

SUMMARY
Mycobacterium tuberculosis (Mtb) possesses an arsenal of virulence factors to evade host immunity. Previously, we showed that the Mtb protein CpsA, which protects Mtb against the host NADPH oxidase, is required in mice during the first 3 weeks of infection but is thereafter dispensable for full virulence. Using flow cytometry, we find that ΔcpsA Mtb is retained in alveolar macrophages, impaired in recruiting and disseminating into monocyte-derived cells, and more likely to be localized in airway cells than wild-type Mtb. The lungs of ΔcpsA-infected mice also have markedly fewer antigen-specific T cells, indicating a delay in adaptive immunity. Thus, we conclude that CpsA promotes dissemination of Mtb from alveolar macrophages and the airways and generation of an adaptive immune response. Our studies of ΔcpsA Mtb show that a more effective innate immune response against Mtb can be undermined by a corresponding delay in the adaptive immune response.
In brief
Grigsby et al. demonstrate that M. tuberculosis CpsA promotes bacterial dissemination from alveolar macrophages to other myeloid cells. While ΔcpsA Mtb is attenuated during the first weeks of infection, it reaches nearly the same burden as wild-type (WT) Mtb, likely due to a delay in adaptive immunity and myeloid cell activation.
Graphical abstract
INTRODUCTION
Despite an extraordinary amount of progress made in the effort to reduce the global burden of tuberculosis (TB), Mycobacterium tuberculosis (Mtb) continues to cause 1.5 million deaths each year, second only to SARS-CoV-2. The rising incidence and deleterious effect of drug resistance on treatment outcomes have made it a global health threat and a public health crisis.1 Bacille Calmette-Guérin (BCG) has been in use for almost a century, but a more effective vaccine has so far eluded research efforts due in part to the incomplete understanding of the anti-mycobacterial immune response and the various mechanisms of Mtb to counteract the immune response.2
The host immune response against Mtb can be divided into two major phases: the innate immune phase, which is insufficient to control Mtb infection, and the adaptive immune phase, which is critical for establishing control and preventing disseminated disease.3 During the innate immune phase, the bacilli replicate relatively unhindered, initially in tissue-resident alveolar macrophages (AMs) and then in additional myeloid cell types that are recruited to the site of infection. Monocyte-derived dendritic cells (DCs) play a key role in transporting Mtb antigens from the lung to the draining lymph node. Their migration to the draining lymph node is delayed during TB relative to other pulmonary infections,4,5 but eventually conventional DCs in the lymph node present antigen to naive T cells to initiate the adaptive immune response. Once antigen-specific T cells arrive in the lungs, the immune system generally controls bacterial replication.
The host-pathogen interactions that underlie bacterial survival and cell-to-cell spread during the innate immune phase of TB are not well understood. In mice, AMs are the predominant cell type infected for the first 2 weeks.6 They exhibit a nuclear factor erythroid 2-related factor 2 (NRF2)-driven antioxidant transcriptional response that correlates with impaired control of Mtb growth.7,8 Based upon the transcriptional responses of the bacilli when they are within AMs, Mtb accesses host iron and fatty acids, experiences minimal oxidative and nitrosative stress, and has a high replicative capacity in this environment.9,10 Moreover, selective depletion of AMs reduces the lung Mtb burden in mice,9,11 supporting the idea that AMs are a particularly permissive niche that facilitate the establishment of infection. However, by 3 weeks post infection (wpi), infected AMs can exhibit a proinflammatory response.12 During this time, infected AMs migrate into the lung parenchyma, and the bacilli diversify their niche by infecting polymorphonuclear leukocytes (PMNs), DCs, and a variety of tissue-resident and recruited macrophage populations. PMNs create a permissive niche for Mtb replication,13–17 as do certain macrophage populations.9,18
The myeloid cells that become infected have been difficult to classify as they are recruited, proliferate, respond to stimuli, and differentiate into macrophages and DCs in the inflammatory environment of the Mtb-infected lung. Broadly, in addition to the AMs, CD11c+ populations, which have been classified as either DCs or macrophages, as well as CD11clow/intermediate populations, previously called “interstitial macrophages” or “recruited macrophages,”4,6,9,12,19 become infected. Recent data suggest a complex picture. Single-cell RNA sequencing data suggest there are at least four distinct Mtb-infected macrophage populations, and their ability to restrict Mtb infection may vary over the course of infection.18 After 4 weeks of infection, a CD11c− population (termed MDC1) was found to be lysosome poor and to be highly infected.20 Other work describes a subset of CD11c+ monocyte-derived macrophage-like cells that by 6 wpi become the predominantly infected cell type, with up to 30% of this population in the lung infected.12 The bacilli within this cell population also appear to experience less stress, suggesting that this CD11c+ macrophage population is more permissive than other monocyte-derived populations.18 Thus, it is not entirely clear which cell populations control intracellular Mtb or how that changes over the course of infection.
We previously demonstrated that the Mtb virulence factor CpsA inhibits NADPH oxidase assembly and generation of reactive oxygen species (ROS) on the Mtb-containing phagosome in bone marrow-derived macrophages (BMDMs).21 ROS are key players in immunity through both direct antimicrobial activity and immunomodulatory mechanism. Apart from its direct microbicidal effects, ROS also act as signaling molecules and have important immunomodulatory effects on the host, including during TB infection.22 One important immunomodulatory role for ROS is in the non-canonical autophagy pathway of LC3-associated phagocytosis (LAP),23,24 which is also inhibited by CpsA.21
When we infected mice with a mutant lacking CpsA, we found that the mutant was highly attenuated 17 days post infection (dpi), demonstrating that it was defective in surviving the innate immune response. Interestingly, despite its marked attenuation early in infection, we found that ΔcpsA Mtb was able to reach bacterial burdens nearly equivalent to WT Mtb by 60 dpi.21 The apparent shift in the ability of ΔcpsA Mtb to survive in vivo corresponds in time to the shift from innate to adaptive immunity, as well as a shift in the cell types infected from AMs to PMNs, DCs, and diverse tissue-resident and recruited macrophages. Thus, by understanding these unusual dynamics, we sought to gain insight into how Mtb evades innate immunity and undergoes cell-to-cell spread. To characterize the immune response elicited by ΔcpsA Mtb, we profiled lung cells of aerosol-infected mice by flow cytometry. In our analysis, we found that ΔcpsA Mtb exhibits a defect in dissemination from AMs to recruited myeloid cells and in egress from the airways into the lung interstitium.
RESULTS
ΔcpsA and ΔcpsA ΔkatG Mtb are delayed in achieving normal bacterial burdens in vivo
We previously reported that the degree of attenuation of ΔcpsA Mtb changed during the course of infection in mice. We repeated these experiments using the previously generated ΔcpsA Mtb.21 When cultured in vitro, Mtb strains can undergo loss of phthiocerol dimycocerosate (PDIM), an important virulence factor. To ensure integrity of the PDIM locus, we first passaged the isolate through mice, and then we analyzed envelop lipids by high-pressure liquid chromatography mass spectrometry (HPLC-MS) and whole-genome sequencing (WGS) to select a clone that made PDIM. As we found previously, ΔcpsA Mtb was markedly attenuated in C57BL/6J mice after aerosol infection at 2 and 3 wpi. However, by 6 wpi, the bacterial burden of mice infected with ΔcpsA Mtb was similar to that of mice infected with WT Mtb (Figure 1A). Complementation of the mutant with cpsA under its endogenous promoter fully rescued its growth defect (Figures S1A and S1B). These infection dynamics were surprising, particularly since during the first 2 weeks, AMs should be the predominant cell type infected, and they are thought to be particularly permissive during initial infection.
Figure 1. ΔcpsA and ΔcpsA ΔkatG Mtb are highly attenuated during initial infection but not at 6 wpi.
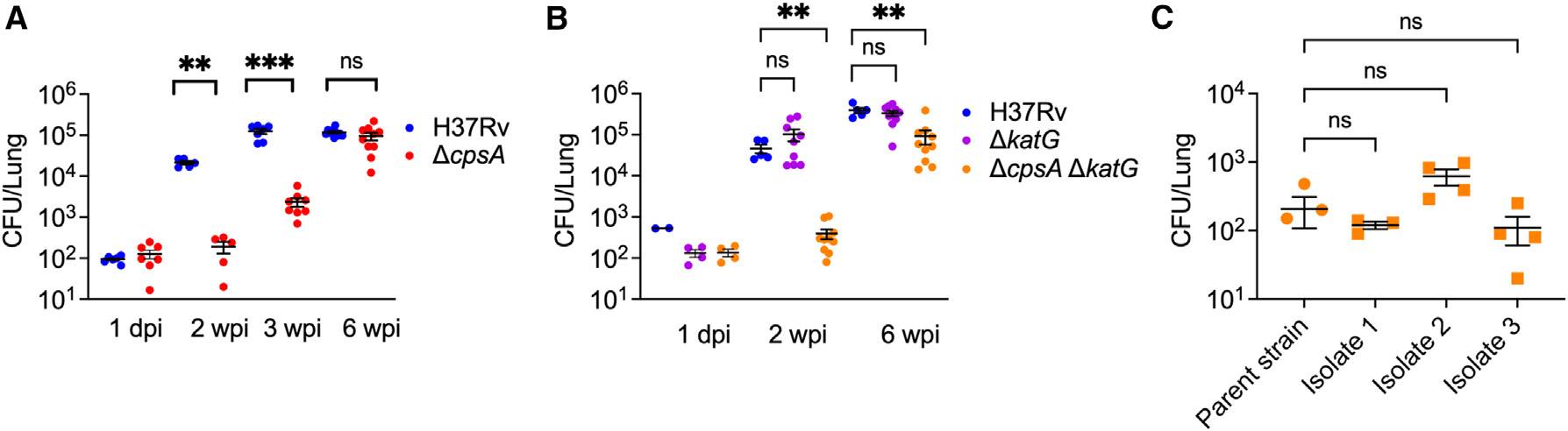
(A–C) C57BL/6J mice were infected with WT (H37Rv), ΔcpsA, ΔcpsA ΔkatG, and mouse-passed Mtb strains (isolates 1, 2, and 3) as indicated with ~100 CFU by aerosol, and CFUs in the lung were enumerated. (A and B) Lungs were harvested 1 dpi and 2, 3, or 6 wpi as indicated. (C) Lungs were harvested at 2 wpi. Each data point represents one mouse. Error bars indicate mean ± SEM. n = 3–11 mice/group from two (A and B) or one (C) independent experiments. ns, not significant; *p < 0.05; **p < 0.01; ****p < 0.0001. Two-way ANOVA with Sidak’s multiple comparisons test (A and B) or Kruskal-Wallis test with Dunn’s multiple comparisons test (C). See also Figures S1 and S2.
We considered a variety of explanations for why ΔcpsA Mtb was specifically attenuated early during infection. Since we previously showed that CpsA inhibits the NADPH oxidase,21 one possibility was that the activity of the NADPH oxidase is higher during initial infection. If this is the case, then strains lacking KatG, a catalase-peroxidase that protects Mtb from ROS,25 should also be attenuated at the same time periods. However, after aerosol infection, ΔkatG Mtb was indistinguishable from WT Mtb in its growth at 2 wpi (Figure 1B). This is consistent with a previous report.25 In contrast to that study, however, we found that ΔkatG Mtb was not attenuated at 6 wpi (Figure 1B).25 This discrepancy may be due to differences in background strain (Erdman versus H37Rv), dose and route of infection (106 colony-forming units [CFU] intravenously versus ~100 CFU by aerosol), or the age and microbiota of the mice. One explanation for the difference in phenotype between the ΔkatG Mtb and ΔcpsA Mtb during the first 2 weeks of the infection is that CpsA protects ΔkatG Mtb by inhibiting ROS generation. We predicted that if this were the case, then ΔcpsA ΔkatG Mtb would be more attenuated than either single-mutant strain. To test this possibility, we generated the double-mutant strain by deleting cpsA from ΔkatG Mtb using mycobacterial recombineering,26 and we verified PDIM production by mass spectrometry (Figure S2). We found that ΔcpsA ΔkatG Mtb exhibited a similar growth pattern to ΔcpsA (Figure 1B); the double mutant was attenuated at 2 wpi, and remarkably, the bacterial burden was close to that of WT Mtb at 6 wpi, though still attenuated. Thus, although mutants lacking CpsA and KatG behave similarly in BMDMs in conferring resistance to the NADPH oxidase,21 they have very distinct phenotypes in vivo. With time, both ΔcpsA Mtb and ΔcpsA ΔkatG Mtb achieve bacterial burdens similar to WT Mtb.
One explanation for our findings is that mutations occur during growth in vivo that allow ΔcpsA Mtb and ΔkatG ΔcpsA Mtb to overcome the loss of CpsA. To determine whether second-site suppressors develop, we extracted genomic DNA from 20 ΔkatG ΔcpsA colonies that were isolated from five different mice at 6 wpi and performed Illumina WGS. Mutations that were present in the parent ΔkatG ΔcpsA strain were excluded from further analysis. We did not find any unique mutations in the mouse-passaged ΔcpsA ΔkatG isolates that, based upon the available literature, could explain reversal of the attenuated phenotype (Table S1). To further rule out the possibility of compensatory mutations, we infected C57BL/6J mice with three of the mouse-passaged ΔcpsA ΔkatG Mtb isolates. 2 weeks after infection, we compared the bacterial burden in the parent ΔcpsA ΔkatG Mtb strain and the three mouse-passaged isolates; the mouse-passaged strains did not exhibit any increase in virulence that would be expected if they had acquired compensatory mutations (Figure 1C). Thus, we conclude that the growth pattern of ΔkatG ΔcpsA Mtb is an inherent property of the strain, not due to second-site suppressor mutations acquired during in vivo growth.
