

Abstract
Tobacco smoke is a complex mixture of chemicals, many of which are toxic and carcinogenic. Hazard assessments of tobacco smoke exposure have predominantly focused on either single chemical exposures or the more complex mixtures of tobacco smoke or its fractions. There are fewer studies exploring interactions between specific tobacco smoke chemicals. Aldehydes such as formaldehyde and acetaldehyde were hypothesized to enhance the carcinogenic properties of the human carcinogen, 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) through a variety of mechanisms. This hypothesis was tested in the established NNK-induced A/J mouse lung tumor model. A/J mice were exposed to NNK (i.p., 0, 2.5, or 7.5 μmol in saline) in the presence or absence of acetaldehyde (0 or 360 ppmv) or formaldehyde (0 or 17 ppmv) for 3 h in a nose-only inhalation chamber and lung tumors were counted 16 weeks later. Neither aldehyde by itself induced lung tumors. However, mice receiving both NNK and acetaldehyde or formaldehyde had more adenomas with dysplasia or progression than those receiving only NNK, suggesting that aldehydes may increase the severity of NNK-induced lung adenomas. The aldehyde co-exposure did not affect the levels of NNK-derived DNA adduct levels. Similar studies tested the ability of a 3 h nose-only carbon dioxide (0, 5, 10, or 15%) co-exposure to influence lung adenoma formation by NNK. While carbon dioxide alone was not carcinogenic, it significantly increased the number of NNK-derived lung adenomas without affecting NNK-derived DNA damage. These studies indicate that the chemicals in tobacco smoke work together to form a potent lung carcinogenic mixture.
Keywords: tobacco, aldehyde, carbon dioxide, co-carcinogenesis, NNK
Graphical Abstract

Introduction
Tobacco smoke is a multisite carcinogen and contains more than 7,000 distinct chemicals.1 Only a small fraction have been evaluated for their potential role in the harmful effects of tobacco products with approximately 70 compounds having well established toxicity and/or carcinogenicity in animal models.2 Less is known about the interactions among these tobacco smoke constituents to cause the overall harmful effects of tobacco use. The complexity of tobacco smoke makes it challenging to identify which compound(s) are responsible for cancer induction. One of the research priorities of the Food and Drug Administration (FDA) Center for Tobacco Products is to reduce the toxicity and carcinogenicity of tobacco and tobacco smoke. The FDA has created the List of Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke.3 Chemicals on this list include the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and two abundant tobacco smoke aldehydes - acetaldehyde and formaldehyde. All three chemicals exhibit harmful effects to both animal models and humans.4–6 It is unknown how regulation of these chemicals will affect the overall carcinogenic activity of tobacco smoke. Important missing information is how these particular constituents impact toxicity and carcinogenicity of tobacco products and smoke.
In this study, we explored the binary interactions between NNK and formaldehyde or acetaldehyde in the well-characterized A/J mouse lung tumor model. While replication of the human exposure scenario in rodents is challenging, in large part due to the anatomical and physiological differences, rodent models are invaluable for investigating the carcinogenic properties of tobacco chemicals. Specifically, they have led to the development of key biomarkers of exposure, metabolism, and toxicity that have been applied to human risk assessment studies linking chemical exposures from tobacco to its harmful effects.7 As such, they can prove useful to assess interactions among chemicals found in the complex mixture of tobacco smoke.
NNK is a lung selective carcinogen present in tobacco smoke and smokeless tobacco products4, 8 and is readily administered via intraperitoneal injection (i.p.).9 NNK is considered “carcinogenic to humans” by the International Agency for Research on Cancer (IARC).4 Acetaldehyde is a rodent carcinogen and a possible human carcinogen (IARC Group 2B).5 It is a rat nasal tumorigen when inhaled.10 Formaldehyde is a human carcinogen.6 It acts locally in rodents causing nasal cavity tumors when inhaled and forestomach tumors when administered in drinking water.6
A/J mice are susceptible to lung tumor formation; a single high dose of NNK generates a reproducibly quantifiable number of lung tumors 16 weeks post-injection, in a model that has been widely used.9, 11, 12 A/J mice are also sensitive to tobacco smoke-induced lung carcinogenesis.13–15 In addition, they also provide an effective means to explore potential interactions between chemicals.16, 17 The single injection protocol eliminates complications of multiple dosing, thereby allowing a focus on lung tumor formation. This is particularly important when investigating interactions between chemicals where one is a locally acting carcinogen or co-carcinogen, like acetaldehyde and formaldehyde, where in a chronic dosing regimen, nasal tumors would dominate and possibly obscure the impact of the co-exposure on lung tumor formation.
The hypothesis for this study was that both acetaldehyde and formaldehyde will enhance the carcinogenic properties of NNK. This hypothesis is based on the knowledge that both aldehydes damage DNA and are mutagens.5, 6 Moreover, they inhibit several repair pathways involved in the protection of cells against NNK-derived DNA damage.18, 19 Furthermore, they are known to induce stimulation of cell proliferation,20, 21, reduce apoptosis22, 23 and alter cell signaling pathways.24–26 To test this hypothesis, mice were exposed to NNK in the presence or absence of inhaled acetaldehyde or formaldehyde. In addition to determining the effect of the co-exposure on tumor yield, we also measured the levels of pulmonary DNA damage resulting from NNK, acetaldehyde, or formaldehyde at early and late time points to provide insight into the mechanism of any interaction between the chemicals. Our studies indicated that formaldehyde and acetaldehyde enhanced the severity of the tumors at early time points. Additional experiments indicated that carbon dioxide increased the number of NNK-derived lung tumors in a dose-dependent manner. The co-exposures had no influence on the levels of NNK-induced DNA damage.
Materials and Methods
Caution: NNK and formaldehyde are human carcinogens (IARC Group 1) and acetaldehyde is a possible human carcinogen (IARC Group 2B), so they should be handled with great caution in a well-ventilated hood and with appropriate personal protective equipment.
Chemicals.
NNK was purchased from Toronto Research Chemicals Inc. (Toronto, Ontario). Acetaldehyde-(2,4-dinitrophenyl)hydrazone (acetaldehyde-DNPH) and formaldehyde-(2,4-dinitrophenyl)hydrazone (formaldehyde-DNPH) were purchased from Supelco Inc. (Bellefonte, PA). XPoSure aldehyde cartridges were purchased from Waters Corporation (Milford, MA). All other chemicals and solvents were acquired from Sigma-Aldrich Chemical Co. (Milwaukee, WI) or Fisher Scientific (Hampton, NH). All solvents used for HPLC and MS analysis were of the purest grade commercially available. Tanks of biological atmosphere air containing 0, 5, 10, and 15% carbon dioxide (v/v) as well as 21% oxygen in nitrogen were purchased from Airgas USA (Roseville, MN).
Animals.
This study was approved by the University of Minnesota Institutional Animal Care and Use Committee. Female A/J mice, 7 weeks of age, were obtained from Jackson Laboratories (Bar Harbor, ME) and housed five mice per cage under standard conditions and sustained on American Institute of Nutrition-76-powdered diet (Research Diets Inc., New Brunswick, NJ). They were randomly divided into treatment groups and acclimated to the facility for one week. The following week the mice were conditioned with an air exposure in the nose-only, flow past cylindrical stainless steel inhalation chamber (In-Tox Products, LLC, Moriarty, NM) for 1.5 h/day for three separate days. The carcinogen exposures were administered the next week when the mice were 9 weeks of age. Body weights were recorded weekly.
Formaldehyde vapors.
A solution of formaldehyde was generated by dissolving paraformaldehyde in a 0.27 mM NaOH solution by heating at 100 °C for 90 min. Formaldehyde gas was delivered to the exposure chamber by passing air through the solution using a stainless steel fritted bubbler at a flow rate of 0.3 LPM (Omega high precision flow meter, Norwalk, CT). The solution was maintained at 21–22 °C with a water bath. The effluent gas was diluted with additional metered air (2.0 L/min) prior to being passed into the exposure chamber.
Acetaldehyde vapors.
Undiluted acetaldehyde was placed into a stoppered test tube and immersed in an ice bath. The desired vapor concentration was achieved by bubbling air through a narrow diameter Teflon tubing immersed in the liquid and diluting the resultant effluent vapor with additional metered air to achieve the desired concentration in the exposure chamber. For the nominal vapor concentrations of 360, 400, and 500 ppmv, the flow rate through the acetaldehyde liquid was 4.3, 4.5, and 5.6 mL/min and the diluting air flow rate was 7.7 LPM. For the nominal concentration of 930 ppmv, the respective flow rates were 5.6 mL/min and 4.6 LPM. The reported concentration is given based on HPLC analysis of samples as described below.
