

Abstract
The mammalian cell cycle is divided into four sequential phases, namely G1 (Gap 1), S (synthesis), G2 (Gap 2), and M (mitosis). Wee1, whose turnover is tightly and finely regulated, is a well-known kinase serving as a gatekeeper for the G2/M transition. However, the mechanism underlying the turnover of Wee1 is not fully understood. Autophagy, a highly conserved cellular process, maintains cellular homeostasis by eliminating intracellular aggregations, damaged organelles, and individual proteins. In the present study, we found autophagy deficiency in mouse liver caused G2/M arrest in two mouse models, namely Fip200 and Atg7 liver-specific knockout mice. To uncover the link between autophagy deficiency and G2/M transition, we combined transcriptomic and proteomic analysis for liver samples from control and Atg7 liver-specific knockout mice. The data suggest that the inhibition of autophagy increases the protein level of Wee1 without any alteration of its mRNA abundance. Serum starvation, an autophagy stimulus, downregulates the protein level of Wee1 in vitro. In addition, the half-life of Wee1 is extended by the addition of chloroquine, an autophagy inhibitor. LC3, a central autophagic protein functioning in autophagy substrate selection and autophagosome biogenesis, interacts with Wee1 as assessed by co-immunoprecipitation assay. Furthermore, overexpression of Wee1 leads to G2/M arrest both in vitro and in vivo. Collectively, our data indicate that autophagy could degrade Wee1—a gatekeeper of the G2/M transition, whereas the inhibition of autophagy leads to the accumulation of Wee1 and causes G2/M arrest in mouse liver.
1. Introduction
The cell cycle refers to the sequence of biological events in the process of cell division [1]. It is well-established as an evolutionarily conserved and sophisticatedly regulated process during cell proliferation and growth, and thus demonstrated to play pivotal roles in apoptosis, tumorigenesis, DNA repair, tissue regeneration, differentiation, and development [2]. The cell cycle is divided into four sequential phases, namely G1 (Gap 1), S (synthesis), G2 (Gap 2), and M (mitosis). Mammalian cell cycle regulation signaling is a multilayered control system that is triggered by external stimuli, prompting the cell to exit its quiescent state (termed as G0) [3,4]. The transitions between stages are tightly regulated by checkpoint quality control systems. Among these, the G2 checkpoint controls the G2/M transition by monitoring the DNA fidelity and revising possible DNA lesions introduced in the DNA synthesis during the S phase [1].
Novel regulatory proteins that control the G2/M transition are continually revealed [2]. For instance, Wee1 is a well-known kinase serving as a mitotic gatekeeper during the G2/M transition to guarantee the completion of DNA replication, the maintenance of genome stability, and the correct activation state of other cell cycle proteins like CDK1 [5]. Wee1 protein levels impact cell cycle progression, and the dysfunction of Wee1 can lead to cell cycle disorder-related syndromes [6]. Therefore, the degradation pathway of Wee1 and the underlying mechanisms have attracted much attention in recent years for their promising application in novel therapeutic target development and drug discoveries [7–10]. Previous reports suggest that the SCF E3 ubiquitin ligase β-TrCP recognizes Wee1, leading to Wee1 degradation through the proteasome machinery [11]. However, whether alternative degradation pathways target Wee1 remains unknown.
Autophagy, the highly-conserved degradation pathway in eukaryotic organisms, maintains cellular homeostasis by eliminating intracellular aggregations, damaged organelles, invasive pathogens, and selective protein substrates [12–14]. ATG5 is an integral part of the ATG5-ATG12-ATG16L1 complex that catalyzes the ATG8 lipidation essential for autophagosome formation and expansion. Deletion of Atg5 in the murine renal proximal tubule causes G2/M arrest via a hitherto unknown mechanism [15]. In the present study, we observed that hepatocyte-specific deletion of Fip200 or Atg7 induces G2/M arrest in the liver of mice, demonstrated that autophagy regulates Wee1 protein degradation, and overexpression of Wee1 promotes G2/M arrest both in vitro and in vivo.
2. Materials and methods
2.1. Animals
All animal experiments were performed following the guidelines provided by the Ethics Committees of Anhui Medical University. LSL spCas9 mice (Strain NO. T002249) were purchased from GemPharmatech (Nanjing, China). All mice had free access to water and food (normal chow diet) and were housed in a specific pathogen-free environment with a 12-h light/12-h dark cycle. Eight-week-old mice were used for experiments and were randomly divided into different groups as needed. Adeno-associated virus, suspended in 200 μL phosphate-buffered saline (PBS), was injected via the tail vein at the dosage of 4e11 vg/mouse. Mice were sacrificed and livers were collected at the indicated time point for subsequent analyses.
2.2. Cells culture, and reagents
HEK293T and HepG2 cells were maintained at 37 °C with 5% CO2 in a cell incubator with complete Dulbecco’s modified Eagle’s medium, high-glucose (DMEM, pH 7.3) (Thermo Scientific, #12800017) supplemented with 10% fetal bovine serum (HyClone, #SH30406.05) and 1% penicillin/streptomycin (Biosharp, #BL505A). Cycloheximide (Selleck, S7418) was dissolved in DMSO and used at the dosage of 300 μM. Chloroquine (Sigma, C6628) was dissolved in PBS and used at the dosage of 200 μM. The cell lysate was prepared using a standard protocol.