CpsA is required for chronic infection
The recovery of ΔcpsA Mtb at 6 wpi suggested that CpsA might only be essential for full virulence during the early, innate phase of infection and dispensable during chronic infection. To test this possibility, we constructed a strain of Mtb that conditionally expresses CpsA by complementing ΔcpsA Mtb with an integrating plasmid containing cpsA under control of a promoter that can be repressed by anhydrotetracycline (ATc) or doxycycline.27 This allowed us to infect mice with Mtb that expressed CpsA until chronic infection was established and then to deplete CpsA by feeding the mice chow containing doxycycline (Figure 2A). We verified that the strain (called TetOFF-CpsA) expressed endogenous levels of CpsA in the absence of ATc and that CpsA expression was reduced by ATc during growth in vitro (Figure 2B). Furthermore, we showed that if mice were given doxycycline immediately post infection, then the TetOFF-CpsA strain was highly attenuated, similar to ΔcpsA Mtb in vivo (Figure S2C).
Figure 2. CpsA is required during chronic infection.
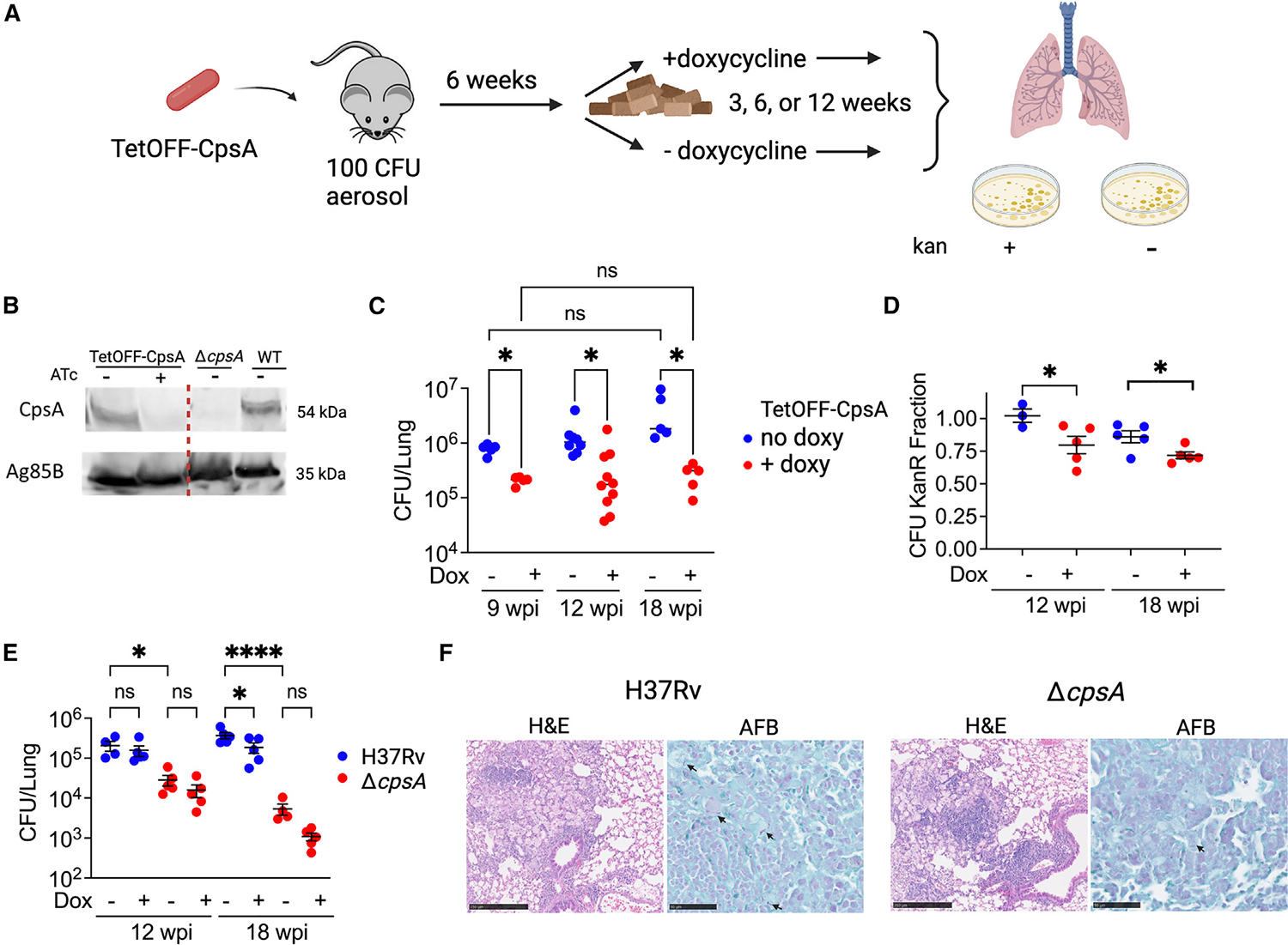
(A) Schematic illustrating the experimental strategy to test the requirement of CpsA during chronic infection.
(B) Immunoblot of CpsA from Mtb whole-cell lysate of TetOFF-CpsA, WT, and ΔcpsA Mtb grown in presence or absence of ATc as indicated. Ag85B served as a loading control. All samples were run on the same gel; the line indicates where the image was cropped to remove irrelevant lanes.
(C) Lung CFUs from mice infected with TetOFF-CpsA and treated as shown in (A).
(D) KanR fraction of bacteria isolated from (C) calculated by dividing CFUs from plates containing kanamycin by CFU counts from plates without antibiotics.
(E and F) Mice were infected with ~100 CFU H37Rv Mtb or ΔcpsA Mtb (without the TetOFF plasmid) and treated with or without doxycycline starting at 6 wpi. (E) Lung CFUs at indicated time points. (F) Representative hematoxylin and eosin (H&E) and acid-fast bacilli stain (AFB) from lungs of mice infected with H37Rv Mtb or ΔcpsA Mtb (without doxy) for 18 weeks. Scale bars correspond to 250 μm (H&E) or 50 μm (AFB). Arrows point to AFB-positive bacilli. (C–E) Each data point represents one mouse. Data bars indicate mean ± SEM. Data consist of 3–10 mice per group from two independent experiments. *p < 0.05; ****p < 0.0001. Two-way ANOVA with Sidak’s multiple comparisons test (C), Student’s t test with Welch’s correction (D), and one-way ANOVA with Sidak’s multiple comparisons test (E). See also Figure S1.
To determine whether CpsA is required during chronic infection, we infected mice with 100 CFU of the TetOFF-CpsA strain by aerosol and fed them normal chow for 6 weeks, allowing the strain to express CpsA while it established chronic infection. At 6 wpi, one group of mice was switched to doxycycline-containing chow to suppress CpsA expression, while the other group continued a normal diet. At 3, 6, and 12 weeks after the initiation of doxycycline treatment, we evaluated the bacterial burden in the two groups. We found that when CpsA was suppressed, the bacterial burden was significantly lower at 3 weeks post treatment (9 wpi) and remained lower through the 12-week post-treatment time point (18 wpi) (Figure 2C). In addition, we assessed CFUs on plates containing kanamycin, the marker on the plasmid that encoded CpsA. We found that nearly all of the isolates from the mice on normal chow retained the plasmid, whereas isolates from mice treated with doxycycline, in which CpsA expression was suppressed, had substantial plasmid loss, demonstrating that there was selective pressure to retain CpsA expression (Figure 2D). As a control, we tested the impact of doxycycline on H37Rv and ΔcpsA Mtb without the TetOFF-CpsA plasmid, and doxycycline had minimal effect on bacterial burden (Figure 2E). These data demonstrate that when the initial adaptive immune response is established in the context of a WT Mtb infection, CpsA plays a role in Mtb survival during chronic infection.
To determine whether CpsA is also required during chronic infection after the mutant established normal bacterial burden at 6 wpi, we compared mice infected with ΔcpsA Mtb to WT Mtb at 12 and 18 wpi. After establishing a normal bacterial burden at 6 wpi, ΔcpsA Mtb was slowly cleared during chronic infection, so that by 18 wpi, the burden of ΔcpsA Mtb was nearly 100-fold less than H37Rv (Figure2E). Furthermore, histopathology revealed less inflammatory cell infiltrate and fewer acid-fast bacilli in the lungs of ΔcpsA-infected mice compared to those of H37Rv-infected mice at 18 wpi (Figure 2F). Combined, these data show that CpsA is required in both acute and chronic infection.
ΔcpsA Mtb induces less inflammation during early infection
Since CpsA was required during both the innate immune phase and chronic infection, we sought to understand why ΔcpsA Mtb was able to achieve a bacterial burden similar to WT Mtb at 6 wpi. To characterize the inflammatory response to infection, initially we infected C57BL/6J mice with ~100 CFU of H37Rv and ΔcpsA Mtb by aerosol and analyzed lung cells by flow cytometry. Adapting previously published gating strategies,6,28 we were able to distinguish inflammatory monocytes, conventional DCs, PMNs, and three macrophage subsets. The macrophage subsets were AMs (Siglec-F+ CD11c+) and two non-AMs (Siglec-F−) populations that differed in their expression of CD11c (CD64+ CD11b+ MHCIIvar CD11c− Ly6C− and CD64+ CD11b+ MHCIIhigh CD11c+), which we refer to CD11c− and CD11c+ non-AMs. These populations are composed of tissue-resident and monocyte-derived macrophages (MDMs; Figure S3). At 3 wpi, ΔcpsA-infected mice had significantly fewer monocytes, CD11c+ non-AMs, and DCs in the lungs compared to mice infected with WT Mtb; the myeloid cell infiltrate in ΔcpsA-infected mice was similar to uninfected mice (Figure 3A). We examined cytokine and chemokine levels in WT and ΔcpsA Mtb-infected mice the preceding week (2 wpi), and there were significantly lower levels of CCL2, CCL3, CCL7, and CXCL10 and a trend toward lower CCL5 in ΔcpsA Mtb-infected mice compared to WT (Figures 3B and S4A). The reduced levels of these chemokines might account for the reduced numbers of monocytes and MDMs in the lungs of ΔcpsA Mtb-infected mice.29 In addition, the AMs, monocytes, CD11c-non-AMs, and PMNs all had reduced expression of MHC II in ΔcpsA Mtb-infected mice compared to WT mice (Figure 3C). (CD11c+ non-AMs and DCs were MHC II positive by definition in our gating strategy.) These findings demonstrated that the attenuation of ΔcpsA Mtb is not explained by increased macrophage recruitment or activation. At 4 wpi, there continued to be significantly fewer CD11c+ non-AMs and DCs in the lungs of ΔcpsA Mtb-infected mice, as well as reduced MHCII expression in all myeloid cells. These alterations in the inflammatory response were restored to WT levels in the complemented strain (Figures 3D and 3E). At 6 wpi, when lung CFUs were equivalent between WT and ΔcpsA Mtb, there was no significant difference in the number of myeloid cells in the lungs (Figure 3F) or their MHC II expression (Figure 3G). Furthermore, histopathology revealed a similar degree of inflammatory cell infiltrate and acid-fast bacilli between the lungs of H37Rv-infected and ΔcpsA-infected mice (Figure 3H). At 6 wpi, cytokine responses in the whole-lung homogenates were also similar (Figures 3I and S4B). Interestingly, mice infected with ΔcpsA Mtb at 6 wpi had less IL-1β and CCL5 and more CCL2, CCL7, and CXCL10 than those infected with WT Mtb (Figure 3I). The reduced IL-1β is in accordance with our previous findings that ΔcpsA Mtb elicits less IL-1β in BMDMs than WT Mtb.21 Elevated levels of CCL2, CCL7, and CXCL10 at 6 wpi may reflect an enhanced drive to recruit monocytes and macrophages so that they reach the same levels in the lungs of ΔcpsA-infected mice as in H37Rv-infected mice. To conclude, in a low-dose aerosol infection, the level of inflammation elicited by ΔcpsA Mtb was markedly reduced during early infection and was comparable to WT Mtb by 6 wpi; this was accompanied initially by lower levels of CCL2, CCL3, CCL7, and CXCL10 at 2 wpi and then higher levels of CCL2, CCL7, and CXCL10 at 6 wpi.
Figure 3. ΔcpsA Mtb elicits a reduced myeloid response early in infection.
