

Abstract
Bacterial resistance to antibiotics poses a significant global challenge for the public sector. Globally, researchers are actively investigating solutions to tackle the issue of bacterial resistance to antibiotics, with Schiff bases standing out as promising contenders in the fight against antimicrobial resistance. This study focused on synthesizing a series of Schiff bases (CA1-CA10) by reacting cinnamaldehyde with various aniline derivatives. Various analytical techniques, such as NMR, FTIR, UV–Vis, elemental analysis, and mass spectrometry, were employed to elucidate the structures of the synthesized compounds. Furthermore, crystal structure of CA8 was obtained using single crystal X-ray spectroscopy. The compounds were subjected to in vitro testing to assess their antibacterial and antifungal properties against eleven bacterial strains and four fungal strains. The results revealed diverse activity levels against the pathogens at varying concentrations, with notable potency observed in compounds CA3, CA4, CA9, and CA10, as indicated by their minimum inhibitory concentrations (MIC) values. The observed activity of the compounds seemed to be influenced by the specific substituents attached to their molecular structure. By conducting computational and molecular docking studies, the electronic properties of the compounds were investigated, further substantiating their potential as effective antimicrobial agents.
Keywords: Bacterial resistance, Antibiotic, Natural products, Cinnamaldehyde, Schiff bases
Graphical abstract
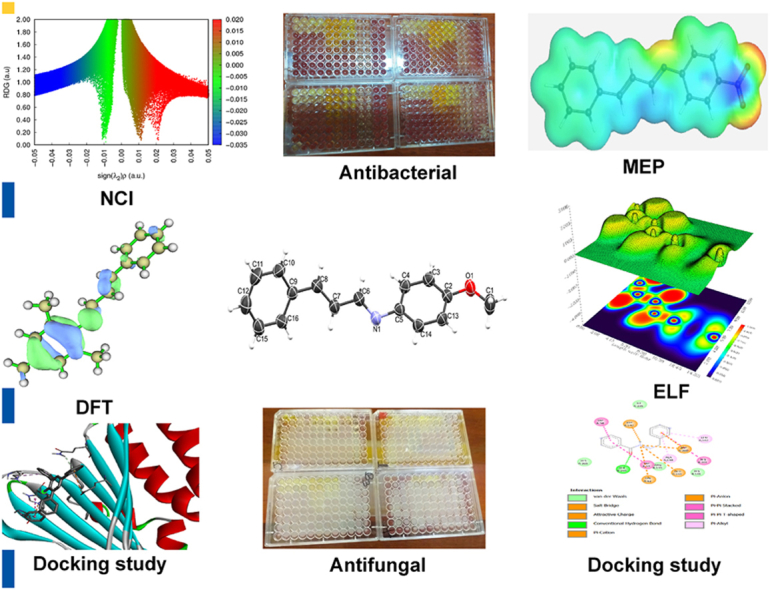
Graphical Abstract
Highlights
-
•
Ten new Schiff base (CA1 to CA10) derivatives containing different substituents were synthesized.
-
•
Detailed spectroscopic and theoretical investigations were carried out.
-
•
All compounds were tested for antibacterial and antifungal activity.
-
•
Compounds (CA3, CA4, CA9, and C10) were identified as the lead compounds.
1. Introduction
Microbial resistance poses a significant threat to both the economy and human survival [[1], [2], [3]]. The excessive and indiscriminate use of antibiotics for both treatment and prevention is a significant factor contributing to this issue [[4], [5], [6]]. The emergence of antibiotic-resistant bacteria at an alarming rate, combined with the limited efficacy of current antibiotics, underscores the urgent need for the development of novel and potent antibacterial medications [[7], [8], [9]]. Failure to address this issue will make it increasingly challenging to treat common infections in the future [[7], [8], [9]]. Moreover, synthetic antibiotics are prone to resistance development, highlighting the importance of exploring natural products for novel antimicrobial compounds [[10], [11], [12]]. Cinnamon, a widely used plant in traditional and modern medicine, has garnered attention for its antimicrobial properties [[13], [14], [15]]. Cinnamaldehyde, a naturally occurring compound derived from cinnamon, has been identified as having pharmacological potential [16]. It demonstrates diverse advantageous properties, including antimicrobial, antitumor, antioxidant, and anti-diabetic activities [[17], [18], [19]]. However, using pure cinnamaldehyde has practical limitations due to its high volatility [20]. As a result, research has primarily focused on cinnamaldehyde derivatives, particularly cinnamaldehyde Schiff bases, and their potential biological applications [21]. Among these derivatives, cinnamaldehyde Schiff base has been extensively studied due to its efficient bioavailability and ease of synthesis [[22], [23], [24]]. Numerous investigations have examined the synthesis and assessment of the antimicrobial efficacy of Schiff bases derived from cinnamaldehyde, as well as their metal complexes. These studies have consistently demonstrated an augmentation in antimicrobial activity [25]. Other researchers have synthesized cinnamaldehyde-amino acid Schiff bases and investigated their antimicrobial potential, demonstrating higher activity against bacteria compared to reference drugs [26]. Similarly, a series of cinnamaldehyde Schiff bases were evaluated for their biological activity against selected pathogens, showing varying degrees of effectiveness [27]. Additionally, silver and zinc complexes derived from symmetrical cinnamaldehyde Schiff bases exhibited superior antimycobacterial activity compared to the free ligand [28]. Earlier research has suggested that the pharmacological properties of Schiff bases can be influenced by the incorporation of substituents with either electron-donating or electron-withdrawing characteristics [29]. Furthermore, the addition of halogens and nitro groups has been shown to increase the lipophilicity of molecules, improving their penetration of lipid membranes and enhancing their biological activities [29,30]. These modifications have demonstrated significant cytotoxicity and antidepressant activity in animal models for Schiff bases [31]. Moreover, the incorporation of halogen and nitro groups as substituents has been observed to augment the antifungal efficacy [32]. Building upon these advancements, this work seeks to explore the impact of substituent variations on the pharmacological characteristics of a novel series of derivatives based on (E)-N-((E)-3-phenylallylidene)aniline, as depicted in Fig. 1.
Fig. 1.
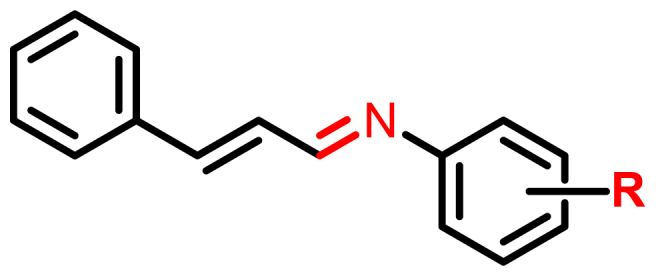
Generic structure of (E)-N-((E)-3-phenylallylidene)aniline derivatives.
2. Results and discussion
2.1. Synthesis and characterization
In this study, a variety of aniline derivatives were reacted with cinnamaldehyde to synthesize Schiff bases with moderate yields (53–91%), as illustrated in Scheme 1. The aniline derivatives encompassed a diverse range of substituents, including halogens, methoxy, nitro, alkyl, and aryl groups, positioned at different locations on the aromatic ring, as depicted in Fig. 2. These substitutions were strategically introduced to incorporate a wide range of electronic and steric effects, potentially enhancing the biological activities of the compounds. Thorough characterization of the synthesized compounds was carried out using multiple spectroscopic techniques, including 1H and 13C NMR, IR spectra, CHN-elemental analysis, and mass spectrometry (detailed information provided in the supporting materials). The IR spectra exhibited a characteristic band in the range of 1673-1600 cm−1, indicating the presence of a –C N group, along with bands at 3001-2800 cm−1 and 1498-1450 cm−1, corresponding to C–H and C C groups within the aromatic ring, respectively. The 1H and 13C NMR spectra showed distinct peaks representing all protons and carbons in the compounds, while the mass spectra exhibited molecular ion peaks (m/z) consistent with the compounds' molecular weights.
Scheme 1.