2.3. Plasmids
One BsmBI site locating at Cre of AAV. TBG.PI.Cre.rBG plasmid (Addgene #107787) was mutated synonymously. The fragment between EcoRI and AscI was replaced by U6-Filler-gRNA scaffold amplified from lentiCRISPR v2 plasmid (Addgene #52961), and then this new plasmid was named AAV. U6.Filler.TBG.Cre. The filler of AAV. U6.Filler.TBG.Cre was replaced by sgRNA for the gene of interest to make gene-targeting plasmids using standard protocols [16]. Fip200 sgRNA, CTC CAT TGA CCA CCA GAA CC, and Atg7 sgRNA, GAA GTT GAA CGA GTA CCG CC were obtained from Sanjana et al. [16]. AAV. TBG.Wee1 was cloned by using the In-fusion strategy. Briefly, the backbone of AAV. TBG.PI.Cre. rBG was amplified by PCR and purified first. The Wee1 was prepared by PCR using cDNA from mouse cell line Hepa 1–6. Linearized backbone and Wee1 ORF fragments were ligated using the In-fusion strategy following the standard protocol (Vazyme, C115). GFP from pLJM1-EGFP (addgene #19319) and Wee1 were cloned into pLJM1 vector (Addgene #91980) using the In-fusion strategy to make pLJM1-Wee1-GFP plasmid.
2.4. Virus generation
Adeno-associated virus serotype 8 packaging and production was performed by OBiO Technology (Shanghai). All plasmids were prepared by using EndoFree Plasmid Maxi Kit (QIAGEN, Germany). Lentivirus was packaged using a standard protocol. Briefly, HEK293T cells were transfected with MD2. G, psPAX2, and transfer plasmid, and the medium containing the virus was harvested at 72 h post-transfection. To obtain stably expressing cultured cell lines, HepG2 cells were selected with puromycin at the dosage of 2 μg/mL.
2.5. Total RNA isolation and quantitative real-time PCR
Total RNA was extracted using Total RNA Extraction Reagent (Vazyme, #R401) and reverse transcribed using HiScript III 1st Strand cDNA Synthesis Kit (gDNA wiper plus) (Vazyme, #R312). Quantitative real-time RT-PCR (qRT-PCR) was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, #Q711) according to the manufacturer’s instructions. The primers used were listed in Supplementary Table 1.
2.6. Immunoblotting assay
Cultured cells or mouse liver were lysed in RIPA lysis buffer supplemented with 1 mM phenylmethanesulfonylfluoride and protease inhibitor cocktail (Biosharp, #BL612A). Total protein concentrations were measured by BCA kit (Biosharp, #BL521A). The supernatant was mixed with SDS loading buffer containing β-mercaptoethanol and boiled at 95 °C for 10 min. An equal amount of total protein for each sample was submitted to run SDS-PAGE and transferred to nitrocellulose membranes followed by Ponceau S staining to monitor the loading amount and a blocking procedure to avoid non-specific binding. Primary and secondary antibodies used in this study are listed as below: rabbit anti-ATG7 (Proteintech, #10088–2-AP), rabbit anti-p62 (Proteintech, #18420–1-AP), rabbit anti-LC3 (Proteintech, #14600–1-AP), mouse anti-β-actin (Proteintech, #66009–1-Ig), rabbit anti-FIP200 (Proteintech, #17250–1-AP), rabbit anti-Wee1 (Zenbio, #863186), rabbit anti-GAPDH (DUONENG-Bio, #AB010301), rabbit anti-Flag (Proteintech, #80010–1-RR), rabbit anti-GST (Proteintech, #10000–0-AP), goat anti-mouse HRP conjugated (Proteintech, #SA00001–1) and goat anti-rabbit HRP conjugated (Proteintech, #SA00001–2). ECL blotting substrates (Abbkine, #BMU101-CN) were used to visualize proteins.
2.7. Immunohistochemistry (IHC) and immunofluorescence (IF) assays
Ki67 IHC staining was supported by Servicebio Tech (Wuhan, China). Briefly, paraffin sections of mouse liver were stained with anti-Ki67 rabbit polyclonal antibody (Servicebio, #GB111141–100) along with Hematoxylin staining nuclei. Images were captured in bright field microscopy. For immunofluorescence analysis, HepG2 cells stably expressing Wee1-GFP were seeded in 35 mm confocal glass bottom dishes (Biosharp, #BS-20-GJM), and incubated at 37 °C overnight. Images were captured by Zeiss LSM800 Confocal System.
2.8. Transcriptomic and proteomic analyses
Liver samples were harvested at the indicated time point from sacrificed mice and delivered to Oebiotech Company (Shanghai, China) for the transcriptomic analysis based on the Illumina Seq method and the proteomic analysis based on the iTRAQ method. The same amount of RNA from 3 mice of each group was pooled to reduce the variation and subjected to RNA-seq with the standard protocol. Raw reads were banked at the National Center for Biotechnology Information’s Sequence Read Archive under accession no. PRJNA881053. The same amount of protein from 3 mice of each group was pooled as well. The protein was digested with trypsin and was subjected to iTRAQ labeling. The LC-MS/MS analyses were performed by a Q-Exactive mass spectrometer (Thermo Scientific, USA) equipped with a Nanospray Flex source (Thermo Scientific, USA). Samples were loaded and separated by a C18 column (15 cm × 75 μm) on an EASY-nLC. Raw data were banked at the iProx database under accession no. PXD043936. The protein hits were classified into three groups based on their profile (related to Fig. 2D) and the criterion of classification was shown in Supplementary Table 2. The upregulated protein hits were further classified into three groups based on their mRNA profile (related to Fig. 2E) and the criterion of classification was shown in Supplementary Table 3.
Fig. 2. Inhibition of autophagy upregulates the protein level of Wee1 but not its mRNA level.