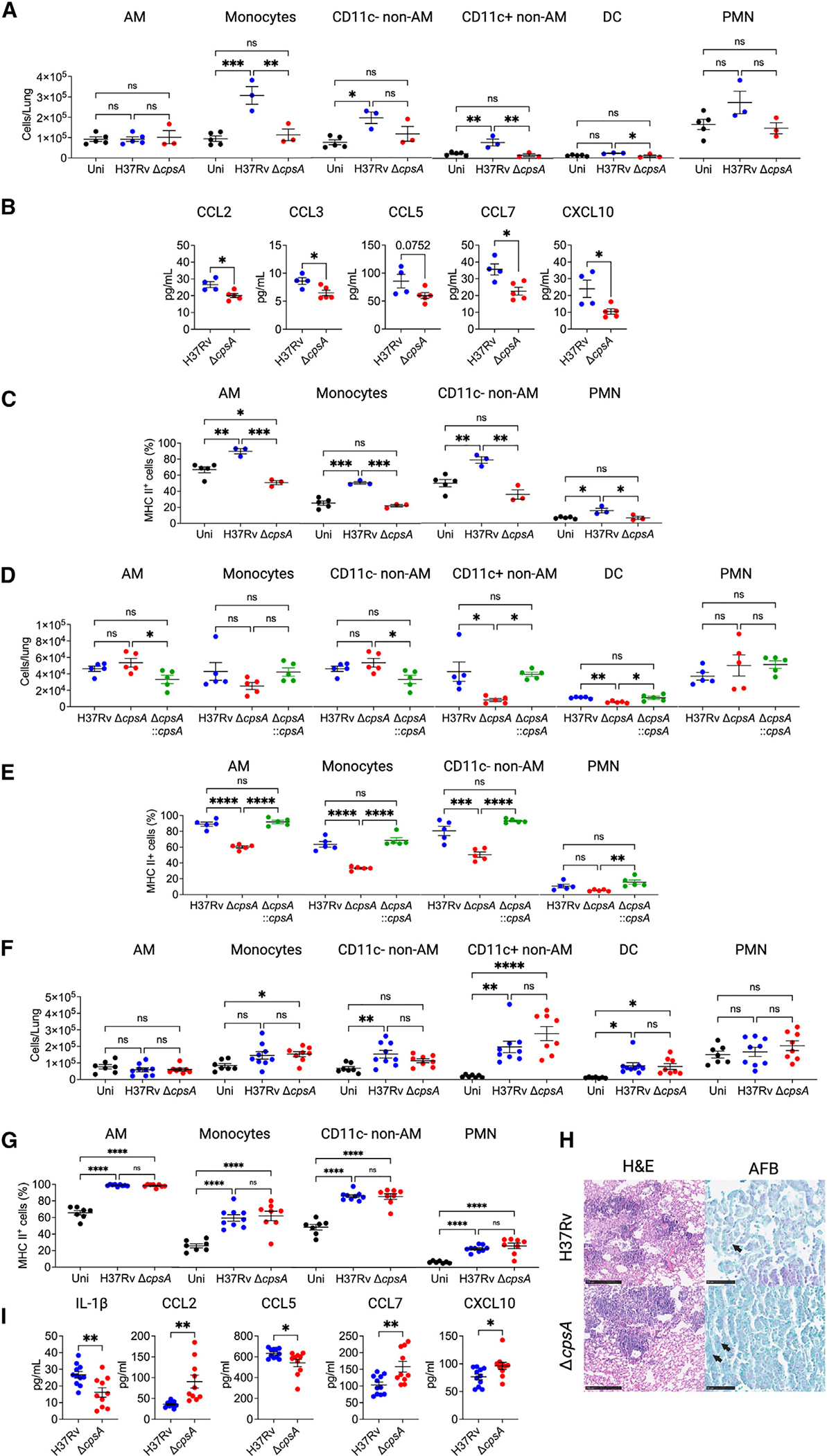
Mice were uninfected or infected with ~100 CFU of H37Rv Mtb or ΔcpsA Mtb for 3 (A–C), 2 (B), or 6 weeks (F–H). Mice were infected with ~30 H37Rv Mtb, ΔcpsA Mtb, or ΔcpsA::cpsA Mtb for 4 weeks (D and E). Lungs were harvested for flow cytometry (A and C–G), cytokine analysis (B and I), and histopathology (H). (A, D, and F) Absolute cell counts of myeloid cell populations. (C, E, and G) MHC II expression shown as the percentage positivity of the indicated cell population. Data consist of seven to nine mice per group from three independent experiments (F–I) or three to five mice per group from one experiment (A–E). Each data point corresponds to one mouse. Error bars indicate mean ± SEM. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. One-way ANOVA with Tukey’s multiple comparisons test (A and C–G) or Student’s t test (B and I). See also Figures S3, S4, and S5.
The increase in ΔcpsA Mtb burden at 6 wpi does not depend on CCR2
The above studies demonstrated that mice infected with ΔcpsA Mtb had fewer monocytes and monocyte-derived cells (both CD11c+ non-AMs and DCs) at 3 weeks of infection, which normalized by 6 wpi. Thus, the ability of ΔcpsA Mtb to achieve near normal bacterial burden at 6 wpi might be explained by its ability to eventually gain access to a permissive monocyte or monocyte-derived population. The recruitment of monocytes and MDMs to the lungs depends upon CCR2, which is required for their egress out of bone marrow.29 To test whether monocyte or monocyte-derived cells are required for a permissive niche for ΔcpsA, we tested whether CCR2 was required for bacterial burden at 6 wpi. We infected Ccr2−/− mice with ~100 CFU ΔcpsA Mtb by aerosol. As expected, there were significantly fewer monocytes and CD11c+ non-AMs in the lungs of Ccr2−/− mice compared to WT mice (Figure S5A). However, there was no difference in the bacterial burden of WT Mtb and ΔcpsA Mtb in the lung of Ccr2 KO mice at 2 or 6 wpi (Figure S5B). Thus, CCR2-dependent recruitment of monocytes and monocyte-derived cells is not required for ΔcpsA Mtb to achieve nearly normal bacterial burdens at 6 wpi.
There is a delayed adaptive immune response to ΔcpsA Mtb
An alternative possibility is that ΔcpsA Mtb, although attenuated, is able to “catch up” to WT Mtb because it elicits a less robust or delayed adaptive immune response, providing a longer window of time for the bacilli to expand while WT Mtb are restricted by the adaptive immune response. To evaluate this possibility, we compared T cell responses to WT Mtb and ΔcpsA Mtb. At 3 wpi, we found that mice infected with ΔcpsA Mtb had significantly fewer CD4 and CD8 T cells in the lung (Figure 4A), as well as fewer NK and γδ T cells, than mice infected with WT Mtb. The mice infected with ΔcpsA Mtb were similar to uninfected mice. The percentage of CD4 and CD8 T cells expressing CD44, a marker of T cell activation, was also significantly lower in mice infected with ΔcpsA Mtb (Figure 4B), compared to WT Mtb-infected mice. Since T cells are just arriving to the site WT Mtb infection at 3 wpi, we looked at 4 wpi, by which time there is a robust adaptive response in WT Mtb-infected mice. At 4 wpi, the adaptive immune response in the lung ΔcpsA Mtb continued to be substantially lower than in mice infected with WT Mtb, and the defect was restored in mice infected with the complemented strain (Figures 4C and 4D). As we had seen with myeloid cell recruitment, by 6 wpi, the mice infected with ΔcpsA Mtb were not different from mice infected with WT Mtb in terms of CD4 T cell, CD8 T cell, NK cell, and γδ T cell number or CD44 expression (Figures 4E and 4F). To further characterize the adaptive immune response, we examined the cytokine profile of CD4 T cells at 4 wpi. All CD4 T cell subsets were lower in ΔcpsA Mtb-infected mice compared to mice infected with WT Mtb or the complemented strain (Figures 4G and S6A). The deficiency was particularly notable for mono- and polyfunctional IFN-γ-expressing T cells, which were found at reduced frequency in ΔcpsA Mtb-infected mice, whereas monofunctional IL-2-expressing T cells were found at a higher frequency (Figure 4H). Fewer IFN-γ-expressing T cells may explain the reduced MCHII expression in myeloid cells (Figure 3C). In addition, using tetramer staining, we found significantly fewer ESAT6- and Ag85-specific CD4 cells and TB10.4-specific CD8 T cells in ΔcpsA Mtb-infected mice, both in terms of absolute numbers and as a percent of total T cells (Figures 4I–4K and S6B). CD4 T cell priming during TB infection is tightly linked to the number of Mtb bacilli in the draining lymph node, with T cell activation occurring after a threshold of >103 bacilli are present.30 To determine whether the defect in T cell arrival in the lungs of ΔcpsA Mtb-infected mice was due to a defect in bacterial dissemination to mediastinal lymph nodes (MLNs), we measured the bacterial burden in MLNs at 4 wpi. While all of the mice infected with ΔcpsA::cpsA had more than 5 × 103 Mtb in the MLNs, none of the ΔcpsA-infected mice had more than 103, and several had no detectable bacilli (Figure 4L). We conclude that the adaptive immune response to ΔcpsA Mtb is delayed beyond that of WT Mtb.
Figure 4. The adaptive immune response is delayed in mice infected with ΔcpsA Mtb.
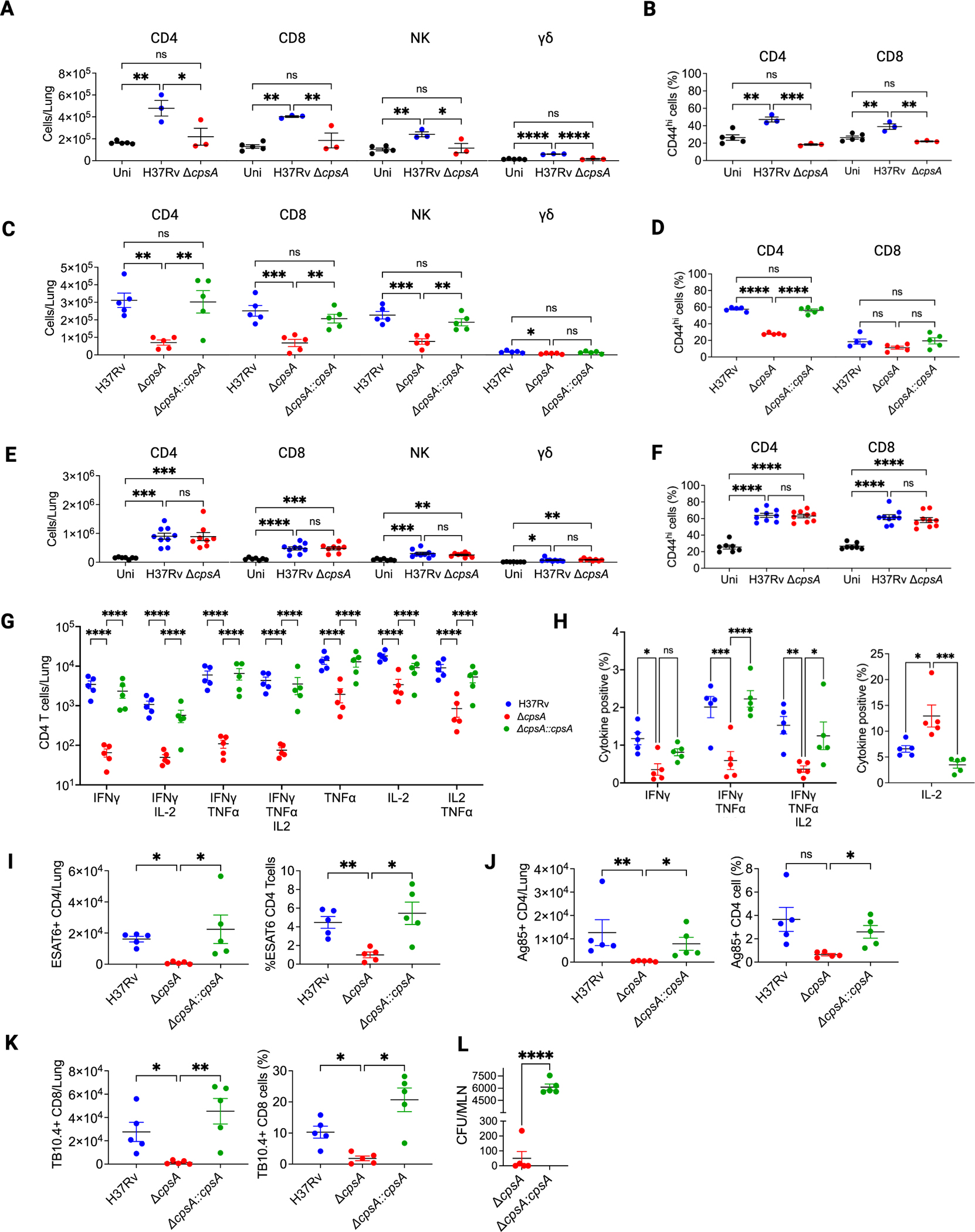
(A–F) Mice were uninfected or infected with 100 CFU of H37Rv or ΔcpsA for 3 (A and B) or 6 weeks (E and F) or ~30 H37Rv, ΔcpsA, or ΔcpsA::cpsA for 4 weeks (C, D, and G–L). Lungs were harvested for flow cytometry (A–K), and mediastinal LNs were analyzed for CFUs (L). (A, C, and E) Absolute cell counts of lymphoid cell populations. (B, D, and F) Percentage of CD44+ CD4 and CD8 T cells.
(G) Absolute counts of polyfunctional CD4 T cells producing TNFα, IFNγ, and IL-2 upon stimulation with PMA and ionomycin were assessed by flow cytometry.
(H) Percentage of CD4 T cells with the indicated cytokine expression.
(I–K) Absolute cell counts and frequencies of tetramer Ag85- or ESAT6-binding CD4 T cells (I–J) and TB10.4-binding CD8 T cells (K) among lung T cells.
(L) CFUs from MLNs at 4 wpi. Data consist of three to five mice per group from one experiment (A–D and G–L) and seven to nice mice per group from three independent experiments (E and F). Each data point corresponds to one mouse. Error bars indicate mean ± SEM. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. One-way ANOVA with Tukey’s multiple comparisons test (A–F), two-way ANOVA with Holm-Sidak’s multiple comparisons test (G) or Tukey’s multiple comparisons test (H), Kruskal-Wallis for absolute cell counts and Brown-Forsythe and Welch ANOVA for percentages (I–K), or Student’s t test (L). Uni, uninfected. See also Figures S3 and S6.
ΔcpsA Mtb induces a muted inflammatory response even when lung bacterial burdens are similar to WT H37Rv
The reduced myeloid and lymphoid infiltrate in the lungs of ΔcpsA-infected mice at 3 wpi might be a consequence of the reduced bacterial burden at that time. In order to compensate for the attenuation of ΔcpsA Mtb, we repeated these studies using a higher inoculum for ΔcpsA Mtb than H37Rv, which allowed us to achieve more equivalent bacterial burdens at 3 wpi in some mice (Figure 5A). For this analysis, we excluded mice infected with H37Rv or ΔcpsA that had lung CFUs outside of the range of 105–106 CFU. Even when bacterial burdens were similar, we found significantly fewer CD11c+ non-AMs and DCs in the lungs of ΔcpsA-infected mice compared to H37Rv-infected mice at 3 wpi (Figure 5B). Expression of MHCII and CD80, a marker of macrophage and DC activation, was significantly lower in myeloid cells from ΔcpsA-infected mice (Figures 5C and 5D). While robust activation of adaptive immunity could be observed in H37Rv-infected mice at 3 wpi in the form of T cell recruitment, ΔcpsA-infected mice showed little activation of adaptive immunity. γδ T cells similarly were reduced in number in the lungs in ΔcpsA-infected mice (Figure 5E). Furthermore, T cells exhibited reduced expression of CD44 in the ΔcpsA-infected mice compared to mice infected with WT Mtb (Figure 5F). In these experiments, which used especially high doses of ΔcpsA, complementation of cpsA restored the cellular inflammatory response to infection (Figure S7). Cytokine analysis of whole-lung homogenate revealed reduced pro-inflammatory cytokines in ΔcpsA-infected mice, including IL-1β, IFN-γ, TNF-α, and IL-17, as well as less CCL2, CCL3, CCL5, CCL7, and CXCL10 (Figure 5G; Figure S4C). There was elevated IFN-β, which often shows a reciprocal relationship with IL1-β, and small changes in several other cytokines (Figure S4C). Thus, during the first 3 weeks of infection, CpsA promotes myeloid and lymphoid cell infiltration and activation in the lungs in response to Mtb infection, and this blunted inflammatory response is not simply a consequence of reduced bacterial burden.