Aldehyde chamber concentrations.
Throughout the exposures, the chamber air was sampled through a modified animal port every 30 min by drawing 50 mL air for formaldehyde and 25 mL air for acetaldehyde through an Xposure aldehyde cartridge (Waters Corporation, Milford, MA) with a 50 mL gas-tight Hamilton syringe. Blanks were obtained by sampling room air. The resultant acetaldehyde-DNPH or formaldehyde-DNPH were quantified by extracting the cartridges with 2 mL acetonitrile and injecting a 50 μL aliquot into a HPLC using a Waters 600 HPLC coupled to a Waters 996 photodiode array detector. A Clarity 5μ Oligo-RP 250 × 4.6 mm (5 μm) column (Thermo Fisher Scientific, Lenexa, KS) provided compound separation using one of two gradients run at 1 mL/min. Mobile phases for both gradients used solvent A: 0.1% trifluoroacetic acid in water and solvent B: 0.1% trifluoroacetic acid in 95% (v/v) acetonitrile:water. For formaldehyde-DNPH, the cartridge eluent was isocratically separated with 40% solvent A and 60% solvent B. The retention times for unreacted DNPH and formaldehyde-DNPH were 5.2 and 7.7 min, respectively. For acetaldehyde-DNPH, the following gradient was employed: After 2 min at 60% solvent A and 40% solvent B, there was a 5 min gradient to 25% solvent A, 75% solvent B, followed by a 2 min gradient to 100% solvent B. After 2 min, the column was returned to the initial conditions. Under these conditions, DNPH and acetaldehyde-DNPH eluted at 10.0 and 16.5 min, respectively. The analyses were performed using Empower software at λ=352 nm for formaldehyde-DNPH and λ=360 nm for acetaldehyde-DNPH using an external standard curve for quantitation. The air concentrations were taken as the assayed moles of acetaldehyde and formaldehyde in the sample air volume, corrected for process dilution. The reported values of the chamber concentration of each aldehyde were taken as the average of all samples for the experiment.
Carcinogen exposures.
For all studies, the mice were sequentially placed into canisters attached to the nose-only 36 port stainless steel inhalation chamber (Intox, Albuquerque, NM). After 1 h of exposure to aldehyde or carbon dioxide, mice were removed from the inhalation chamber and given a single i.p. injection of 0, 2.5, 5.0, or 7.5 μmol NNK in saline (0.2 mL), immediately returned to the inhalation chamber for an additional 2 h, and then removed from the chamber and returned to their cages, such that the total exposure time to the inhaled gas was 3 h. The exposures occurred in batches with an equal number of each NNK treatment group exposed to air, acetaldehyde, formaldehyde, or air containing 5, 10, or 15% carbon dioxide at any one time. The specific experiments are outlined below.
Study 1.
Groups of 30 mice were exposed to one 3 h of air, 363 ± 7 ppmv acetaldehyde, or 16.7 ± 6.5 ppmv of formaldehyde and 0, 2.5, or 7.5 μmol of NNK. The exposures were done in three batches (10 mice per NNK dose in each batch). At 4, 24, or 96 h post-NNK injection, groups of 5 mice per treatment group were euthanized with carbon dioxide, after which tissues were removed and flash frozen for DNA isolation. The remaining 15 mice per group were euthanized with carbon dioxide 16 weeks following the exposure. The lungs were removed, tumors counted, and the tissues were placed in formalin for histopathological analysis.
Study 2.
Groups of 21 mice were exposed to one 3 h of 0, 403 ± 20, 520 ± 7, or 929 ± 26 ppmv acetaldehyde and given i.p. 0, 2.5, or 7.5 μmol of NNK in saline as described in Study 1. Four additional groups of 20 mice were given i.p. injections of 0, 2.5, 5.0, or 7.5 μmol NNK without placement in the exposure chamber. As previously, the exposures were done in three batches (7 mice per NNK dose in each batch). All mice were euthanized with carbon dioxide 16 weeks following the exposure. The lungs were removed and slightly inflated by slowly infusing 0.5 mL of 10% formalin in phosphate buffer through the trachea into the lungs with a syringe prior to placement in formalin for histopathological analysis. Surface tumors were counted prior to placement in formalin.
Study 3.
Groups of 20 mice were exposed to one 3 h of 0 or 358 ± 34 ppmv acetaldehyde or 18 ± 2 ppmv formaldehyde and 7.5 μmol NNK. The exposures were performed in 2 batches with 10 mice per time point included in each batch. After 32, 44, or 56 weeks, the mice were euthanized with carbon dioxide. The lungs were removed, and tumors were measured, counted, and analyzed by histopathology as described above.
Study 4.
Groups of 20 mice were exposed to one 3 h of air containing 0, 5, 10, or 15% carbon dioxide and 0, 2.5, or 7.5 μmol of NNK. Three additional groups of 20 mice were given 0, 2.5, or 7.5 μmol NNK without placement in the exposure chamber. Another three groups of 20 mice were conditioned in the exposure chamber (1.5 h/day of compressed air for three separate days) one week prior to receiving 0, 2.5, or 7.5 μmol NNK. After 16 weeks, the mice were euthanized with carbon dioxide. The lungs were removed and tumors were measured, counted, and analyzed by histopathology as described above.
Study 5.
Groups of 10 mice received one 3 h of air containing 10% carbon dioxide or air alone and 0, 2.5, or 7.5 μmol NNK. At 4 and 96 h post-NNK injection, groups of 5 mice were euthanized with carbon dioxide. Tissues were removed and flash frozen for DNA isolation.
Histopathology.
Tissue processing and histopathological analyses were performed by the Comparative Pathology Shared Resource, University of Minnesota Masonic Cancer Center. Formalin-fixed left lung lobes were embedded in paraffin using standard methods, and 4 μm sections were cut and stained with hematoxylin and eosin prior to counting and classifying tumors in the sections using light microscopy by an ACVP-board certified veterinary pathologist. Lung tumors were classified as adenomas, adenomas with dysplasia, adenomas with progression, and adenocarcinomas based on our experience with the A/J model, on previous reports,27–30 and on the recommendations of the Mouse Models of Human Cancers Consortium.31 Adenomas are discrete expansive masses comprised of monomorphic or well differentiated cells. An adenoma with dysplasia has increased numbers of cells (10 or more), often present in a discrete focus, with hyperchromatic nuclei and nuclear size greater than those found in the predominant cell type in the adenoma. An adenoma with progression has a distinct and discrete focus (or foci) of cells arranged in a papillary or acinar (gland like) pattern consistent with the early stages of the development of carcinomas arising within adenomas described by Foley et al. 1991.27 Adenocarcinomas are expansile masses, often locally infiltrative at the margins and sometimes invading small or large airways, comprised of cells arranged in papillary or acinar patterns with regional variations in growth pattern, and exhibiting cytological atypia and increased mitoses.31 Representative images are shown in Figure S1.
DNA Adduct studies.
Lung DNA was isolated using the Puregene protocol from Qiagen (Valencia, CA) with two modifications. First, the incubation with proteinase K was done overnight at room temperature. Second, all washes of the precipitated DNA were done with isopropyl alcohol instead of ethanol to prevent aldehyde contamination. The purified samples were dissolved in 10 mM TrisHCl/5 mM MgCl2, aliquoted for the individual DNA analyses, and stored at −20 °C.
Methyl DNA adducts.
Levels of 7-methylguanine (7-mG) and O6-methylguanine (O6-mG) were determined in 25 μg DNA with an established LC-MS/MS method.32 Guanine concentrations were measured according to published methods33 and used to normalize the amount of DNA adducts in each sample.
Pyridyloxobutyl DNA adducts.
Levels of 7-[4-(3-pyridyl)-4-oxobut-1-yl]-guanine (7-pobG), O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (O6-pobdG), and O2-[4-(3-pyridyl)-4-oxobut-1-yl]-2´-deoxythymidine (O2-pobdT) were measured in enzyme hydrolysates of DNA (100 μg) by published LC-MS/MS methods and expressed relative to the 2´-deoxyguanosine concentrations in the samples.34, 35
Pyridylhydroxylbutyl DNA adducts.