Synthesis of the compounds CA1-CA10; i = CH3OH/HCOOH, ii = RT/4h
Fig. 2.

Structures of the synthesized compounds.
2.2. Crystal structure
Several attempts were made to obtain single crystal of the compounds suitable for data collection, but unsuccessful except that of CA8 which was obtained by slow evaporation in methanol after 72 h as a yellow crystal with a dimension of 0.123 × 0.123 × 0.11 mm3. Compound CA8 crystalizes in the orthorhombic crystal system with a space group of pbca. Similarly, the cell parameters are a = 7.2137(4) Å, b = 6.3051(3) Å, and c = 57.046(3) Å. The summary of the crystallographic data is presented in Table S1, and the crystal structure is shown in Fig. 3.
Fig. 3.

Crystal structure of CA8.
2.3. Biological study
2.3.1. Antibacterial activity
The synthesized compounds (CA1–CA10) were evaluated for their antimicrobial activity against Gram-positive bacteria (Staphylococcus aureus (sa), Enterococcus faecalis (ef), Staphylococcus agalactiae (sg), Micrococcus luteus (ml), and Bacillus agri (ba)) and Gram-negative bacteria (Escherichia coli (ec), Salmonella typhi (st), Pseudomonas aeruginosa (pa), Klebsiella pneumoniae (kp), and Acinetobacter baumannii (ab)) using the broth microdilution method (supplementary information) [33]. The results are summarized in Table 1. In general, the compounds exhibited moderate to significant antimicrobial activity, with MIC values ranging from 0.13 to 8 mg/mL. Notably, CA3, CA4, CA9, and CA10 demonstrated broad antimicrobial activity with lower MIC values. Comparatively, the substituted compounds showed lower MIC values than the parent compound (CA7), with CA3 and CA4 exhibiting the lowest MIC values against all tested bacteria. Particularly noteworthy was the activity against Gram-negative bacteria, specifically K. pneumoniae, where CA3 and CA4 displayed MIC values of 0.13 mg/mL (Table 1). However, the compounds generally exhibited weaker activity against Gram-positive bacteria compared to the positive control (ciprofloxacin).
Table 1.
Minimum Inhibitory Concentration (mg.mL−1) results of compounds on selected bacteria isolatesa.
| Entry | Bacteria |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ec | st | ef | sa | pa | kp | sg | ac | ml | ab | |
| CA1 | 4 | 4 | 8 | 8 | 0.5 | 0.13 | 8 | 0.5 | 2 | 8 |
| CA2 | 4 | 8 | 8 | 4 | 1 | 0.13 | 8 | 0.5 | 2 | 8 |
| CA3 | 1 | 1 | 4 | 0.5 | 0.5 | 0.13 | 4 | 0.5 | 2 | 2 |
| CA4 | 1 | 2 | 0.25 | 0.5 | 0.5 | 0.13 | 4 | 0.5 | 0.25 | 0.5 |
| CA5 | 8 | 8 | 8 | 8 | 1 | 8 | 8 | 0.5 | 2 | 8 |
| CA6 | 4 | 8 | 8 | 8 | 8 | 8 | 8 | 0.5 | 2 | 8 |
| CA7 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 4 |
| CA8 | 0.5 | 4 | 8 | 8 | 8 | 8 | 8 | 4 | 4 | 2 |
| CA9 | 0.25 | 2 | 0.5 | 4 | 2 | 0.5 | 2 | 2 | 2 | 0.5 |
| CA10 | 0.13 | 0.25 | 2 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.13 | 1 |
| Ciprob | 0.25 | <0.13 | 0.13 | <0.13 | <0.13 | 0.13 | 0.25 | <0.13 | 0.25 | 0.25 |
Furthermore, the response of the organisms to the compounds varied, showing concentration-dependent susceptibility. The significant activity observed for CA3 and CA4, with lower MIC values, can be attributed to the presence of alkyl and aryl substituents, respectively [34,35]. These substituents play a crucial role in the development and modification of the bioactivity of organic molecules. An alkyl substituent on the aromatic ring enhances binding affinity to receptors, while the introduction of more aryl groups increases binding affinity, resulting in enhanced biological potency, as observed in CA3. Additionally, electron-withdrawing groups such as –NO2, -Cl, -Br, or –I create localized electron-deficient sites that promote optimal interactions between the compounds and the active sites of receptors [36,37].
2.3.2. Antifungal activity
To assess the antifungal potential of the synthesized compounds, four different fungal species, including Candida albicans (ca), Candida tropicalis (ct), Candida glabrata (cg), and Candida dublumiansis (cd), were subjected to antifungal activity testing (supplementary information). The minimum inhibitory concentration (MIC) values were determined using the broth microdilution method, and the results are summarized in Table 2. The compounds exhibited varying degrees of activity, ranging from moderate to significant, following a similar trend observed in the antibacterial activity results. The MIC values ranged from 0.13 to 4 mg/mL.
Table 2.
Minimum Inhibitory Concentration (mgmL−1)a results of compounds on selected Fungi Isolates.
| Entry | Fungi |
|||
|---|---|---|---|---|
| ca | ct | cg | cd | |
| CA1 | 4 | 4 | 0.5 | 1 |
| CA2 | 2 | 1 | 0.5 | 1 |
| CA3 | 2 | 0.25 | 0.25 | 1 |
| CA4 | 1 | 1 | 0.25 | 0.5 |
| CA5 | 2 | 2 | 1 | 1 |
| CA6 | 8 | 1 | 2 | 1 |
| CA7 | 8 | 4 | 2 | 4 |
| CA8 | 8 | 0.5 | 1 | 0.5 |
| CA9 | 2 | 1 | 0.13 | 0.5 |
| CA10 | 0.13 | 0.25 | 0.5 | 0.5 |
| Nysb | 0.25 | 0.25 | 0.13 | 0.25 |
Overall, CA3, CA4, CA9, and CA10 demonstrated higher activity compared to the other compounds. For instance, CA3 exhibited MIC values of 2, 0.25, 0.25, and 1 mg/mL against ca, ct, cg, and cd, respectively. Similarly, CA4 displayed MIC values of 1, 1, 0.25, and 0.5 mg/mL against the same fungal species. CA9 exhibited MIC values of 2, 1, 0.13, and 0.5 mg/mL on these fungi. Among the lead compounds, CA10 exhibited the most potent antifungal activity, with MIC values of 0.13, 0.25, 0.5, and 0.5 mg/mL against ca, ct, cg, and cd, respectively. It is worth noting that the positive control (Nystatin) outperformed the lead compounds against all tested fungi, except where CA10 displayed a lower MIC value of 0.13 mg/mL.
The superior antifungal activity of CA10 compared to CA3, CA4, CA9, and the other compounds can be attributed to the presence of a strong electron-withdrawing nitro group on the aromatic ring, which exerts a significant influence on the biological activity.
2.4. Structure-activity relationship
The synthesized compounds (CA1-CA10) were evaluated for their antimicrobial and antifungal activities, and the results presented in Table 1, Table 2 allow us to analyze the structure-activity relationship (SAR) of these compounds. SAR analysis provides valuable insights into the relationship between the structural features and the observed activity, offering guidance for future modifications to enhance activity.
Starting with the parent compound (CA7), which consists of two aromatic rings without any substituents, the introduction of cinnamaldehyde was expected to improve its antibacterial and antifungal activity based on the known properties of cinnamaldehyde itself. Indeed, CA7 exhibited activity against both bacteria and fungi.
Moving on to the derivatives, namely CA1, CA2, CA5, CA6, and CA8, it was observed that these compounds demonstrated moderate enhancements in antibacterial activity compared to CA7. The alteration in the structural framework significantly influenced the biological activity. Notably, the presence of alkyl, nitro, aryl, and halogen substituents led to increased activity. The type and position of the substituents also played a crucial role in determining the activity. Aryl substituents improved the activity of CA3, while alkyl substituents decreased the activity in CA4. The addition of nitro substituents at the C-4 position of the aromatic ring enhanced both antibacterial and antifungal activity in CA10. Furthermore, the combination of chlorine (-Cl) and bromine (-Br) substituents increased the activity in CA9.
Overall, the SAR analysis highlights the impact of structural modifications on the antimicrobial and antifungal activities of the synthesized compounds. These findings provide valuable insights for future optimization and development of more potent compounds with enhanced biological activity.
2.4.1. Mechanism of action (MOA)
The mechanism of action (MOA) of the synthesized compounds can be inferred based on their antimicrobial and antifungal activities and the structural features of the compounds.
2.4.1.1. Parent compound (CA7)
The parent compound, CA7, showed activity against both bacteria and fungi. Since CA7 consists of two aromatic rings without any substituents, its MOA may involve interference with essential microbial processes or structures. It could potentially disrupt cell membranes, inhibit key enzymes, or interfere with nucleic acid synthesis, leading to the observed antimicrobial and antifungal effects.
2.4.1.2. Derivatives with alkyl, nitro, aryl, and halogen substituents
The derivatives (CA1, CA2, CA5, CA6, and CA8) that exhibited moderate enhancements in antibacterial activity compared to CA7 contain various alkyl, nitro, aryl, and halogen substituents. These modifications introduce electron-withdrawing or electron-donating groups, which can affect the compounds' interactions with microbial targets. The MOA of these compounds may involve interactions with specific cellular receptors, enzymes, or structural components, leading to inhibition of microbial growth or metabolism.
2.4.1.3. Compound CA3
CA3, which showed improved activity with aryl substituents, may have a MOA related to the specific interactions between the aryl group and microbial targets. The aryl group could enhance binding affinity to receptors or enzymes involved in essential microbial processes, thereby disrupting their function, and inhibiting microbial growth.
2.4.1.4. Compound CA4
In contrast to CA3, CA4 exhibited decreased activity with alkyl substituents. This suggests that the presence of alkyl groups may interfere with the compound's ability to interact effectively with microbial targets. The MOA of CA4 could involve hindered binding to specific receptors or enzymes, leading to reduced antimicrobial and antifungal activity.
2.4.1.5. Compound CA10
CA10, with nitro substituents at the C-4 position of the aromatic ring, displayed enhanced antimicrobial and antifungal activity. The nitro group is known to have electron-withdrawing properties, which can influence the reactivity and interactions of the compound. The MOA of CA10 may involve the nitro group interacting with specific microbial targets, potentially disrupting essential cellular processes or structures, leading to antimicrobial effects.
2.4.1.6. Compound CA9
CA9, with a combination of chlorine and bromine substituents, exhibited increased activity. Halogen substituents can introduce localized electron-deficient sites, which may enhance the binding affinity and interactions of the compound with microbial targets. The MOA of CA9 could involve optimal binding and inhibition of specific enzymes or processes crucial for microbial growth.
2.5. DFT study and chemical reactivity parameters
To gain a deeper understanding of the lead compounds CA3, CA4, CA9, and CA10, which exhibited favorable biological activity, further computational investigations were conducted. The optimized ground state geometry of these compounds was obtained using the Gaussian 09 software at the M06-2x/6-311G(d,p) level of theory (supplementary information). The structures of the compounds are depicted in Fig. 4. Various computational techniques were employed to provide additional insights into the performance, stability, and reactivity of the lead compounds. These techniques included the analysis of non-covalent interactions (NCI), molecular electrostatic potential (MEP), conceptual density functional theory (CDFT), and electron localization function (ELF). The NCI analysis allowed for the examination of intermolecular interactions and the identification of non-covalent bonding patterns within the lead compounds. This analysis provided crucial information regarding the stability and binding characteristics of the compounds, shedding light on their potential mode of action.
Fig. 4.