Figure 5. ΔcpsA Mtb induces less inflammation than WT Mtb.
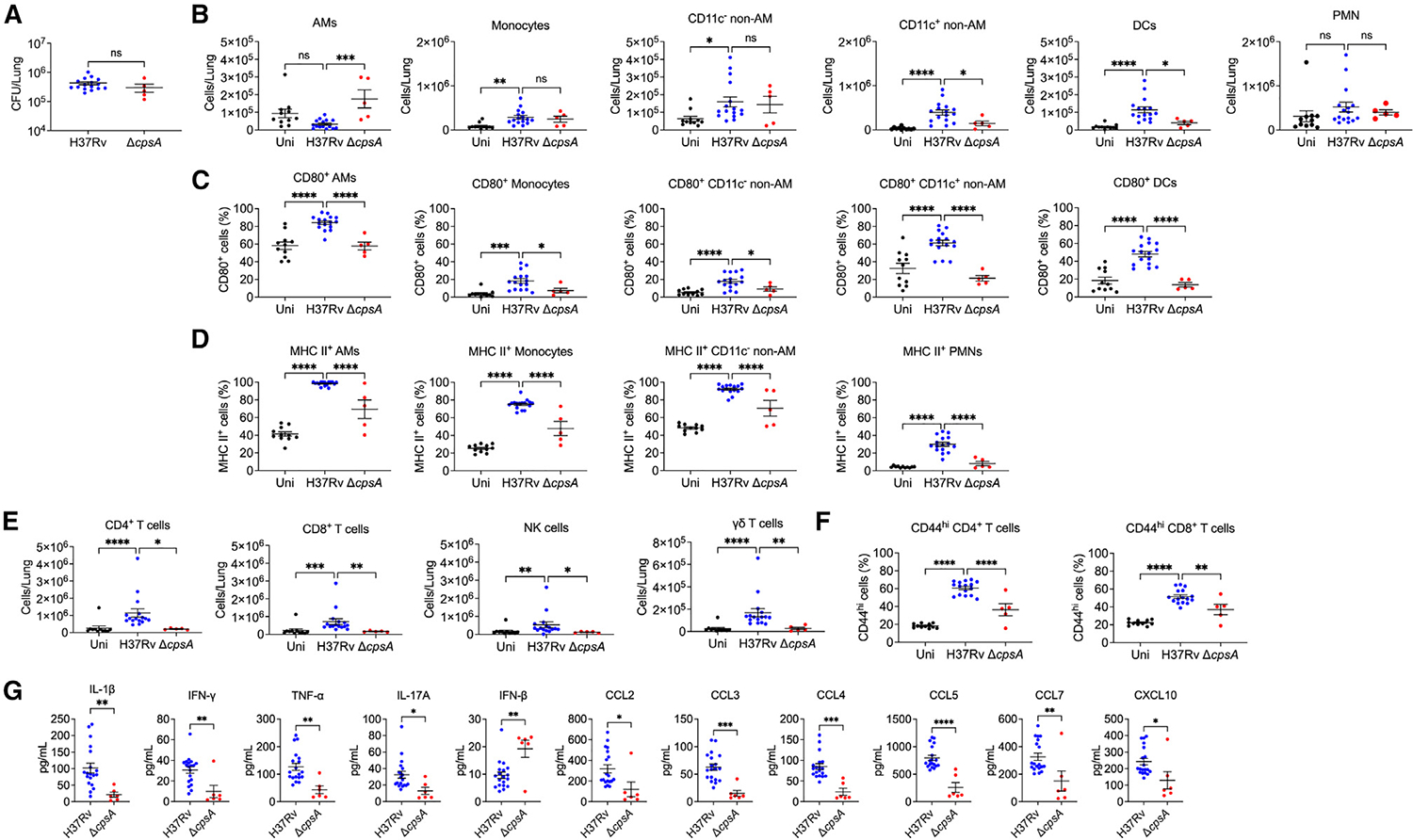
(A–F) Mice were uninfected or infected with H37Rv (CFU ~1,500) or ΔcpsA (CFU ~7,000) for 3 weeks, and lungs were analyzed for CFUs (A) and flow cytometry
(B–F). Absolute cell counts of (B) myeloid and (E) lymphoid cell populations were quantified. (C) CD80, (D) MHC II, and (F) CD44 expression is expressed as the percentage positivity of the indicated cell population.
(G) Cytokines measured by Luminex of whole-lung homogenate from mice. Data consist of 5–16 mice per group from at least two independent experiments. Each data point corresponds to one mouse. Error bars indicate mean ± SEM. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Mann-Whitney test (A), one-way ANOVA with Dunnett’s multiple comparisons test (B–D and F) or Kruskal-Wallis test with Dunn’s multiple comparisons test (E), and Student’s t test (G). Uni, uninfected. See also Figures S3, S4, and S6.
ΔcpsA Mtb is retained in alveolar macrophages
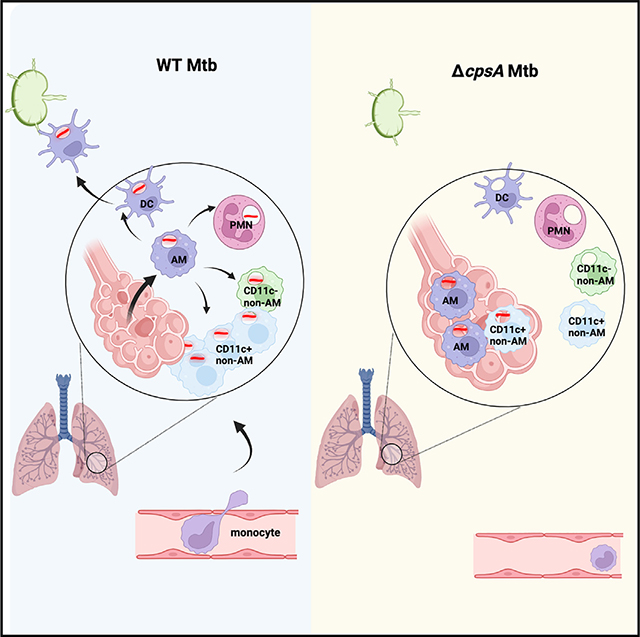
Since Mtb is known to infect different myeloid cells over the course of infection, we sought to investigate the cells infected by ΔcpsA Mtb in vivo. We were able to detect infected cells in the lungs by flow cytometry because the strains expressed GFP (Figure S8). To examine the distribution of infected cells, we analyzed the mice infected with the higher dose of ΔcpsA Mtb to achieve relatively similar CFUs at 3 wpi (CFU in Figure 5A). For this analysis, we excluded mice infected with H37Rv or ΔcpsA Mtb with CFUs outside of the range of 105–106. As has been previously published,6 at 3 wpi with WT Mtb, CD11c+ non-AMs and PMNs were the most prominently infected cell populations in terms of the fraction of the total infected cell population, and there were very few infected AMs. However, the distribution of GFP-positive cells in ΔcpsA-infected mice was markedly skewed toward AMs at a time when the infection had already shifted out of AMs into non-AMs and PMNs in H37Rv-infected mice (Figure 6A). Mice infected with ΔcpsA Mtb had significantly more infected AMs and fewer infected monocytes, non-AMs, DCs, and PMNs (Figures 6B and 6C). Thus, ΔcpsA Mtb exhibits a defect in dissemination out of AMs into recruited myeloid cells. To determine whether ΔcpsA Mtb can grow in AMs, we harvest AMs from uninfected mice and compared the ability of WT, ΔcpsA, and the complemented strain to grow intracellularly. In contrast to the attenuation of ΔcpsA in BMDMs,21 ΔcpsA Mtb grew similarly to WT Mtb and the complemented strain in AMs (Figure 6D). Since we previously showed that CpsA inhibits the NADPH oxidase (NOX2),21 we suspected that NADPH oxidase may play a role in the altered cell tropism of ΔcpsA Mtb. We compared WT and Nox2−/− mice infected with WT and ΔcpsA Mtb. Again, we excluded mice that had lung CFUs outside of the range of 105–106 (Figure 6E). Flow cytometry of lung cells from Nox2−/− mice at 3 wpi revealed that Nox2−/− mice infected with ΔcpsA Mtb had an intermediate phenotype between that of WT-infected WT mice and ΔcpsA-infected WT mice in terms of the fraction of infection found in AMs and CD11c+ non-AMs (Figure 6F). Therefore, we conclude that ΔcpsA Mtb has a defect in disseminating from AMs into other myeloid populations, and this appears to depend, in part, on NADPH oxidase.
Figure 6. ΔcpsA fails to efficiently spread from AMs to other myeloid populations.
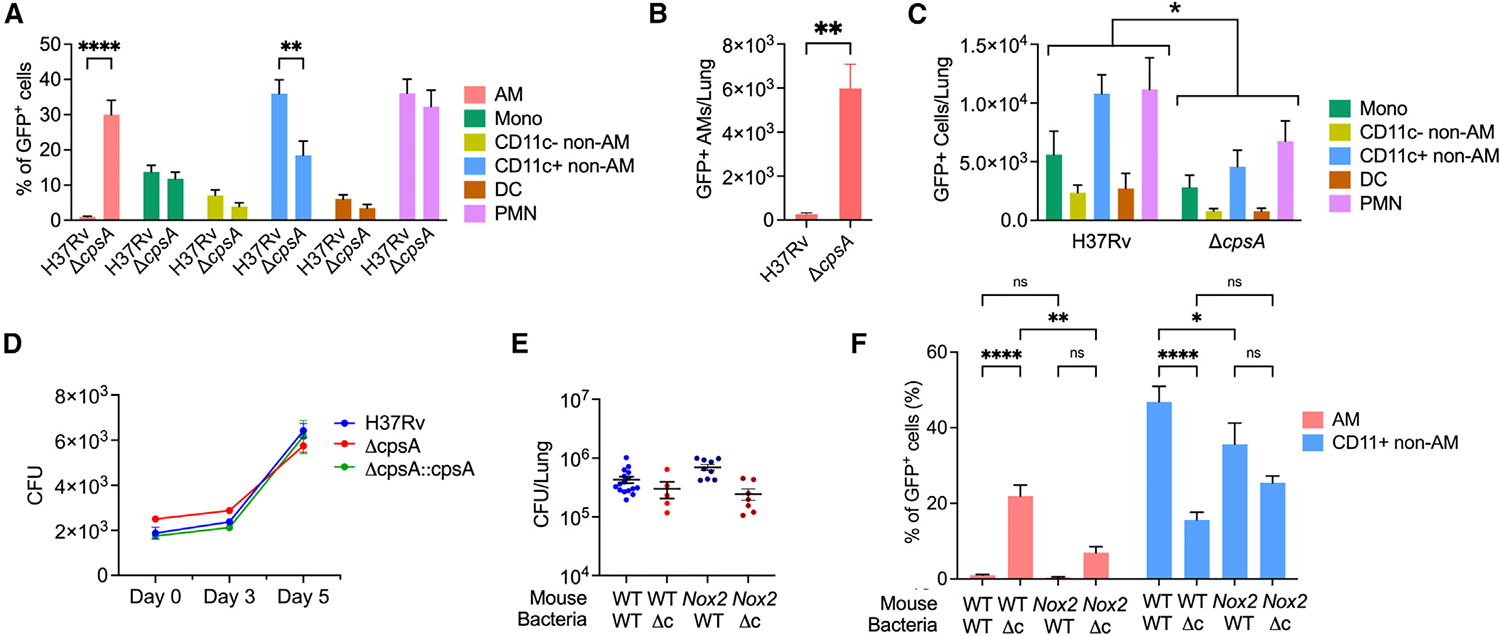
(A–C, E, and F) WT (C57BL/6) or Nox2−/− mice were infected with ~1,500 CFU GFP-expressing H37Rv or ~7,000 GFP-expressing ΔcpsA Mtb for 3 weeks, after which lungs were harvested for flow cytometry. (A and F) Percentage of GFP-positive cells of the indicated cell population compared to total GFP-positive cells from WT or Nox2−/− mice.
(B and C) Number of GFP-positive cells of the indicated cell population from WT mice.
(D) AMs were infected ex vivo with indicated strains, and CFUs were enumerated at indicated times.
(E) CFUs from lungs of WT and Nox2−/− mice.
(F) Percentage of GFP+ AMs and CD11c+ non-AMs in the total infected (GFP-positive) population from WT and Nox2−/− mice. Bars indicate mean ± SEM. Data consist of 6–16 mice per group from at least two independent experiments. ns, not significant; *p < 0.05; **p < 0.01; ****p < 0.0001. Two-way ANOVA with Sidak’s multiple comparisons test (A and F), Student’s t test with Welch’s correction (B), and two-way ANOVA (C). Δc indicates ΔcpsA. See also Figures S3 and S8.