Levels of 7-[4-(3-pyridyl)-4-hydroxybut-1-yl]-guanine (7-phbG), O6-[4-(3-pyridyl)-4-hydroxybut-1-yl]-2′-deoxyguanosine (O6-phbdG), and O2-[4-(3-pyridyl)-4-hydroxybut-1-yl]-2´-deoxythymidine (O2-phbdT) were measured in enzyme hydrolysates of DNA (100 μg) by published LC-MS/MS methods and expressed relative to the 2´-deoxyguanosine concentrations in the samples.36
Acetaldehyde DNA adducts as N2-ethyl-2´-deoxyguanosine (N2-ethyldG).
The levels of N2-ethyldG were determined in enzyme hydrolysates, following sodium cyanoborohydride reduction of DNA (20 μg), by LC-MS/MS using a published protocol and are expressed relative to the 2´-deoxyguanosine concentration of the sample.37
Formaldehyde-DNA adducts as N6-methyl-2´-deoxyadenosine (N6-methyldA).
The levels of N6-methyldA were determined using an adaptation of a published method.38 Briefly, 300 μL of 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) and 5 mM magnesium chloride buffer, pH 7.0, containing [15N5]N6-methyl-dAdo (50 fmol) and sodium cyanoborohydride (6 mg) were added to DNA solutions (20 μg). After overnight digestion at room temperature with 25 units DNase I (recombinant, from Pichia pastoris), 25 units of DNase I, 2 mUnits phosphodiesterase I (type II, from Crotalus adamanteus venom), and another 25 units alkaline phosphatase (recombinant, from Pichia pastoris) were added. The samples were incubated at 37 °C for 60 min. Enzymes were removed by centrifugation using a Microcon (Millipore Sigma, Burlington, MA) filter (MW cutoff of 10,000). A 10–15 μL aliquot was removed from each sample after the filtration step for dG analysis according to published methods.37 Adducts were enriched using a solid-phase extraction (SPE) cartridge (Strata-X 33 μm, 30 mg/1 ml, Phenomenex, Torrance, CA). A 50% methanol fraction was collected. The dried samples were reconstituted in 10 μL LCMS grade water.
LC-MS/MS for the analysis of the N2-ethyldG adducts.
Six μL of the reconstituted hydrolysis solution was injected in an UltiMate 3000 UHPLC System (Thermo Fisher Scientific, Waltham, MA) with a 250 × 0.5 mm Luna C18 100Å column (Phenomenex, Torrance, CA) coupled to a TSQ Vantage (Thermo Fisher Scientific, Waltham, MA) triple quadrupole mass spectrometer. The solvents used were 15 mM ammonium acetate and methanol. The solvent elution program was a 10 μL/min gradient from 5% to 35% methanol in 30 min, followed by a column wash and re-equilibration. The ESI source was set in the positive ion mode as follows: voltage, 3.0 kV; heated ion transfer tube, 300 °C. The collision energy was 12 eV. Adducts were quantified by MS/MS with selected reaction monitoring (SRM) at m/z 296 → m/z 180 ([M + H]+ → [BH]+) for N2-ethyldG, m/z 301 → m/z 185 for [15N5]N2-ethyldG, m/z 266 → m/z 150 ([M + H]+ → [BH]+) for N6-methyldA, and m/z 271 → m/z 150 for [15N5]N6-methyldA.
LC-MS/MS for the analysis of the N6-methyldA adduct.
One μL was injected in an UltiMate 3000 RSLCnano UPLC (Thermo Fisher Scientific, San Jose, CA) system equipped with a 5 μL injection loop. Separation was performed with a capillary column (75 μm ID, 20 cm length, 10 μm orifice) created by hand packing a commercially available fused-silica emitter (New Objective, Woburn, MA) with 5 μm Luna C18 bonded separation media (Phenomenex, Torrance, CA). The flow rate was 1000 nL/min for 5 min at 2% CH3CN and 98% 5 mM ammonium acetate, then decreased to 300 nL/min followed by a linear gradient of 0.75 %/min over 20 min. The column was then washed and re-equilibrated using a flow rate of 1000 nL/min. The injection valve was switched at 5.5 min to remove the sample loop from the flow path during the gradient. Mass spectrometric data was acquired with an Orbitrap Elite hybrid ion trap-orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Positive mode electrospray ionization was used under nanospray conditions (300 nL/min) using a source voltage of 2.2 kV, and the capillary temperature was 350 °C. The S-Lens RF level setting was 60%. SRM mass chromatograms were acquired with a quadrupole isolation window of m/z 1.6, HCD fragmentation of 30 %, resolution setting of 15,000, AGC settings of 2 × 105, and maximum injection times of 100 ms. The adduct was quantified with SRM at m/z 266 → m/z 150 ([M + H]+ → [BH]+) for N6-methyldA and m/z 271 → m/z 150 for [15N5]N6-methyldA.
Biostatistical methods.
We tested the effects of aldehydes or carbon dioxide combined with NNK on total tumor counts as well as counts of tumors per histopathology category using over-dispersed Poisson regression model in R. The regression models include the doses of NNK and aldehyde/carbon dioxide and their interaction terms as predictors. They were adjusted for batches when batch effects were tested as significant. If there were additive effects of aldehyde/carbon dioxide and NNK on tumor occurrence, their synergistic effect was tested using the delta method following VanderWeele and Knol.39
Results
Justification for exposure doses.
Previous studies with NNK demonstrated that there is a biphasic linear dose-response curve for tumor formation in female A/J mice so we used doses of NNK on the linear portion of the curve (2.5 or 7.5 μmol NNK, i.p. in saline).16 Literature reports indicated that 500 ppmv acetaldehyde and 15 ppmv formaldehyde were predicted to produce a comparable exposure in mice as experienced by smokers.40 To ensure that the aldehydes were present in the lung at the same time as NNK, we chose to expose the mice to the aldehyde for 1 h prior to the NNK injection and then continued the aldehyde exposure for 2 h more (Figure 1). A 3 h exposure to 500 ppmv acetaldehyde or 15 ppmv formaldehyde is estimated to deliver a total dose of 9000 μg (204 μmol) acetaldehyde or 135 μg (4.5 μmol) formaldehyde to the mouse lung.40 The amount of NNK reaching the lungs will be less than the total dose administered because NNK is primarily metabolized in the liver when given by i.p. injection41 so the lungs are likely experiencing a higher dose of aldehyde than NNK under our experimental conditions. NNK-derived methyl DNA adducts are highest at 4 h post-injection.16 In the experiments, we measured the aldehyde concentrations in the exposure chambers and the actual amounts that the mice were exposed to are reported in the sections below.
Figure 1.
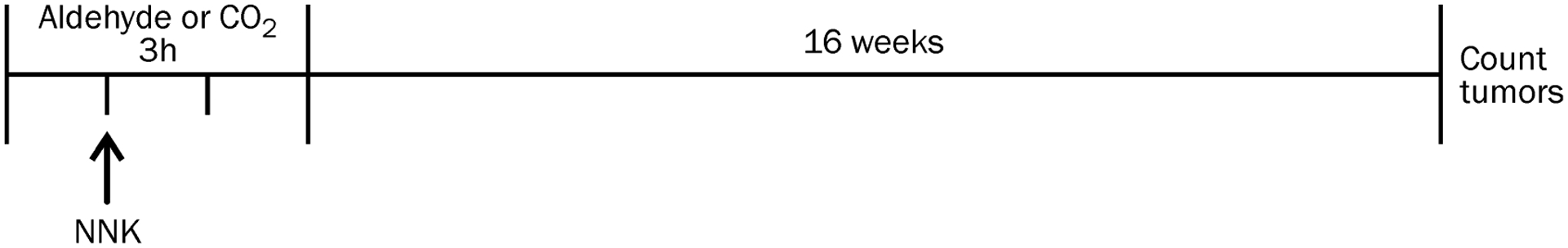
Experimental design of the exposures to NNK and aldehydes or carbon dioxide.
Co-exposure of acetaldehyde or formaldehyde enhanced the tumorigenic effect of NNK.
A small but significant increase in lung adenomas was observed when mice were exposed to both NNK and inhaled formaldehyde or acetaldehyde (Figure 2, Table S1). Neither aldehyde was tumorigenic by itself under these exposure conditions. Histopathological analysis of the lung tumors revealed a significant increase in the number of adenomas with dysplasia or progression in mice exposed to NNK and either aldehyde (Figure 3, Table S2), indicating that the co-exposure led to an increase in the severity of tumor type. The interaction between both aldehydes and NNK to cause increased numbers of adenomas with dysplasia or progression was significant (p < 0.01).