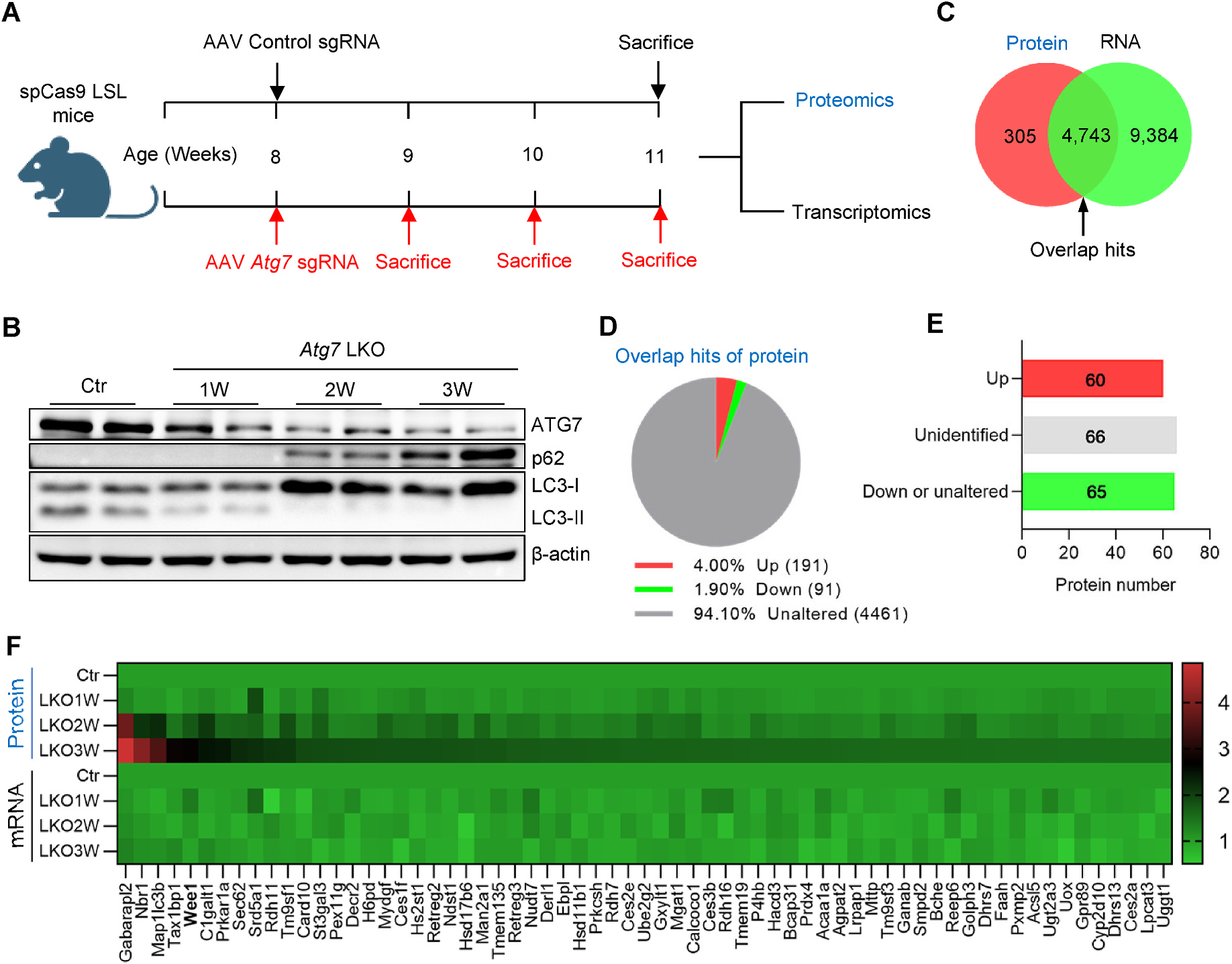
(A) The graphic view of the experimental setting, briefly, the liver samples were collected from control and Atg7 LKO mice at the indicated time and subjected to proteomics and transcriptomics analyses. (B) Western blotting for ATG7, p62, LC3, and beta-actin to confirm the knockout efficiency. (C) Venn graph for the detected hits of proteomics and transcriptomics. (D) Proteins of the overlapping hits from (C) were classified into three groups based on their profile with the time of autophagy inhibition. (E) The protein hits from the Up group of (D) were further classified into three groups based on their mRNA profile. (F) The detailed information of 65 protein hits from the Down or unaltered group of (E) was presented in the heatmap.
2.9. Co-immunoprecipitation assay
The detailed co-immunoprecipitation protocol was described previously [17]. Briefly, HEK293T cells transfected with GST-LC3 and Wee1-Flag plasmids were lysed in mild lysis buffer supplemented with 1 mM phenylmethanesulfonylfluoride and protease inhibitor cocktail (Biosharp, #BL612A). Supernatants were incubated with GST-agarose beads (Thermo Scientific, #16100) overnight at 4 °C. Beads were rinsed five times before 95 °C boiled for 10 min and then submitted to the immunoblotting analysis for coprecipitated proteins.
2.10. Cell cycle distribution analysis by flow cytometry (FCM) analysis
Cell cycle distribution was assessed based on the FCM method reported previously [18]. Briefly, cultured cells were harvested, washed twice with cold PBS, and then fixed with 70% ethanol overnight. Before being subjected to the flow cytometer apparatus, cells were resuspended and permeabilized in PBS containing 0.2% Triton X-100 and DNAs were stained with propidium iodide (Sangon Biotech, #A601112–0020) for 30 min on ice.
2.11. Statistical analysis
Data were expressed as mean ± SEM. Statistical significance was evaluated using unpaired two-tailed Student’s t-test and among more than two groups by one-way ANOVA analysis of variance. Statistical differences were considered significant at p < 0.05, dramatically significant at p < 0.01, and so forth.