ΔcpsA-infected cells are preferentially retained in the airways
Since previous studies have suggested that infected AMs migrate out of the airways into the lung interstitium before disseminating to other myeloid cell populations,6 we wondered if AMs infected with ΔcpsA Mtb were defective in migrating out of the airways. To examine the airway localization of infected cells, we infected mice with GFP-expressing WT and ΔcpsA Mtb. Again, we infected mice with a higher inoculum of ΔcpsA Mtb than WT Mtb to compensate for the attenuation of ΔcpsA Mtb. At 3 wpi, we instilled a fluorescently tagged αCD45 antibody intratracheally to label airway-localized leukocytes and then analyzed lung cells by flow cytometry (Figure 7A).31 First, we examined whether there were differences in the myeloid populations in the airways. In uninfected mice, approximately 60% of the airway cells were AMs, with monocytes, non-AMs, DCs, and PMNs all contributing more minor fractions to the airway (CD45+) cells. During infection with WT Mtb, AMs made up less than 20% of the total airway myeloid cell population, and there was a greater contribution from monocytes, CD11c+ non-AMs, DCs, and PMNs than in uninfected mice. Mice infected with ΔcpsA Mtb exhibited an intermediate phenotype, with AMs representing ~40% of the total cells and a significantly lower percentage of CD11c+ non-AMs in the airways compared to WT Mtb, as well as trends toward fewer monocytes and DCs (Figure 7B).
Figure 7. ΔcpsA-infected cells are preferentially retained in the airways.
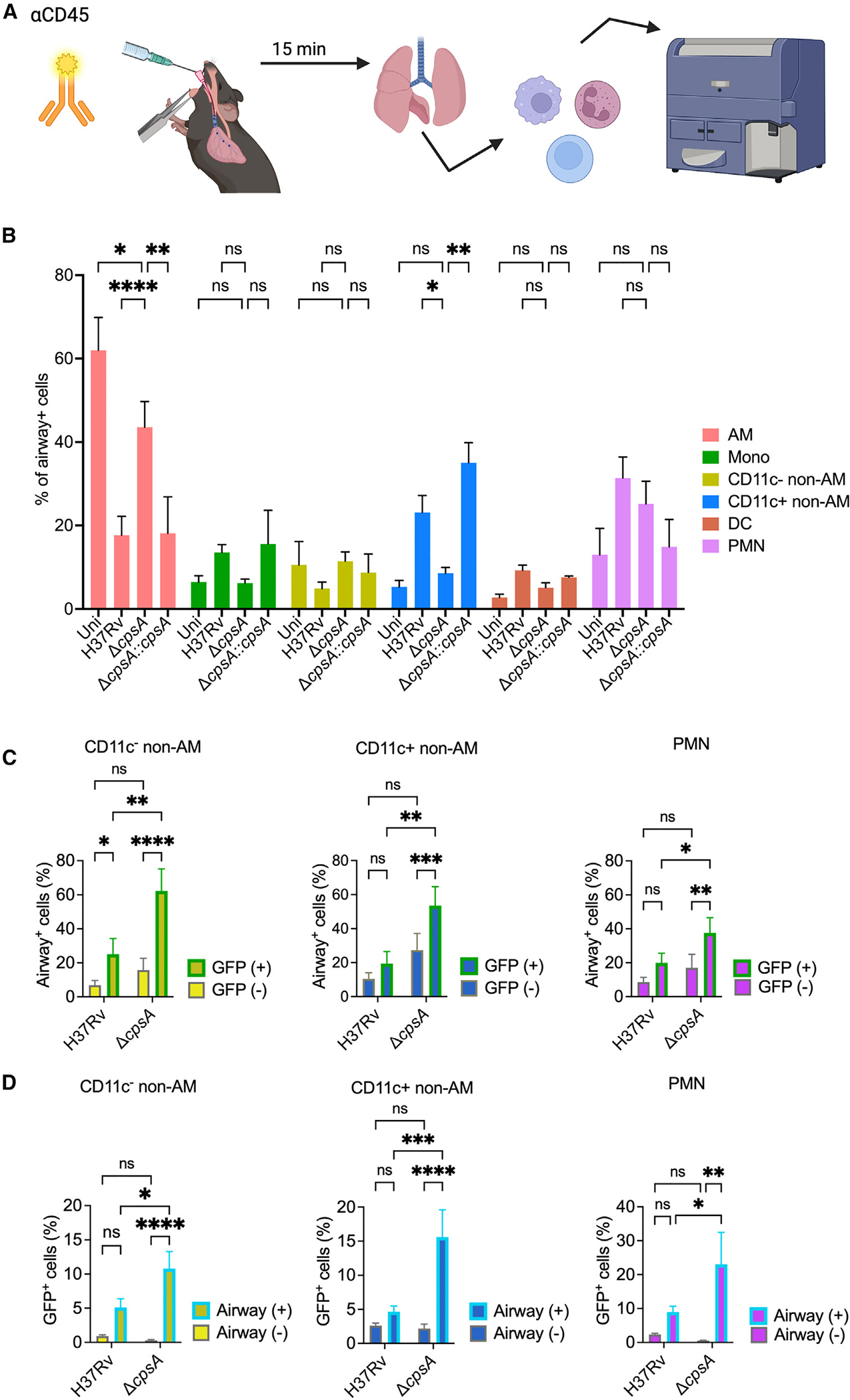
(A) Schematic of experimental strategy for airway cell labeling of mice that were uninfected (uni) or infected with 1,500 CFU H37Rv or 7,000 CFU ΔcpsA Mtb. Prior to euthanasia at 3 wpi, they were treated with intratracheal fluorescently tagged anti-CD45 antibody, followed by flow cytometry.
(B) Distribution of airway-positive cells expressed as the percentage of airway-positive cells of the indicated cell population.
(C) Percentage of infected (GFP+) and uninfected (GFP−) myeloid cells that were positive for CD45 labeling, indicating their airway localization.
(D) Percent of airway-positive and airway-negative myeloid cells that were infected (GFP+). (B–D) Data consist of 7–17 mice per group from five independent experiments, except for the complemented strain in (B), which is from three mice from one experiment. Error bars indicate mean ± SEM. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. Two-way ANOVA with Dunnett’s multiple comparisons test (B) or two-way ANOVA or mixed effects analysis (C and D). Uni, uninfected. See also Figures S3 and S8.
Next, we examined whether there were differences in the airway localization of Mtb-infected cells. In 10 out of 11 mice infected with WT Mtb, we could not detect Mtb-infected AMs in the airways at this time point. In contrast, we found that the majority of ΔcpsA-infected mice had infected AMs in the airway with 56% ± 36% of the infected AMs CD45+. There were also significantly more ΔcpsA-infected non-AMs and PMNs in the airway than WT-infected macrophages (Figure 7C). We also examined what percent of the airway-localized cell populations were infected, and we found that significantly more non-AMs and PMNs in the airways of ΔcpsA-infected mice were infected than corresponding populations in the airways of WT Mtb-infected mice (Figure 7D). Overall, our findings show that the distribution of myeloid cells in the airway is significantly different between mice infected with WT Mtb and those infected with ΔcpsA Mtb, with enhanced retention of AMs in the airways. In addition, ΔcpsA Mtb-infected myeloid cells are preferentially airway localized compared to those infected with WT Mtb.
DISCUSSION
A mechanistic understanding of the initial events that enable Mtb to establish infection is critical to the development of interventions to generate protection, such as trained immunity and protective vaccines. Recent work has revealed the complexity and dynamic changes that occur in the cellular niche during the first month of infection, but the host-pathogen interactions underlying these changes are not well understood. Our data point to a critical role for CpsA in mediating cell-to-cell spread of Mtb from AMs to a broader population of myeloid cells during the earliest steps of Mtb infection. Since ΔcpsA Mtb is highly attenuated early during infection, the reduced cellular response that we observed might simply have reflected the reduced bacterial burden. By using a higher inoculum of ΔcpsA Mtb compared to WT Mtb, we were able to obtain WT and ΔcpsA Mtb-infected mice with bacterial burdens within the same order of magnitude, thus allowing us to compensate for differences in bacterial burden at least in part. Even with this compensation, ΔcpsA Mtb still recruited fewer CD11c+ non-AMs and DCs to the lungs, and ΔcpsA Mtb was preferentially retained in AMs. Furthermore, the CD11c+ non-AMs infected with ΔcpsA Mtb were more likely to be localized in the airways compared to those infected with WT Mtb. Previous reports indicated that infected AMs migrate out of the airways before Mtb disseminates into other myeloid populations.6 Our data show that recruited macrophages are preferentially being infected by ΔcpsA Mtb in the airways. Based on our combined findings, we conclude that CpsA mediates dissemination of Mtb from AMs and into recruited macrophages and the lung interstitium.
How does CpsA promote early bacterial dissemination? It is likely that the NADPH oxidase is involved since previous work showed that CpsA inhibits the NADPH oxidase,21 and here, we found that the retention of ΔcpsA Mtb in AMs was partially reversed in Nox2−/− mice. We also found that in the first 2 weeks of infection, ΔcpsA Mtb elicited less CCL2, CCL3, CCL5, CCL7, and CXCL10 in total lung homogenate than WT Mtb (Figure 3B). Even when mice had relatively similar bacterial burdens at 3 wpi, these chemokines were lower in mice infected with ΔcpsA Mtb than WT Mtb (Figure 5G). In addition, previously, we found that Ccl2, Ccl3, Ccl5, and Ccl7 were all induced to lower levels in BMDMs 4 h after ex vivo infection, when there is no differences in bacterial burdens (see Figure S5C in Köster et al.21). Thus, a failure of ΔcpsA Mtb to elicit robust chemokine responses likely contributes to its failure to recruit myeloid cells in which to disseminate. Interestingly, ex vivo ΔcpsA Mtb is not attenuated in AMs (Figure 6D), while it is attenuated in BMDMs.21 Thus, we think another reason that it fails to efficiently disseminate is that it does not grow effectively in the recruited myeloid populations. Since the NADPH oxidase is required to control ΔcpsA Mtb in BMDMs,21 NADPH oxidase may play a role by restricting growth of the mutant once it gains access to recruited macrophages. Lastly, IL-1β has been shown to be important for the dissemination of Mtb infection out of AMs,6 so it is notable that we found lower IL-1β levels in the lungs of mice infected with ΔcpsA Mtb both when we adjusted the inoculum to achieve similar bacterial burdens at 3 wpi (Figure 3G) and at 6 wpi when the inflammatory responses are similar between mice infected with WT and ΔcpsA Mtb (Figure 5G). In addition, previously, we showed that ΔcpsA Mtb elicits less IL-1β in macrophages ex vivo (see Figure S5D in Köster et al.21). Reduced IL-1β may be connected to the ability of CpsA to inhibit the NADPH oxidase, since NADPH oxidase activity has been shown to inhibit IL-1β production in the lungs.32 Thus, CpsA may also promote the dissemination of WT Mtb by enhancing IL-1β levels. In conclusion, the failure of ΔcpsA Mtb to efficiently disseminate from AMs and the airways into other macrophages in the interstitium is likely multifactorial. The mutant elicits impaired chemokine responses leading to reduced recruitment of macrophages; reduced IL-1β may contribute to the failure to exit the airways; and once ΔcpsA Mtb is phagocytosed by recruited macrophages, they are better able to control its replication than WT Mtb.
There are few other bacterial mutants that have been investigated in terms of their early infection dynamics. Mtb mutants defective in PDIM have been proposed to selectively recruit more restrictive macrophages in mouse infections.33 In mice infected with ΔcpsA Mtb, all myeloid cell types were reduced in the lungs, suggesting that the mutant does not selectively recruit more restrictive macrophages. In addition, even in Ccr2 KO mice, ΔcpsA Mtb was still attenuated early and able to eventually reach bacterial burdens similar to WT Mtb (Figure S5). Since the populations that we defined by flow cytometry contain subpopulations, it is still possible that CpsA impacts a specific subpopulation, altering the balance between permissive and restrictive macrophages. NuoG is another virulence factor that, like CpsA, reduces ROS levels and has been shown to be attenuated during chronic infection. However, NuoG impairs T cell priming and recruitment to the lungs,34–36 while CpsA appears to have the opposite effect. Finally, ΔesxA Mtb, which is defective in the ESX-1 type VII secretion system, has been shown to have a defect exiting the AM population into interstitial macrophages similar to ΔcpsA Mtb.6 Both ΔcpsA Mtb and ΔesxA Mtb are attenuated in macrophages ex vivo as well as during the innate immune phase of infection, and both elicit less IL-1β from the host.21,37–39 Thus, both ΔcpsA Mtb and ΔesxA Mtb may be impaired in IL-1β production and controlled in recruited macrophages, leading to their delay in eliciting the inflammatory response necessary for dissemination.
Our observations provide a mechanism by which ΔcpsA Mtb reaches nearly the same infectious burden in the lung as WT Mtb despite its initial attenuation. At 3 wpi, in addition to reduced myeloid cell recruitment, there were also substantially fewer CD4 and CD8 T cells. This defect persisted at 4 wpi, when the adaptive immune response is well underway in WT Mtb-infected mice. Activation of adaptive immunity by Mtb has been shown to depend on antigen production in the lymph nodes and not the lungs.30 Since migration of infected DCs to the lymph nodes is essential for priming naive T cells,30,40 the delayed adaptive immune response is likely caused by the series of events that we documented during the innate immune response: retention of ΔcpsA Mtb in AMs, reduced infection of DCs, and retention of infected cells in the airways, leading to delayed arrival of Mtb in the MLN. Consistent with this idea, strains that exhibit an earlier shift into recruited macrophages and DCs cells are associated with greater, earlier activation of CD4 T cells.41 In addition, the reduced bacterial load in the lungs will also contribute to lower T cell responses. One could envision alternative possibilities. For example, CpsA could impact the ability of DCs to present antigen. However, previously, we showed that CpsA does not affect the ability of infected DCs to activate CD4 T cells in vitro,42 so we consider this a less likely possibility. Instead, CpsA appears to indirectly promote T cell responses by promoting dissemination of Mtb into recruited myeloid cells. The reduced IFN-γ-producing, antigen-specific T cell responses likely account for the diminished CD80 and MHC II expression that we saw in the myeloid populations. Thus, the AMs and the small fraction of other myeloid cells that are responding to the infection remain less activated and more permissive for longer during infection with ΔcpsA Mtb compared to WT Mtb. In analogy to a race, WT Mtb starts out with an enormous lead, but it encounters substantial headwinds that result in its standstill, giving ΔcpsA Mtb time to “catch up.” Once adaptive immune responses develop against ΔcpsA Mtb, then it experiences similar headwinds, and it is defective, relative to WT Mtb, in persistence. Although, it should be noted that since we did not examine the adaptive immune response later during infection, it is possible that it is not only delayed in the context of the ΔcpsA Mtb infection but also impaired thereafter.
