetz M. O. Molecular Mechanism of Action of Microtubule-Stabilizing Anticancer Agents. Science 2013, 339 (6119), 587–590. 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- Burnouf D. Y.; Olieric V.; Wagner J.; Fujii S.; Reinbolt J.; Fuchs R. P. P.; Dumas P. Structural and Biochemical Analysis of Sliding Clamp/Ligand Interactions Suggest a Competition Between Replicative and Translesion DNA Polymerases. J. Mol. Biol. 2004, 335 (5), 1187–1197. 10.1016/j.jmb.2003.11.049. [DOI] [PubMed] [Google Scholar]
- Altieri A. S.; Kelman Z. DNA Sliding Clamps as Therapeutic Targets. Front. Mol. Biosci. 2018, 5, 87 10.3389/fmolb.2018.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijffels G.; Dalrymple B.; Kongsuwan K.; Dixon N. Conservation of Eubacterial Replicases. IUBMB Life 2005, 57 (6), 413–419. 10.1080/15216540500138246. [DOI] [PubMed] [Google Scholar]
- Robinson A.; Causer R. J.; Dixon N. E. Architecture and Conservation of the Bacterial DNA Replication Machinery, an Underexploited Drug Target. Curr. Drug Targets 2012, 13 (3), 352–372. 10.2174/138945012799424598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple B. P.; Kongsuwan K.; Wijffels G.; Dixon N. E.; Jennings P. A. A Universal Protein-Protein Interaction Motif in the Eubacterial DNA Replication and Repair Systems. Proc. Natl. Acad. Sci. U.S.A. 2001, 98 (20), 11627–11632. 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulye M.; Singh M. I.; Jain V. From Processivity to Genome Maintenance: The Many Roles of Sliding Clamps. Genes 2022, 13 (11), 2058 10.3390/genes13112058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.; Fujii S.; Gruz P.; Nohmi T.; Fuchs R. P. P. The β Clamp Targets DNA Polymerase IV to DNA and Strongly Increases Its Processivity. EMBO Rep. 2000, 1 (6), 484–488. 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel O. Pivotal Role of the β-Clamp in Translesion DNA Synthesis and Mutagenesis in E. Coli Cells. DNA Repair 2002, 1 (9), 703–708. 10.1016/S1568-7864(02)00106-4. [DOI] [PubMed] [Google Scholar]
- Georgescu R. E.; Yurieva O.; Kim S.-S.; Kuriyan J.; Kong X.-P.; O’Donnell M. Structure of a Small-Molecule Inhibitor of a DNA Polymerase Sliding Clamp. Proc. Natl. Acad. Sci. U.S.A. 2008, 105 (32), 11116–11121. 10.1073/pnas.0804754105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z.; Whittell L. R.; Wang Y.; Jergic S.; Liu M.; Harry E. J.; Dixon N. E.; Beck J. L.; Kelso M. J.; Oakley A. J. Discovery of Lead Compounds Targeting the Bacterial Sliding Clamp Using a Fragment-Based Approach. J. Med. Chem. 2014, 57 (6), 2799–2806. 10.1021/jm500122r. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Whittell L. R.; Wang Y.; Jergic S.; Ma C.; Lewis P. J.; Dixon N. E.; Beck J. L.; Kelso M. J.; Oakley A. J. Bacterial Sliding Clamp Inhibitors That Mimic the Sequential Binding Mechanism of Endogenous Linear Motifs. J. Med. Chem. 2015, 58 (11), 4693–4702. 10.1021/acs.jmedchem.5b00232. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Wang Y.; Whittell L. R.; Jergic S.; Liu M.; Harry E.; Dixon N. E.; Kelso M. J.; Beck J. L.; Oakley A. J. DNA Replication Is the Target for the Antibacterial Effects of Nonsteroidal Anti-Inflammatory Drugs. Chem. Biol. 2014, 21 (4), 481–487. 10.1016/j.chembiol.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Kling A.; Lukat P.; Almeida D. V.; Bauer A.; Fontaine E.; Sordello S.; Zaburannyi N.; Herrmann J.; Wenzel S. C.; Konig C.; Ammerman N. C.; Barrio M. B.; Borchers K.; Bordon-Pallier F.; Bronstrup M.; Courtemanche G.; Gerlitz M.; Geslin M.; Hammann P.; Heinz D. W.; Hoffmann H.; Klieber S.; Kohlmann M.; Kurz M.; Lair C.; Matter H.; Nuermberger E.; Tyagi S.; Fraisse L.; Grosset J. H.; Lagrange S.; Muller R. Targeting DnaN for Tuberculosis Therapy Using Novel Griselimycins. Science 2015, 348 (6239), 1106–1112. 