3. Results and discussion
3.1. Autophagy deficiency in mouse liver causes G2/M arrest
A series of core proteins comprise the mammalian autophagy machinery, and their deficiency blocks the autophagic progression at different stages [19]. Atg5 knockout in the murine renal proximal tubule is demonstrated to cause cell cycle G2/M arrest via an unknown mechanism [15]. To study the mechanism underlying the regulation of autophagy in G2/M transition, we utilized an adeno-associated virus in LSL spCas9 mice to specifically delete autophagy genes in the liver [20] (Fig. 1A). As expected, the deletion of Fip200 led to increased hepatic levels of the autophagic cargo protein p62 and autophagic receptor protein LC3 (Fig. 1B), suggesting that the autophagic flux was successfully inhibited. Consistent with our reports and others previously [21, 22], Fip200 deletion leads to hepatomegaly (Fig. 1C). The mRNA levels of G2/M arrest markers [23], such as Aurka, Foxm1, and Ccnb2, were significantly increased (Fig. 1D), indicating that autophagy deficiency leads to G2/M arrest in mouse liver.
Fig. 1. Autophagy deficiency in mouse liver induces G2/M arrest.
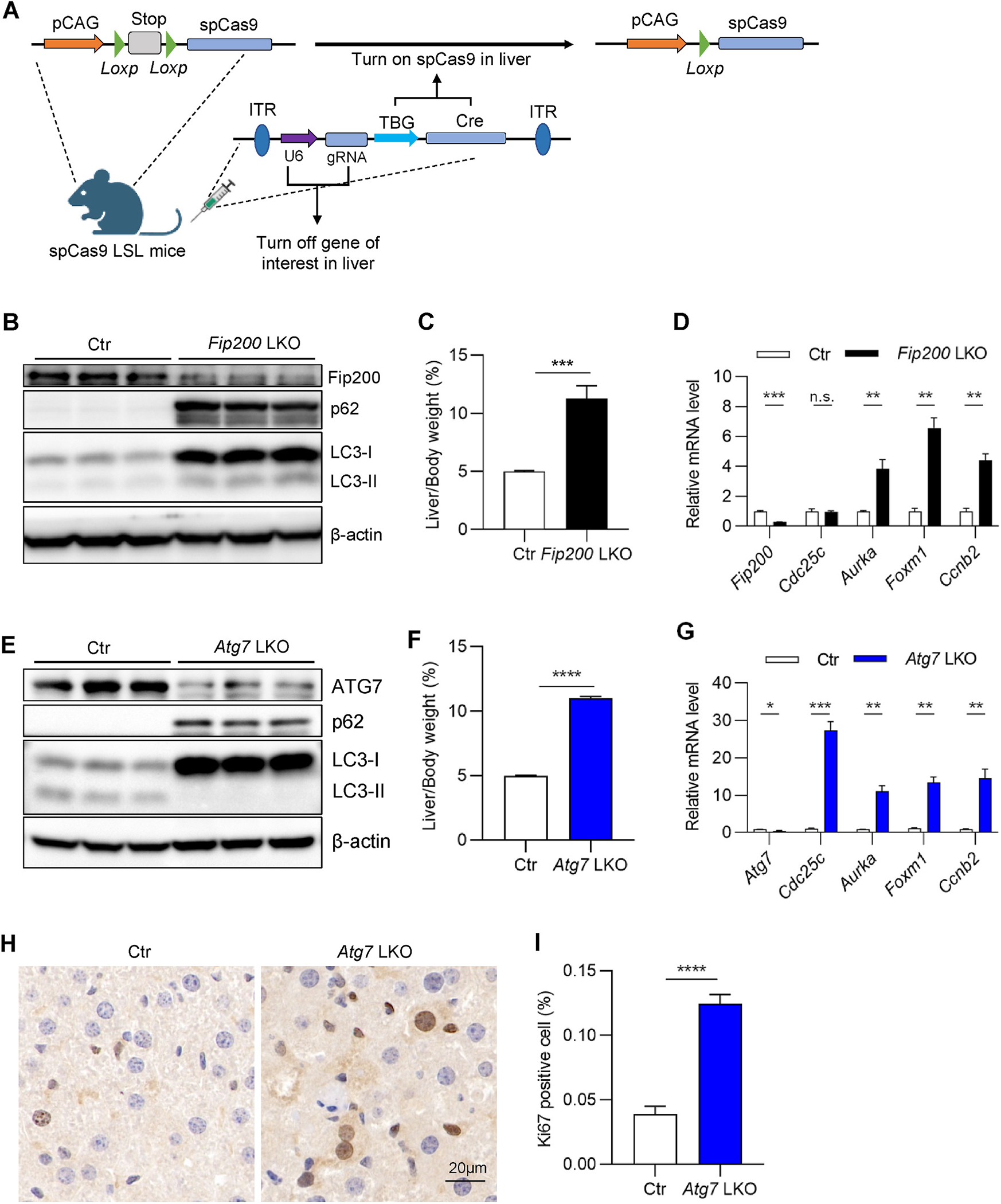
(A) The graphic view of liver-specific gene knockout mice generation by combining adeno-associated virus serotype 8 expressing sgRNA and liver-specific Cre and conditional spCas9 mice. (B–D) Liver samples were collected from LSL spCas9 mice injected with either control or Fip200 sgRNA AAV for 3 weeks. (B) Western blotting for FIP200, p62, LC3, and beta-actin. (C) Liver/body weight ratio (n = 3–5/group). (D) Quantitative RT-PCR analysis for Fip200 and G2/M arrest markers. (E–I) Liver samples were collected from LSL spCas9 mice injected with either control or Atg7 sgRNA AAV for 3 weeks. (E) Western blotting for ATG7, p62, LC3, and beta-actin. (F) Liver/body weight ratio (n = 3–5/group). (G) Quantitative RT-PCR analysis for Atg7 and G2/M arrest markers (n = 3–5/group). (H) Representative images of Ki67 staining for the liver. (I) Quantification of (H) (n = 9/group). The statistical significance was examined by Student’s t-test. n. s. indicates p > 0.05, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001 and **** indicates p < 0.0001.