One strategy to enhance protection against Mtb is to overcome the delay in CD4 T cell responses that is characteristic of the immune response to TB.43 BCG vaccination enhances Mtb dissemination into recruited macrophages and subsequently T cell activation.44 Moreover, the contained Mtb infection model, which consists of injection of Mtb into the dermis of the ear, enhances recruitment of Mtb-specific T cells to the lungs, which is also associated with enhanced activation of AMs and reduced Mtb burden.45 However, while these strategies result in reduced Mtb burden, they do not provide sterilizing immunity. Alternative strategies to promote innate clearance, for example, through trained immunity are also areas of active investigation. Importantly, the delay in the activation of adaptive immunity elicited by ΔcpsA Mtb shows that even strains that are highly attenuated in the face of innate immunity can establish infection due to a corresponding longer delay in the adaptive immune response. This observation has important implications for ongoing efforts to design novel interventions to prevent tuberculosis. Our studies highlight the challenge of enhancing innate immunity to confer protection; unless innate immune responses are sterilizing, they will likely alter the dynamics of the immune response to Mtb but may be insufficient to protect against infection.
Limitations of the study
This study examines the effect of CpsA in the H37Rv background, which may differ in terms of host-pathogen interactions from circulating clinical strains. For example, some hypervirulent strains of the Beijing/W lineage in particular have been found to elicit distinct immune responses compared to the commonly used laboratory strains. Our infection model uses C57BL/6J mice, which provide a useful, genetically tractable model that has yielded important information about the earliest events in Mtb infection. However, there are differences in granuloma architecture and the course of infection compared to humans and non-human primates. Finally, although we were able to achieve similar bacterial burdens between WT Mtb and ΔcpsA Mtb infections at 3 weeks post infection to examine responses independent of burden, the unequal initial inoculum may have altered infection dynamics in ways that are difficult to predict. Finally, we focused our studies on the first 6 weeks of infection, so how CpsA influences the immune response during chronic infection was not addressed.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jennifer A. Philips (philips.j.a@wustl.edu).
Materials availability
Mycobacterium tuberculosis strains and plasmids generated in the study are available upon request. Transfer of the M. tuberculosis strains will require that the receiving individual have appropriate biosafety approval to work with them.
Data and code availability
Flow cytometry data reported in this paper will be shared by the lead contact upon request. Whole-genome sequencing data has been deposited at Genome Sequence Archive and is publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Purified anti-mouse CD16/32, Clone 93 | Biolegend | Cat#101302; RRID:AB_312801 |
| BD OptiBuild BUV395 Hamster Anti-Mouse CD80, Clone 16-10A1 | BD Biosciences | Cat#740246; RRID:AB_2739993 |
| Brilliant Violet 421 anti-mouse I-A/I-E, Clone M5/114.15.2 | Biolegend | Cat#107632; RRID:AB_2650896 |
| Brilliant Violet 510 anti-mouse CD11c, Clone N418 | Biolegend | Cat#117353; RRID:AB_2562016 |
| Brilliant Violet 711 anti-mouse/human CD11b, Clone M1/70 | Biolegend | Cat#101242; RRID:AB_2563310 |
| BD OptiBuild BV786 Mouse Anti-Mouse CD64 a and b Alloantigens, Clone X54-5/7.1 | BD Biosciences | Cat#741024; RRID:AB_2740644 |
| BD Pharmingen PE Rat Anti-Mouse Siglec-F, Clone E50-2440 | BD Biosciences | Cat#552126; RRID:AB_394341 |
| PE/Cyanine7 anti-mouse Ly-6G, Clone 1A8 | Biolegend | Cat#127618; RRID:AB_1877262 |
| Alexa Fluor 700 anti-mouse CD45, Clone 30-F11 | Biolegend | Cat#103128; RRID:AB_493715 |
| APC/Cyanine7 anti-mouse Ly-6C, Clone HK1.4 | Biolegend | Cat#128026; RRID:AB_10640120 |
| BD Horizon BUV395 Mouse Anti-Mouse NK-1.1, Clone PK136 | BD Biosciences | Cat#564144; RRID:AB_2738618 |
| Brilliant Violet 421 anti-mouse TCR γ/δ, Clone GL3 | Biolegend | Cat#118120; RRID:AB_2562566 |
| Brilliant Violet 711 anti-mouse CD19, Clone 6D5 | Biolegend | Cat#115555; RRID:AB_2565970 |
| PE anti-mouse/human CD44, Clone IM7 | Biolegend | Cat#103007; RRID:AB_493686 |
| PE/Cyanine7 anti-mouse CD8a, Clone 53-6.7 | Biolegend | Cat#100722; RRID:AB_312761 |
| APC anti-mouse CD4, Clone GK1.5 | Biolegend | Cat#100412; RRID:AB_312696 |
| APC/Cyanine7 anti-mouse TCR β chain, Clone H57-597 | Biolegend | Cat#109220; RRID:AB_893624 |
| Brilliant Violet 650 anti-mouse CD45, Clone 30-F11 | Biolegend | Cat#103151; RRID:AB_2565884 |
| Rabbit anti-CpsA | 21 | N/A |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa 647 | Thermo Fisher | Cat#A-21245; RRID:AB_2535813 |
| Rabbit anti-Ag85B | Joel Ernst | N/A |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher | Cat#A32731; AB_2633280 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| M. tuberculosis H37Rv | William R Jacobs Jr | N/A |
| M. tuberculosis ΔkatG | William R Jacobs Jr | N/A |
| M. tuberculosis ΔcpsA | (24,26) | N/A |
| M. tuberculosis ΔcpsA ΔkatG | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Purina 5053 Rodent Chow with 2,000 ppm Doxycycline | Research Diets | Cat#C15020601i |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | Cat#D4527 |
| Collagenase D | Roche | Cat#COLLD-RO |
| Trucount Absolute Counting Tubes | BD Biosciences | Cat#340334 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat#CO-RO |
| 4–20% Mini-PROTEAN TGX Precast Protein Gels | Bio-Rad | Cat#4561094 |
| 10x Tris/Glycine/SDS | Bio-Rad | Cat#1610732 |
| 10x Tris/Glycine Buffer for Western Blots and Native Gels | Bio-Rad | Cat#1610734 |
| Restore Western Blot Stripping Buffer | Thermo Scientific | Cat#21059 |
| Nitrocellulose/Filter Paper Sandwich, 0.2 μm, 8.3 × 7.3 cm | Invitrogen | Cat#LC2000 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cytokine & Chemokine Convenience 26-Plex Mouse ProcartaPlex Panel 1 | Invitrogen | Cat#EPXR260-26088-901 |
| IFN alpha Mouse ProcartaPlex Simplex Kit | Invitrogen | Cat#EPX01A-26027-901 |
| IFN beta Mouse ProcartaPlex Simplex Kit | Invitrogen | Cat#EPX01B-26044-901 |
| DuoSet ELISA kits for IL-1 β | R&D Systems | Cat#DY401 |
|
| ||
| Deposited data | ||
|
| ||
| Whole-genome sequencing data; ΔcpsA katG parent strain and mouse passaged isolates | Genome Sequence Archive | GSA: CRA013548 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: WT: C57BL/6J | The Jackson Laboratory | Cat#000664 |
| Mouse: Nox2−/−: B6.129S-Cybbtm1Din/J | Mary Dinauer | N/A |
| Mouse: Ccr2−/−: B6.129S4-Ccr2tm1Ifc/J | The Jackson Laboratory | Cat#004999 |
|
| ||
| Oligonucleotides | ||
|
| ||
| 5’-CGGGCCAACCTGGGATGC-3’ | IDT | N/A |
| 5’-GCAGGCTCGCGTAGGAATCATCC-3’ | IDT | N/A |
| 5’-GAACTGCTCGCCTTCACCTTCC-3’ | IDT | N/A |
| 5’-AACCGCGTGGCATGGCCGGCTGCA-3’ | IDT | N/A |
| 5’-TGCTCATCGGGCTGGACTCG-3’ | IDT | N/A |
| 5’-ACTCGATGGCGGTGGT GAACG-3’ | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pGMCK-TetOFF-cpsA | This paper | N/A |
| pKP617 cpsA | 21 | N/A |
| pMV261 GFP | Christina Stallings | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | GraphPad | N/A |
| FlowJo | BD Biosciences | N/A |
| FACSDiva | BD Biosciences | N/A |
| BioRender | BioRender | N/A |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Bacterial strains and plasmids
M. tuberculosis (Mtb) H37Rv strain was from William R Jacobs Jr (AECOM). All mutants were generated in H37Rv. ΔcpsA Mtb was generated using mycobacterial recombineering as described previously.21,26 ΔkatG Mtb was provided by William R Jacobs Jr (AECOM). ΔcpsA ΔkatG Mtb was constructed using mycobacterial recombineering to knockout cpsA from the ΔkatG Mtb. Cloning strategy and screening PCR are illustrated in Figure S1. Single colonies were screened by PCR of genomic DNA using the following pairs of primers: 5′ cloning junction 5′-CGGGCCAACCTGGGATGC-3’ & 5′-GCAGGCTCGCGTAGGAATCATCC-3’; 3′ cloning junction 5′-GAACTGCTCGCCTTCACCTTCC-3’ & 5′-AACCGCGTGGCATGGCCGGCTGCA-3’; deleted cpsA 5v-TGCTCATCGGGCTG GACTCG-3’ & 5′-ACTCGATGGCGGTGGTGAACG-3’. Verified transformants were screened for PDIM production by whole-genome sequencing and HPLC-MS. Construction of the cpsA complementing plasmid was described previously.21 The plasmid used to regulate CpsA expression, pGMCK-TetOFF-cpsA, was constructed by MultiSite Gateway recombination using the procedures published previously.46 In this plasmid, transcription of cpsA is mediated by a promoter that can be repressed by a reverse TetR,47 which was also cloned into pGMCK-TetOFF-cpsA. Reverse TetRs are activated by tetracyclines so that addition of anhydrotetracycline or doxycycline reduces transcription of, in this case, cpsA. The cpsA-containing entry plasmid required for the Gateway recombination was purchased from GenScript. Mtb was cultured in Middlebrook 7H9 (BD Biosciences) with BBL OADC enrichment (BD Biosciences), 0.05% Tween 80 (Fisher), and 0.2% glycerol (Sigma). ΔcpsA Mtb was cultured with 50 μg/mL hygromycin (Gold Biotechnology). Mtb expressing GFP or CpsA TetOFF were cultured with 25 μg/mL kanamycin (IBI Scientific). The GFP plasmid was provided by Christina Stallings (WUSM).
Mice
All mice were in the C57BL/6J background. WT (000664) and Ccr2 KO (004999) mice were from The Jackson Laboratory and bred in house in a specific pathogen-free animal barrier facility at Washington University. Nox2−/− mice were provided by Mary Dinauer (WUSM). Infections were performed with mice of both sexes, aged 8–14 weeks old. All procedures were done in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) of Washington University School of Medicine.
Alveolar macrophage isolation
Mice were euthanized with 1 mL isoflurane instilled in a cotton ball. The trachea was exposed, incised with a small horizontal cut, and cannulated with an 18G catheter. 8 washes with 1 mL wash buffer (ice-cold PBS, 2 mM EDTA, 1% FBS) were performed. Cells were washed and seeded in alveolar macrophage media [DMEM with 10% L929 cell supernatant, 10% FBS, 1 mM pyruvate, and 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES)] and allowed to adhere for 2 h prior to infection. AMs were pooled from three male and three female B/6 mice for the ex vivo infections described below.
METHOD DETAILS
Mouse infections
Mtb was cultured to mid-log phase (OD600 0.6–0.8), and a single-cell suspension was generated through sequential low-speed centrifugation at 800 rpm until stabilization of OD600 of the supernatant. Mice were infected by aerosol using a Glas-Col inhalation exposure system. Harvested tissues were homogenized using Bio-Gen PRO200 tissue homogenizer (PRO Scientific) and plated on Middlebrook 7H11 supplemented with BBL OADC enrichment (BD Biosciences) for CFU enumeration. In most cases, homogenized samples were passed through a 70 μm cell strainer (BD Biosciences) before plating. CFU were enumerated after 2–3 weeks of incubation at 37°C. For tet-mediated CpsA depletion, mice were fed a diet consisting of Purina 5053 chow containing 2000 ppm doxycycline (Research Diets).
Flow cytometry
Flow cytometry was done as described previously.28 Briefly, mouse lungs were perfused with sterile PBS (Thermo Fisher) before harvesting. Lungs were digested at 37°C with 75 U/mL DNase I (Millipore Sigma) and 0.63 mg/mL collagenase D (Millipore Sigma). The cell suspension was washed with PBS containing 2% heat-inactivated FBS (Thermo Fisher) and 2 mM EDTA (Corning). Staining for surface markers was done at 4°C for 20 min. The following reagents were used for staining: anti-CD16/32 Fc Block (93, Biolegend), LIVE/DEAD Fixable Blue Dead Cell Stain (Thermo Fisher), BUV395 anti-CD80 (16–10A1, BD Biosciences), BV421 anti-I-A/I-E (M5/114.15.2, Biolegend), BV510 anti-CD11c (N418, Biolegend), BV711 anti-CD11b (M1/70, Biolegend), BV786 anti-CD64 (X54–5/7.1, BD Biosciences), PE anti-Siglec-F (E50–2440, BD Biosciences), PE-Cy7 anti-Ly6G (1A8, Biolegend), Alexa 700 anti-CD45 (30-F11, Biolegend), APC-Cy7 anti-Ly6C (HK1.4, Biolegend), BUV395 anti-NK1.1 (PK136, BD Biosciences), BV421 anti-TCR γδ (GL3, Biolegend), BV711 anti-CD19 (6D5, Biolegend), PE anti-CD44 (IM7, Biolegend), PE-Cy7 anti-CD8α (53–6.7, Biolegend), APC anti-CD4 (GK1.5, Biolegend), APC-Cy7 anti-TCR β (H57–597, Biolegend). After staining, cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 30 min at RT, and data was acquired on an LSR Fortessa X-20 flow cytometer (BD Biosciences) with 355 nm, 405 nm, 488 nm, 561 nm, and 640 nm lasers running BD FACSDiva software. Flow cytometry data was compensated manually post-acquisition and analyzed using FlowJo software (BD Biosciences). Gating strategies are depicted in Figure S3. Channel voltages were set using single-stained cell controls. Gates for GFP were set using cells from uninfected mice, and gates for CD80, CD44, and intratracheally administered CD45 were set using fluorescence minus one (FMO) controls. Absolute cell counts were calculated using Trucount tubes (BD Biosciences).