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- Wijffels G.; Dalrymple B. P.; Prosselkov P.; Kongsuwan K.; Epa V. C.; Lilley P. E.; Jergic S.; Buchardt J.; Brown S. E.; Alewood P. F.; Jennings P. A.; Dixon N. E. Inhibition of Protein Interactions with the β2 Sliding Clamp of Escherichia Coli DNA Polymerase III by Peptides from β2-Binding Proteins. Biochemistry 2004, 43 (19), 5661–5671. 10.1021/bi036229j. [DOI] [PubMed] [Google Scholar]
- Wijffels G.; Johnson W. M.; Oakley A. J.; Turner K.; Epa V. C.; Briscoe S. J.; Polley M.; Liepa A. J.; Hofmann A.; Buchardt J.; Christensen C.; Prosselkov P.; Dalrymple B. P.; Alewood P. F.; Jennings P. A.; Dixon N. E.; Winkler D. A. Binding Inhibitors of the Bacterial Sliding Clamp by Design. J. Med. Chem. 2011, 54 (13), 4831–4838. 10.1021/jm2004333. [DOI] [PubMed] [Google Scholar]
- Wolff P.; Oliéric V.; Briand J. P.; Chaloin O.; Dejaegere A.; Dumas P.; Ennifar E.; Guichard G.; Wagner J.; Burnouf D. Y. Structure-Based Design of Short Peptide Ligands Binding onto the E. Coli Processivity Ring. J. Med. Chem. 2011, 54 (13), 4627–4637. 10.1021/jm200311m. [DOI] [PubMed] [Google Scholar]
- Gui W.-J.; Lin S.-Q.; Chen Y.-Y.; Zhang X.-E.; Bi L.-J.; Jiang T. Crystal Structure of DNA Polymerase III β Sliding Clamp from Mycobacterium Tuberculosis. Biochem. Biophys. Res. Commun. 2011, 405 (2), 272–277. 10.1016/j.bbrc.2011.01.027. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Kelso M. J.; Beck J. L.; Oakley A. J. Structural and Thermodynamic Dissection of Linear Motif Recognition by the E. Coli Sliding Clamp. J. Med. Chem. 2013, 56 (21), 8665–8673. 10.1021/jm401118f. [DOI] [PubMed] [Google Scholar]
- Wolff P.; Amal I.; Oliéric V.; Chaloin O.; Gygli G.; Ennifar E.; Lorber B.; Guichard G.; Wagner J.; Dejaegere A.; Burnouf D. Y. Differential Modes of Peptide Binding onto Replicative Sliding Clamps from Various Bacterial Origins. J. Med. Chem. 2014, 57 (18), 7565–7576. 10.1021/jm500467a. [DOI] [PubMed] [Google Scholar]
- McGrath A. E.; Martyn A. P.; Whittell L. R.; Dawes F. E.; Beck J. L.; Dixon N. E.; Kelso M. J.; Oakley A. J. Crystal Structures and Biochemical Characterization of DNA Sliding Clamps from Three Gram-Negative Bacterial Pathogens. J. Struct. Biol. 2018, 204 (3), 396–405. 10.1016/j.jsb.2018.10.008. [DOI] [PubMed] [Google Scholar]
- André C.; Martiel I.; Wolff P.; Landolfo M.; Lorber B.; Silva da Veiga C.; Dejaegere A.; Dumas P.; Guichard G.; Oliéric V.; Wagner J.; Burnouf D. Y. Interaction of a Model Peptide on Gram Negative and Gram Positive Bacterial Sliding Clamps. ACS Infect. Dis. 2019, 5 (6), 1022–1034. 10.1021/acsinfecdis.9b00089. [DOI] [PubMed] [Google Scholar]
- André C.; Veillard F.; Wolff P.; Lobstein A.-M.; Compain G.; Monsarrat C.; Reichhart J.-M.; Noûs C.; Burnouf D. Y.; Guichard G.; Wagner J. E. Antibacterial Activity of a Dual Peptide Targeting the Escherichia Coli Sliding Clamp and the Ribosome. RSC Chem. Biol. 2020, 1 (3), 137–147. 10.1039/D0CB00060D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsarrat C.; Compain G.; André C.; Engilberge S.; Martiel I.; Oliéric V.; Wolff P.; Brillet K.; Landolfo M.; Silva da Veiga C.; Wagner J.; Guichard G.; Burnouf D. Y. Iterative Structure-Based Optimization of Short Peptides Targeting the Bacterial Sliding Clamp. J. Med. Chem. 2021, 64 (23), 17063–17078. 10.1021/acs.jmedchem.1c00918. [DOI] [PubMed] [Google Scholar]
- A. Baumannii (PDB ID: 6AP4), E. Cloacae (PDB ID: 6AMQ), P. Aeruginosa (PDB ID: 6AMS), K. Pneumoniae (PDB ID: 6P81), S. Aureus (PDB ID: 7EVP), S. Pyogenes (PDB ID: 2AVT).