The optimized geometry of the lead compounds visualized using Mercury [38].
The MEP analysis provided a visual representation of the electrostatic potential distribution on the molecular surfaces of the lead compounds. By studying the MEP, valuable information about the reactivity and potential sites of chemical reactions within the compounds could be obtained. This analysis allowed for the identification of regions with high electron density, indicating potential sites for interactions with other molecules or receptors.
CDFT, a conceptual density functional theory approach, was employed to explore the electronic structure and reactivity of the lead compounds. By analyzing various conceptual DFT descriptors such as chemical potential, hardness, softness, and electrophilicity, a comprehensive understanding of the compounds' reactivity and stability could be achieved. These descriptors provided insights into the compounds' ability to donate or accept electrons, their resistance to oxidation or reduction, and their overall chemical reactivity.
Furthermore, the ELF analysis was conducted to investigate the electron density distribution and electron localization within the lead compounds. This analysis allowed for the identification of regions of electron localization, providing valuable information about the stability and electronic structure of the compounds. It also aided in understanding the nature of chemical bonding within the compounds and the potential for electron delocalization or localization.
By employing these computational techniques, a comprehensive analysis of the lead compounds CA3, CA4, CA9, and CA10 was conducted. The results obtained from the NCI, MEP, CDFT, and ELF analyses helped establish connections and correlations between the experimental findings and the stability, reactivity, and electronic properties of the compounds. This computational investigation provided further insights and a more detailed understanding of the lead compounds, contributing to the overall knowledge and potential development of these compounds for future applications.
2.5.1. Non-covalent interaction (NCI)
NCI plays crucial roles in biological systems, contributing to compound identification, macro molecules stabilization, and the specificity and efficiency of enzymic reactions. These interactions happens during formation and depend on factors such as the properties of the interacting groups or atoms, their distances, and the surrounding entity [39]. NCI also have significant implications in crystal engineering and the construction of multicomponent structures [40]. The analysis of these interactions can be facilitated by utilizing the NCI index, which provides both qualitative and quantitative information. This index employs a parameter called “s" to identify regions in space where the reduced density gradient (RDG) approaches zero, forming distinct troughs. These RDG regions, known as isosurfaces, exhibit well-defined density values (ρ) resulting from small gradients along each of them. Weak interactions are characterized when both “s" and ρ indicate low values. Different types of interactions are represented by different colors in the index: red represents steric repulsion, green indicates van der Waals attractive forces, and blue signifies hydrogen bonding. Intense colors indicate stronger interactions. The strength of the interaction is measured by the index ρ, assigning a trough with the highest value as the strongest interaction. The eigenvalue λ2 encompasses all forces involved [29]. Among the compounds analyzed, CA3 exhibits the most distinct troughs, which can be attributed to its higher number of aromatic rings compared to other compounds. Intra-molecular interactions involving these aromatic rings are possible in CA3. Furthermore, compounds containing electron-withdrawing groups display less defined troughs, indicating a higher level of volatility Fig. 5.
Fig. 5.

Two-dimensional reduce density gradient scatter maps for the leading compounds CA3, CA4, CA9, and CA10.
In the three-dimensional representation (Fig. 6), we can visually examine the locations of these interactions on each compound. The core region of each aromatic ring is depicted by elongated red isosurfaces, indicating the closure of the ring structure. Moreover, the extended sheets, characterized by a combination of red and green colors, reveal the presence of both van der Waals repulsive and attractive forces [41]. Additionally, in the compounds that contain electron-withdrawing groups, a specific type of atomic interaction line (AIL) called C–H⋯H can be observed. However, no AILs are observed in the other compounds (Fig. 7).
Fig. 6.