Recently, multiple studies suggest that autophagic proteins can function independently of autophagy [24]. To test whether the effect of Fip200 deletion on G2/M transition is due to autophagy or the protein per se, we deleted Atg7—another critical autophagy gene in mouse liver using the same strategy as Fip200. The deletion of Atg7 led to p62 and LC3-I accumulation, indicating successful inhibition of autophagy (Fig. 1E). Like Fip200 LKO mice, Atg7 LKO mice also display hepatomegaly (Fig. 1F). In line with the results of Fip200 knockout, Atg7 knockout robustly increases the expression level of G2/M arrest markers (Fig. 1G). The deletion of Atg7 leads to higher numbers of Ki67-positive cells (Fig. 1H–I), which may reflect higher cell proliferation activity and/or more cells arrested in the G2/M transition. Taken together, inhibiting autophagy in mouse liver leads to G2/M arrest.
3.2. Inhibition of autophagy upregulates the protein level of Wee1 but not its mRNA level
To explore the mechanism underlying the effect of autophagy deficiency on G2/M transition, we performed proteomic and transcriptomic analysis on liver samples from Control (Ctr) or Atg7 LKO mice at one, two, and three weeks after AAV administration (Fig. 2A). As expected, p62 and LC3-I accumulated gradually after Atg7 deletion (Fig. 2B). Collectively, we detected 5048 proteins and 14,127 mRNAs (Fig. 2C). Next, we analyzed the hits detected in both proteomics and transcriptomics to decipher how autophagy regulates these targets. By classifying the 4743 overlapping hits based on protein expression profile, we found that 4461 were unaltered, 91 decreased, and 191 increased in Atg7 LKO, relative to Ctr liver (Fig. 2D).
To help decipher how proteins that are directly degraded by autophagy could induce G2/M arrest, as seen with autophagy inhibition, we categorized the 191 protein hits into three groups based on their mRNA profile, namely upregulated (Up), downregulated and unaltered (Down or unaltered), and unidentified (Fig. 2E). Presumably, the 65 hits whose protein levels increased but whose mRNA levels were either decreased or unaltered are candidate degradation targets of autophagy (Fig. 2F). As expected, top hits included autophagic proteins such as Gabarapl2, Nbr1, Map1lc3b (LC3), and Tax1bp1 [5,6] (Fig. 2F). Surprisingly, the fifth hit is Wee1—the gatekeeper of the cell cycle G2/M transition (Fig. 2F). In summary, inhibition of autophagy upregulates the Wee1 protein levels, but mRNA abundance is unaffected.
3.3. Wee1 interacts with autophagic protein LC3 and its turnover is regulated by autophagy
Since inhibition of autophagy increases protein levels of Wee1 (Fig. 2F), we tested whether the activation of autophagy could decrease the abundance of Wee1 protein. Serum starvation has been widely used to induce autophagy [25,26]. As predicted, serum starvation downregulated levels of the autophagic protein p62 (Fig. 3A). Meanwhile, Wee1 protein levels rapidly reduced with serum starvation (Fig. 3A). To further confirm the role of autophagy in Wee1 degradation, we conducted the CHX chase assay in the absence or presence of the autophagy inhibitor chloroquine (CQ). The half-life of Wee1 protein levels increased with chloroquine treatment (Fig. 3B–C), indicating Wee1 as a likely target of autophagic degradation. Interestingly, in line with starvation-induced Wee1 degradation, starvation stimulated the translocation of Wee1 from the nucleus to the cytosol (Fig. 3D), allowing for the possible association of Wee1 with autophagy machinery. Previous work has demonstrated that LC3 is involved in the nuclear export of Lamina—a nuclear protein that can be transported to the cytosol and degraded by autophagy [27]. To test the possible interaction of Wee1 with the autophagic machinery, we performed a co-immunoprecipitation assay by exogenously coexpressing Flag-tagged Wee1 and GST-tagged LC3 in HEK293T cells. Wee1 coprecipitated with GST-LC3 but not GST alone, suggesting a physical interaction between Wee1 and LC3 (Fig. 3E). Whether the interaction between Wee1 and LC3 is necessary for the nuclear export of Wee1 requires further investigation. In summary, Wee1 is regulated by autophagy and it interacts with the autophagy receptor LC3.
Fig. 3. Wee1 interacts with autophagic receptor LC3 and its turnover is regulated by autophagy.