Figure 2.
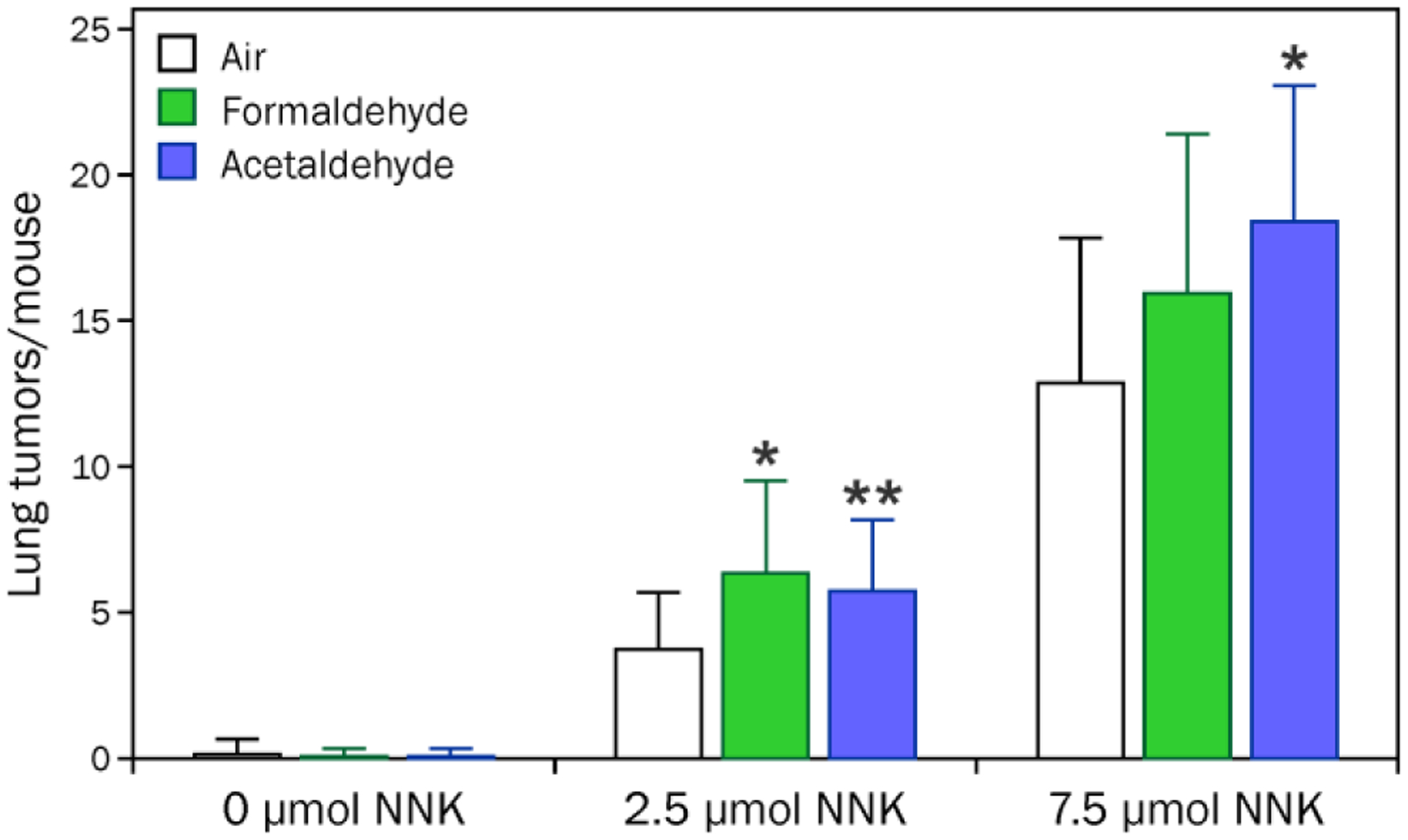
Acetaldehyde or formaldehyde had a small effect on the number of pulmonary adenomas in NNK-treated A/J mice. Groups of 15 mice were treated with 0, 2.5, or 7.5 μmol NNK (i.p. in saline) in the presence or absence of 3 h of inhaled formaldehyde (16.5 ± 6.5 ppmv) or acetaldehyde (363 ± 7 ppmv). After 16 weeks, the mice were sacrificed and lung tumors were counted. *Significantly different from air alone, p < 0.01. **Significantly different from air alone, p < 0.05.
Figure 3.

Histopathological analysis of the pulmonary tumors indicated that formaldehyde and acetaldehyde had (A) no effect on the number of adenomas produced by NNK and (B) significantly increased the number of NNK-induced pulmonary adenomas with dysplasia or progression in A/J mice. Groups of 15 mice were treated with 0, 2.5, or 7.5 μmol NNK (i.p. in saline) in the presence or absence of 3 h of inhaled formaldehyde (16.5 ± 6.5 ppmv) or acetaldehyde (363 ± 7 ppmv). After 16 weeks, the mice were sacrificed and lungs were preserved with formalin for histopathological analysis. *The index of interaction between formaldehyde and NNK was 1.18, p = 0.003 and between acetaldehyde and NNK was 1.36, p = 0.003.
A second experiment was performed with 0, 2.5, 5.0, or 7.5 μmol NNK and increasing concentrations of acetaldehyde (0, 403, 512, and 929 ppmv). In this experiment, there was no effect of acetaldehyde on the number of NNK-induced lung adenomas/mouse (Table S3). Histopathological analysis of the tumors from the mice treated with 7.5 μmol NNK indicated that the number of adenomas with dysplasia or progression were significantly elevated when NNK was combined with acetaldehyde (p = 0.006; Figure 4, Table S4). However, there was no dose response with increasing concentration of acetaldehyde.
Figure 4.