Image of three-dimensional isosurfaces for non-covalent interactions of the leading compounds CA3, CA4, CA9, and CA10.
Fig. 7.

Image of atomic interaction lines and bond critical points between atoms in each of the lead compounds CA3, CA4, CA9, and CA10.
2.5.2. Molecular electrostatic potential (MEP)
MEP is a valuable chemical identifier used to study viable reactive sites on compounds. It quantifies variations in electronegativity around these sites, providing insights into their nature. These variations are depicted in an electron density map, represented by a range of colors (Fig. 8).
Fig. 8.

Electron density map for the molecular electrostatic potential (MEP) of the leading compounds CA3, CA4, CA9, and CA10.
The order of reducing electron densities, from blue to red, carries significant meaning. A blue color indicates a nucleophilic attack, while a red color signifies an electrophilic attack [42,43]. In our observations, we found that electrons are predominantly concentrated on the nitrogen atom of the imine moiety, gradually decreasing in concentration on the substituted aromatic rings in CA3 and CA4. The presence of electron-withdrawing moieties leads to a lower electron concentration in C9 and C10. By assigning atomic numbering to the imine moieties of each compound (Fig. 9), we can see that the nitrogen atom in CA4 exhibits a higher electron concentration compared to the nitrogen atoms in the other compounds (Table 3). As expected, this moiety also displays a polarization trend.
Fig. 9.

Structures showing the atomic numbering of imine moieties of the leading compounds CA3, CA4, CA9, and CA10.
Table 3.
Molecular electrostatic potential values (−1 × 10−3) on the imine moieties of Schiff bases.
| Atom | CA3 | CA4 | CA9 | CA10 |
|---|---|---|---|---|
| N9/42 | 323 | 381 | 335 | 346 |
| C10/43 | −20.4 | −98.2 | −64.8 | −69.9 |
2.5.3. Conceptual density functional theory (CDFT)
CDFT serves as an alternative chemical descriptor to Molecular Electrostatic Potential (MEP) for measuring reactive sites. CDFT offers additional capabilities, such as detecting sites prone to radical attacks and quantifying various parameters including chemical potential, ionization potential (IP), electron affinity (EA), electrophilicity index (ω), nucleophilicity index (N), chemical hardness (η), global softness (δ), electronegativity (χ), and energy differences (Table 4). Analyzing these properties becomes crucial in structure-activity relationship studies for molecular docking analyses, as it helps identify sites where reactive oxygen species (ROS) can be neutralized through radical attacks. The resistance of an atom to charge transfer (CT) is assessed through η [44,45], while the ability of an atom or group of atoms to accept electrons is measured by δ [46]. Predicting the stabilization energies of compounds aids in estimating their biological activity, with the parameter ω playing a significant role in this analysis [[47], [48], [49], [50]]. Nucleophilicity index (N) quantifies the electrophilic and nucleophilic behavior of organic molecules [51]. EA gauges the electron-accepting capacity of a molecule, releasing energy when an electron transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) [45]. On the other hand, the ionization potential (IP) measures the electron-donating ability of a compound, representing its capability to remove an electron from its HOMO [52]. Larger energy gaps between HOMO and LUMO result in more stable compounds [53]. While earlier studies suggested that electrons occupying the HOMO level would automatically be excited to the LUMO level, recent research has shown that the electron's position in atomic space and the molecular contour also influence excitations [54]. Nonetheless, HOMO-LUMO energy gaps continue to play a crucial role in electron excitations [53]. Comparing para-substituted electron-withdrawing groups with ortho/meta-substituted electron-donating groups, we noted lower stability, higher IP, higher EA, higher χ, softer δ, greater ω, lower N, and lower μ in the former. Consequently, substituted electron-withdrawing moieties enhance biological activities, such as the para-substituted NO2 moiety. This enhancement results from the strong electron-withdrawing nature of the NO2 group, which increases the compound's lipophilicity and improves penetration of lipid membranes [29]. While initially, we observed localized electron distribution across the molecules (Fig. 10), excitations cause electrons to become highly delocalized toward the –C N moiety (Fig. 11). Similarly, in CA10, electrons exhibit high delocalization toward the NO2 group.
Table 4.
Comparisons of the energies for highest occupied molecular orbitals, lowest unoccupied molecular orbitals, energy gaps and ionization potentials for the lead compounds.
| Compound | HOMO (eV) | LUMO (eV) | ΔE (eV) | Ionization Potential (eV) | Electron Affinity (A) | Electronegativity (χ) | Hardness (η) | Softness (δ) (1 × 10−2) | Electrophilicity index (ω) | Nucleophilicity index (N) | Chemical Potential (μ) (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CA3 | −7.153 | −1.430 | 5.723 | 6.595 | 0.514 | 3.554 | 6.081 | 1.644 | 1.039 | 3.696 | −3.554 |
| CA4 | −7.273 | −1.247 | 6.026 | 6.953 | 0.198 | 3.575 | 6.755 | 1.480 | 0.946 | 3.642 | −3.575 |
| CA9 | −7.378 | −1.713 | 5.665 | 7.251 | 0.788 | 4.019 | 6.463 | 1.547 | 1.250 | 3.242 | −4.019 |
| CA10 | −7.555 | −2.219 | 5.336 | 7.614 | 1.243 | 4.428 | 6.372 | 1.569 | 1.539 | 2.930 | −4.428 |
Fig. 10.

Highest occupied molecular orbitals (HOMO) diagram of the leading compounds CA3, CA4, CA9, and CA10.
Fig. 11.

Lowest unoccupied molecular orbitals (HUMO) diagram of the leading compounds CA3, CA4, CA9, and CA10.
The Fukui Function, a descriptor utilized to assess the reactivity and selectivity of specific atomic sites on a molecule, was employed in this study using CDFT measurements. The Fukui Function is defined using equations (1)–(4) as follows[55]:
| (1) |
The charge density difference and related data are presented in Table 5. The Fukui Function can be calculated using the following equations[55]:
| (2) |
Table 5.
Conceptual density functional theory parameters for the cinnamaldehyde derives Schiff base compounds.
| CA3 | ||||
|---|---|---|---|---|
| Atom | f⁻ | f⁺ | f⁰ | CDD |
| N42 | 0.0461 | 0.0657 | 0.0559 | 0.0195 |
| C43 | 0.0276 | 0.0604 | 0.0440 | 0.0328 |
| CA4 | ||||
| N9 | 0.0629 | 0.0850 | 0.0739 | 0.0222 |
| C10 | 0.0347 | 0.0651 | 0.0499 | 0.0304 |
| CA9 | ||||
| N9 | 0.0488 | 0.0692 | 0.0590 | 0.0204 |
| C10 | 0.0299 | 0.0718 | 0.0509 | 0.0419 |
| CA10 | ||||
| N9 | 0.0659 | 0.0481 | 0.0570 | −0.0177 |
| C10 | 0.0274 | 0.0655 | 0.0465 | 0.0381 |
for a nucleophilic attack[55].
| (3) |
for an electrophilic attack[55].
| (4) |
for a radical attack.
The charge density difference (CCD) is the difference between f+() and f-() [56]. The congruence between the quantitative values for reactivity and MEP analyses prompted us to investigate radical attacks. However, the outcomes of the radical attack analysis did not align entirely with our observations in the biological study. Surprisingly, the highest probability for a radical attack was observed on N9 in CA4, which contrasts with our initial expectations.
2.5.4. Electron localization function (ELF)
By employing a reference electron with equivalent spin and Fermi hole in proximity, it becomes possible to ascertain the chance of identifying an electron pair. Through this approach, the spatial localization of these electron pairs can be described, with higher localization of the reference electron indicated by lower probabilities. Extracting information about the spatial electron localization is made feasible through the utilization of Becke and Edgecombe's dimensionless scalar electron localization function (ELF), which enables analysis based on the kinetic energy density of the electrons using equation 4 and 5 shown below.
| (4) |
| (5) |
Equation 5 reveals a description of the kinetic energy density of a homogeneous electron gas with the spin density locally equal to ρσ (r) where the ELF is restricted to the range 0 ≤ η ≤ 1. If η = 1 then the electron is completely localized, and if η = , a homogeneous electron gas-like pair probability (electron delocalization) results. A value close to zero implies borders between electron pairs [57]. Molecular and atomic shell structures is clarified through ELF as it provides a topological interpretation of molecular space, which aids in identifying bonds, lone pairs, and core regions [58]. By analyzing the C–N–C plane, we observed insignificant differences in electron localizations between CA3 and CA4, although electrons do appear to be slightly more localized in the region 12.44–12.45 Bohr in the former along the x-direction. However, electrons are significantly more localized in the region 4.15–14.51 Bohr in the CA9 and CA10 along the x-direction. When comparing C-9 to C-10, we noted a greater localization along the x-direction in the region 4.14–6.22 Bohr in the former. However, electrons are more localized in the latter along the x-direction in the region 6.22–10.37 Bohr, Fig. 12.
Fig. 12.