- Jöst C.; Nitsche C.; Scholz T.; Roux L.; Klein C. D. Promiscuity and Selectivity in Covalent Enzyme Inhibition: A Systematic Study of Electrophilic Fragments. J. Med. Chem. 2014, 57 (18), 7590–7599. 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
- Hunter J. C.; Gurbani D.; Ficarro S. B.; Carrasco M. A.; Lim S. M.; Choi H. G.; Xie T.; Marto J. A.; Chen Z.; Gray N. S.; Westover K. D. In Situ Selectivity Profiling and Crystal Structure of SML-8–73–1, an Active Site Inhibitor of Oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. U.S.A. 2014, 111 (24), 8895–8900. 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob C. G.; Upadhyay A. K.; Donner P. L.; Nicholl E.; Addo S. N.; Qiu W.; Ling C.; Gopalakrishnan S. M.; Torrent M.; Cepa S. P.; Shanley J.; Shoemaker A. R.; Sun C. C.; Vasudevan A.; Woller K. R.; Shotwell J. B.; Shaw B.; Bian Z.; Hutti J. E. Novel Modes of Inhibition of Wild-Type Isocitrate Dehydrogenase 1 (IDH1): Direct Covalent Modification of His315. J. Med. Chem. 2018, 61 (15), 6647–6657. 10.1021/acs.jmedchem.8b00305. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Itoh T.; Hori T.; Kato A.; Anami Y.; Yoshimoto N.; Yamamoto K. Identification of the Histidine Residue in Vitamin D Receptor That Covalently Binds to Electrophilic Ligands. J. Med. Chem. 2018, 61 (14), 6339–6349. 10.1021/acs.jmedchem.8b00774. [DOI] [PubMed] [Google Scholar]
- Paulussen F. M.; Schouten G. K.; Moertl C.; Verheul J.; Hoekstra I.; Koningstein G. M.; Hutchins G. H.; Alkir A.; Luirink R. A.; Geerke D. P.; van Ulsen P.; den Blaauwen T.; Luirink J.; Grossmann T. N. Covalent Proteomimetic Inhibitor of the Bacterial FtsQB Divisome Complex. J. Am. Chem. Soc. 2022, 144 (33), 15303–15313. 10.1021/jacs.2c06304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga S.; Shirai H.; Kobori M.; Hirayama N. Use of Amino Acid Composition to Predict Ligand-Binding Sites. J. Chem. Inf. Model. 2007, 47 (2), 400–406. 10.1021/ci6002202. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Bai J.; Bandla C.; Garcia-Seisdedos D.; Hewapathirana S.; Kamatchinathan S.; Kundu D. J; Prakash A.; Frericks-Zipper A.; Eisenacher M.; Walzer M.; Wang S.; Brazma A.; Vizcaino J. A. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50 (D1), D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry proteomics raw data have been deposited to the ProteomeXchange Consortium via the PRIDE49 partner repository with the following project accession number PXD042384 and 10.6019/PXD042384.