Intracellular cytokine staining
For T cell stimulation, cells from Mtb-infected lung samples resuspended in complete media (RPMI-1640 + 10% FBS) were stimulated with phorbol myristate acetate (30 ng/mL, Sigma Aldrich) plus ionomycin (1 μg/mL) and incubated for 1 h at 37° C, 5% CO2. After the initial incubation, Golgistop (containing Monensin, BD Biosciences) and Golgiplug (containing Brefeldin A, BD Biosciences) were added to the cell suspension and further incubated for 4 h. Cells were washed once, stained with surface antibodies, fixed in 4% PFA, and permeabilized with BD Perm/Wash buffer as per manufacturers protocol. Permeabilized cells were stained with BV785 anti-IFNγ (XMG1.2, Biolegend), BV510 anti-TNFα (MP6-XT22, Biolegend), and APC-Cy7 anti-IL-2 (JES6–5H4. BD Pharmingen) antibodies for 30 m, and then washed and resuspended in PBS, followed by acquisition on an LSR Fortessa X-20 flow cytometer. Data was analyzed using FlowJo software. A hierarchical gating strategy was used to first select single-cell CD4 T cells. Gates for cytokine-expressing CD4 T cells in samples stimulated with PMA + ionomycin were set using FMO controls. A Boolean combination strategy was used to estimate polyfunctional T-cells producing two or more cytokines.
Tetramer staining
Tetramer staining was done prior to surface staining with antibodies. PE-conjugated MHC class II tetramers ESAT64–17 (I-A(b) QQWNFAGIEAAASA), PE-Ag85b280–294 (I-A(b) FQDAYNAAGGHNAVF) and BV421-conjugated MHC class I tetramer TB10.44–11 (H-2K(b) IMYNYPAM), obtained from NIH Tetramer Core Facility, were used to detect Mtb-specific CD4 and CD8 T cells. Lung single cell suspensions were incubated with 100 nM of freshly prepared Dasatinib (Sigma-Aldrich) for 30 m at 37°C in a CO2 incubator. The cells were washed with FACS buffer and incubated with saturating concentrations of MHC tetramers. For MHCII tetramer staining, the cells were incubated with tetramers at 37°C for 30 min in a CO2 incubator. For MHCI tetramer staining, the cells were incubated with TB10.44–11 tetramer for 30 m at 4°C. Cells were then washed and stained with surface antibodies.
Labeling of airway cells
Labeling of airway cells was done as described previously.31 Mice were injected intraperitoneally with 200 μL of PBS containing 10 mg/mL ketamine and 1 mg/mL xylazine. Once the mice were fully anesthetized, a 20G 1” catheter containing a 0.75 mm fiber optic cable connected to a light source was inserted into the trachea. The fiber optic cable was then withdrawn from the catheter. 50 μL of PBS with 14 μg/mL BV650 αCD45 (30-F11, Biolegend) was introduced into the catheter. Air was blown into the catheter using a 1 mL syringe to distribute the antibody through the airways. After mice recovered for 15 min, lungs were harvested and processed for flow cytometry.
Alveolar macrophage infection
Single cell suspensions of Mtb in DMEM complete with 10% L929 were added to 25,000 macrophages in 96 well plates at an MOI of 5, and plates were spun for 5 min at 51×g. The MOI was verified by plating the inoculum. At 4 hpi, macrophages were washed 3 times with DMEM to remove extracellular bacteria. To enumerate CFU, at specified time points macrophages were lysed with 0.06% sodium dodecyl sulfate (SDS) in water and serially diluted in PBS. The cell lysates were plated on 7H11 agar plates supplemented with OADC and glycerol, and CFU were counted after 14–21 days.
Western blotting
Western blotting was done as described previously.21 Briefly, Mtb was cultured to mid-log phase, diluted to OD600 0.1, and then cultured for 3 days to reach an OD600 of 0.8. For ATc induction, Mtb was cultured with 500 ng/mL ATc. To generate a whole-cell lysate, cultures were pelleted and resuspended in lysis buffer consisting of 25 mM Tris HCl (Sigma; pH 7.5), 150 mM NaCl (Sigma), 0.6% SDS (Invitrogen), 1mM PMSF (Sigma), and cOmplete protease inhibitor cocktail (Roche). Cells were lysed by bead beating with zirconium beads (BioSpec Products). Lysates were mixed with 5x loading buffer (50% glycerol, 10% SDS, 0.5 M DTT, and 0.25% bromophenol blue) and heated at 85°C for 10 min. SDS-PAGE was done using a 4–20% Mini-PROTEAN TGX Precast Protein Gel (BIO-RAD) and PageRuler Prestained Protein Ladder (Thermo Fisher). Protein bands were transferred to a Novex nitrocellulose membrane (Thermo Fisher) using a wet transfer with Tris/glycine buffer containing 10% methanol (BIO-RAD) overnight at 25V at 4°C. The membrane was blocked using PBST (PBS 0.05% Tween 20 (Fisher) with 3% BSA (Bio Basic)). The membrane was probed with PBST +1:2000 rabbit αCpsA anti-serum,21 followed by 1:5000 goat mAb αrabbit Alexa 647 (Thermo Fisher). After imaging, the membrane was stripped with Restore Western Blot Stripping Buffer (Thermo Fisher), then re-probed, blocked, and re-probed with 1:1000 rabbit αAg85B antibody (provided by Joel Ernst), followed by 1:5000 goat mAb αrabbit Alexa 488. The membrane was imaged using a ChemiDoc MP Imaging System (BIO-RAD) and analyzed using iBright Analysis Software.
Whole-genome sequencing
To extract genomic DNA (gDNA), Mtb was grown to an OD600 of 0.8, then cultured for an additional 24 h with 1% glycine. The culture was pelleted and resuspended in GTE buffer [25mM Tris-HCl (Sigma) pH 8.0.10mM EDTA (Fisher), 50mM glucose (Sigma)]. Cells were lysed with 1 mg/mL lysozyme (Sigma) overnight at 37°C, and samples were treated with 1.5% SDS (Invitrogen), 3 mg/mL proteinase K (Thermo Fisher), and heating (55°C), followed by addition of 1 M NaCl (Sigma), 1.6% CTAB (Sigma), and heating (60°C). gDNA was isolated by extracting twice in an equal volume of 24:1 chloroform:isoamyl alcohol (Sigma). gDNA was precipitated in isopropanol (Sigma), washed with 70% ethanol, air-dried, and then resuspended in TE buffer (Corning). gDNA was submitted to the Genome Technology Access Center at the McDonnel Genome Institute (WUSM) for KAPA Hyper PCR-Free automated WGS library prep, followed by Illumina NovaSeq6000 S4 XP (2×150) sequencing. Sequencing data was aligned to the H37Rv reference genome (NCBI), and variants were identified using mpileup. Variant data was analyzed using Integrated Genomics Viewer (Broad Institute) and annotated using Variant Effect Predictor (Ensembl).
Mtb lipid extraction and HPLC-MS
Mtb lipids were extracted and analyzed by HPLC-MS as described previously.21 Briefly, Mtb was plated on Middlebrook 7H11 and incubated for ~3 weeks. The bacterial lawn was pelleted, boiled for 20 min, pelleted to remove excess liquid, and weighed in order to analyze the same amount of biomass for each strain tested. The Mtb pellet was sonicated in 2:1 chloroform:methanol (Sigma) containing 10 μg/mL triarachidin (Cayman Chemical) as an internal standard. After sonication, samples were centrifuged, and the organic phase was collected. Sonication was repeated with fresh chloroform:methanol and triarachidin, followed by centrifugation and collection of the organic phase. The fractions from both extractions were combined and desiccated under nitrogen gas. Lipids were resuspended in chloroform:methanol and submitted for HPLC-MS to the Mass Spectrometry and Lipidomics Core of Washington University. Analysis was conducted on a Scientific TSQ Vantage mass spectrometer (Thermo Fisher) with a Thermo Accela UPLC operated by Xcalibur software. Separation of lipid was achieved by a 100 × 2.1 mm (2.7-μm particle size) Ascentis C-8 column (Supelco) at a flow rate of 260 μL/min as described previously.48 The electrospray ionization (ESI) mass spectra were acquired in both positive-ion and negative-ion modes, ranging from m/z 400–3,000, at a scan rate of scan/2 s, and the ESI mass spectra of each of the major lipid classes were obtained.
Cytokine analysis
Lung homogenate was pelleted twice at 13,000×g for 5 min at 4°C. The supernatant was filtered using a 0.22μm MultiScreen-GV filter plate (EMD Millipore). Filtrates were submitted to the Immunomonitoring Laboratory (IML) of the Center for Human Immunology and Immunotherapy Programs (CHiiPs) at Washington University for analyses using the IFN-α and IFN-β simplex kits (Thermo Fisher) and the Cytokine & Chemokine Convenience 26-Plex Mouse ProcartaPlex Panel 1 kit (Thermo Fisher) and the Luminex FLEXMAP 3D platform (Invitrogen).
Histopathology
Lungs were harvested and fixed in 10% neutral buffered formalin (Fisher) for 3–4 days. Fixed lungs were then washed with PBS and dehydrated with serial incubation in 30%, 50%, and then 70% ethanol. Lungs were embedded, sectioned, and stained at the Pulmonary Morphology Core at Washington University. Dehydrated lungs were embedded in paraffin and sectioned at 5 μm using an RM2255 Fully Automated Microtome (Leica). Sections were collected onto charged microscope slides from a water flotation bath at 40°C. Slides were allowed to dry before staining. Sections were de-paraffinized with xylene; serially rehydrated in 100% alcohol, 95% alcohol, and then 70% alcohol; and washed briefly with distilled water. For H&E staining, sections were stained in Mayer hematoxylin solution, washed in warm running tap water, rinsed in distilled water, rinsed in 95% alcohol, and counter-stained in eosin Y solution. After staining, sections were dehydrated through 95% alcohol and then 100% alcohol, cleared through xylene, and mounted with Cytoseal 60 (Epredia). For AFB staining, sections were stained with Carbol Fuchsin and Fast Green Stain using an Acid Fast Bacillus Stain Kit (Epredia). Slides were imaged at the Alafi Neuroimaging Laboratory at Washington University using a NanoZoomer 2.0-HT Whole-Slide Imaging System (Hamamatsu) with a 40× objective.
QUANTIFICATION AND STATISTICAL ANALYSIS
In vivo data is from mice randomly sorted into each experimental group. No blinding was performed during animal experiments. Statistical differences were calculated using Prism (GraphPad Software) and details are available in the figure legends. For two groups with normal distributions, unpaired Student’s t test was used. In cases where the two groups appeared to have non-normal distributions, Mann-Whitney test was used. For more than two groups with normal distributions, one-way ANOVA with Tukey’s multiple comparisons test or Dunnett’s multiple comparisons test was used to compare one variable, and two-way ANOVA with Sidak’s multiple comparisons test was used to compare two variables. For more than two groups with non-normal distributions, Kruskal-Wallis test with Dunn’s multiple comparisons test was used. When used, center values and error bars represent mean ± SEM. Figures were created using BioRender.
Supplementary Material
Highlights.
CpsA promotes recruitment and activation of lung myeloid cells during Mtb infection
CpsA promotes dissemination of Mtb from alveolar macrophages and the airways
In the absence of CpsA, there is a delay in the adaptive immune response to Mtb
Our study shows how enhanced innate control of Mtb is offset by delayed adaptive immunity
ACKNOWLEDGMENTS
The following sources provided funding and support: NIAID R01 AI130454 (JAP), F31 AI152321 (SJG), P01 AI143575 (DS), and the Bursky Center for Human Immunology and Immunotherapy Programs at Washington University, Immunomonitoring Laboratory. Christina Stallings provided the GFP plasmid. William R. Jacobs provided ΔkatG Mtb. Mary Dinauer provided the Nox2−/− mice. We thank members of the Bursky Center for Human Immunology and Immunotherapy Programs (CHiiPs; WUSM), Immunology & Pathology Flow Cytometry & Fluorescence Activated Cell Sorting Core Facility (WUSM), Division of Comparative Medicine (WUSM), Pulmonary Morphology Core (WUSM), Alafi Neuroimaging Center (WUSM), and Genome Technology Access Center at the McDonnel Genome Institute (WUSM). We thank the NIH tetramer core facility for providing the PE-conjugated Ag85b and ESAT6 MHCII tetramers and the BV421-conjugated TB10.4 tetramers. We also thank Sharmila Nair, Rachel Kinsella, Andrew Roth, and members of the Philips lab for their expertise and discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113607.