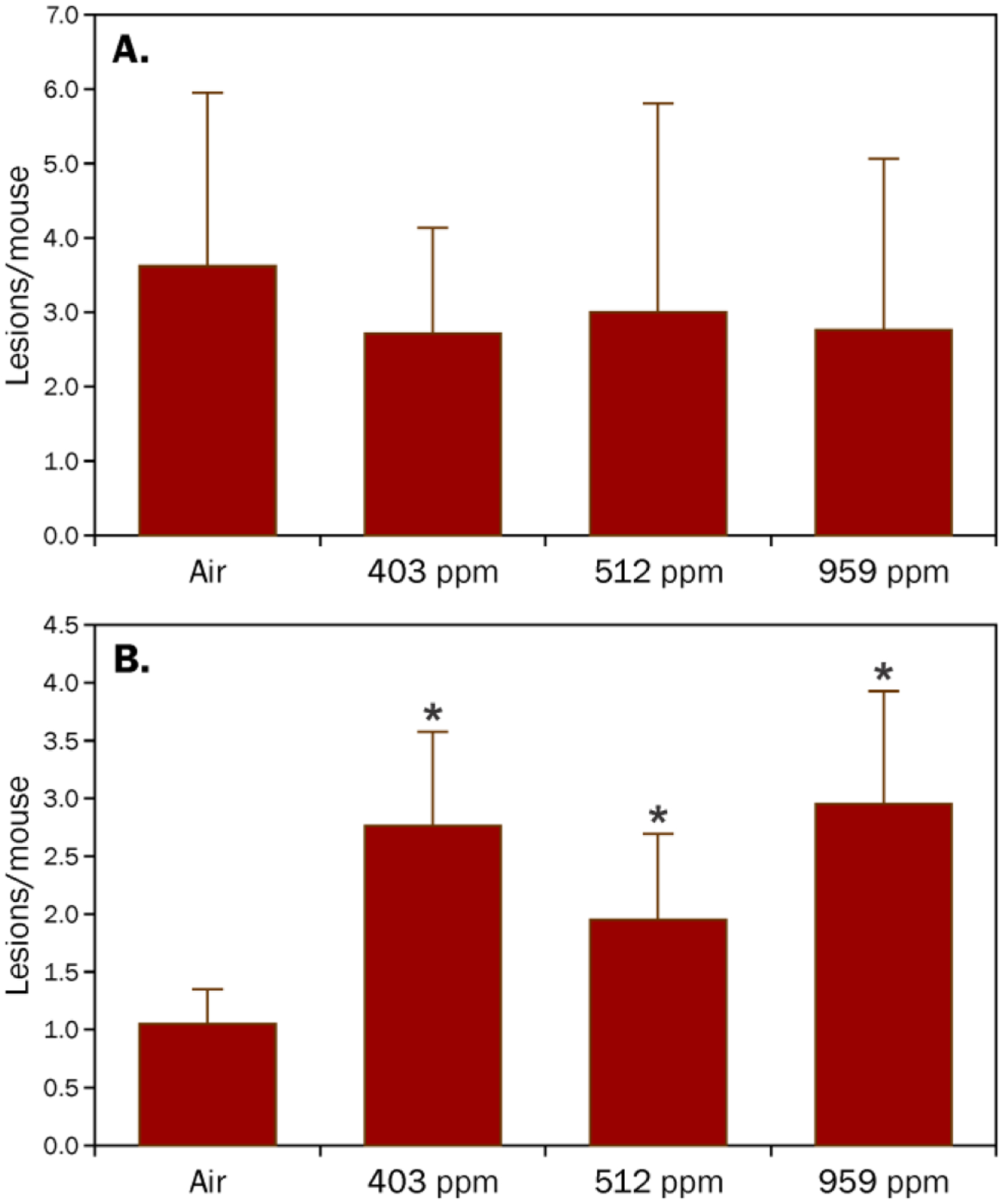
Histopathological analysis of the pulmonary tumors indicated that increasing concentrations of acetaldehyde had (A) no effect on the number of adenomas produced by NNK and (B) significantly increased the number of NNK-induced pulmonary adenomas with dysplasia or progression in A/J mice. Groups of 21 mice were treated with 7.5 μmol NNK (i.p. in saline) in the presence of 3 h of acetaldehyde (0, 403 ± 44, 510 ± 44, or 929 ± 46 ppmv). After 16 weeks, the mice were sacrificed and lung adenomas were counted followed by preservation for histopathological analysis. *Over-dispersed Poisson regression analysis indicated a significant additive effect of acetaldehyde on the number of NNK-derived adenomas with dysplasia or progression, p = 0.006
To determine whether the acetaldehyde or formaldehyde-derived effects were enhanced at later time points, groups of 20 mice were exposed to 7.5 μmol NNK in the absence or presence of a 3 h exposure to 18.4 ± 2.1 ppmv formaldehyde or 358 ± 38 ppmv acetaldehyde and were sacrificed 32, 44, or 56 weeks post exposure. The tumor number and size increased with time with the tumor count plateauing at 44 weeks (Table S5). Histopathological analysis of a subset of the lungs indicated that there was no treatment difference in the distribution of the adenoma types at these later time points (data not shown).
The co-exposure showed no influence on the levels of NNK- or aldehyde-derived DNA adducts.
Possible enhanced tumorigenicity as a result of alterations in the levels of NNK-derived DNA damage was assessed by measuring the levels of NNK-derived DNA adducts at 4, 24, and 96 h after NNK exposure using established LC-MS/MS assays. NNK is metabolically activated to DNA methylating and pyridyloxobutylating intermediates (Scheme 1). Methyl hydroxylation results in the formation of pyridyloxobutyl DNA adducts, 7-pobdG, O2-pobdT, and O6-pobdG.42–44 Methylene hydroxylation generates a DNA methylating agent, which reacts with DNA to form methyl DNA adducts, 7-mG and O6-mG.16, 33, 45–47 NNK is also reduced to 4-methylnitrosamino-1-(3-pyridyl)-1-butanol (NNAL).48 α-Hydroxylation of NNAL to DNA reactive metabolites generates DNA methyl and pyridylhydroxybutyl DNA adducts (7-phbdG, O2-phbdT, and O6-phbdG).49 There was no impact of either acetaldehyde or formaldehyde on the levels of any of the NNK-derived DNA adducts measured (Tables S6–S8).
Scheme 1.

Pathways leading to DNA adducts in NNK-treated mice.
The levels of aldehyde-induced DNA damage were also quantified. Acetaldehyde DNA adducts were detected as N2-ethyldG following NaBH3CN reduction. Similarly, formaldehyde DNA adducts were measured as N6-methyldA adducts following NaBH3CN reduction. The levels of these adducts were not significantly elevated above background levels in any of the treatment groups (Table S9).
Carbon dioxide enhances the lung tumorigenic effect of NNK in A/J mice.
In initial studies, exposure of the mice to air while they were in the chamber appeared to affect the number of tumors/mouse observed in the NNK-treated mice (Study 1, Table S10). To test whether this enhancement was due to stress of the exposure chamber or slightly elevated levels of carbon dioxide with lower air flow rates, groups of 20 mice were exposed to 0, 2.5, or 7.5 μmol NNK as well as 3 h exposure to 0, 5, 10, or 15% carbon dioxide. Control groups included mice that were exposed to NNK without being placed in the chamber or underwent the chamber training sessions the week prior to the experiment but were not placed in the chamber the day of NNK exposure. While there was a small effect caused by the training in the exposure chamber, carbon dioxide caused a dose dependent increase in the number of NNK-derived lung adenomas with the maximal enhancement occurring at 10% carbon dioxide (Figure 5 and Tables S10 and S11). The synergistic interaction between carbon dioxide and NNK leading to more lung adenomas was highly significant (p = 1 × 10−14). Carbon dioxide had no significant influence on the number of adenomas with dysplasia or progression (data not shown). It also had no effect on NNK-derived methyl DNA adducts (Table S12).
Figure 5.
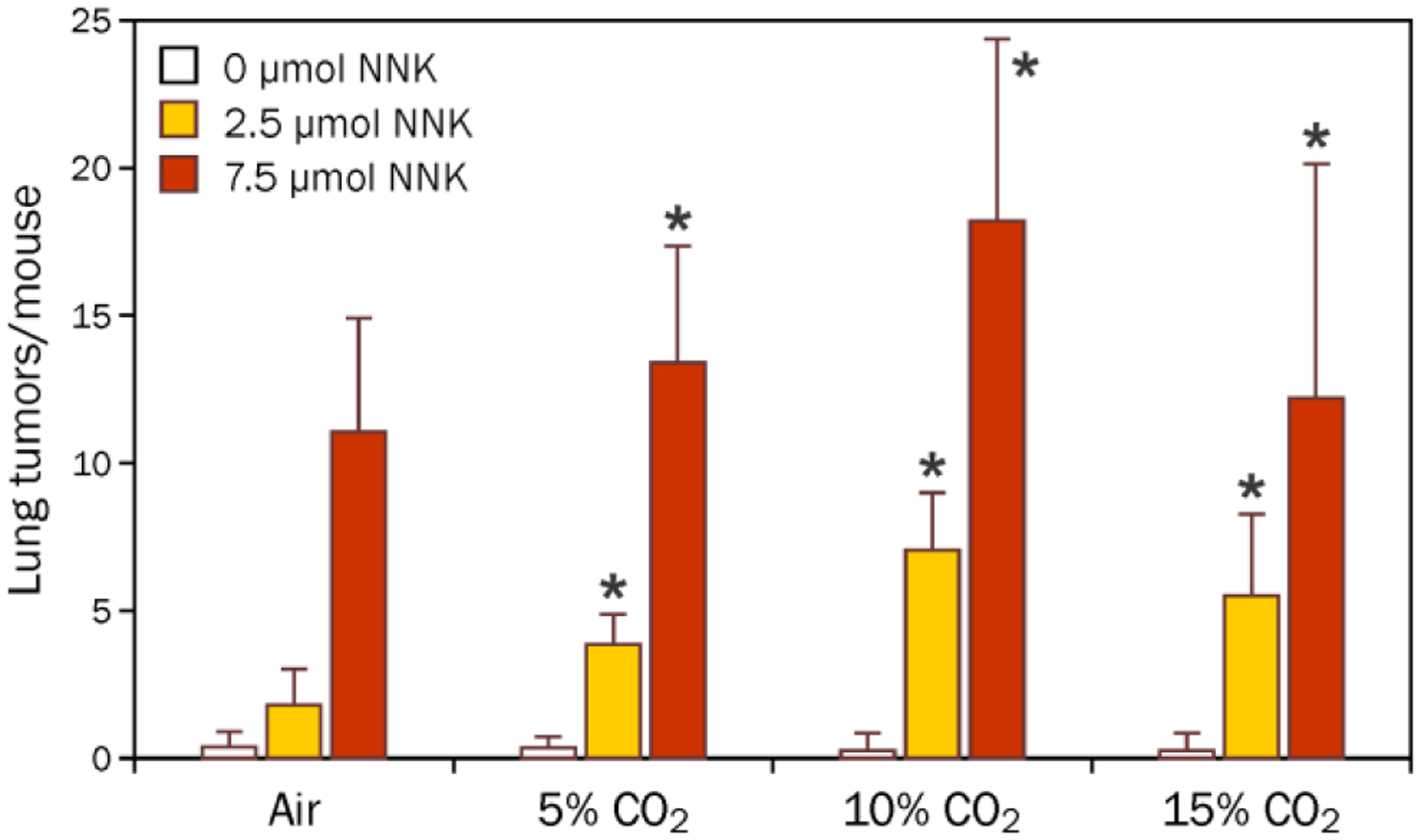
Carbon dioxide enhanced the number of pulmonary adenomas/mouse caused by NNK. Groups of 20 mice were treated with 0, 2.5 or 7.5 μmol NNK (i.p. in saline) and 3 h of 0, 5, 10, or 15% carbon dioxide. After 16 weeks, they were sacrificed and lung adenomas were counted. *The interaction between NNK and carbon dioxide was significant, p = 0.002.