Electron localization function (ELF) maps diagram of the leading compounds CA3, CA4, CA9, and CA10.
2.6. Molecular docking study
In this study, four compounds were chosen for a docking study against specific target proteins involved in the regulation of Gram-positive bacteria, Gram-negative bacteria, and fungi. The target proteins were bacterial gyrase (PDB: 3G75), Phosphopantetheine Adenylyltransferase (PDB: 6ckw), and cytochrome P450 14a-sterol demethylase (CYP51) (PDB: 1ea1), representing the respective pathogens (supplementary information).
To evaluate the inhibitory activity of the selected compounds, a comparison was made with reference drugs used in the study: ciprofloxacin for Gram-positive and Gram-negative bacteria, and nystatin for fungi. The inhibitory activity of the compounds was assessed by calculating the binding affinity, expressed in kcal/mol, against the target proteins.
Among the selected compounds, CA3 exhibited the lowest binding affinity values of −7.6 kcal/mol against bacterial gyrase (PDB: 3G75), −7.8 kcal/mol against Phosphopantetheine Adenylyltransferase (PDB: 6ckw), and −7.6 kcal/mol against cytochrome P450 14a-sterol demethylase (CYP51) (PDB: 1ea1) (supplementary information, Table S2). These lower values indicate that CA3 had a more potent inhibitory effect on the target pathogens compared to the other compounds (CA4, CA9, and CA10). The increased potency of CA3 can be attributed to the substituent attached to the parent compound, as depicted in Fig. 13, Fig. 14, Fig. 15.
Fig. 13.

2D and 3D structures showing the interaction of compound CA3 with the bacterial gyrase complex.
Fig. 14.

2D and 3D structures showing the interaction of compound CA3 with Phosphopantetheine Adenylyltransferase complex.
Fig. 15.