REFERENCES
- 1.WHO (2021). Global Tuberculosis Report 2021 (World Health Organization; ). https://www.who.int/publications/i/item/9789240037021. [Google Scholar]
- 2.Davenne T, and McShane H (2016). Why don’t we have an effective tuberculosis vaccine yet? Expert Rev. Vaccines 15, 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandra P, Grigsby SJ, and Philips JA (2022). Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 20, 750–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf AJ, Linas B, Trevejo-Nuñez GJ, Kincaid E, Tamura T, Takatsu K, and Ernst JD (2007). Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179, 2509–2519. [DOI] [PubMed] [Google Scholar]
- 5.Lai R, Jeyanathan M, Afkhami S, Zganiacz A, Hammill JA, Yao Y, Kaushic C, and Xing Z (2018). CD11b + Dendritic Cell–Mediated Anti–Mycobacterium tuberculosis Th1 Activation Is Counterregulated by CD103 + Dendritic Cells via IL-10. J. Immunol. 200, 1746–1760. [DOI] [PubMed] [Google Scholar]
- 6.Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, Sherman DR, Gerner MY, and Urdahl KB (2018). Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 24, 439–446.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavalett L, Rodriguez H, Ortega H, Sadee W, Schlesinger LS, and Barrera LF (2017). Alveolar macrophages from tuberculosis patients display an altered inflammatory gene expression profile. Tuberculosis 107, 156–167. [DOI] [PubMed] [Google Scholar]
- 8.Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, Diercks AH, and Aderem A (2019). Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response. Sci. Immunol 4, eaaw6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang L, Nazarova EV, Tan S, Liu Y, and Russell DG (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med. 215, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pisu D, Huang L, Grenier JK, and Russell DG (2020). Dual RNA-Seq of Mtb-Infected Macrophages In Vivo Reveals Ontologically Distinct Host-Pathogen Interactions. Cell Rep. 30, 335–350.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, van Deventer SJ, and van der Poll T (2001). Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J. Immunol. 166, 4604–4611. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, Boyce S, Powers J, Baer C, Sassetti CM, and Behar SM (2020). CD11cHi monocyte-derived macrophages are a major cellular compartment infected by Mycobacterium tuberculosis. PLoS Pathog. 16, e1008621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, and Stallings CL (2015). Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovewell RR, Baer CE, Mishra BB, Smith CM, and Sassetti CM (2021). Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. 14, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, et al. (2017). Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2, 17072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corleis B, Korbel D, Wilson R, Bylund J, Chee R, and Schaible UE (2012). Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol. 14, 1109–1121. [DOI] [PubMed] [Google Scholar]
- 17.Filio-Rodríguez G, Estrada-García I, Arce-Paredes P, Moreno-Altamirano MM, Islas-Trujillo S, Ponce-Regalado MD, and Rojas-Espinosa O (2017). In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a Guinea pig model. Innate Immun. 23, 625–637. [DOI] [PubMed] [Google Scholar]
- 18.Pisu D, Huang L, Narang V, Theriault M, Lê-Bury G, Lee B, Lakudzala AE, Mzinza DT, Mhango DV, Mitini-Nkhoma SC, et al. (2021). Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J. Exp. Med. 218, e20210615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norris BA, and Ernst JD (2018). Mononuclear cell dynamics in M. tuberculosis infection provide opportunities for therapeutic intervention. PLoS Pathog. 14, e1007154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng W, Chang IC, Limberis J, Budzik JM, Zha BS, Howard Z, Ernst JD Mycobacterium tuberculosis Resides in Lysosome-Poor Monocyte-Derived Lung Cells during Persistent Infection. Research Square. 2023. 10.21203/rs.3.rs-3049913/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, Grigsby SJ, Mittal E, Park HS, Jones V, et al. (2017). Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 114, E8711–E8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olive AJ, and Sassetti CM (2018). Tolerating the unwelcome guest; How the host withstands persistent Mycobacterium tuberculosis. Front. Immunol. 9, 2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan J-L, Tan H, Peng J, et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Köster S, Upadhyay S, and Philips JA (2018). Why macrophages cannot LAP up TB. Autophagy 14, 552–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng VH, Cox JS, Sousa AO, MacMicking JD, and McKinney JD (2004). Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302. [DOI] [PubMed] [Google Scholar]
- 26.Murphy KC, Papavinasasundaram K, and Sassetti CM (2015). Mycobacterial recombineering. Methods in molecular biology. Clifton, N.J.) 1285, 177–199. [DOI] [PubMed] [Google Scholar]
- 27.Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, and Schnappinger D (2005). Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huynh JP, Lin CC, Kimmey JM, Jarjour NN, Schwarzkopf EA, Bradstreet TR, Shchukina I, Shpynov O, Weaver CT, Taneja R, et al. (2018). Bhlhe40 is an essential repressor of IL-10 during Mycobacterium tuberculosis infection. J. Exp. Med. 215, 1823–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serbina NV, and Pamer EG (2006). Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317. [DOI] [PubMed] [Google Scholar]
- 30.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, and Ernst JD (2008). Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 205, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunlap MD, Howard N, Das S, Scott N, Ahmed M, Prince O, Rangel-Moreno J, Rosa BA, Martin J, Kaushal D, et al. (2018). A novel role for C–C motif chemokine receptor 2 during infection with hypervirulent Mycobacterium tuberculosis. Mucosal Immunol. 11, 1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chao W-C, Yen C-L, Hsieh C-Y, Huang Y-F, Tseng Y-L, Nigrovic PA, and Shieh C-C (2017). Mycobacterial infection induces higher interleukin-1β and dysregulated lung inflammation in mice with defective leukocyte NADPH oxidase. PLoS One 12, e0189453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, Cosma CL, and Ramakrishnan L (2014). Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR, Porcelli SA, and Briken V (2007). Mycobacterium tuberculosis nuoG Is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 3, e110–e0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blomgran R, Desvignes L, Briken V, and Ernst JD (2012). Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller JL, Velmurugan K, Cowan MJ, and Briken V (2010). The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 6, e1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, and Sherman DR (2003). Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J. Infect. Dis. 187, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, and Brown EJ (2008). ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 10, 1866–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, and Anes E (2010). Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 12, 1046–1063. [DOI] [PubMed] [Google Scholar]
- 40.Samstein M, Schreiber HA, Leiner IM, Susac B, Glickman MS, and Pamer EG (2013). Essential yet limited role for CCR2(+) inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. Elife 2, e01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zha BS, Desvignes L, Fergus TJ, Cornelius A, Cheng TY, Moody DB, and Ernst JD (2022). Bacterial Strain-Dependent Dissociation of Cell Recruitment and Cell-to-Cell Spread in Early M. tuberculosis Infection. mBio 13, e0133222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Köster S, Klevorn T, Papavinasasundaram K, Sassetti CM, Portal-Celhay C, and Philips JA (2018). Consequence of enhanced LC3-trafficking for a live, attenuated M. tuberculosis vaccine. Vaccine 36, 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grace PS, and Ernst JD (2016). Suboptimal Antigen Presentation Contributes to Virulence of Mycobacterium tuberculosis In Vivo. J. Immunol. 196, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delahaye JL, Gern BH, Cohen SB, Plumlee CR, Shafiani S, Gerner MY, and Urdahl KB (2019). Cutting Edge: Bacillus Calmette–Guérin–Induced T Cells Shape Mycobacterium tuberculosis Infection before Reducing the Bacterial Burden. J. Immunol. 203, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nemeth J, Olson GS, Rothchild AC, Jahn AN, Mai D, Duffy FJ, Delahaye JL, Srivatsan S, Plumlee CR, Urdahl KB, et al. (2020). Contained Mycobacterium tuberculosis infection induces concomitant and heterologous protection. PLoS Pathog. 16, e1008655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnappinger D, O’Brien KM, and Ehrt S (2015). Construction of Conditional Knockdown Mutants in Mycobacteria. In Methods in Molecular Biology (Springer; ), pp. 151–175. [DOI] [PubMed] [Google Scholar]
- 47.Kim JH, O’Brien KM, Sharma R, Boshoff HIM, Rehren G, Chakraborty S, Wallach JB, Monteleone M, Wilson DJ, Aldrich CC, et al. (2013). A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. USA 110, 19095–19100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flentie KN, Stallings CL, Turk J, Minnaard AJ, and Hsu FF (2016). Characterization of phthiocerol and phthiodiolone dimycocerosate esters of M. tuberculosis by multiple-stage linear ion-trap MS. J. Lipid Res. 57, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Flow cytometry data reported in this paper will be shared by the lead contact upon request. Whole-genome sequencing data has been deposited at Genome Sequence Archive and is publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Purified anti-mouse CD16/32, Clone 93 | Biolegend | Cat#101302; RRID:AB_312801 |
| BD OptiBuild BUV395 Hamster Anti-Mouse CD80, Clone 16-10A1 | BD Biosciences | Cat#740246; RRID:AB_2739993 |
| Brilliant Violet 421 anti-mouse I-A/I-E, Clone M5/114.15.2 | Biolegend | Cat#107632; RRID:AB_2650896 |
| Brilliant Violet 510 anti-mouse CD11c, Clone N418 | Biolegend | Cat#117353; RRID:AB_2562016 |
| Brilliant Violet 711 anti-mouse/human CD11b, Clone M1/70 | Biolegend | Cat#101242; RRID:AB_2563310 |
| BD OptiBuild BV786 Mouse Anti-Mouse CD64 a and b Alloantigens, Clone X54-5/7.1 | BD Biosciences | Cat#741024; RRID:AB_2740644 |
| BD Pharmingen PE Rat Anti-Mouse Siglec-F, Clone E50-2440 | BD Biosciences | Cat#552126; RRID:AB_394341 |
| PE/Cyanine7 anti-mouse Ly-6G, Clone 1A8 | Biolegend | Cat#127618; RRID:AB_1877262 |
| Alexa Fluor 700 anti-mouse CD45, Clone 30-F11 | Biolegend | Cat#103128; RRID:AB_493715 |
| APC/Cyanine7 anti-mouse Ly-6C, Clone HK1.4 | Biolegend | Cat#128026; RRID:AB_10640120 |
| BD Horizon BUV395 Mouse Anti-Mouse NK-1.1, Clone PK136 | BD Biosciences | Cat#564144; RRID:AB_2738618 |
| Brilliant Violet 421 anti-mouse TCR γ/δ, Clone GL3 | Biolegend | Cat#118120; RRID:AB_2562566 |
| Brilliant Violet 711 anti-mouse CD19, Clone 6D5 | Biolegend | Cat#115555; RRID:AB_2565970 |
| PE anti-mouse/human CD44, Clone IM7 | Biolegend | Cat#103007; RRID:AB_493686 |
| PE/Cyanine7 anti-mouse CD8a, Clone 53-6.7 | Biolegend | Cat#100722; RRID:AB_312761 |
| APC anti-mouse CD4, Clone GK1.5 | Biolegend | Cat#100412; RRID:AB_312696 |
| APC/Cyanine7 anti-mouse TCR β chain, Clone H57-597 | Biolegend | Cat#109220; RRID:AB_893624 |
| Brilliant Violet 650 anti-mouse CD45, Clone 30-F11 | Biolegend | Cat#103151; RRID:AB_2565884 |
| Rabbit anti-CpsA | 21 | N/A |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa 647 | Thermo Fisher | Cat#A-21245; RRID:AB_2535813 |
| Rabbit anti-Ag85B | Joel Ernst | N/A |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher | Cat#A32731; AB_2633280 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| M. tuberculosis H37Rv | William R Jacobs Jr | N/A |
| M. tuberculosis ΔkatG | William R Jacobs Jr | N/A |
| M. tuberculosis ΔcpsA | (24,26) | N/A |
| M. tuberculosis ΔcpsA ΔkatG | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Purina 5053 Rodent Chow with 2,000 ppm Doxycycline | Research Diets | Cat#C15020601i |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | Cat#D4527 |
| Collagenase D | Roche | Cat#COLLD-RO |
| Trucount Absolute Counting Tubes | BD Biosciences | Cat#340334 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat#CO-RO |
| 4–20% Mini-PROTEAN TGX Precast Protein Gels | Bio-Rad | Cat#4561094 |
| 10x Tris/Glycine/SDS | Bio-Rad | Cat#1610732 |
| 10x Tris/Glycine Buffer for Western Blots and Native Gels | Bio-Rad | Cat#1610734 |
| Restore Western Blot Stripping Buffer | Thermo Scientific | Cat#21059 |
| Nitrocellulose/Filter Paper Sandwich, 0.2 μm, 8.3 × 7.3 cm | Invitrogen | Cat#LC2000 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cytokine & Chemokine Convenience 26-Plex Mouse ProcartaPlex Panel 1 | Invitrogen | Cat#EPXR260-26088-901 |
| IFN alpha Mouse ProcartaPlex Simplex Kit | Invitrogen | Cat#EPX01A-26027-901 |
| IFN beta Mouse ProcartaPlex Simplex Kit | Invitrogen | Cat#EPX01B-26044-901 |
| DuoSet ELISA kits for IL-1 β | R&D Systems | Cat#DY401 |
|
| ||
| Deposited data | ||
|
| ||
| Whole-genome sequencing data; ΔcpsA katG parent strain and mouse passaged isolates | Genome Sequence Archive | GSA: CRA013548 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: WT: C57BL/6J | The Jackson Laboratory | Cat#000664 |
| Mouse: Nox2−/−: B6.129S-Cybbtm1Din/J | Mary Dinauer | N/A |
| Mouse: Ccr2−/−: B6.129S4-Ccr2tm1Ifc/J | The Jackson Laboratory | Cat#004999 |
|
| ||
| Oligonucleotides | ||
|
| ||
| 5’-CGGGCCAACCTGGGATGC-3’ | IDT | N/A |
| 5’-GCAGGCTCGCGTAGGAATCATCC-3’ | IDT | N/A |
| 5’-GAACTGCTCGCCTTCACCTTCC-3’ | IDT | N/A |
| 5’-AACCGCGTGGCATGGCCGGCTGCA-3’ | IDT | N/A |
| 5’-TGCTCATCGGGCTGGACTCG-3’ | IDT | N/A |
| 5’-ACTCGATGGCGGTGGT GAACG-3’ | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pGMCK-TetOFF-cpsA | This paper | N/A |
| pKP617 cpsA | 21 | N/A |
| pMV261 GFP | Christina Stallings | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | GraphPad | N/A |
| FlowJo | BD Biosciences | N/A |
| FACSDiva | BD Biosciences | N/A |
| BioRender | BioRender | N/A |