Discussion
These studies demonstrate that gas phase chemicals in tobacco smoke can enhance the pulmonary tumorigenic activity of NNK. Acetaldehyde and formaldehyde increased the severity of NNK-derived adenomas in the A/J mouse lung whereas carbon dioxide increased the numbers of lung adenomas caused by NNK. The impact of all co-exposures was larger at the lower NNK dose.
There are multiple mechanisms by which the aldehydes could enhance the carcinogenic properties of NNK. First, they could directly contribute to the mutagenic burden of the co-exposure through the formation of genotoxic DNA adducts. Both acetaldehyde and formaldehyde are mutagenic.5, 6 They react directly with DNA to form multiple mono-addition adducts as well as DNA-DNA and DNA-protein crosslinks,5, 6, 50 resulting in point mutations and chromosomal aberrations.5, 6 In this current study, the levels of N2-ethyldG and N6-methyldA did not increase over their endogenous levels in lung DNA from mice treated with acetaldehyde or formaldehyde, respectively. This observation is comparable to another report in which mice were exposed to 10 ppm [13C2H2]formaldehyde for 1 or 5 days (6 h/day); there was no detectable formation of exogenous formaldehyde adducts in lung DNA following either exposure.51 The method of DNA adduct analysis used in both studies excludes the detection of more complex DNA modifications that are known to be formed by formaldehyde and acetaldehyde.5, 6, 50, 52, 53 Therefore, we cannot eliminate the possibility that exogenous aldehyde-derived DNA damage occurs in the lungs. Additionally, the aldehyde exposures in our study were not tumorigenic by themselves, indicating that any mutations caused by the aldehydes were not sufficient to initiate lung adenomas on their own. It is possible that they are causing genetic changes that, when combined with NNK-derived mutations, lead to a more advanced adenoma. Sequence analysis of the tumor DNA will be required to explore this possibility.
Another way that acetaldehyde and formaldehyde can function as co-carcinogens is through the inhibition of NNK-derived DNA adduct repair. Both aldehydes inhibit O6-alkylguanine DNA alkyltransferase (AGT) in cell models,18 an important repair pathway for mutagenic O6-mG and O6-pobG formed from NNK.17, 54, 55 Our data indicates that there was no impact of either acetaldehyde or formaldehyde co-exposure on the levels of any of the NNK-derived DNA adducts measured at 4 or 96 h after NNK exposure (Tables S6–S8). There are at least two possible explanations for this result. First, the in vivo concentrations of aldehydes reaching the lungs may be much lower than the concentrations used in the cell-based studies. Second, O6-mG repair is blocked in NNK-treated mouse lung by the presence of the pyridyloxobutylation pathway,16, 17 preventing the detection of any additional repair inhibition.
A third possible mechanism by which the aldehydes could affect the carcinogenic properties of NNK is through alterations in NNK metabolism, leading to increases in NNK-derived DNA damage and reduction of NNAL-derived DNA adducts. If this situation occurred, the overall levels of NNK-derived DNA damage would be elevated in the mice receiving the aldehyde co-exposure or the ratio of the NNK-derived pyridyloxobutyl DNA adducts to NNAL-derived pyridylhydroxylbutyl DNA adducts would be altered. However, the aldehyde co-exposures had no influence on the levels of 7-mG, a measure of overall DNA methylation by NNK, or on the levels of NNK-derived pyridyloxobutyl and NNAL-derived pyridylhydroxybutyl DNA adducts (Tables S6–S8). Therefore, this possible mechanism was discounted.
Several nongenotoxic mechanisms require additional experimentation. It is possible that the co-carcinogenic properties of formaldehyde and acetaldehyde derive from their toxicity and subsequent stimulation of cell proliferation,20, 21 which would increase the initiating potential of NNK. Both acetaldehyde and formaldehyde promote cell transformation in C3H/10T1/2 cell cultures.56 These aldehydes also alter gene expression in ways that could enhance tumor initiation by NNK. For example, both acetaldehyde and formaldehyde reduce apoptotic activity.22, 23 Acetaldehyde increases matrix metalloproteinase-9 activity and promotes cell invasion in HepG2 cells.26 Finally, both acetaldehyde and formaldehyde stimulate histone H3 phosphorylation via MAPK signaling pathways,24, 25 so they may alter the lung cell epigenome contributing to the carcinogenic properties of NNK. All of these mechanisms could explain why the aldehyde co-exposure increases the stage of adenoma detected 16 weeks following NNK exposure. Further experiments are required to explore their role in the co-carcinogenic effects of acetaldehyde and formaldehyde.
Carbon dioxide is present in the gas phase of tobacco smoke; chemical analysis of tobacco smoke indicates the presence of 12.5% carbon dioxide.57 Carbon dioxide is also a metabolite of acetaldehyde and formaldehyde;6 the concentration of carbon dioxide formed from the exposures in our studies is expected to be well-below the concentration of carbon dioxide normally exhaled by mice. Previous studies demonstrated that carbon dioxide has acute inflammatory effects in mouse lungs and is thought to be a major contributor to the inflammatory effects of tobacco smoke.58, 59 Studies in cell lines indicated that carbon dioxide increases the mitogenic effects of NNK through the stimulation of autocrine and protein kinase C-dependent pathways.60 Carbon dioxide is also known to alter gene expression and endogenous metabolism as a result of acidosis.61 In our study, carbon dioxide significantly amplified the pulmonary tumorigenesis of NNK without significantly impacting the levels of NNK-derived DNA damage. More studies will be necessary to understand the mechanism of this interaction.
In conclusion, we demonstrated that three gas phase components of tobacco smoke enhance the tumorigenic properties of the lung-specific carcinogen, NNK. Formaldehyde and acetaldehyde enhanced the severity of the tumors at early time points and carbon dioxide increased the number of tumors generated. The mechanism of these co-carcinogenic interactions requires further exploration. These studies indicate that tobacco smoke chemicals work together to form a potent carcinogenic mixture. Future studies will apply this experimental approach to explore the contribution of other tobacco smoke chemicals to the harmful properties of tobacco smoke.
Supplementary Material
Acknowledgements
The authors thank Mr. Bob Carlson for his assistance with the figures and manuscript preparation. They also thank Ms. Beverly Wuertz for her assistance with tumor counting and Anna M. Haynes, Atlanta Bidinger, Beth Bonillo, and Ebisie Deressa for their assistance with animal care.
Funding
This work was supported by a grant from the United States Food and Drug Administration (FDA) Center for Tobacco Products (CTP) and the National Cancer Institute under award number R01 CA-184987 to LAP. The content is solely the responsibility of the authors and does not necessarily represent the official views of the FDA or National Institutes of Health. The Masonic Cancer Center Analytical Biochemistry and Comparative Pathology Shared Resources are funded in part by the National Cancer Institute [P30 CA-077598].