2D and 3D structures showing the interaction of compound CA3 with cytochrome P450 14a-sterol demethylase complex.
According to a reference study by Semire et al., 2023 [59], the compound with the lowest binding affinity is considered to possess the best inhibiting capacity. In this case, ciprofloxacin, and nystatin (reference compounds) exhibited greater potency in inhibiting bacterial gyrase (PDB: 3G75), Phosphopantetheine Adenylyltransferase (PDB: 6ckw), and cytochrome P450 14a-sterol demethylase (CYP51) (PDB: 1ea1) compared to the selected compounds (CA3, CA4, CA9, and CA10).
The residues and types of non-bonding interactions involved in the binding of the compounds to the target proteins are presented in Table S2. These interactions play a crucial role in the stability and specificity of the compound-protein complexes and further contribute to the inhibitory activity.
Overall, the docking study provided insights into the inhibitory potential of the selected compounds against specific target proteins involved in bacterial and fungal infections. The comparison with reference drugs and the analysis of binding interactions helped evaluate the potency and understand the underlying mechanisms of action of the compounds.
In summary, the key findings of the study highlight the potency of compound CA3 in inhibiting the target pathogens, the influence of substituents on the inhibitory activity, the comparison of the selected compounds with reference drugs, and the identification of non-bonding interactions involved in compound-protein binding. These findings contribute to the understanding of the potential of the selected compounds for therapeutic applications and provide directions for further optimization and development.
3. Conclusion
Cinnamaldehyde-derived Schiff bases (CA1-CA10) were synthesized and characterized using various analytical techniques such as NMR, IR, elemental-CHN analysis, mass spectrometry, melting point, and solubility. The spectro-chemical analysis confirmed the formation of the compounds with their respective chemical structures. Additionally, the crystal structure of CA8 is reported. The synthesized compounds were then tested for their antibacterial and antifungal activity against selected Gram-positive and Gram-negative bacteria, as well as four fungi species, using the broth microdilution method. The results showed varying degrees of activity, ranging from moderate to high, compared to the control, in a concentration-dependent manner. CA3, CA4, CA9, and CA10 exhibited particularly high activity against both bacteria and fungi, with MIC values ranging from 0.13 to 2 mg/mL for some of the tested isolates. In addition, computational analyses were performed to investigate the electronic properties, reactivity, and stability of the lead compounds. Molecular docking studies revealed the interactions between the selected compounds and the target proteins. Among the compounds, CA3 showed the highest binding affinity against Bacterial gyrase (PDB: 3G75), Phosphopantetheine Adenylyltransferase (PDB: 6ckw), and cytochrome P450 14a-sterol demethylase (CYP51) (PDB: 1ea1). Although CA3 demonstrated good inhibitory activity against the pathogens, it was less potent than the reference drugs. The type of substituent attached to the compounds significantly influenced their activity. Aryl substituents improved the activity of CA3, while alkyl substituents decreased the activity of CA4. Nitro substituents at the C-4 position of the aromatic nucleus enhanced the antibacterial and antifungal activity of CA10, and the combination of -Cl and -Br groups increased the activity of CA9. These findings provide valuable insights for future research, highlighting the potential of cinnamaldehyde-derived Schiff bases as antibacterial and antifungal agents. The structure-activity relationship and the impact of different substituents on the compounds' efficacy contribute to our understanding of their mode of action and guide further optimization efforts.
4. Experimental
4.1. General procedure for the synthesis of the compounds
The synthesis of (E)-N-((E)-3-phenylallylidene)aniline Schiff bases was carried out following modified procedures described in the literature [60,61]. In a nutshell, cinnamaldehyde (0.5 g, 3.85 mmol, 1eq) was dissolved in 10 mL of methanol and reacted individually with the corresponding solutions of aniline derivatives (3.85 mmol, 1eq) in 10 mL of methanol. A small amount of formic acid (three drops) was added as a catalyst, and the mixture was stirred at room temperature for 4 h. The progress of the reaction was monitored using thin-layer chromatography (TLC) with a hexane and ethyl acetate (9:1) solvent system, until the reaction was complete, or the starting materials were fully converted. The resulting precipitates were filtered, washed with methanol and then ether, and dried using CaCl2 to obtain the Schiff bases (CA1–CA10) as the final products. The confirmation of compound formation was accomplished through various characterization techniques.
4.1.1. (Z)-4-Iodo-N-((E)-3-phenylallylidene)aniline (CA1)
Yield: (1.18 g, 83 %); light yellow; m.p. 103–105 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.38 (1H, d, J = 8.8 Hz), 7.72 (2H, d, J = 8.2 Hz), 7.67 (2H, d, J = 6.8 Hz), 7.42 (2H, d, J = 7.4 Hz), 7.38 (2H, d, J = 16.5 Hz), 7.15 (1H, dd, J = 8.9 & 15.9 Hz), 7.02 (2H, d, J = 8.2 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 162.54, 151.08, 144.72, 137.84, 135.31, 129.63, 128.89, 128.17, 127.58, 123.23, 90.73. Selected IRATR: (ʋ, cm−1): 2800, 1640, 1450; CHN Anal. Calculated for C15H12IN; C, 54.08; H, 3.63; N, 4.20; found: C, 54.05; H, 3.62; N, 4.18; HRMS-ESI m/z [M+H]+ = 334.0096 (Calculated for C15H12IN, = 334.0093).
4.1.2. (Z)-5-Chloro-2-iodo-N-((E)-3-phenylallylidene)aniline (CA2)
Yield: (0.96 g, 63 %); brown solid; m.p. 113–115 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.25 (1H, d, J = 8.9 Hz), 7.87 (1H, d, J = 8.4 Hz), 7.72 (2H, d, J = 6.8 Hz), 7.44 (4H, m, J = 5.2 & 9.5 Hz), 7.20 (2H, t, J = 1.9 & 10.6 Hz), 7.03 (1H, d, J = 1.9 & 6.4 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 164.67, 154.15, 146.23, 139.80, 135.09, 134.01, 129.92, 128.93, 128.80, 128.07, 127.82, 127.64, 126.63, 118.66, 93.20. Selected IRATR: (ʋ, cm−1): 2700, 1650, 1500; CHN Anal. Calculated for C15H11ClIN; C, 49.01; H, 3.02; N, 3.81; found: C, 49.00; H, 3.04; N, 3.83; HRMS-ESI m/z [M+H]+ = 367.9702 (Calculated for C15H11ClIN, = 367.9703).
4.1.3. (Z)-5’-phenyl-N-((E)-3-phenylallylidene)-[1,1’,3’,1’’-terphenyl]-2’-amine (CA3)
Yield: (1.12 g, 78 %); yellow solid; m.p. 137–140 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 7.81 (1H, d, J = 3.2 Hz), 7.76 (2H, d, J = 7.3 Hz), 7.60 (2H, s), 7.54 (2H, d, J = 5.2 Hz), 7.46 (6H, m, J = 7.6 Hz), 7.37 (8H, m, J = 7.5 Hz), 7.28 (2H, brt, J = 7.0 Hz), 6.89 (2H, brs); 13C NMR (DMSO‑d6, 125 MHz): δ 166.76, 147.94, 144.00, 139.51, 136.20, 134.93, 133.37, 129.91, 129.60, 128.87, 128.80, 127.94, 127.57, 127.28, 126.67, 126.62. Selected IRATR: (ʋ, cm−1): 3024, 1705, 1612, 1481; CHN Anal. Calculated for C33H25N; C, 91.00; H, 5.79; N, 3.22; found: C, 90.96; H, 5.77; N, 3.21; HRMS-ESI m/z [M+H]+ = 436.2050 (Calculated for C33H25N, = 436.2065).
4.1.4. (Z)-2,4,6-Trimethyl-N-((E)-3-phenylallylidene)aniline (CA4)