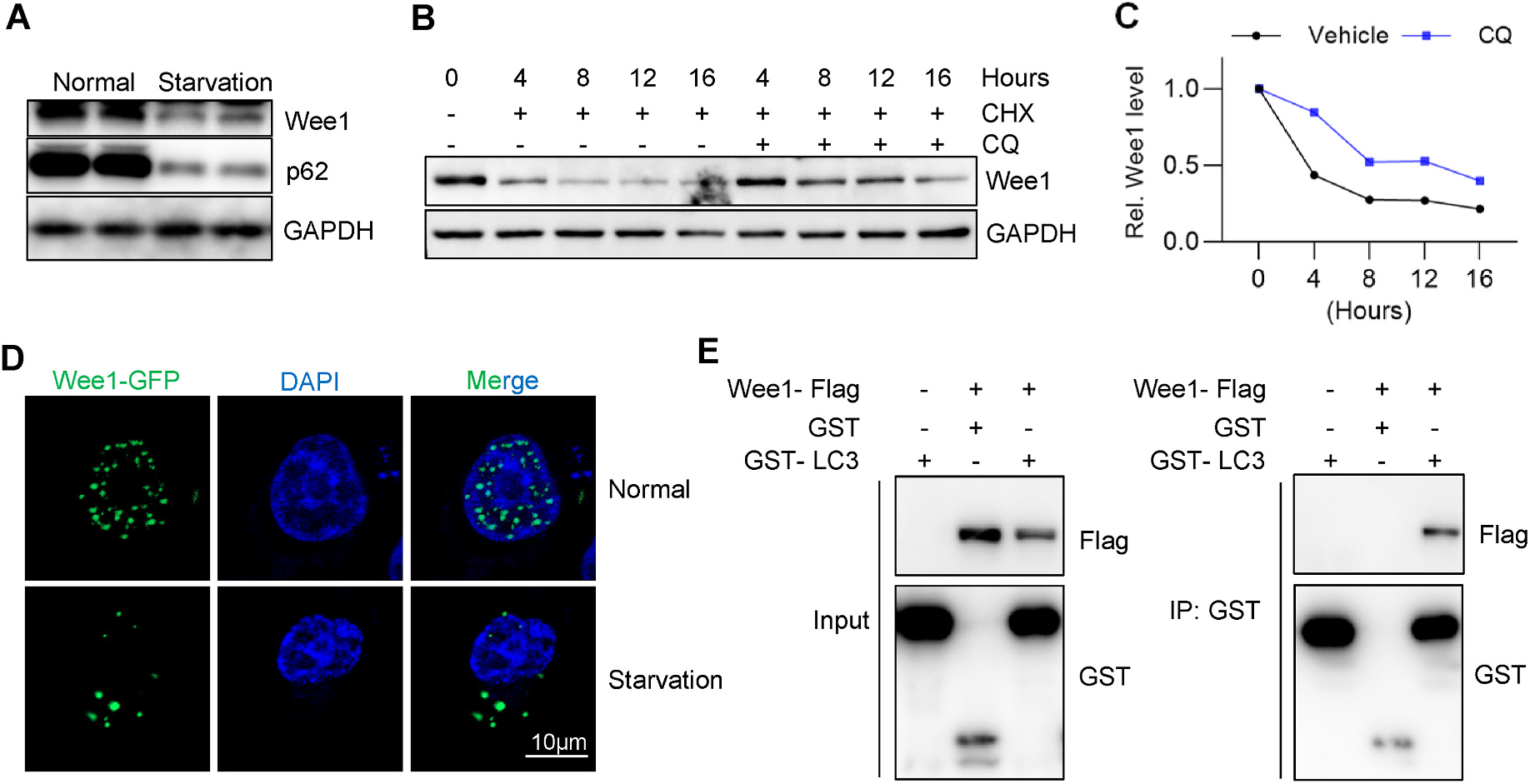
(A) Cultured HepG2 cells were serum starved for 2 h and subjected to immunoblotting assay. (B) Cycloheximide (CHX) chase assay was performed in the absence or presence of autophagy inhibitor chloroquine (CQ). Briefly, 300 μM of CHX and 200 μM of CQ were used, and the cells were harvested for western blotting at the indicated time. GAPDH was used as the loading control for the immunoblotting assay. (C) Quantification of (B). (D) HepG2 cells stably expressing Wee1-GFP were either treated with normal or serum-free medium for 2 h and then fixed for DAPI staining, followed by confocal imaging. (E) GST co-immunoprecipitation assay demonstrated the interaction between Wee1 and the autophagic receptor LC3. Wee1-Flag, GST, and GST-LC3 plasmids were transfected into HEK 293T cells as indicated, and cells were harvested for IP assay after 36 h.
3.4. Overexpression of Wee1 causes G2/M arrest
To mimic autophagy inhibition-induced Wee1 accumulation causing G2/M arrest, we stably overexpressed Wee1 in HepG2 cells (Fig. 4A). As expectedly, mRNA levels of G2/M arrest markers were upregulated in overexpressed cells (Fig. 4B). FACS-based cell cycle distribution assay showed that the proportion of cells retained in the G2/M phase increased significantly from 12.8% in the control group to 19.1% in the Wee1-overexpressing group (Fig. 4C and D). These results indicated that the G2/M transition was interrupted upon Wee1 overexpression in vitro.
Fig. 4. Overexpression of Wee1 causes G2/M arrest.