Footnotes
Supporting Information. Table S1 shows the tumor yields in mice treated with NNK in the presence or absence of acetaldehyde or formaldehyde. Table S2 presents the histologically characterized lung adenomas in mice treated with the presence or absence of acetaldehyde or formaldehyde. Table S3 shows the tumor yields in mice treated with NNK in the presence or absence of increasing concentrations of acetaldehyde. Table S4 shows the histologically characterized lung adenomas in mice treated with 7.5 μmol NNK in the presence or absence of increasing concentrations of acetaldehyde. Table S5 shows the lung tumor yields in mice treated with NNK in the presence or absence of acetaldehyde or formaldehyde 32, 44, or 56 weeks after exposure. Tables S6 – S8 shows the levels of NNK-derived DNA adducts in lungs of NNK-treated mice with co-exposure to air, formaldehyde, or acetaldehyde. Table S9 shows the aldehyde-derived DNA adducts. Table S10 shows the impact of the exposure chamber on the NNK-derived lung tumor formation. Table S11 shows the effect of the percentage of carbon dioxide on NNK-derived tumors. Table S12 shows the effect of carbon dioxide on NNK-derived methyl DNA adducts. Figure S1 displays representative histopathological images of NNK-derived lung tumors.
Conflict of Interest Statement: None declared.
References
- (1).Rodgman A, and Perfetti TA (2016) The Chemical Components of Tobacco and Tobacco Smoke. 2nd Edition ed., pp. CRC press, Boca Raton. [Google Scholar]
- (2).United States Surgeon General. (2010) How Tobacco Smoke Causes Disease: The Biology and Behavorial Basis for Smoking-Atributable Disease. pp. United States Department of Health and Human Services, Rockville, MD. [Google Scholar]
- (3).U.S. Food and Drug Administration. (2012) Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke; Established List. Federal Register 77, 20034–20037. [Google Scholar]
- (4).International Agency for Research on Cancer. (2007) Smokeless tobacco and tobacco-specific nitrosamines. Vol. 89, pp. IARC, Lyon, France. [Google Scholar]
- (5).International Agency for Research on Cancer. (1999) Acetaldehyde, In Re-evaluation of some organic chemicals, hydrazine and hydrogen peroxide pp 319–335, IARC, Lyon, France. [Google Scholar]
- (6).International Agency for Research on Cancer. (2012) Formaldehyde, In A Review of Human Carcinogens. F. Chemical Agents and Related Occupations pp 401–436, IARC, Lyon, France. [Google Scholar]
- (7).Hecht SS (2008) Progress and challenges in selected areas of tobacco carcinogenesis. Chem. Res. Toxicol 21, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hecht SS (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chemical Research in Toxicology 11, 559–603. [DOI] [PubMed] [Google Scholar]
- (9).Morse MA, Amin SG, Hecht SS, and Chung FL (1989) Effects of aromatic isothiocyanates on tumorigenicity, O6-methylguanine formation, and metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in A/J mouse lung. Cancer Res 49, 2894–2897. [PubMed] [Google Scholar]
- (10).Woutersen RA, Appelman LM, Garderen-Hoetmer A, and Feron VJ (1986) Inhalation toxicity of acetaldehyde in rats. III. Carcinogenicity study. Toxicology 41, 213–231. [DOI] [PubMed] [Google Scholar]
- (11).Adkins J, van Stee JW, Simmons JE, and Eustis SL (1986) Oncogenic response of strain A/J mice to inhaled chemicals. J. Toxicol. Environ. Health 17, 311–322. [DOI] [PubMed] [Google Scholar]
- (12).Ross JA, Nelson GB, Wilson KH, Rabinowitz JR, Galati A, Stoner GD, Nesnow S, and Mass MJ (1995) Adenomas induced by polycyclic aromatic hydrocarbons in strain A/J mouse lung correlate with time-integrated DNA adduct levels. Cancer Res 55, 1039–1044. [PubMed] [Google Scholar]
- (13).Gordon T, and Bosland M (2009) Strain-dependent differences in susceptibility to lung cancer in inbred mice exposed to mainstream cigarette smoke. Cancer Lett 275, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Witschi H (2007) Tobacco smoke-induced lung cancer in animals -A challenge to toxicology (?). Int. J. Toxicol 26, 339–344. [DOI] [PubMed] [Google Scholar]
- (15).Stinn W, Berges A, Meurrens K, Buettner A, Gebel S, Lichtner RB, Janssens K, Veljkovic E, Xiang Y, et al. (2013) Towards the validation of a lung tumorigenesis model with mainstream cigarette smoke inhalation using the A/J mouse. Toxicology 305, 49–64. [DOI] [PubMed] [Google Scholar]
- (16).Peterson LA, and Hecht SS (1991) O6-Methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res 51, 5557–5564. [PubMed] [Google Scholar]
- (17).Peterson LA, Thomson NM, Crankshaw DL, Donaldson EE, and Kenney PJ (2001) Interactions between methylating and pyridyloxobutylating agents in A/J mouse lungs: implications for 4-(methylnitrosamino)-1-(3-pyridyl)-1- butanone-induced lung tumorigenesis. Cancer Res 61, 5757–5763. [PubMed] [Google Scholar]
- (18).Grafstrom RC, Dypbukt JM, Sundqvist K, Atzori L, Nielsen I, Curren RD, and Harris CC (1994) Pathobiological effects of acetaldehyde in cultured human epithelial cells and fibroblasts. Carcinogenesis 15, 985–990. [DOI] [PubMed] [Google Scholar]
- (19).Grafstrom RC, Curren RD, Yang LL, and Harris CC (1986) Aldehyde-induced inhibition of DNA repair and potentiation of N-nitrosocompound-induced mutagenesis in cultured human cells. Prog. Clin. Biol. Res 209A, 255–264. [PubMed] [Google Scholar]
- (20).Maronpot RR, Miller RA, Clarke WJ, Westerberg RB, Decker JR, and Moss OR (1986) Toxicity of formaldehyde vapor in B6C3F1 mice exposed for 13 weeks. Toxicology 41, 253–266. [DOI] [PubMed] [Google Scholar]
- (21).Homann N, Karkkainen P, Koivisto T, Nosova T, Jokelainen K, and Salaspuro M (1997) Effects of acetaldehyde on cell regeneration and differentiation of the upper gastrointestinal tract mucosa. J. Natl. Cancer Inst 89, 1692–1697. [DOI] [PubMed] [Google Scholar]
- (22).Saad MA, Kuo SZ, Rahimy E, Zou AE, Korrapati A, Rahimy M, Kim E, Zheng H, Yu MA, et al. (2015) Alcohol-dysregulated miR-30a and miR-934 in head and neck squamous cell carcinoma. Mol Cancer 14, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tyihak E, Bocsi J, Timar F, Racz G, and Szende B (2001) Formaldehyde promotes and inhibits the proliferation of cultured tumour and endothelial cells. Cell Prolif 34, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lee YJ, and Shukla SD (2007) Histone H3 phosphorylation at serine 10 and serine 28 is mediated by p38 MAPK in rat hepatocytes exposed to ethanol and acetaldehyde. Eur J Pharmacol 573, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yoshida I, and Ibuki Y (2014) Formaldehyde-induced histone H3 phosphorylation via JNK and the expression of proto-oncogenes. Mutat Res 770, 9–18. [DOI] [PubMed] [Google Scholar]
- (26).Hsiang CY, Wu SL, Chen JC, Lo HY, Li CC, Chiang SY, Wu HC, and Ho TY (2007) Acetaldehyde induces matrix metalloproteinase-9 gene expression via nuclear factor-kappaB and activator protein 1 signaling pathways in human hepatocellular carcinoma cells: Association with the invasive potential. Toxicol Lett 171, 78–86. [DOI] [PubMed] [Google Scholar]
- (27).Foley JF, Anderson MW, Stoner GD, Gaul BW, Hardisty JF, and Maronpot RR (1991) Proliferative lesions of the mouse lung: progression studies in strain A mice. Exp Lung Res 17, 157–168. [DOI] [PubMed] [Google Scholar]
- (28).Dagne A, Melkamu T, Schutten MM, Qian X, Upadhyaya P, Luo X, and Kassie F (2011) Enhanced inhibition of lung adenocarcinoma by combinatorial treatment with indole-3-carbinol and silibinin in A/J mice. Carcinogenesis 32, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Estensen RD, Jordan MM, Wiedmann TS, Galbraith AR, Steele VE, and Wattenberg LW (2004) Effect of chemopreventive agents on separate stages of progression of benzo[alpha ]pyrene induced lung tumors in A/J mice. Carcinogenesis 25, 197–201. [DOI] [PubMed] [Google Scholar]