Yield: (1.22 g, 88 %); brown solid; m.p. 108–113 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.00 (1H, d, J = 8.7 Hz), 7.66 (2H, d, J = 7.1 Hz), 7.40 (3H, m, J = 7.5 Hz), 7.28 (1H, d, J = 16.0 Hz), 7.17 (1H, dd, J = 8.7 & 16.0 Hz), 6.84 (2H, s), 2.20 (3H, s), 2.01 (6H, s); 13C NMR (DMSO‑d6, 125 MHz): δ 164.89, 148.79, 143.92, 135.40, 132.15, 129.63, 129.06, 129.02, 128.56, 128. 46, 128.14, 127.65, 127.62, 126.31, 20.39, 18.01. Selected IRATR: (ʋ, cm−1): 3047 2898, 1632, 1475; CHN Anal. Calculated for C18H19N; C, 86.70; H, 7.68; N, 5.60; found: C, 86.68; H, 7.69; N, 5.59; HRMS-ESI m/z [M+H]+ = 250.1604 (Calculated for C18H19N, = 250.1596).
4.1.5. (Z)-4-Chloro-2-iodo-N-((E)-3-phenylallylidene)aniline (CA5)
Yield: (0.86 g, 57 %); yellow solid; m.p. 123–125 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.21 (1H, d, J = 8.9 Hz), 7.93 (1H, d, J = 1.7 Hz), 7.71 (2H, d, J = 6.9 Hz), 7.48 (1H, dd, J = 1.7 & 8.4 Hz), 7.43 (4H, m, J = 7.5 & 9.5 Hz), 7.20 (1H, dd, J = 9.0 & 16.0 Hz), 7.10 (1H, d, J = 8.4 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 163.96, 151.88, 145.78, 137.38, 135.13, 130.06, 129.84, 129.29, 128.91, 127.76, 127.73, 119.63, 95.97. Selected IRATR: (ʋ, cm−1): 2887, 1623, 1458; CHN Anal. Calculated for C15H11ClIN; C, 49.01; H, 3.02; N, 3.81; found: C, 49.01; H, 3.01; N, 3.80; HRMS-ESI m/z [M+H]+ = 368.9741 (Calculated for C15H11ClIN, = 368.6200).
4.1.6. (Z)-4-Chloro-N-((E)-3-phenylallylidene)aniline (CA6)
Yield: (1.02 g, 73 %); light yellow; m.p. 93–96 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.39 (1H, d, J = 8.9 Hz), 7.93 (2H, d, J = 7.0 Hz), 7.40 (6H, m, J = 8.0 Hz), 7.22 (2H, d, J = 8.3 Hz), 7.15 (1H, dd, J = 8.9 & 16.0 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 162.62, 150.23, 144.68, 135.31, 130.10, 129.62, 129.03, 128.88, 128.15, 127.57, 122.59. Selected IRATR: (ʋ, cm−1): 2984, 1629, 1463; CHN Anal. Calculated for C15H12ClN; C, 74.53; H, 5.00; N, 5.79; found: C, 74.51; H, 4.97; N, 5.73; HRMS-ESI m/z [M+H]+ = 242.0733 (Calculated for C15H12ClN, = 242.0737).
4.1.7. (Z)-N-((E)-3-phenylallylidene)aniline (CA7)
Yield: (1.16 g, 78 %); brown solid; m.p. 88–96 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.39 (1H, d, J = 3.2 Hz), 7.66 (2H, d, J = 7.1 Hz), 7.37 (4H, m, J = 6.9 & 14.3 Hz), 7.16 (5H, m, J = 7.3, 7.9 & 14.6 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 160.63, 143.30, 129.38, 129.16, 128.62, 128.24, 127.20, 120.51, 20.27. Selected IRATR: (ʋ, cm−1): 2864, 1673, 1496; CHN Anal. Calculated for C15H13N; C, 86.92; H, 6.32; N, 6.76; found: C, 86.92; H, 6.32; N, 6.76; HRMS-ESI m/z [M+H]+ = 207.9358 (Calculated for C15H13N, = 207.1048).
4.1.8. (Z)-4-Methoxy-N-((E)-3-phenylallylidene)aniline (CA8)
Yield: (1.10 g, 73 %); yellow solid; m.p. 120–123 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.41 (1H, d, J = 8.7 Hz), 7.65 (2H, d, J = 7.3 Hz), 7.42 (2H, t, J = 7.4 & 14.4 Hz), 7.36 (1H, d, J = 7.1 Hz), 7.29 (1H, d, J = 16.0 Hz), 7.23 (2H, d, J = 8.6 Hz), 7.14 (1H, dd, J = 8.8 & 15.9 Hz), 6.96 (2H, d, J = 8.6 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 159.52, 157.91, 144.23, 142.86, 135.60, 129.27, 128.86, 128.63, 127.36, 122.18, 114.39, 55.24. Selected IRATR: (ʋ, cm−1): 2953, 1600, 1498; CHN Anal. Calculated for C16H15NO; C, 80.98; H, 6.37; N, 5.90; found: C, 80.96; H, 6.35; N, 5.87; HRMS-ESI m/z [M+H]+ = 238.1230 (Calculated for C15H15NO, = 238.1233).
4.1.9. (Z)-2-Bromo-4-chloro-N-((E)-3-phenylallylidene)aniline (CA9)
Yield: (0.75 g, 53 %); yellow solid; m.p. 130–136 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.29 (1H, d, J = 8.9 Hz), 7.77 (1H, d, J = 2.0 Hz), 7.71 (2H, d, J = 6.4 Hz), 7.46 (5H, m, J = 2.1, 8.7 & 16.2 Hz), 7.21 (1H, dd, J = 5.8 & 16.0 Hz), 7.18 (1H, d, J = 8.4 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 164.57, 149.20, 146.03, 135.10, 131.77, 130.07, 129.90, 128.92, 128.68, 127.80, 127.78, 121.09, 118.53. Selected IRATR: (ʋ, cm−1): 3001, 1600, 1463; CHN Anal. Calculated for C15H11BrClN; C, 56.19; H, 3.46; N, 4.37; found: C, 56.15; H, 3.45; N, 4.35; HRMS-ESI m/z [M+H]+ = 319.9833 (Calculated for C15H11BrClN, = 319.9842).
4.1.10. (Z)-4-Nitro-N-((E)-3-phenylallylidene)aniline (CA10)
Yield: (1.42 g, 91 %); orange solid; m.p. 127–132 °C; 1H NMR (DMSO‑d6, 500 MHz): δ 8.40 (1H, d, J = 8.9 Hz), 8.24 (2H, d, J = 8.7 Hz), 7.70 (2H, d, J = 6.5 Hz), 7.46 (4H, m, J = 7.7 & 16.1 Hz), 7.35 (2H, d, J = 8.6 Hz), 7.19 (1H, dd, J = 9.0 & 15.9 Hz); 13C NMR (DMSO‑d6, 125 MHz): δ 165.10, 157.47, 157.46, 146.69, 144.79, 135.08, 130.08, 129.97, 127.88, 127.83, 127.82, 124.90, 121.69. Selected IRATR: (ʋ, cm−1): 2908, 1627, 1504; CHN Anal. Calculated for C15H12N2O2; C, 71.42; H, 4.79; N, 11.10; found: C, 71.39; H, 4.77; N, 11.07; HRMS-ESI m/z [M+H]+ = 253.0972 (Calculated for C15H12N2O2, = 253.0977).
Data availability
Crystal information data: CCDC no: 2311363 revealed the crystallographic data for CA8 and can be assessed from the Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk/data_request/cif.
The other relevant supplementary data to this article can be found online at … … … …
CRediT authorship contribution statement
Ibrahim Waziri: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Monsuru T. Kelani: Writing – review & editing, Validation, Formal analysis, Data curation. Mariam O. Oyedeji-Amusa: Writing – review & editing, Methodology, Investigation, Formal analysis, Data curation. Abel K. Oyebamiji: Writing – review & editing, Visualization, Software, Methodology, Investigation, Formal analysis, Data curation. Louis-Charl C. Coetzee: Writing – review & editing, Visualization, Software, Methodology, Formal analysis. Alfred J. Muller: Writing – review & editing, Validation, Supervision, Resources, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors express their gratitude to the Department of Chemical Sciences, University of Johannesburg, for providing access to the analytical instruments necessary for this research. Dr. I. Waziri extends appreciation to the University of Johannesburg, URC for the prestigious award of the Postdoctoral Research Fellowship.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2024.e26632.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Dhingra S., Rahman N.A.A., Peile E., Rahman M., Sartelli M., Hassali M.A., Islam T., Islam S., Haque M. Front. Public Health. 2020;8 doi: 10.3389/fpubh.2020.535668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Razzaque M.S. Front. Public Health. 2021;8 doi: 10.3389/fpubh.2020.629120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waziri I., Isa M.A., Sonopo M., Williams D.B.G., Muller A. Bioorg. Med. Chem. Lett. 2021;52 doi: 10.1016/j.bmcl.2021.128381. [DOI] [PubMed] [Google Scholar]
- 4.Manyi-Loh C., Mamphweli S., Meyer E., Okoh A. Molecules. 2018;23:795. doi: 10.3390/molecules23040795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood M.J., Moellering R.C., Jr. Clin. Infect. Dis. 2003;36:S2. doi: 10.1086/344655. [DOI] [PubMed] [Google Scholar]
- 6.Argudín M.A., Deplano A., Meghraoui A., Dodémont M., Heinrichs A., Denis O., Nonhoff C., Roisin S. Antibiotics. 2017;6:12. doi: 10.3390/antibiotics6020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaman S.B., Hussain M.A., Nye R., Mehta V., Mamun K.T., Hossain N. Cureus. 2017;9 doi: 10.7759/cureus.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tagliabue A., Rappuoli R. Front. Immunol. 2018;9:1068. doi: 10.3389/fimmu.2018.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spellberg B., Guidos R., Gilbert D., Bradley J., Boucher H.W., Scheld W.M., Bartlett J.G., Edwards J., Jr., America I.D.S.o. Clin. Infect. Dis. 2008;46:155. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 10.Martelli G., Giacomini D. Eur. J. Med. Chem. 2018;158:91. doi: 10.1016/j.ejmech.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Hobson C., Chan A.N., Wright G.D. Chem. Rev. 2021;121:3464. doi: 10.1021/acs.chemrev.0c01214. [DOI] [PubMed] [Google Scholar]