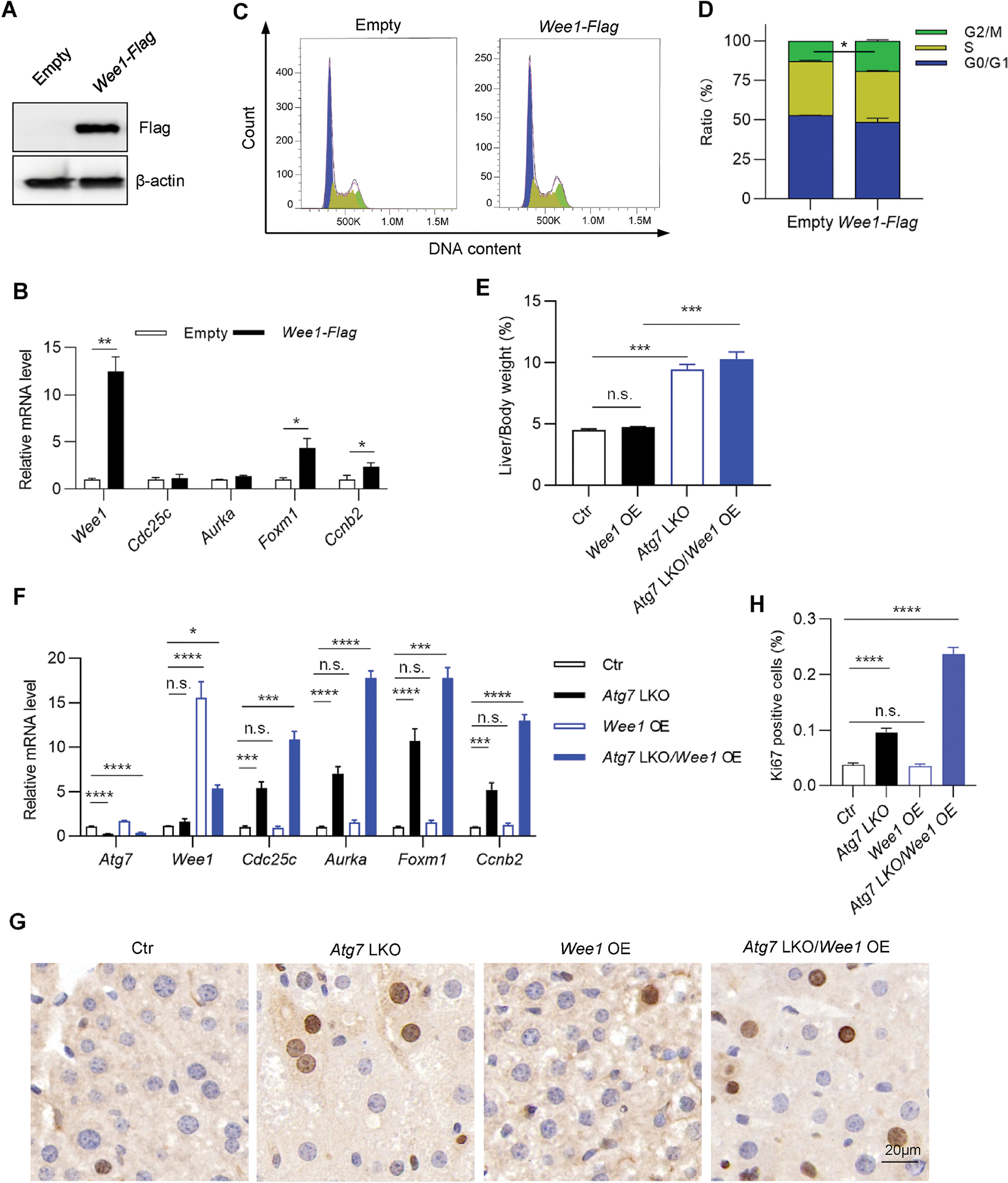
(A–D) HepG2 cells stably expressing empty vector or Wee1-Flag were subjected to immunoblotting assay for overexpression efficiency detection (A) and to quantitative RT-PCR for the detection of G2/M arrest markers (B). Cell cycle distribution was examined through propidium iodide (PI) staining based on FACS analysis (C and D). (E–H) Eight-week-old LSL spCas9 male mice were tail vein injected with either AAV. TBG.Wee1, AAV. TBG.Atg7 sgRNA, or both, and sacrificed for liver collection at three weeks after AAV administration. (E) Liver/body weight ratio (n = 4–5/group). (F) Quantitative RT-PCR for G2/M arrest markers (n = 4/group). (G) Representative Ki67 staining images. (H) Quantification of (G) (n = 9/group). The statistical significance was examined by Student’s t-test. n. s. indicates p > 0.05, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001 and **** indicates p < 0.0001.
To test the effects of hepatic Wee1 overexpression in vivo, we injected LSL spCas9 mice with either AAV. TBG.Wee1, AAV. TBG.Atg7sg, or both, and collected liver tissues after three weeks. Interestingly, hepatic overexpression of Wee1 did not cause hepatomegaly or G2/M arrest (Fig. 4E and F). One possible explanation is that hepatocytes have a long turnover time in vivo, and the proportion of proliferating cells is low at the basal level [28]. Along these lines, Ki67 staining showed that the proportion of proliferating cells in the control mouse liver was only 0.03% (Fig. 4G and H). Atg7 LKO increased the proportion of proliferating cells by 2.6-fold (Fig. 4G and H), however overexpression of Wee1 on the Atg7 LKO background led to a 2.5-fold increase in the fraction of proliferating cells, relative to Atg7 LKO alone (Fig. 4H), which might suggest that more proliferating cells were arrested at the G2/M transition stage. In line with this observation, overexpression of Wee1 on the Atg7 LKO background also further upregulated the expression levels of G2/M arrest markers (Fig. 4F). Taken together, our data indicate that autophagy could degrade Wee1, a gatekeeper of G2/M transition, and inhibition of autophagy leads to the accumulation of Wee1 that contributes to G2/M arrest in mouse liver.
Supplementary Material
Acknowledgments
This work was supported by Nature Science Foundation of Anhui Province (2108085MC81 to A.H., 2208085QH248 to Y.C.), Key Projects of Nature Science for Universities in Anhui Province (2022AH050642 to Y.Y.), and NIH grants (K01 DK137050 to D.F.).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbrc.2023.08.010.