- (30).Song JM, Qian X, Molla K, Teferi F, Upadhyaya P, Sullivan O, Luo X, and Kassie F (2015) Combinations of indole-3-carbinol and silibinin suppress inflammation-driven mouse lung tumorigenesis by modulating critical cell cycle regulators. Carcinogenesis 36, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, Fraire AE, Gabrielson EW, Gunning WT, et al. (2004) Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res 64, 2307–2316. [DOI] [PubMed] [Google Scholar]
- (32).Peterson LA, Urban AM, Vu CC, Cummings ME, Brown LC, Warmka JK, Li L, Wattenberg EV, Patel Y, et al. (2013) Role of aldehydes in the toxic and mutagenic effects of nitrosamines. Chem. Res. Toxicol 26, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hecht SS, Trushin N, Castonguay A, and Rivenson A (1986) Comparative tumorigenicity and DNA methylation in F344 rats by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N-nitrosodimethylamine. Cancer Res 46, 498–502. [PubMed] [Google Scholar]
- (34).Thomson NM, Mijal RS, Ziegel R, Fleischer NL, Pegg AE, Tretyakova N, and Peterson LA (2004) Development of a quantitative liquid chromatography/electrospray mass spectrometric assay for a mutagenic tobacco-specific nitrosamine-derived DNA adduct, O6-[4-oxo-4-(3-pyridyl)butyl]-2’-deoxyguanosine. Chemical Research in Toxicology 17, 1600–1606. [DOI] [PubMed] [Google Scholar]
- (35).Lao Y, Villalta PW, Sturla SJ, Wang M, and Hecht SS (2006) Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol 19, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Upadhyaya P, Kalscheuer S, Hochalter JB, Villalta PW, and Hecht SS (2008) Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol 21, 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sobh A, Loguinov A, Stornetta A, Balbo S, Tagmount A, Zhang L, and Vulpe CD (2019) Genome-wide crispr screening identifies the tumor suppressor candidate OVCA2 as a determinant of tolerance to acetaldehyde. Toxicol Sci 169, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang M, Cheng G, Villalta PW, and Hecht SS (2007) Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol 20, 1141–1148. [DOI] [PubMed] [Google Scholar]
- (39).Schmidt AF, Groenwold RH, Knol MJ, Hoes AW, Nielen M, Roes KC, de Boer A, and Klungel OH (2014) Exploring interaction effects in small samples increases rates of false-positive and false-negative findings: results from a systematic review and simulation study. J. Clin. Epidemiol 67, 821–829. [DOI] [PubMed] [Google Scholar]
- (40).Cassee FR, de Burbure CY, Rambali B, Vleeming W, van de KA, van Steeg H, Fokkens PH, van Amsterdam JG, Dormans JA, et al. (2008) Subchronic inhalation of mixtures of cigarette smoke constituents in Xpa−/−p53+/− knock-out mice: a comparison of intermittent with semi-continuous exposure to acetaldehyde, formaldehyde, and acrolein. Food Chem. Toxicol 46, 527–536. [DOI] [PubMed] [Google Scholar]
- (41).Weng Y, Fang C, Turesky RJ, Behr M, Kaminsky LS, and Ding X (2007) Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res 67, 7825–7832. [DOI] [PubMed] [Google Scholar]
- (42).Wang M, Cheng G, Sturla SJ, McIntee EJ, Villalta PW, Upadhyaya P, and Hecht SS (2003) Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chemical Research in Toxicology 16, 616–626. [DOI] [PubMed] [Google Scholar]
- (43).Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P, and Wang M (2004) Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with DNA. Chem. Res. Toxicol 17, 588–597. [DOI] [PubMed] [Google Scholar]
- (44).Wang L, Spratt TE, Liu XK, Hecht SS, Pegg AE, and Peterson LA (1997) Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol 10, 562–567. [DOI] [PubMed] [Google Scholar]
- (45).Belinsky SA, White CM, Boucheron JA, Richardson FC, Swenberg JA, and Anderson M (1986) Accumulation and persistence of DNA adducts in respiratory tissue of rats following multiple administrations of the the tobacco specific carcinogen 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res 46, 1280–1284. [PubMed] [Google Scholar]
- (46).Belinsky SA, Foley JA, White CM, Anderson MW, and Maronpot RR (1990) Dose-response relationship between O6-methylguanine formation in Clara cells and induction of pulmonary neoplasia in the rat by NNK. Cancer Res 50, 3772–3780. [PubMed] [Google Scholar]
- (47).Murphy SE, Palomino A, Hecht SS, and Hoffmann D (1990) Dose-response study of DNA and hemoglobin adduct formation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res 50, 5446–5452. [PubMed] [Google Scholar]
- (48).Peterson LA (2009) Molecular mechanisms of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung carcinogenesis. Advances in Molecular Toxicology 3, 117–160. [Google Scholar]
- (49).Hecht SS, Jordan KG, Choi CI, and Trushin N (1990) Effects of deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in A/J mice. Carcinogenesis 11, 1017–1020. [DOI] [PubMed] [Google Scholar]
- (50).Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, and Hecht SS (2000) Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol 13, 1149–1157. [DOI] [PubMed] [Google Scholar]
- (51).Lu K, Collins LB, Ru H, Bermudez E, and Swenberg JA (2010) Distribution of DNA Adducts Caused by Inhaled Formaldehyde Is Consistent with Induction of Nasal Carcinoma but Not Leukemia. Toxicological Sciences 116, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Cheng G, Shi Y, Sturla SJ, Jalas JR, McIntee EJ, Villalta PW, Wang M, and Hecht SS (2003) Reactions of formaldehyde plus acetaldehyde with deoxyguanosine and DNA: formation of cyclic deoxyguanosine adducts and formaldehyde cross-links. Chem. Res. Toxicol 16, 145–152. [DOI] [PubMed] [Google Scholar]
- (53).Brooks PJ, and Zakhari S (2014) Acetaldehyde and the genome: beyond nuclear DNA adducts and carcinogenesis. Environ Mol Mutagen 55, 77–91. [DOI] [PubMed] [Google Scholar]
- (54).Sandercock LE, Hahn JN, Li L, Luchman HA, Giesbrecht JL, Peterson LA, and Jirik FR (2008) Mgmt deficiency alters the in vivo mutational spectrum of tissues exposed to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Carcinogenesis 29, 866–874. [DOI] [PubMed] [Google Scholar]
- (55).Urban AM, Upadhyaya P, Cao Q, and Peterson LA (2012) Formation and repair of pyridyloxobutyl DNA adducts and their relationship to tumor yield in A/J mice. Chem. Res. Toxicol 25, 2167–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Parfett CL (2003) Combined effects of tumor promoters and serum on proliferin mRNA induction: a biomarker sensitive to saccharin, 2,3,7,8-TCDD, and other compounds at minimal concentrations promoting C3H/10T1/2 cell transformation. J Toxicol Environ Health A 66, 1943–1966. [DOI] [PubMed] [Google Scholar]
- (57).Norman V (1977) An overview of the vapor phase, semivolatile and nonvolatile components of cigarette smoke. Recent Adv. Tobacco Res 3, 28–58. [Google Scholar]
- (58).Schwartz L, Guais A, Chaumet-Riffaud P, Grevillot G, Sasco AJ, Molina TJ, and Abolhassani M (2010) Carbon dioxide is largely responsible for the acute inflammatory effects of tobacco smoke. Inhalation toxicology 22, 543–551. [DOI] [PubMed] [Google Scholar]
- (59).Abolhassani M, Guais A, Chaumet-Riffaud P, Sasco AJ, and Schwartz L (2009) Carbon dioxide inhalation causes pulmonary inflammation. Am. J. Physiol. Lung Cell Mol. Physiol 296, L657–L665. [DOI] [PubMed] [Google Scholar]
- (60).Schuller HM (1994) Carbon dioxide potentiates the mitogenic effects of nicotine and its carcinogenic derivative, NNK, in normal and neoplastic neuroendocrine lung cells via stimulation of autocrine and protein kinase C-dependent mitogenic pathways. Neurotoxicology 15, 877–886. [PubMed] [Google Scholar]
- (61).Guais A, Brand G, Jacquot L, Karrer M, Dukan S, Grevillot G, Molina TJ, Bonte J, Regnier M, et al. (2011) Toxicity of carbon dioxide: a review. Chem. Res. Toxicol 24, 2061–2070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.