- 12.Lewis K. Nat. Rev. Drug Discov. 2013;12:371. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 13.Rao P.V., Gan S.H. 2014. Evidence-Based Complementary and Alternative Medicine 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ribeiro-Santos R., Andrade M., Madella D., Martinazzo A.P., Moura L.d.A.G., de Melo N.R., Sanches-Silva A. Trends Food Sci. Technol. 2017;62:154. [Google Scholar]
- 15.Akrami S., Amin M., Saki M. Mol. Biol. Rep. 2021;48:2583. doi: 10.1007/s11033-021-06309-w. [DOI] [PubMed] [Google Scholar]
- 16.Doyle A.A., Stephens J.C. Fitoterapia. 2019;139 doi: 10.1016/j.fitote.2019.104405. [DOI] [PubMed] [Google Scholar]
- 17.Shreaz S., Wani W.A., Behbehani J.M., Raja V., Irshad M., Karched M., Ali I., Siddiqi W.A., Hun L.T. Fitoterapia. 2016;112:116. doi: 10.1016/j.fitote.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 18.Patel D., Prasad S.K., Kumar R., Hemalatha S. Asian Pac. J. Trop. Biomed. 2012;2:320. doi: 10.1016/S2221-1691(12)60032-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibi A.A., Kyuka C.K. Trends in Pharmaceutical Sciences. 2022;8:263. [Google Scholar]
- 20.Huang Y., Ho S. J. Stored Prod. Res. 1998;34:11. [Google Scholar]
- 21.Wang H., Jiang M., Sun F., Li S., Hse C.-Y., Jin C. Molecules. 2018;23:3027. doi: 10.3390/molecules23113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H., Jiang M., Li S., Hse C.-Y., Jin C., Sun F., Li Z. R. Soc. Open Sci. 2017;4 doi: 10.1098/rsos.170516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma C., Quraishi M., Alfantazi A., Rhee K.Y. Int. J. Biol. Macromol. 2021;184:135. doi: 10.1016/j.ijbiomac.2021.06.049. [DOI] [PubMed] [Google Scholar]
- 24.Wang H., Jian M., Li S., Hse C.Y., Jin C., Sun F., Li Z. R. Soc. Open Sci. 2017;4 doi: 10.1098/rsos.170516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kudrat-E-Zahan M., Hossen M.F., Ansary R.H., Zamir R., Asraf M.A. Thematics Journal of Chemistry. 2018;2:55. [Google Scholar]
- 26.Wei Q.-Y., Xiong J.-J., Jiang H., Zhang C., Ye W. Int. J. Food Microbiol. 2011;150:164. doi: 10.1016/j.ijfoodmicro.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 27.Hossain M.S., Roy P.K., Zakaria C., Kudrat-E-Zahan M. Int. J. Chem. Stud. 2018;6:19. [Google Scholar]
- 28.Amaral T.C., Miguel F.B., Couri M.R., Corbi P.P., Carvalho M.A., Campos D.L., Pavan F.R., Cuin A. Polyhedron. 2018;146:166. [Google Scholar]
- 29.Waziri I., Kelani M.T., Oyedeji-Amusa M.O., Oyebamiji A.K., Coetzee L.-C.C., Adeyinka A.S., Muller A.J. J. Mol. Struct. 2023;1276 [Google Scholar]
- 30.Waziri I., Yusuf T.L., Akintemi E., Kelani M.T., Muller A. J. Mol. Struct. 2023;1273 [Google Scholar]
- 31.Sadia M., Khan J., Naz R., Zahoor M., Shah S.W.A., Ullah R., Naz S., Bari A., Mahmood H.M., Ali S.S. J. King Saud Univ. Sci. 2021;33 [Google Scholar]
- 32.Marzi M., Farjam M., Kazeminejad Z., Shiroudi A., Kouhpayeh A., Zarenezhad E. J. Chem. 2022;2022:1. doi: 10.1155/2022/4584846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oyedeji-Amusa M., Van Vuuren S. South Afr. J. Bot. 2020;133:83. [Google Scholar]
- 34.Jia B., Ma Y.-m., Liu B., Chen P., Hu Y., Zhang R. Front. Chem. 2019;7:837. doi: 10.3389/fchem.2019.00837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munir A., Ayaz S., Shah A., Kokab T., Iftikhar F.J. Am. J. Phys. Chem. 2021;10:45. [Google Scholar]
- 36.Feng Z.-Q., Yang X.-L., Ye Y.-F., Hao L.-Y. Bull. Kor. Chem. Soc. 2014;35:1121. [Google Scholar]
- 37.Hagmann W.K. J. Med. Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 38.Macrae C.F., Sovago I., Cottrell S.J., Galek P.T., McCabe P., Pidcock E., Platings M., Shields G.P., Stevens J.S., Towler M. J. Appl. Crystallogr. 2020;53:226. doi: 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye G., Cheng J.-G., Yan J.-Q., Sun J., Matsubayashi K., Yamauchi T., Okada T., Zhou Q., Parker D.S., Sales B.C. Phys. Rev. B. 2016;94 [Google Scholar]
- 40.Ashfaq M., Tahir M.N., Munawar K.S., Behjatmanesh-Ardakani R., Kargar H. J. Mol. Struct. 2022;1261 [Google Scholar]
- 41.Aslam B., Wang W., Arshad M.I., Khurshid M., Muzammil S., Rasool M.H., Nisar M.A., Alvi R.F., Aslam M.A., Qamar M.U. Infect. Drug Resist. 2018;11:1645. doi: 10.2147/IDR.S173867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kostova I., Trendafilova N., Momekov G. J. Inorg. Biochem. 2005;99:477. doi: 10.1016/j.jinorgbio.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 43.Salih R.H.H., Hasan A.H., Hussen N.H., Hawaiz F.E., Hadda T.B., Jamalis J., Almalki F.A., Adeyinka A.S., Coetzee L.-C.C., Oyebamiji A.K. J. Mol. Struct. 2023;1282 [Google Scholar]
- 44.Roy R.K., Krishnamurti S., Geerlings P., Pal S. J. Phys. Chem. 1998;102:3746. [Google Scholar]
- 45.Chattaraj P.K., Giri S. Annual Reports Section" C"(Physical Chemistry) 2009;105:13. [Google Scholar]
- 46.Chattaraj P.K., Roy D.R. Chem. Rev. 2007;107:PR46. [Google Scholar]
- 47.Parr R.G., Pearson R.G. J. Am. Chem. Soc. 1983;105:7512. [Google Scholar]
- 48.Parr R.G., Szentpály L.v., Liu S. J. Am. Chem. Soc. 1999;121:1922. [Google Scholar]
- 49.Zhan C.-G., Nichols J.A., Dixon D.A. J. Phys. Chem. 2003;107:4184. [Google Scholar]
- 50.Pearson R.G. Proc. Natl. Acad. Sci. USA. 1986;83:8440. doi: 10.1073/pnas.83.22.8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rad A.S., Ayub K. J. Alloys Compd. 2016;678:317. [Google Scholar]
- 52.Domingo L.R., Pérez P. Org. Biomol. Chem. 2011;9:7168. doi: 10.1039/c1ob05856h. [DOI] [PubMed] [Google Scholar]
- 53.Lewis D., Ioannides C., Parke D. Xenobiotica. 1994;24:401. doi: 10.3109/00498259409043243. [DOI] [PubMed] [Google Scholar]
- 54.Hasan M.M., Bithe S.A., Neher B., Ahmed F. J. Mol. Model. 2022;28:59. doi: 10.1007/s00894-022-05048-4. [DOI] [PubMed] [Google Scholar]
- 55.Fukui K. Springer; 2006. Orientation and Stereoselection; p. 1. [Google Scholar]
- 56.AlRabiah H., Muthu S., Al-Omary F., Al-Tamimi A.-M., Raja M., Muhamed R.R., El-Emam A.A.-R. Macedonian Journal of Chemistry and Chemical Engineering. 2017;36:59. [Google Scholar]
- 57.Tsirelson V., Stash A. Chem. Phys. Lett. 2002;351:142. [Google Scholar]
- 58.Feixas F., Matito E., Poater J., Solà M. Chem. Soc. Rev. 2015;44:6434. doi: 10.1039/c5cs00066a. [DOI] [PubMed] [Google Scholar]
- 59.Semire B., Afolabi S.O., Latona D.F., Oyebamiji A.K., Adeoye M.D., Owonikoko A.D., Oyebamiji O.M., Abdulsalami I.O., Odunola O.A. Chemistry Africa. 2023:1. [Google Scholar]
- 60.Hosny S., Ragab M.S., Abd El-Baki R.F. Sci. Rep. 2023;13:1502. doi: 10.1038/s41598-023-28402-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuldasheva N., Acikyildiz N., Akyuz M., Yabo-Dambagi L., Aydin T., Cakir A., Kazaz C. J. Mol. Struct. 2022;1270 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystal information data: CCDC no: 2311363 revealed the crystallographic data for CA8 and can be assessed from the Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk/data_request/cif.
The other relevant supplementary data to this article can be found online at … … … …