References
- [1].Uzbekov R, Prigent C, A journey through time on the discovery of cell cycle regulation, Cells (2022) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jamasbi E, Hamelian M, Hossain MA, Varmira K, The cell cycle, cancer development and therapy, Mol. Biol. Rep. 49 (2022) 10875–10883. [DOI] [PubMed] [Google Scholar]
- [3].Heber-Katz E, Zhang Y, Bedelbaeva K, Song F, Chen X, Stocum DL, Cell cycle regulation and regeneration, Curr. Top. Microbiol. Immunol. 367 (2013) 253–276. [DOI] [PubMed] [Google Scholar]
- [4].Fischer M, Schade AE, Branigan TB, Muller GA, DeCaprio JA, Coordinating gene expression during the cell cycle, Trends Biochem. Sci. 47 (2022) 1009–1022. [DOI] [PubMed] [Google Scholar]
- [5].Ghelli A Luserna di Rora, C. Cerchione, G. Martinelli, G. Simonetti, A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target, J. Hematol. Oncol. 13 (2020) 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Szmyd R, Niska-Blakie J, Diril MK, Renck Nunes P, Tzelepis K, Lacroix A, van Hul N, Deng LW, Matos J, Dreesen O, Bisteau X, Kaldis P, Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality, Oncogene 38 (2019) 998–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serpico AF, D’Alterio G, Vetrei C, Della Monica R, Nardella L, Visconti R, Grieco D, Wee1 rather than Plk1 is inhibited by AZD1775 at therapeutically relevant concentrations, Cancers 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li Z, Pinch BJ, Olson CM, Donovan KA, Nowak RP, Mills CE, Scott DA, Doctor ZM, Eleuteri NA, Chung M, Sorger PK, Fischer ES, Gray NS, Development and characterization of a Wee1 kinase degrader, Cell Chem. Biol. 27 (2020) 57–65 e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liang L, He YJ, Wang HQ, Zhou H, Xiao L, Ye M, Kuang YJ, Luo SQ, Zuo YN, Feng PF, Yang CY, Cao WJ, Liu TH, Roy M, Xiao XJ, Liu J, The Wee1 kinase inhibitor MK1775 suppresses cell growth, attenuates stemness and synergises with bortezomib in multiple myeloma, Br. J. Haematol. 191 (2020) 62–76. [DOI] [PubMed] [Google Scholar]
- [10].Fallah Y, Demas DM, Jin L, He W, Shajahan-Haq AN, Targeting WEE1 inhibits growth of breast cancer cells that are resistant to endocrine therapy and CDK4/6 inhibitors, Front. Oncol. 11 (2021), 681530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H, M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 4419–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dikic I, Elazar Z, Mechanism and medical implications of mammalian autophagy, Nat. Rev. Mol. Cell Biol. 19 (2018) 349–364. [DOI] [PubMed] [Google Scholar]
- [13].Levine B, Kroemer G, Biological functions of autophagy genes: a disease perspective, Cell 176 (2019) 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cao W, Li J, Yang K, Cao D, An overview of autophagy: mechanism, regulation and research progress, Bull. Cancer 108 (2021) 304–322. [DOI] [PubMed] [Google Scholar]
- [15].Li H, Peng X, Wang Y, Cao S, Xiong L, Fan J, Wang Y, Zhuang S, Yu X, Mao H, Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis, Autophagy 12 (2016) 1472–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening, Nat. Methods 11 (2014) 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang Y, Ma F, Liu Z, Su Q, Liu Y, Liu Z, Li Y, The ER-localized Ca(2+)-binding protein calreticulin couples ER stress to autophagy by associating with microtubule-associated protein 1A/1B light chain 3, J. Biol. Chem. 294 (2019) 772–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Maadi H, Soheilifar MH, Wang Z, Analysis of cell cycle by flow cytometry, in: Wang Z (Ed.), Cell-Cycle Synchronization: Methods and Protocols, Springer US, New York, NY, 2022, pp. 183–195. [DOI] [PubMed] [Google Scholar]
- [19].Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC, Regulation of mammalian autophagy in physiology and pathophysiology, Physiol. Rev. 90 (2010) 1383–1435. [DOI] [PubMed] [Google Scholar]
- [20].Wang X, Xu BL, Chen XW, Acute gene inactivation in the adult mouse liver using the CRISPR-Cas9 technology, STAR protoc. 2 (2021), 100611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ni H-M, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, Ding W-X, Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury, Toxicol. Sci. 127 (2012) 438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].He A, Chen X, Tan M, Chen Y, Lu D, Zhang X, Dean JM, Razani B, Lodhi IJ, Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation, Molecular Cell, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].He F, Antonucci L, Yamachika S, Zhang Z, Taniguchi K, Umemura A, Hatzivassiliou G, Roose-Girma M, Reina-Campos M, Molina AD, NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly, J. Hepatol. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Galluzzi L, Green DR, Autophagy-independent functions of the autophagy machinery, Cell 177 (2019) 1682–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].De S, Das S, Sengupta S, Involvement of HuR in the serum starvation induced autophagy through regulation of Beclin1 in breast cancer cell-line, MCF-7, Cell. Signal. 61 (2019) 78–85. [DOI] [PubMed] [Google Scholar]
- [26].Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, Liu B, Chang C, Zhou T, Lippincott-Schwartz J, Liu W, Deacetylation of nuclear LC3 drives autophagy initiation under starvation, Mol. Cell 57 (2015) 456–466. [DOI] [PubMed] [Google Scholar]
- [27].Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, Catanzaro JM, Ricketts MD, Lamark T, Adam SA, Marmorstein R, Zong WX, Johansen T, Goldman RD, Adams PD, Berger SL, Autophagy mediates degradation of nuclear lamina, Nature 527 (2015) 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yano K, Choijookhuu N, Ikenoue M, Fidya, Fukaya T, Sato K, Lee D, Taniguchi N, Chosa E, Nanashima A, Hishikawa Y, Spatiotemporal expression of HMGB2 regulates cell proliferation and hepatocyte size during liver regeneration, Sci. Rep. 12 (2022), 11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.